
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast microwave-assisted synthesis of Li-stuffed garnets and insights into Li diffusion from muon spin spectroscopy†
Marco
Amores
a,
Thomas E.
Ashton
a,
Peter J.
Baker
b,
Edmund J.
Cussen
*c and
Serena A.
Corr
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: serena.corr@glasgow.ac.uk
bISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
cWestCHEM, Department of Pure and Applied Chemistry, The University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK
First published on 17th December 2015
Abstract
Lithium-stuffed garnets attract huge attention due to their outstanding potential as solid-state electrolytes for lithium batteries. However, there exists a persistent challenge in the reliable synthesis of these complex functional oxides together with a lack of complete understanding of the lithium-ion diffusion mechanisms in these important materials. Addressing these issues is critical to realizing the application of garnet materials as electrolytes in all solid-state lithium-ion batteries. In this work, a cubic phase garnet of nominal composition Li6.5Al0.25La2.92Zr2O12 is synthesized through a microwave-assisted solid-state route for the first time, reducing considerably the reaction times and heating temperatures. Lithium-ion diffusion behavior is investigated by electrochemical impedance spectroscopy (EIS) and state-of-art muon spin relaxation (μSR) spectroscopy, displaying activation energies of 0.55 ± 0.03 eV and 0.19 ± 0.01 eV respectively. This difference arises from the high inter-grain resistance, which contributes to the total resistance in EIS measurements. In contrast, μSR acts as a local probe providing insights on the order of the lattice, giving an estimated value of 4.62 × 10−11 cm2 s−1 for the lithium diffusion coefficient. These results demonstrate the potential of this lithium-stuffed garnet as a solid-state electrolyte for all-solid state lithium-ion batteries, an area of growing interest in the energy storage community.
1 Introduction
Increasing global population, global warming and depletion of fossil fuels have resulted in a growing need for sustainable and clean energy sources. Renewable energy resources such as wind power, solar or hydroelectricity, for example, are variable with time and it is desirable to develop efficient methods to store energy from such activities. The lithium ion battery is an excellent established energy storage system, providing one of the highest gravimetric and volumetric energy densities.1 However, there are safety concerns with the use of liquid electrolytes in conventional lithium ion batteries.2 Typically, this liquid electrolyte consists of a lithium salt, commonly LiPF6, dissolved in an organic solvent mixture such as ethylene carbonate and dimethyl carbonate. These carbonate-based solvents are highly flammable, with flash points below 30 °C,3 while LiPF6 can undergo autocatalytic decomposition to form PF5, which reacts readily with water and with the carbonate solvent itself above 60 °C.4 Moreover, the use of lithium metal as an anode can provide its own drawbacks due to the possible formation of lithium dendrites.5 The use of a liquid electrolyte not only compromises the safety of the battery but also the performance by limiting the voltage window in which the battery can operate. Therefore, finding an alternative to liquid electrolytes is crucial in order to match the demands of new technologies, such as electric vehicles, which require greater energy densities.Solid-state electrolytes are a suitable alternative to liquid electrolytes and among them lithium-stuffed garnets represent one of the most promising class of materials due to their outstanding properties, including their high Li+ conductivities of about 10−3 S cm−1,6 chemical stability for high voltage Li cathodes and high electrochemical decomposition voltages of up to 6 V versus elemental lithium.7 In addition to that, the metal cations present within the lithium stuffed garnets may also act as flame retardants.8
Conventional garnets are described by the formula A3B3C2O12, where cations A are tetrahedrally coordinated by oxygen, B cations are surrounded by 12 oxygens and C cations are accommodated in octahedral sites. Lithium ions settle on A sites, while B and C sites can be occupied by a wide range of different metals. Depending on the valence state of the metal ions present in the B and C sites, additional lithium ions can be inserted into the structure in order to compensate the ionic charges in the crystal, leading to complex lithium distributions and crystallographic order/disorder transitions which effectively affects the lithium ion conduction through the garnet.9 These extra lithium ions can be accommodated within six octahedral and two trigonal prismatic free interstitial sites, in addition to the three fully occupied tetragonal interstitial sites on a conventional Li3B3C2O12 garnet.
These lithium-stuffed garnets generally crystallize in a face centered cubic structure in the space group Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d,10 except for the higher lithium containing Li7LaM2O12 (M = Sn, Zr, Hf) garnets which crystallize in a tetragonal structure in the space group I41/acd at room temperature.11–13 However, the Li7La3Zr2O12 garnet family reported by Murugan et al.14 displays high ionic conductivities near 10−3 S cm−1 and low activation energies for lithium ion diffusion around 0.30 eV. It was found later, that Al contamination from the alumina crucibles plays an important role in the stabilization of the cubic Li7La3Zr2O12 phase. This arises from sintering at high temperatures for long times.15 Stabilization of the cubic phase of lithium-rich garnets is not the only benefit of Al insertion within the garnet structure. Formation of lithium vacancies for facile diffusion of lithium15 and formation of an amorphous Li–Al–O phase which aids lithium diffusion between grains16 has also been reported. Thus, stabilization of the cubic phase is crucial in this type of materials.
d,10 except for the higher lithium containing Li7LaM2O12 (M = Sn, Zr, Hf) garnets which crystallize in a tetragonal structure in the space group I41/acd at room temperature.11–13 However, the Li7La3Zr2O12 garnet family reported by Murugan et al.14 displays high ionic conductivities near 10−3 S cm−1 and low activation energies for lithium ion diffusion around 0.30 eV. It was found later, that Al contamination from the alumina crucibles plays an important role in the stabilization of the cubic Li7La3Zr2O12 phase. This arises from sintering at high temperatures for long times.15 Stabilization of the cubic phase of lithium-rich garnets is not the only benefit of Al insertion within the garnet structure. Formation of lithium vacancies for facile diffusion of lithium15 and formation of an amorphous Li–Al–O phase which aids lithium diffusion between grains16 has also been reported. Thus, stabilization of the cubic phase is crucial in this type of materials.
Lithium garnets are not only candidate materials for electrolytes in lithium-ion batteries but they are versatile ceramics which can be also employed as insertion electrodes, as recently reported for a carbon-coated Li3Nd3W2O12 garnet.17 This enables multiple uses of lithium garnets within lithium batteries, making the necessity for finding more reliable, greener synthetic routes to their fabrication vital. Most of the reported lithium-containing garnets have been synthesized by conventional solid-state routes employing long reaction times and high temperatures,14,18,19 most recently by sol–gel chemistry (presenting lower lithium conductivities)20,21 and even in the form of nanowires obtained through electrospinning techniques.22 Nowadays, the implementation of microwave irradiation during solid-state synthesis is emerging as an alternative preparative approach, enabling reductions in reaction times and heating temperatures, due to the strong absorption of microwave radiation by the starting materials.23
Based on these facts, we present a fast, reliable microwave-assisted approach for the synthesis of Al-doped LLZ garnet with a nominal composition of Li6.5Al0.25La2.92Zr2O12. The lithium ion diffusion behavior has been examined by both EIS and μSR. μSR studies have enabled the examination of the local dynamics of lithium ions with no interferences extrinsic to the material itself as occurs in traditional EIS analyses. This provides us a local probe to examine dynamic behavior in powered samples, without the need for contacts or pellets. We demonstrate this synthetic method as a viable, significantly faster, alternative route to cubic phase Li-stuffed garnets and we highlight the importance of multiple techniques for the examination of Li diffusion processes in solid state electrolytes.
2 Experimental
2.1 Synthesis, composition, microstructure and structural characterization
All reagent grade chemicals employed were purchased from the following suppliers and used without further purification unless otherwise noted: LiOH·H2O (98%) and La2O3 (99%) from Sigma-Aldrich and Al(NO3)3·9H2O (98–102%) and ZrO2 (99.978%) from Alfa Aesar.For the synthesis of the Li6.5Al0.25La2.92Zr2O12 garnet, stoichiometric quantities of La2O3 (previously dried at 900 °C for 24 h), ZrO2, Al(NO3)3·9H2O and a 10% excess of LiOH·H2O were ball milled for 30 min at a vibrational frequency of 20 Hz in a stainless steel jar. Subsequently, the fine powder was pelleted at 3 tonnes under uniaxial pressure. The pelleted material was heated at 700 °C for 6 hours in a 2.45 GHz CEM Phoenix hybrid microwave furnace composed of three silicon carbide walls surrounding the sample cavity in order to decompose the starting materials. Subsequently, the material was reground and pelletized for a second heat treatment carried out in air at 900 °C for 6 h in the same microwave furnace. Finally, the lithium garnet was ground and pelletized for a last annealing of 1 hour at 1000 °C in the same hybrid microwave furnace. During each heat treatment, the bottom of the alumina crucibles were covered with mother powder in order to avoid any accidental Al contamination and the heating rate was held at 2 °C min−1 to reduce lithium evaporation. Chemical composition was determined via Induced Coupled Plasma-Mass Spectroscopy (ICP-MS) analyses performed on an Agilent 7700 ICP-MS instrument. Approximately 6 mg of the sample was dissolved in 50 mL of 2% HNO3 solution in deionized water for measurements.
Scanning Electron Microscopy (SEM) images were acquired with a Phillips XL30 ESEM microscope, using a carbon-taped sample holder and a gold coating step to avoid any charge process. Energy-Dispersive X-Ray (EDX) spectra were recorded using an Oxford Instruments Energy 250 energy dispersive spectrometer system attached to the SEM device. Copper tape was employed as a standard for calibration and the voltage of the incident beam was 20 keV.
Powder X-Ray Diffraction (PXRD) studies were performed using a PANalytical X'Pert PRO Diffractometer using Cu-Kα radiation in the 2θ range 15–85° with a nominal scan rate of 53.975 seconds per step and a step size of 0.016° at room temperature. Neutron Powder Diffraction (NPD) patterns were acquired using HRPD at the ISIS pulsed neutron and muon source at the Rutherford Appleton Laboratory, UK. The sample, ca. 1 g, was placed in an 11 mm diameter cylindrical vanadium can and loaded into the beam line. The data were collected at room temperature. The broad incident pulsed neutron flux was narrowed in a 100 K methane moderator giving a peak flux at λ = 2 Å, prior to sample scattering. Data were collected in the backscattering detector, bank 1 (2θ = 168.33°), and the 90° detector, bank 2 (2θ = 89.58°), over a time-of-flight region of 30–130 ms and were corrected for absorption. Rietveld refinements of PXRD and NPD patterns were performed with the Generalized Structure Analysis System (GSAS), along with the graphical user interface EXPGUI, by means of a least squares approach.26,27
2.2 EIS measurements
EIS AC measurements were performed on a Solartron 1260 Impedance Analyzer in the frequency range of 1–106 Hz and a temperature range between RT and 250 °C in 50 °C intervals. Lithium stuffed garnet powders were cold-pressed under uniaxial pressure at 5 tonnes and the resultant pellets were heated for 6 hours at 900 °C. In order to enhance the connection between the pellet and the electrodes, a suspension of 0.5–5 μm platinum particles in n-butyl acetate was prepared and a few drops of this suspension were deposited on both surfaces of the pelletized material. The electrodes consisted of square pieces of a 0.025 mm thickness platinum foil, connected through a 0.127 mm diameter platinum wire to the device.2.3 μSR spectroscopy
μSR studies were performed using the EMU instrument at the ISIS pulsed muon facility. The sample, ca. 1.5 g, was packed into a disk of 30 mm diameter and 1.5 mm thickness and sealed in a titanium sample holder where the front window was made of 25 μm thickness titanium foil. 3.2 MeV spin-polarised positive muons were implanted into the sample and the outcoming positrons were detected by 96 scintillator segments grouped in two circular arrays. The temperature was controlled up to 420 K by a hot stage attached to a closed cycle refrigerator and the measurements were acquired at three different applied longitudinal magnetic fields (0, 5 and 10 G). A 20 G transverse magnetic field was also applied for the initial asymmetry calibration.3 Results and discussion
3.1 Microwave-assisted solid-state synthesis and Al-doping
The Al-doped LLZ cubic garnet with nominal composition Li6.5Al0.25La2.92Zr2O12 was prepared by a microwave-assisted solid-state method. The absorption of microwave irradiation in our approach is facilitated by the presence of precursors in the reaction that can effectively absorb microwave radiation, transforming it into heat. Specifically, LiOH·H2O has been reported as a super accelerator for microwave heated ceramic synthesis.28 Dramatically enhanced absorption of microwave radiation has been reported for ZrO2 at temperatures above 800 °C.29 Initially, the microwave irradiation is absorbed by the SiC walls of the microwave furnace releasing thermoradiation as a blackbody, heating the precursor materials. In a second stage, it is believed that the ZrO2 increases its microwave absorption as the temperature increases, driving the reaction towards completion.The PXRD pattern of the as-synthesised Li6.5Al0.25La2.92Zr2O12 garnet, shown in Fig. 1, reveals a highly crystalline single phase garnet, corresponding to the cubic form of the LLZ garnet with Iad space group (see also ESI Fig. S1†). The lattice parameter obtained from Rietveld refinement was found to be 12.9744(1) Å, which is in excellent agreement with the reported cubic LLZ garnet [Rwp = 0.1346, Rp = 0.0976, χ2 = 1.674 and RF2 = 0.0515].6 The use of this synthetic approach along with the intentional Al-doping resulted in faster reactions (up to 36 times) and a 230 °C reaction temperature decrease compared with the original synthesis reported by Murugan et al.14 Compared to the faster solid-state synthesis of Al-doped LLZ reported in the literature,16 our approach is still up to four times faster in the final annealing step without any La2Zr2O7 or LaAlO3 impurities. To demonstrate the robustness of this method, the Al-doped LLZ garnet of nominal composition Li6.25Al0.25La2.92Zr2O12 was also prepared (see ESI, Fig. S2†). For the same times and temperatures employed, it is clear that with a conventional solid-state approach the garnet is not fully formed, while with our microwave-synthetic approach a pristine cubic garnet is obtained.
 | ||
| Fig. 1 Rietveld refinement to (a) PXRD and (b) backscattered NPD data collected on Li6.5Al0.25La2.92Zr2O12. Fits were in excellent agreement with the cubic LLZ garnet in the (Iad) space group.24 (b) For the NPD data, a 2% LiOH impurity is also detected.25 | ||
The lithium ion highly conductive cubic phase was stabilized by intentional Al-doping. Over the different Al concentrations tested, it was found that at least 0.25 mol per formula unit are required to readily form a single cubic phase, while concentrations above this result in the formation of LaAlO3 or La2Zr2O7 by-products. A partial replacement of lanthanum at concentrations greater than 0.15 mol Al was also detected, leading to the formation of a LaAlO3 impurity. Therefore, the nominal stoichiometry employed was Li6.5Al0.25La2.92Zr2O12 in order to prevent any LaAlO3 perovskite formation. This is also the lithium content at which maximum conductivity is observed.30 PXRD diffraction patterns for the different stoichiometries are given in the ESI, Fig. S3.†
The chemical composition of the Al-doped garnet was determined by ICP-MS analyses which indicates that the Al content of the sample had been accurately controlled by our synthetic approach. The analysis gave a stoichiometry of Li6.3(2)Al0.237(7)La3.06(6)Zr2.00(1)O12, in good agreement with the target stoichiometry. No other major metal impurities were detected as possible contaminants, for example from the ball milling procedure. NPD studies of the sample after the muon experiment (Fig. 1(b), refinement details available in the ESI Fig. S4†) revealed an excellent fit to the cubic phase, with a small (∼2%) impurity phase, identified as LiOH.25 The stoichiometry extracted from Rietveld refinement was Li6.3(2)Al0.23La2.91Zr2.02(2)O12 which is very close to the nominal stoichiometry employed. In addition, EDX analyses indicated a stoichiometry for Al, La and Zr of Al0.17(4)La2.9(2)Zr2.0(2).
The neutron diffraction data were fitted using the established model for Li-stuffed garnets that is with a disordered occupancy of Li across tetrahedral, 24d, and large distorted octahedral, 96h, sites. Due to the small quantity of Al present, it is not possible to unambiguously identify the site for this cation within the structure. From consideration of the cation radii and from previous reports, we have elected to place the Al on the 24d site with the stoichiometry derived from the ICP analysis.24 The Li occupancies across the two sites were allowed to refine freely, i.e. with no compositional restraint on the Li content. The Zr was also allowed to freely refine. The La content was fixed at the target composition, an assignment strongly supported by the ICP and EDX analyses. On the basis of this refinement, this analysis yielded a refined composition of Li6.3(2)Al0.23La2.91Zr2.02(2)O12 which is very close to the nominal stoichiometry employed. Taken in conjunction with ICP and EDX, the stoichiometry of Li6.5Al0.25La2.92Zr2O12 is within the estimated standard deviation of each measurement. We are therefore confident that the garnet phase does have the target stoichiometry.
To avoid any potential moisture uptake by the Li-stuffed garnets, samples are stored in a glovebox under argon and transported for measurement in glass vials under argon. Issues with moisture uptake have been reported for Li garnets previously.31 When this occurs to a significant extent, there are gross changes in the diffraction profile and changes in space group symmetry. We observe a relatively minor change (<0.5%), exhibited purely in the lattice parameter from the fitted neutron diffraction [13.02051(5) Å] data compared to original X-ray diffraction data collected prior to the muon experiments, where the sample was briefly and unavoidably exposed to air after measurement. The neutron diffraction data indicate that the sample has the target stoichiometry, within the precision of this high resolution diffraction measurement. We believe the <0.5% change in the cell parameter is due to the slight uptake of water by our sample. This may have occurred inadvertently during sample cool-down after muon measurements were taken. In order to be consistent, the same sample was used for neutron diffraction studies. This absorption of water can occur either by direct insertion of water molecules into the garnet structure or through a H+/Li+ exchange mechanism as previously reported.32,33 Since our refined Li content from neutron data is in agreement with both the nominal stoichiometry and ICP analysis, any potential H+/Li+ exchange must be limited suggesting that in our case it is water molecules that are directly inserted into the structure which cause the observed small lattice expansion.
3.2 Lithium ion diffusion: EIS and μSR spectroscopy studies
The lithium ion diffusion was studied by EIS and μSR spectroscopy at different temperatures in order to understand the lithium ion mobility in this garnet material.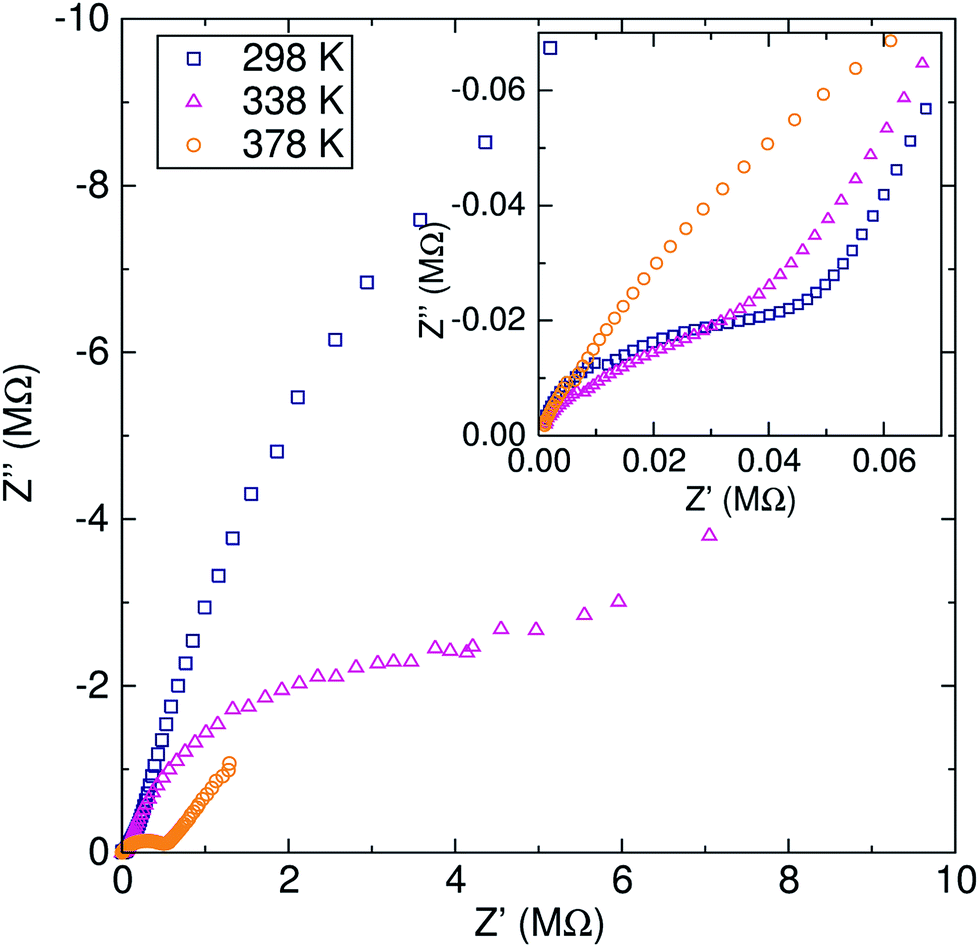 | ||
| Fig. 2 Nyquist plot of the impedance for the Li6.5Al0.25La2.92Zr2O12 garnet at 298 K (blue squares), 338 K (pink triangles) and 378 K (orange circles). Inset highlights the first semicircle at high frequency due to intra-grain resistance to lithium diffusion. | ||
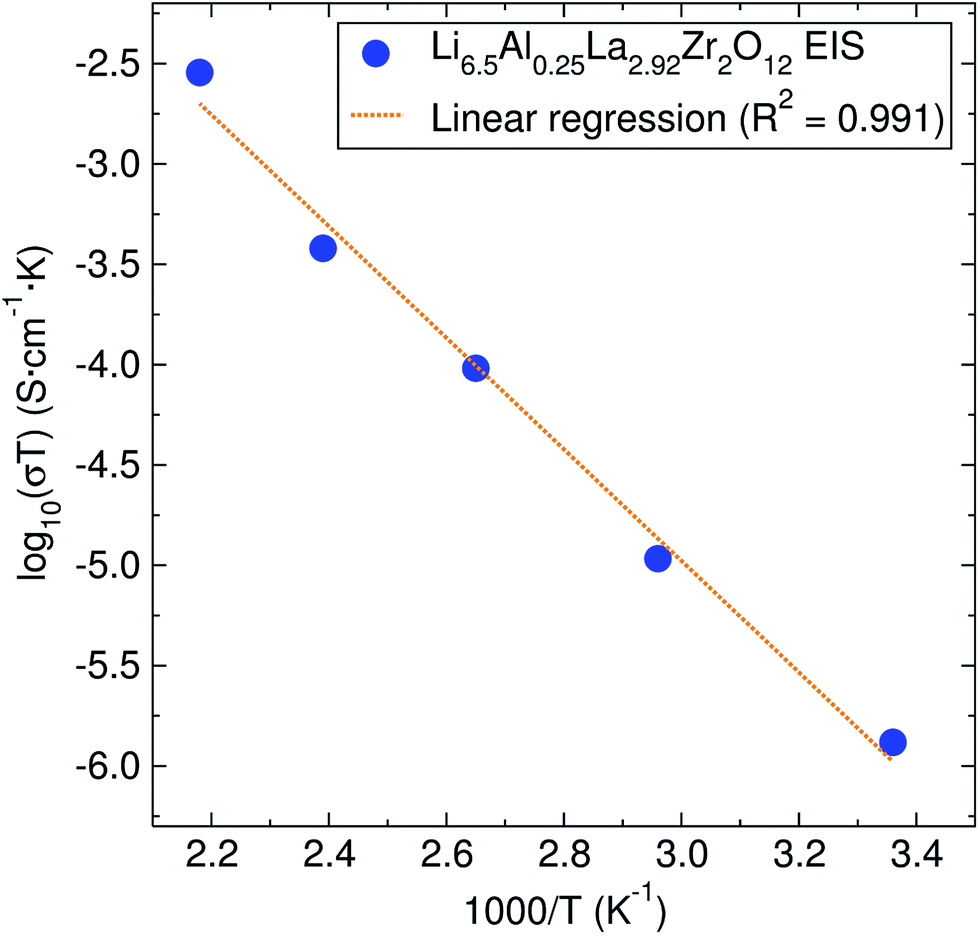 | ||
| Fig. 3 Arrhenius plot of the conductivity coefficient for the Li6.5Al0.25La2.92Zr2O12 garnet. The calculated activation energy from it is 0.55 ± 0.03 eV. | ||
The activation energy calculated from EIS experiments was 0.55 ± 0.03 eV. This value is higher than that reported for related cubic phase LLZ garnets of similar composition (∼0.3–0.4 eV).6,14 For our material, which has a relative density of 81.5%, this increase in the activation energy may be due to our lower sintering temperature and time, which has previously been suggested in the literature.35,36
The EIS technique is widely employed for evaluating the lithium ion conductivity in lithium-stuffed garnets.6 However, it is not without its drawbacks, leading to different values reported for the lithium ion conductivity. Variations in sintering temperatures,35 density of the pellet,36 electrodes employed (from platinum deposited particles to sputtered and melted gold) or the use of hot-pressing techniques16 can greatly affect the experimental values. Specifically, for a similar range of Al-doped cubic LLZ garnets, the values of the activation energy for the lithium ion conduction can vary from 0.26 eV, where the material was hot-pressed and sputtered with gold electrodes,16 to 0.56 eV for a pellet of lower relative density.37 These discrepancies are more related to the measurement experimental set up and the microstructure of the sample [SEM images of samples are shown in the ESI (Fig. S5†)], rather than to the intrinsic nature of the material itself.
To obtain the fluctuation rate of the muons due to lithium ion diffusion, the muon decay asymmetry data were fitted using Keren's analytic generalization of the Abragam function appropriate for μSR,41 multiplied by a temperature-independent relaxation rate fixed for each sample:
| Pz(t) = exp[−Γ(Δ,ν,ωL)t] | (1) |
The Γ(Δ,ν,ωL) term refers to the muon polarization after subtracting a fixed background component. The Δ parameter corresponds to the static width of the local field distribution at the disordered sites, ν is the field fluctuation rate and ωL is the Larmor precession frequency in the applied magnetic field.
The data were fit to the Keren function for each temperature at three longitudinal applied magnetic fields (0, 5 and 10 G) parallel to the initial muon spin polarization and are shown in Fig. 4. By applying different longitudinal applied magnetic fields, interactions between the muons and any local nuclear magnetic field distribution can be decoupled. In addition, the use of three longitudinal applied magnetic fields adds more constraints to the fitting process, resulting in more reliable fits. The calculated ν and Δ values are plotted in Fig. 5 as a function of temperature.
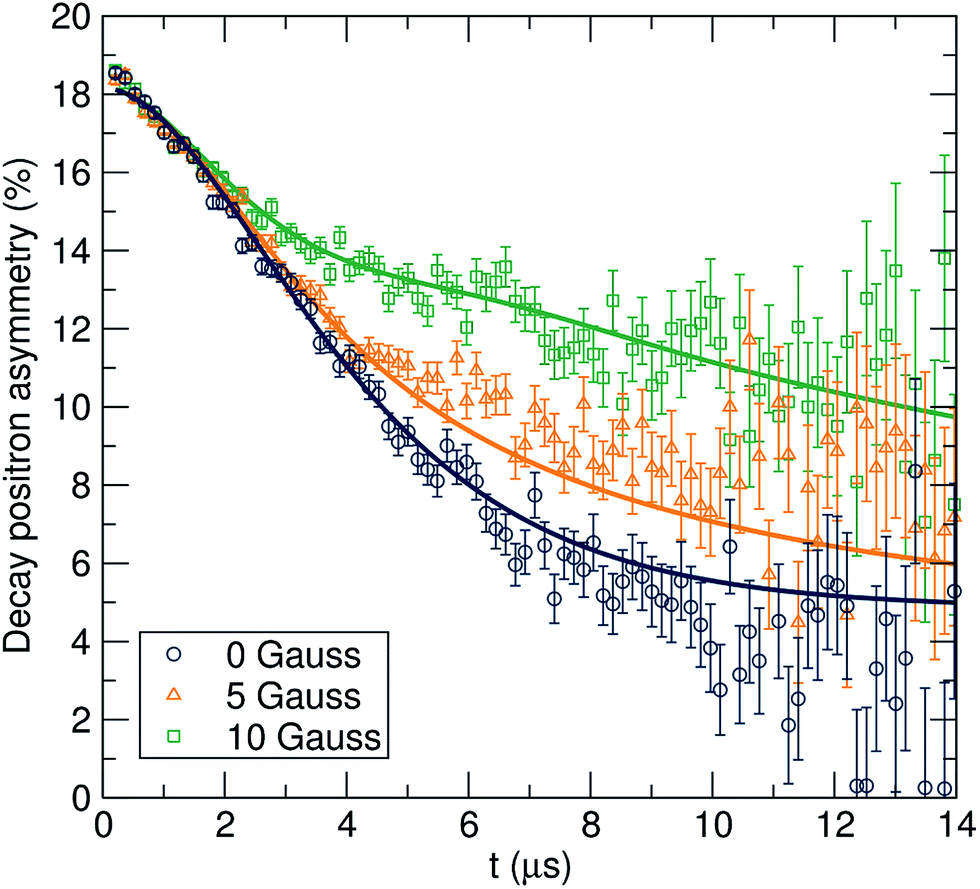 | ||
| Fig. 4 μSR raw data collected at room temperature at zero field (circles) and applied fields of 5 G (triangles) and 10 G (squares), fit using the Keren function for the Li6.5Al0.25La2.92Zr2O12 garnet. | ||
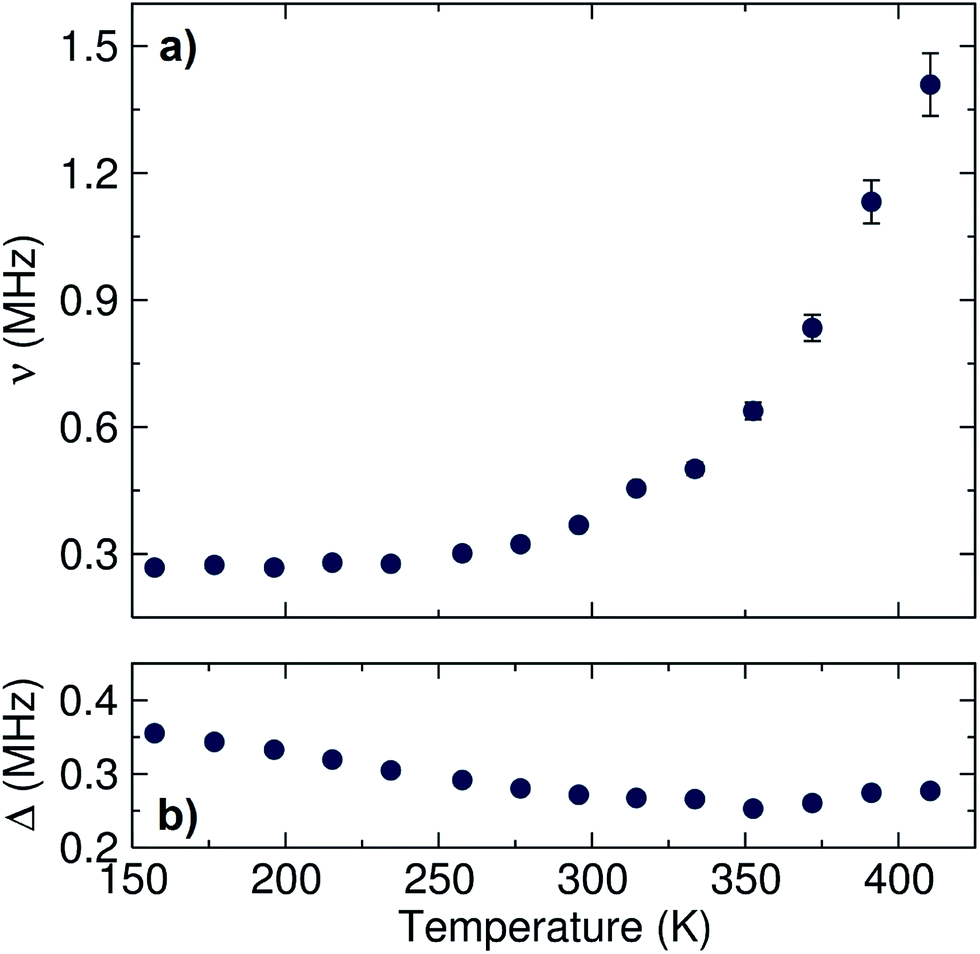 | ||
| Fig. 5 Temperature dependence of (a) ν and (b) Δ values obtained for the fits to the Keren function for the Li6.5Al0.25La2.92Zr2O12 garnet measured from 150 K to 420 K. | ||
A plateau region is observed followed by an exponential increase in the fluctuation rate, due to the thermally activated lithium ion diffusion above 290 K. The Δ plot shows a progressively smooth decrease as the static lithium ions become mobile, reducing their contribution to the static width for the local field distribution. In order to calculate the lithium ion diffusion coefficient from the fluctuation rate, corresponding to the lithium ion jump rate between neighboring sites, eqn (2) can be applied:42
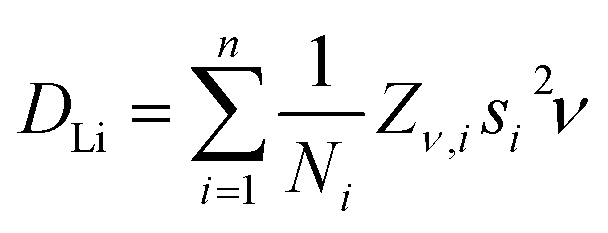 | (2) |
 | ||
| Fig. 6 Simplified crystal structure of an Al-doped LLZ garnet showing both possible Li positions. The lithium ions are drawn in blue, showing tetrahedral environment for the 24d site explicitly. Zr(V) ions in green and La(III) ions given in magenta. Al-dopant ions are shown as an orange fraction in 24d sites. | ||
From these pathways, the calculated lithium diffusion coefficient is 4.62 × 10−11 cm2 s−1, which is greater than that reported for a non-doped tetragonal LLZ garnet (10−14 cm2 s−1) obtained through NMR studies.44 The activation energy required for lithium ion diffusion is then calculated from an Arrhenius plot of either the fluctuation rate or the diffusion coefficient (Fig. 7) and is found to be 0.19 ± 0.01 eV. This value is in excellent agreement with that reported by Nozaki et al. of ca. 0.20 eV for the related LiLaZrNbO family.45
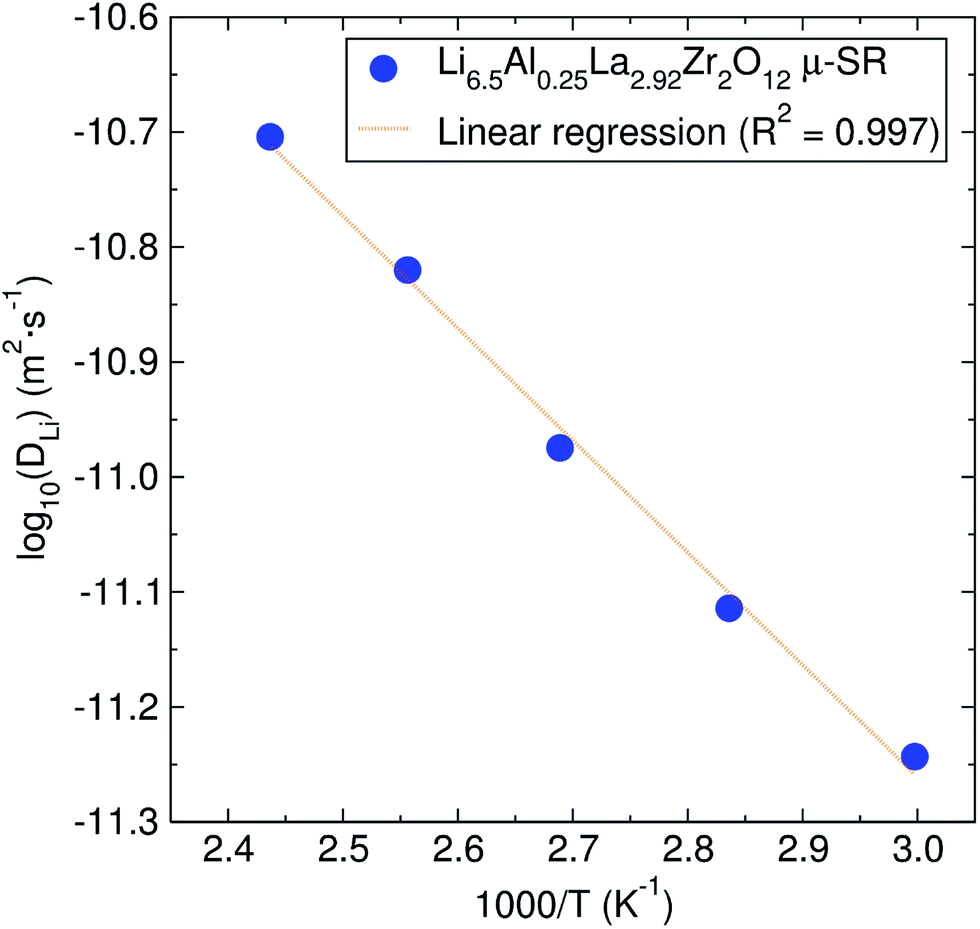 | ||
| Fig. 7 Arrhenius plot of the diffusion coefficient calculated from μSR for the Li6.5Al0.25La2.92Zr2O12 garnet. The calculated activation energy from it is 0.19 ± 0.01 eV. | ||
We have calculated the mobile Li-ion density to be 21.7%, similar to a value reported for Li6.75La3Zr1.75Nb0.25O12 of 15%.45
We can also estimate a DLi of 1.44 × 10−11 cm2 s−1 from EIS by using the Nernst–Einstein equation (eqn (3)):
 | (3) |
The difference in the activation energy between μSR and EIS values arise from the different resistance contributions to the measurements. In EIS, resistance to lithium ion diffusion through grain-boundaries contributes to the total resistance of the sample, increasing the activation energy required for lithium ion conduction. On the other hand, μSR acts as a local probe sensing mostly intra-grain diffusion for lithium ions, not being altered by other extrinsic interferences. These results encourage the necessity of a better understanding of the sintering process employed for these materials in order to find an optimal reduction in grain boundaries, without the use of high temperatures and long times.47
4 Conclusions
The Al-doped LLZ Li6.5Al0.25La2.92Zr2O12 cubic garnet has been synthesized by a fast microwave-assisted solid-state approach, demonstrated for the first time. The highly crystalline cubic phase was obtained at much lower heating temperatures and shorter reaction times compared with any conventional solid-state methods previously reported. ICP-MS analyses and NPD studies confirm the stoichiometry of the cubic sample. The lithium ion diffusion has been studied using EIS and μSR, showing a high inter-grain resistance and low intra-grain activation energy of 0.19 ± 0.01 eV, demonstrating the potential of these materials as solid-state electrolytes for lithium-ion batteries. This marks a new approach to accessing high quality garnets in a fast, clean manner and also illustrates the use of μSR as an excellent local probe for the elucidation of Li diffusion in these materials.Acknowledgements
The authors gratefully acknowledge technical support from Mr Michael Beglan. The authors also thank the STFC for beamtime allocation, the EPSRC (EP/K029290/1 and EP/N001982/1) for funding, the University of Glasgow for support and the School of Chemistry for use of its facilities and PhD studentship funding.References
- F. Putois, J. Power Sources, 1995, 57, 67–70 CrossRef CAS.
- R. Spotnitz and J. Franklin, J. Power Sources, 2003, 113, 81–100 CrossRef CAS.
- L. Vogdanis, B. Martens, H. Uchtmann, F. Hensel and W. Heitz, Makromol. Chem., 1990, 191, 465–472 CrossRef CAS.
- S. Sloop, J. Pugh, S. Wang, J. Kerr and K. Kinoshita, Electrochem. Solid-State Lett., 2001, 4, A42–A44 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- Y. Gao, X. Wang, W. Wang, Z. Zhuang, D. Zhang and Q. Fang, Solid State Ionics, 2010, 181, 1415–1419 CrossRef CAS.
- U. Eisele, T. Koehler, S. Hinderberger, B. Kozinsky and A. Logeat, Lithium ion-conducting garnet-like compounds, US Patent App. 14/233,940, 2014.
- E. J. Cussen, J. Mater. Chem., 2010, 20, 5167–5173 RSC.
- E. J. Cussen, Chem. Commun., 2006, 412–413 RSC.
- J. Percival, E. Kendrick, R. I. Smith and P. R. Slater, Dalton Trans., 2009, 5177–5181 RSC.
- J. Awaka, N. Kijima, H. Hayakawa and J. Akimoto, J. Solid State Chem., 2009, 182, 2046–2052 CrossRef CAS.
- J. Awaka, N. Kijima, K. Kataoka, H. Hayakawa, K.-i. Ohshima and J. Akimoto, J. Solid State Chem., 2010, 183, 180–185 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- C. A. Geiger, E. Alekseev, B. Lazic, M. Fisch, T. Armbruster, R. Langner, M. Fechtelkord, N. Kim, T. Pettke and W. Weppner, Inorg. Chem., 2011, 50, 1089–1097 CrossRef CAS PubMed.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS.
- R. Satish, V. Aravindan, W. C. Ling, J. B. Goodenough and S. Madhavi, Adv. Energy Mater., 2014, 4, 1301715 Search PubMed.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- M. P. O'Callaghan and E. J. Cussen, Solid State Sci., 2008, 10, 390–395 CrossRef.
- K. Tadanaga, H. Egawa, A. Hayashi, M. Tatsumisago, J. Mosa, M. Aparicio and A. Duran, J. Power Sources, 2015, 273, 844–847 CrossRef CAS.
- I. Kokal, M. Somer, P. H. L. Notten and H. T. Hintzen, Solid State Ionics, 2011, 185, 42–46 CrossRef CAS.
- T. Yang, Z. D. Gordon, Y. Li and C. K. Chan, J. Phys. Chem. C, 2015, 119, 14947–14953 CAS.
- J. Wang, J. Binner, B. Vaidhyanathan, N. Joomun, J. Kilner, G. Dimitrakis and T. Cross, J. Am. Ceram. Soc., 2006, 89, 1977–1984 CrossRef CAS.
- H. Buschmann, J. Dolle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienle and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC.
- S. L. Mair, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1978, 34, 542–547 CrossRef.
- A. Larson and R. V. Dreele, Los Alamos National Laboratory Report LAUR 86-748, 1994 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- W. Hennig, W. Voss, J. Dubowik, J. Piche, R. Griffiths, R. Kovar and H. Kua, Novel microwave susceptor composition and method for making same, WO Patent App. PCTUS1991001782, 1991.
- J. Lasri, P. D. Ramesh and L. Schächter, J. Am. Ceram. Soc., 2000, 83, 1465–1468 CrossRef CAS.
- T. Thompson, A. Sharafi, M. D. Johannes, A. Huq, J. L. Allen, J. Wolfenstine and J. Sakamoto, Adv. Energy Mater., 2015, 5, 1500096 Search PubMed.
- Y. Wang and W. Lai, J. Power Sources, 2015, 275, 612–620 CrossRef CAS.
- G. Larraz, A. Orera and M. L. Sanjuan, J. Mater. Chem. A, 2013, 1, 11419–11428 CAS.
- G. Larraz, A. Orera, J. Sanz, I. Sobrados, V. Diez-Gomez and M. L. Sanjuan, J. Mater. Chem. A, 2015, 3, 5683–5691 CAS.
- V. Thangadurai and W. Weppner, J. Power Sources, 2005, 142, 339–344 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, J. Electrochem. Soc., 2008, 155, A90–A101 CrossRef CAS.
- C. Yang, L. Yi-Qiu and G. Xiang-Xin, Chin. Phys. B, 2013, 22, 078201 CrossRef.
- R. Djenadic, M. Botros, C. Benel, O. Clemens, S. Indris, A. Choudhary, T. Bergfeldt and H. Hahn, Solid State Ionics, 2014, 263, 49–56 CrossRef CAS.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238–6245 CAS.
- M. Månsson and J. Sugiyama, Phys. Scr., 2013, 88, 068509 CrossRef.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Phys. Rev. Lett., 2009, 103, 147601 CrossRef PubMed.
- A. Keren, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10039–10042 CrossRef CAS.
- An Introduction to Solid State Diffusion, ed. R. J. B. G. Dienes, Academic Press, San Diego, 1988 Search PubMed.
- M. Klenk and W. Lai, Phys. Chem. Chem. Phys., 2015, 17, 8758–8768 RSC.
- A. Kuhn, S. Narayanan, L. Spencer, G. Goward, V. Thangadurai and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094302 CrossRef.
- H. Nozaki, M. Harada, S. Ohta, I. Watanabe, Y. Miyake, Y. Ikedo, N. H. Jalarvo, E. Mamontov and J. Sugiyama, Solid State Ionics, 2014, 262, 585–588 CrossRef CAS.
- M. M. Ahmad, RSC Adv., 2015, 5, 25824–25829 RSC.
- N. Janani, C. Deviannapoorani, L. Dhivya and R. Murugan, RSC Adv., 2014, 4, 51228–51238 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08107f |
| This journal is © The Royal Society of Chemistry 2016 |