
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoelectrochemical water splitting in an organic artificial leaf†
Serkan
Esiner
a,
Robin E. M.
Willems
a,
Alice
Furlan
a,
Weiwei
Li
ab,
Martijn M.
Wienk
ac and
René A. J.
Janssen
*ac
aMolecular Materials and Nanosystems & Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: r.a.j.janssen@tue.nl
bBeijing National Laboratory for Molecular Sciences, Organic Solids Laboratory, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P.R. China
cDutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ Eindhoven, The Netherlands
First published on 3rd November 2015
Abstract
Photoelectrochemical water splitting is demonstrated in an organic artificial leaf composed of a triple junction polymer solar cell for light absorption and charge generation and low-overpotential catalytic electrodes for hydrogen and oxygen evolution. For small area solar cells (<0.1 cm2), a solar to hydrogen conversion efficiency of 5.4% is obtained using RuO2 catalysts. Using earth-abundant NiMoZn and Co3O4 catalysts for hydrogen and oxygen evolution, the efficiency is 4.9%. For larger area (1.7 cm2) solar cell devices the solar to hydrogen efficiency with RuO2 catalysts reduces to 3.6% as a consequence of an increased overpotential for water splitting. This shifts the operating point of the water splitting device beyond the maximum power point of the solar cell and reduces the photocurrent.
Introduction
Storage of solar energy is important to counterbalance the intermittency of solar electricity supply and demand. Capturing solar energy in chemical bonds of molecular fuels is most effective in terms energy density and the successful construction of a direct artificial system for efficient solar fuel generation is an important challenge for science and engineering. Solar fuels are attracting considerable attention recently and solutions are emerging on how this can be achieved.1,2 Solar to chemical energy conversion requires three concerted steps: (1) absorption of light, (2) creation of charges (electrons and holes) with an appropriate chemical potential to enable (3) catalytic chemical reactions in which the charges are used to oxidize and reduce compounds in endothermic reactions such as the splitting of water and the reduction of carbon dioxide.1 To enable solar energy production in yields exceeding the energy conversion of natural photosynthesis (typically <1%) with cheap and abundant materials is a tremendous challenge.3 Photoelectrochemical (PEC) water splitting requires a theoretical potential of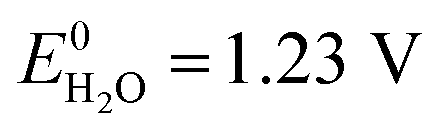 , but in practice occurs at a potential (VH2O) higher than
, but in practice occurs at a potential (VH2O) higher than 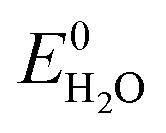 due to overpotential losses (η) occurring at the electrodes
due to overpotential losses (η) occurring at the electrodes  . Depending on the type of the electrodes and catalysts, the electrolyte, and the current density, overpotentials for both hydrogen and oxygen evolution vary and the electrolysis potential lies typically in the range of 1.4–1.9 V.4,5
. Depending on the type of the electrodes and catalysts, the electrolyte, and the current density, overpotentials for both hydrogen and oxygen evolution vary and the electrolysis potential lies typically in the range of 1.4–1.9 V.4,5
The principle of solar energy driven water splitting has previously been described using inorganic solar cells with suitable catalysts for hydrogen and oxygen production. Early publications include the monolithic photoelectrochemical water splitting devices of Lin et al.6 based on a triple stack amorphous silicon solar cell with RuO2 and Pt electrodes and of Turner et al.7 based on tandem GaAs/InGaP2 tandem photocathode and a Pt electrode. One of the most efficient water splitting devices was published by Licht et al.,8 who reached a solar to hydrogen conversion efficiency of ηSTH = 18.3% with a Si/Al0.15Ga0.85As tandem cell in combination with Pt and RuO2 electrodes for hydrogen and oxygen evolution. Based on a similar concept using a GaInP/GaInAs dual junction cell in an optical concentrator system with a polymer electrolyte membrane electrolyser Wittstadt et al. demonstrated solar water splitting with 18% efficiency in an integrated system.9
More recently, Nocera et al.10 reported ηSTH = 2.5% for a wireless stand-alone device based on a triple junction solar cell with earth-abundant nickel–molybdenum–zinc (NiMoZn) and cobalt oxide cubane (CoO) catalysts for hydrogen and oxygen evolution. van de Krol et al. reached ηSTH = 4.9% based on a semi-transparent tungsten doped bismuth vanadate (W:BiVO4) photoanode that was optically and electrically connected to an amorphous silicon tandem solar cell with cobalt phosphate and Pt catalysts.11 Grätzel et al. have reported ηSTH = 12.3% based on two series connected lead perovskite solar cells and a NiFe layered double hydroxide for hydrogen and oxygen evolution.12
We were interested to explore the feasibility of an organic artificial leaf by combining organic solar cells with suitable catalysts for water splitting. Organic and polymer solar cells produce electrical power directly by converting sunlight. The best devices reach power conversion efficiencies in excess of 10%.13–15 Examples of photoelectrochemical water splitting via organic absorber layers are, however, scarce and have not yet reached high efficiencies.16–18 Designing an organic artificial leaf capable of producing hydrogen directly from sunlight requires the selection and optimization of a number of parameters. To minimize losses, the solar cell should operate close to the maximum power point and for efficient water splitting the maximum power point voltage (Vmax) of the cell should be designed such that its value matches with the potential for water splitting, VH2O, under the relevant working conditions, related to choice of electrodes, electrolyte, and current density. With a required Vmax ≈ VH2O = 1.4–1.9 V, single junction or series connected tandem polymer solar cells are generally not sufficient for the water splitting reaction to take place. However, a series connected triple junction polymer solar cell can provide the required potential at its maximum power point as we have shown recently.16
An important aspect of photoelectrochemical water splitting devices is the selection of appropriate catalysts for oxygen and hydrogen evolution reactions. The operating potential during water splitting mainly depends on this selection. The best catalysts in terms of lowering the overpotential are based on precious transition metals or their oxides. Many earth-abundant catalysts require higher overpotentials,19–22 but extensive efforts are directed to overcome this limitation.19–23 Besides overpotential, the compatibility of the hydrogen and oxygen evolution catalysts with each other in the selected electrolyte is very important. In general, catalysts operate better in highly acidic or highly alkaline media compared to more neutral pH conditions. Highly acidic or alkaline conditions, however, affect the stability of the catalyst and the sealing of an integrated photoelectrochemical device when kept in contact with the electrolyte for a long time. Preferably, the hydrogen and oxygen evolution catalysts are formed from earth-abundant materials and should be able to operate at near neutral pH conditions.
Here we present the design and characterization of a photoelectrochemical artificial organic leaf that integrates an organic triple junction solar cell with catalysts for hydrogen and oxygen evolution. We demonstrate photoelectrochemical water splitting with a solar to hydrogen conversion efficiency of 5.4% with low-overpotential ruthenium oxide (RuO2) catalysts and of 4.9% efficiency with earth-abundant cobalt oxide (Co3O4) and NiMoZn catalysts. To ensure a low overpotential, the catalyst to solar cell area ratio is about 15–20. In an alternative third configuration we use a larger area triple junction solar cell with RuO2 similar sized catalyst surface areas, resulting in ηSTH = 3.6%.
Results and discussion
Organic absorber layers
For solar to hydrogen conversion we use a triple junction polymer solar cell composed of one wide band gap and two identical narrow band gap sub cells (Fig. 1), fabricated by solution processing as described previously.24 The wide band gap layer is a blend of poly[[9-(1-octylnonyl)-9H-carbazole-2,7-diyl]-2,5-thiophenediyl-2,1,3-benzothiadiazole-4,7-diyl-2,5-thiophenediyl] (PCDTBT) and [6,6]-phenyl-C71-butyric acid methyl ester ([70]PCBM), while the narrow band gap layers are blends of poly[[2,5-bis(2-hexyldecyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl]-alt-[3′,3′′-dimethyl-2,2′:5′,2′′-terthiophene]-5,5′′-diyl] (PMDPP3T) and [6,6]-phenyl-C61-butyric acid methyl ester ([60]PCBM). The recombination layers that connect the different sub cells use a thin film of ZnO nanoparticles covered with a pH neutral poly(3,4-ethylene dioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) layer. This cell provides a Voc above 2.0 V, which is sufficient for photoelectrochemical water splitting. The energy level diagram is shown in Fig. 1. | ||
| Fig. 1 Device layout of triple junction organic solar cell, the organic compounds used in the small band gap (PMDPP3T:[60]PCBM) and wide band gap (PCDTBT:[70]PCBM) photoactive layers, and schematic energy level diagram (close to open circuit) showing the operation of the triple junction cell coupled to the water splitting reactions. | ||
Catalysts and electrolytes
Presently, ruthenium oxide is the best oxygen evolution catalyst employed in the water splitting process.25–27 The electrocatalytic properties of RuO2 have been studied for a significant time,28 not only for oxygen evolution but also as employing the oxide as a hydrogen evolution catalyst.29,30 Ruthenium oxide can be deposited on various substrates mainly via two methods: electrodeposition or thermal decomposition.28 As indium tin oxide (ITO) is commonly employed as electrode in polymer solar cells, it is convenient to apply the catalyst layers directly onto ITO to create a monolithic device. Even though it is possible to electrodeposit RuO2 on ITO,31,32 we observed that the electrodeposited layers were not very stable when used for oxygen evolution. Another method for depositing ruthenium oxide is thermal decomposition of RuCl3 from an aqueous solution of RuCl3, followed by annealing at elevated temperatures.25 In our experiments, the RuO2 catalyst on transparent conductive substrates tended to delaminate in time especially during active gas evolution periods, but attached better to metal substrates.In this study, ruthenium oxide was deposited onto titanium substrates through thermal decomposition of RuCl3.25 The details are described in the Experimental section. The procedure gives reproducible and stable performance over a couple of hours. The activity of RuO2 both as oxygen and hydrogen evolution catalysts is shown in a Tafel plot in Fig. 2. RuO2 on a Ti substrate gives an overpotential of less than 130 mV for hydrogen evolution and less than 315 mV for oxygen evolution for current densities up to 10 mA cm−2 in a 1.0 M KOH electrolyte. RuO2 is actually a remarkably good electrocatalyst for hydrogen evolution as a result of an activation that occurs under reductive conditions and that is considered as an intrinsic property of the oxide.28,33
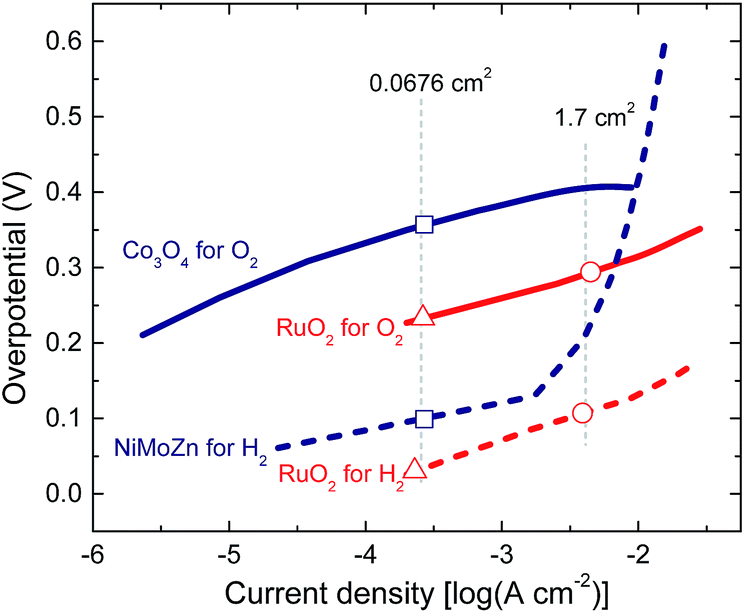 | ||
| Fig. 2 Tafel plots of RuO2 in 1.0 M of KOH and of Co3O4 and NiMoZn in 0.1 M potassium borate (KBi) at pH 9.2. The markers indicate the expected overpotentials during the operation of the small scale RuO2/RuO2 PEC cell (△), the small scale Co3O4/NiMoZn PEC cell (□), and the large scale RuO2/RuO2 PEC cell (○). | ||
For the selection of earth-abundant catalysts, we considered that choice for the catalyst for oxygen evolution is more important than the catalyst for hydrogen evolution, because the overpotentials for oxygen are generally higher. One of the earth-abundant oxygen evolution catalysts, which has been extensively studied lately, is cobalt oxide. Two well-known forms of this oxide are cobalt borate (CoBi)34 and cobalt phosphate (CoPi).35 These catalysts are deposited electrochemically and have been used for photoelectrochemical water splitting.10,11 Cobalt oxide can also be deposited on conductive surfaces in the form of nanoparticles for oxygen evolution.36 In this method the synthesized nanoparticles are dispersed in methanol and deposited on ITO through drop casting and thermal annealing (see Experimental section for details). The cobalt oxide catalyst can operate in an electrolyte of 0.1 M potassium borate (KBi) at pH 9.2, which is crucial for the catalyst stability. The electrochemical activity of cobalt oxide nanoparticles is shown in Fig. 2. The catalyst gives an overpotential up to 410 mV for oxygen evolution for current densities up to 10 mA cm−2. This performance is comparable to the CoBi and CoPi catalysts.34,35 Due to reproducibility and ease of processing, cobalt oxide nanoparticles were selected as the oxygen evolution catalyst for the desired water splitting device.
The selection of a suitable earth-abundant hydrogen evolution catalyst also requires ease of processing, reproducibility and ability to operate in an electrolyte of near neutral pH conditions. Nickel based hydrogen evolution catalysts have been investigated and used for hydrogen evolution.37–40 Among the available options of hydrogen evolution catalysts, many nickel compounds are used under harsh conditions with pH values above 13 or below 1.37–40 However, the NiMoZn catalyst developed by Nocera et al.10 was shown to work well in KBi electrolyte where Co3O4 nanoparticles can also operate. Hence, the NiMoZn alloy was selected as the hydrogen evolution catalyst. The catalyst is made by electrodeposition onto a nickel substrate (see Experimental section for details).10 The Tafel plot for NiMoZn in 0.1 M KBi for hydrogen evolution shows that the catalyst requires very low overpotentials of about 100 mV for current densities up to 1 mA cm−2, but that the overpotential increases significantly when the current density is increased.
Photoelectrochemical water splitting with RuO2/RuO2 catalysts at low current densities
For achieving a high ηSTH, a larger catalyst surface area provides smaller current densities and hence smaller overpotential. A triple junction polymer solar cell with an active area of 0.0676 cm2 was connected to RuO2 catalysts for oxygen and hydrogen evolution submerged in a 1.0 M KOH electrolyte. The catalyst areas of 1.1 and 1.3 cm2, respectively, were obtained by drop casting of the RuCl3 precursor solution. Fig. 3a and b show the time evolution of the voltage and current density of photoelectrochemical water splitting device under illumination by simulated AM 1.5G light. The three curves in Fig. 3a correspond to the J–V characteristics of the triple junction cell measured just before, during, and directly after a 20 min water splitting experiment. The power conversion efficiency (PCE) of the solar cell just before the water splitting experiment is ∼6.7% with a Vmax = 1.44 V. The PCE of 6.7% is less than the previously reported value of 9.6%,24 mainly because the batch of PCDTBT polymer used in this study gave inferior performance. | ||
| Fig. 3 (a, c, e) J–V curves of the triple junction solar cells before, during and after water splitting measurement of 20 min. (b, d, f) Simultaneous measurement of operating voltage and current density of the solar cell during photoelectrochemical water splitting, (a, b) 0.0676 cm2 solar cell connected to RuO2/RuO2 catalysts in 1.0 M KOH. (c, d) 0.0676 cm2 solar cell connected to Co3O4/NiMoZn catalysts in 0.1 M KBi. (e, f) 1.7 cm2 solar cell connected to RuO2/RuO2 catalysts in 1.0 M KOH. The light source is not chopped and the electrolyte is not stirred during measurements. The inset in panel (f) shows the hydrogen and oxygen evolution from the RuO2 catalysts on the Ti substrates. | ||
The voltage and current density measured during water splitting follow the J–V curve of the solar cell until the operating point stabilizes (Fig. 3a). The stabilization takes roughly 15 min and it is mainly due to charging of the double layer on the catalyst surfaces and back reaction of the reaction intermediates on or in the neighbourhood of the catalysts (Fig. 3b).41 In this specific case it takes longer because of the very low current density on the catalyst surfaces. After reaching stabilization, a slight decrease in the operating current density is observed for the following 5 min. The decrease is attributed mainly to the degradation of the solar cell during operation, as evidenced from the solar cell performance just after the water splitting measurements (Fig. 3a). Solar to hydrogen conversion efficiency can also be affected by the slow degradation of the RuO2 catalysts because it is known that the electrolyte used (1.0 M KOH) does not allow for stable catalyst performance in the long run.42 In our experiments, however, the decrease in catalyst performance over time turned out to be marginal.
Fig. 3b shows that the stable operation takes place at around 1.49 V at a solar cell current density of 4.40 mA cm−2. At an operating potential of Vop = 1.49 V, the total overpotential for hydrogen and oxygen evolution is 0.26 V, in excellent agreement with the value expected from the Tafel plots for the corresponding current densities on the catalyst surfaces of 0.03 V and 0.23 V for hydrogen and oxygen evolution reactions, respectively (shown with the △ symbols in Fig. 2). It is important to highlight that, the operating point during water splitting is very close to the maximum power point of the solar cell (Vmax = 1.44 V), which enhances the efficiency of the PEC water splitting device. The solar cell efficiency in the operating point is estimated as Vop × Jop/Pin = 6.6%, close to the maximum PCE of 6.7%. Assuming 100% Faradaic efficiency, the photocurrent in the operating point Jop (4.40 mA cm−2) allows to estimate the solar to hydrogen efficiency as: ηSTH = 1.23 × Jop/Pin = 5.4%. Separate electrochemical experiments in which evolved gassed were collected, showed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 volume ratio of O2 to H2 and Faradaic efficiencies higher than 88%. In this PEC device with low current densities at the electrodes, hydrogen and oxygen bubbles during water splitting were visualized using a high resolution camera.
:2 volume ratio of O2 to H2 and Faradaic efficiencies higher than 88%. In this PEC device with low current densities at the electrodes, hydrogen and oxygen bubbles during water splitting were visualized using a high resolution camera.
Photoelectrochemical water splitting with Co3O4/NiMoZn catalysts at low current densities
To realize solar to hydrogen conversion with the earth-abundant catalysts, a 0.0676 cm2 triple junction polymer solar cell was combined with Co3O4 and NiMoZn catalysts with surface areas tuned to 1 cm2. Fig. 3c shows the J–V curves of the solar cell just before, during, and after 20 min of water splitting with Co3O4/NiMoZn catalysts submerged in a 0.1 M KBi electrolyte at pH = 9.2. The simultaneous measurement of the transient current density and voltage during water splitting is shown in Fig. 3d. In this case the stabilization of the operating point takes about 3 min. We attribute the reason of shorter stabilization period to the different nature of catalysts and the electrolyte. Another reason can be the difference between the geometrical and actual surface areas of the catalysts. The RuO2 catalysts are placed on very rough titanium substrates while both Co3O4 and NiMoZn layers are deposited on smoother surfaces. Higher roughness would mean higher surface area, which will require more time to form the double layer on the catalyst surfaces. During 20 min of PEC water splitting, a slight decrease in the operating current density is observed together with a slight increase in operating voltage. These are mainly due to the degradation of the solar cell that is more prominent in the case of this specific solar cell (Fig. 3c).The triple junction solar cell combined with earth-abundant catalysts had a PCE of 6.5%. The operating point of the water splitting device stabilized at Vop = 1.56 V. This operating voltage is again close to Vmax = 1.45 V. The Vop = 1.56 V suggests a total overpotential of 0.33 V, which is less than the value of ∼0.46 V expected from the Tafel plots. The difference might be related to different concentrations of dissolved hydrogen and oxygen in the electrolyte during Tafel plot measurements. After 15 min we find Jop = 3.98 mA cm−2, providing a PCE of 6.1% and ηSTH = 4.9% in the operating point.
A larger area organic leaf
For a more realistic estimation of ηSTH, the catalyst surface areas should be in the same range as the solar cell surface. Therefore, we constructed an organic leaf with a larger area triple junction solar cell integrated with RuO2/RuO2 catalyst for hydrogen and oxygen evolution on Ti substrates (Fig. 4a). The solar cell area was 1.7 cm2 and the catalyst surface areas are ∼1.2 cm2 each. To reduce the potential drop over the ITO front surface of this larger area solar cell, the current was collected by a boundary metal contact (Fig. 4b, left). This metal border also defines the nominal solar cell area. After completion, the solar cell was sealed with a second glass plate and a modified epoxy resin (Fig. 4b, middle) and then titanium substrates with the RuO2 catalysts are attached at the back side with a glue and graphite conductive adhesive (Fig. 4b, right). | ||
| Fig. 4 (a) Schematic device layout of the organic artificial leaf. (b) Photographs of the actual device, from left to right: front side, back side before, and back side after applying the water splitting electrodes and catalysts. | ||
Fig. 3e shows that the l.7 cm2 triple junction solar cell has a PCE of 6.1%, which is somewhat less than the PCEs of the small area cells (compare panels (a) and (c) in Fig. 3) due to a slight reduction in short-circuit current and fill factor. For PEC water splitting, the triple junction solar cell was coupled to the two RuO2 catalysts for oxygen and hydrogen evolution in 1.0 M KOH electrolyte. Evolution of hydrogen and oxygen was easily observed by the eye (Fig. 3f). Fig. 3e shows the J–V-voltage characteristics measured during water splitting. After 15 min operation, the operating point of the artificial leaf is Vop = 1.67 V and Jop = 2.94 mA cm−2. The current stabilization in this configuration takes less than a minute and is much faster than for the smaller area cells due to the high current density on the catalyst surfaces. After stabilization, the operating point does not significantly change over the course of a 20 min measurement (Fig. 3f). The slight decrease in current density can be attributed to the degradation of the solar cell (Fig. 3e).
The maximum power point of this specific solar cell is at Vmax = 1.40 V while the operating point during water splitting is now at Vop = 1.67 V. The latter is a direct consequence of the increased current density experienced by the catalysts and results in higher overpotentials both for hydrogen and oxygen evolution (see ○ markers in Fig. 2) and a higher operating potential. The total overpotential of 0.44 V in the operating point is 0.04 V higher than the value expected from the Tafel plots (Fig. 2). At Vop = 1.67 V, the power output of the solar cell is 4.91 mW cm−2, which is significantly less than the maximum power of 6.05 mW cm−2 that can be delivered by the cell. As Vop is now significantly larger than Vmax, the photocurrent is reduced considerably to Jop = 2.94 mA cm−2. As a result, ηSTH is 3.6%. The loss in ηSTH from 5.4% to 3.6% by increasing the solar cell area is significant, and solely due to the increased overpotential. This results in a significant reduction of the operating current for this particular triple junction cell. This demonstrates that designing an efficient artificial leaf requires a subtle balance between Vop and Vmax. For Vop > Vmax, a significant loss in photocurrent can be expected. When Vop < Vmax, the photocurrent density actually is increased but because the efficiency of a solar cell is always a trade-off between current density and voltage, operating a cell too far below from the maximum power point represents an avoidable loss.
We also constructed a large area organic artificial leaf with earth abundant catalysts. In this case the triple junction polymer solar cell with a ∼1.2 cm2 area was integrated with Co3O4 and NiMoZn catalysts operating in 0.1 M KBi electrolyte. The 20 min water splitting experiments revealed a ηSTH = 1.3% (Fig. S1, ESI†). This significantly lower efficiency is partially due to a large operating voltage of Vop = 1.83 V (Fig. S2, ESI†), but also due to faster degradation of this specific solar cell used in this experiment (Fig. S1, ESI†).
The stability of the RuO2/RuO2 and Co3O4/NiMoZn catalysts used in the PEC water splitting devices were tested with two-electrode measurements at applied potentials of 1.65 V and 1.85 V for 20 min. The applied potentials were selected with respect to the operating potentials of large area artificial leafs. The results (Fig. S3, ESI†) show that both catalyst couples do not show substantial degradation on this time scale. The catalyst stability is important for future and long term application. Several investigations have addressed the stability of the catalysts under the conditions used in this work. The intrinsic activity and stability of RuO2 for oxygen and hydrogen evolution reactions in alkaline electrolytes has been described in detail, showing that dissolution of RuO2 during oxygen evolution is limiting the stability but that it is table during hydrogen evolution.43 For Co3O4 nanoparticle a high electrocatalytic stability in alkaline conditions has been reported for oxygen evolution.44 Likewise, the NiMoZn electrode in 0.1 M KBi at pH = 9.2 showed no appreciable degradation during more than 150 h operation.45
A further level of integration is shown in Fig. 5, where a larger area organic leaf is fabricated with dual RuO2 catalysts electrodeposited on ITO. At this level of integration it is no longer possible to measure J–V characteristics, but the evolution of hydrogen and oxygen evidences the functionality of the device.
 | ||
| Fig. 5 Artificial organic leaf under illumination based on a triple junction organic solar, with RuO2 covered ITO electrodes for hydrogen (right) and oxygen (left) evolution. | ||
Conclusions
We have demonstrated efficient photoelectrochemical solar to hydrogen conversion using an organic artificial leaf based on an organic triple junction solar cell and transition metal and metal oxide electrocatalysts. A solar to hydrogen conversion efficiency of ηSTH = 5.4% was obtained with RuO2 catalysts and ηSTH = 4.9%with earth-abundant Co3O4/NiMoZn catalysts. In these examples the overpotential was kept low, by a large catalyst to solar cell catalyst to solar cell area ratio of ∼15. For a lower surface area ratio (∼0.7), ηSTH is reduced to 3.6%, mainly because the 0.18 V increase in overpotential that originates from the ∼20 times higher electrocatalytic current density moves the operating point of the artificial leaf too far away from the maximum power point of the solar cell and results is a significant decrease in photocurrent density.This work demonstrates that for efficient artificial leafs, balancing the nature and surface area of the catalysts with the materials used in the solar cell is crucial. In this respect organic solar cells offer an advantage for designing photoelectrochemical water splinting devices because a wide choice in organic semiconductors is available, which enables tuning the maximum power point voltage (Vmax) to coincide with the operating point (Vop) determined by the thermodynamic potential for water splitting and the overpotentials defined by the catalysts. By optimizing these parameters, a significant progress in the performance of organic artificial leaves can be achieved. Further improvements of the present system should also focus on improving the stability of triple junction polymer solar cells and the catalysts.
Experimental
Materials
All commercial chemicals were used as received. Cobalt(II) acetate tetrahydrate (99.999% trace metals basis), nickel foil (thickness 0.125 mm, ≥99.9%), potassium tetraborate tetrahydrate (≥99.0%), and sodium molybdate dihydrate (≥99.5%) were obtained from Sigma-Aldrich. Graphite conductive adhesive (aqueous based, 20 Ω in−2 at 0.001 in thickness), nickel(II) sulfamate hydrate, and zinc chloride (anhydrous, 98+%) were obtained from Alfa Aesar. Ruthenium(III) chloride (35–40% Ru) was obtained from Acros Organics. PCDTBT was obtained from 1-Material. [60]PCBM (purity ∼ 99%) and [70]PCBM (purity ∼ 95%) were obtained from Solenne BV. PMDPP3T was prepared as described previously.24 DELO-KATIOBOND® LP655 light-/UV-curing adhesive was obtained from DELO Industrial Adhesives. Platinum was obtained from Drijfhout. Water is purified in a Millipore system and has a resistance of at least 18 MΩ.RuO2 catalysts
RuO2 was prepared through thermal decomposition of RuCl3. RuCl3 was dissolved in ultra-pure water to a concentration of 0.2 M and 200 μL of this solution was placed onto a pre-cleaned and air plasma treated titanium substrate to form a catalyst area of about 1.3 cm2. The substrate was then dried on a 90 °C hot plate for 20 min and then oxidized in a 350 °C oven for 3 h. The same procedure was used for oxygen and hydrogen evolution RuO2 catalysts. X-ray diffraction (XRD) confirmed the formation of crystalline RuO2 on Ti (Fig. S4, ESI†).Co3O4 nanoparticles
The procedure for the synthesis of the Co3O4 nanoparticles was adapted from literature.36 Under constant magnetic stirring at 45 °C, cobalt(II) acetate tetrahydrate (0.5 g) was dissolved in a mixture of ultra-pure water (2 mL) and ethanol (23 mL). After 15 min, ammonia (25+%, 3.3 mL) was added dropwise. The reaction mixture was heated to 80 °C and kept there for 3 h under reflux to enable the formation of the nanoparticles. To this crude product mixture, acetone (100 mL) was added to start precipitation. To improve separation, the mixture was centrifuged for 20 min at 2000 rpm. After decanting the solvent, methanol (12 mL) and acetone (120 mL) were added to the precipitate, followed by centrifugation at 2000 rpm for 20 min. The precipitated particles were redispersed in methanol (>25 mL). The Co3O4 nanoparticles were characterized with UV-vis spectroscopy and transmission electron microscopy and found to be 3–5 nm in size (Fig. S5 and S6, ESI†).Co3O4 electrode
A solution of the Co3O4 nanoparticles (250 μL, circa 3 mg mL−1) was placed onto a pre-heated ITO coated glass slide at 110 °C to form a catalyst area of around 1 cm2. After 5 min, the covered ITO slide is heated with a heat gun with a power of 2000 W for several minutes to improve binding of the nanoparticles to the substrate. The Co3O4 electrode was characterized with scanning electron microscopy (SEM), energy-dispersive X-ray (EDX), and XRD (Fig. S7–S9, ESI†).NiMoZn electrode
The procedure for the preparation of a NiMoZn electrode was adapted from literature.10 In ultra-pure water (100 mL), nickel(II) sulfamate hydrate (1.309 g), sodium molybdate dihydrate (0.460 g), zinc chloride (6 mg), sodium pyrophosphate (3.460 g), and sodium bicarbonate (7.5 g) were dissolved under constant magnetic stirring. To circa 30 mL of this solution, a few drops of hydrazine hydrate were added just before deposition. The substrate, a nickel foil, was pretreated in diluted sulfuric acid at −2.0 V versus Ag/AgCl for 3 min, without iR correction. Electrodeposition was carried out in a one-compartment electrochemical cell for 60 min at 0.0775 A cm−2versus Ag/AgCl. Two nickel electrodes were used as working and counter electrode. After deposition, the catalyst films on the counter electrode were allowed to leach overnight in 10 M KOH. The overpotential of the NiMoZn electrode is significantly smaller than that of the Ni foil (Fig. S10, ESI†).Electrochemical measurements
Tafel plots were constructed from the cyclic voltammetry (CV) measurements performed in the stationary current mode with 50 mV steps taking 300 s per step. Two cycles were performed for each catalyst and the second cycle was used to form the Tafel plots. In all cases, a Pt disk and Ag/AgCl electrode were used respectively as counter electrode and reference electrode. A three-compartment cell was used during measurements, where the compartments for working and counter electrodes are separated with a fine porosity glass frit. The compartment with the reference cell was connected to the working electrode compartment using a Luggin capillary. For Co3O4, the measuring range was selected starting above 0.60 V versus Ag/AgCl, for NiMoZn beneath −0.7 V. Owing to a different electrolyte and different catalyst performance, these values were selected as above 0.25 V and below −0.95 V when RuO2 is used as oxygen and hydrogen evolution catalysts respectively. The electrolytes were not stirred during measurements.Tafel plots were constructed by characterizing each catalyst in a three electrode CV measurement. The measured potential (Emeas) was converted into the overpotential η via: η = Emeas + Eref. vs. RHE − E0 − iRu, where Eref. vs. RHE is the potential difference between the reference and the reversible hydrogen electrode (RHE), E0 is water oxidation or hydrogen reduction potential (1.23 V or 0.00 V), i is the current and Ru the uncompensated resistance between the working and the reference electrode. The value for Ru was determined by electrochemical impedance spectroscopy. The measurement was done in the frequency range from 10 kHz to 100 mHz. All measurements were done with respect to an Ag/AgCl (3 M KCl) reference electrode in media with different pH such that: Eref. vs. RHE = 0.210 + 0.059 × pH.
Device preparation
The small area triple junction solar cells were prepared by first spin casting PEDOT:PSS (Clevios® P VP AI 4083, H. C. Starck) in air onto pre-cleaned glass substrates with indium tin oxide (ITO) patterns (Naranjo Substrates). Then, the PEDOT:PSS layer was dried at 140 °C for 10 min. On top of the dried PEDOT:PSS the PCDTBT:[70]PCBM blend was spin cast in nitrogen atmosphere. The PCDTBT active layer was dried on a hot plate for 10 min at 70 °C. Afterwards, the ZnO, pH-neutral PEDOT and PMDPP3T:[60]PCBM layers were spin cast sequentially to form the middle cell. This process was then repeated to form the back cell. For optimal performance, the ZnO layer was deposited in nitrogen atmosphere, while pH-neutral PEDOT and PMDPP3T:[60]PCBM layers were spin cast in air. The triple junction devices were completed by thermal evaporation of 1 nm LiF and 100 nm Al at 3 × 10−7 mbar.The front cell was spin cast from a warm solution of PCDTBT and [70]PCBM (1:4 w/w) in chlorobenzene at 7 mg mL−1 polymer concentration. The middle and the back cells were spin cast from a solution of PMDPP3T and [60]PCBM (1:3 w/w) in chloroform containing 7.5% (v/v) o-DCB at 4 mg mL−1 polymer concentration. ZnO nanoparticles of ∼5 nm diameter were spin cast from a solution of 10 mg mL−1 ZnO in isopropanol (IPA). pH-neutral PEDOT was prepared by diluting Neutral pH PEDOT NT5/CH03311/BH from Agfa with ultra-pure water at a 1:1 volume ratio and adding 0.2 mL/mL IPA to improve the wetting on ZnO nanoparticles. The solution was then filtered with a 5.0 μm Whatman Puradisc FP30 syringe filter.
The large area solar cell was manufactured by first etching away a stripe of ITO from a full-ITO covered substrate to form two separate electrode areas. The etching was performed using zinc dust and hydrochloric acid. Afterwards the substrates were cleaned thoroughly and a 100 nm of aluminum frame was evaporated to improve charge collection and specify the solar cell area. Deposition of the remaining layers was performed as explained above. The back electrode of LiF (1 nm) and Al (100 nm) was evaporated in such a way that it makes contact with the other half of the ITO. As a result, both holes and electrons can be collected from the ITO layers.
The large area stand-alone device was prepared by first sealing the solar cell inside a nitrogen atmosphere with a UV-curing resin. This resin was allowed to cure for 15 min under a 365 nm UV lamp. Subsequently, the titanium substrates with RuO2 catalysts were glued at the back side of the solar cell onto the glass cover using a two-component glue (Bison-Kombi Snel®). These catalyst layers were connected to the positive and negative poles of the solar cell with graphite conductive adhesive.
Characterization
The characterization of the solar cells was first done inside the glove-box with nitrogen atmosphere. A Keithley 2400 source-measurement unit was used to measure current density to voltage (J–V) characteristics of the devices. The illumination was carried out with ∼100 mW cm−2 white light from a tungsten-halogen lamp filtered by a Schott GG385 UV filter and a Hoya HMC 80A 72 mm daylight filter. No mismatch correction was performed. The measurements were performed inside a glove box with a nitrogen atmosphere. The triple junction devices were exposed to UV illumination (with a Spectroline EN-160L/F 365 nm lamp from Spectronics Corporation) for about 10 min to provide an ohmic contact between the ZnO and pH-neutral PEDOT layers before being measured. To prevent parasitical charge collection due to the high lateral conductivity of pH-neutral PEDOT, triple junction devices were measured with a mask slightly smaller than the actual device area, which is determined by the overlap of the ITO and Al electrodes.Solar to hydrogen conversion efficiencies were determined using a home-built setup. As the water splitting experiments took place in air, the solar cells were placed in a nitrogen filled box and connected to the catalysts through external cables. The solar cell was illuminated with white-light from a tungsten-halogen lamp (∼100 mW cm−2) filtered by a Schott GG385 UV filter and a Hoya HMC 80A 72 mm daylight filter. The solar cell was placed such that the generated short-circuit current in this setup corresponded to the short-circuit current measured inside the glove box, which corresponds to AM 1.5G power standards. A Keithley 2600 source-measurement unit was used for simultaneous measurement of current and voltage during water splitting.
The small-area (0.0676 cm2) solar cells provide low current densities on the catalyst surfaces (∼1.2 cm2), which makes it difficult to observe the hydrogen and oxygen bubbles during water splitting with by eye. Using a high resolution camera during water splitting traces of bubbles that are not apparent to eye were easily visualized. For the RuO2/RuO2 catalysts movements of tiny gas bubbles were observed, while in the case of Co3O4/NiMoZn catalysts, bubbles growing on the Co3O4 catalyst surface were seen. Water splitting experiments with the stand-alone large area device were made by placing the device inside a glass container filled with the electrolyte and taking into account for the AM 1.5G illumination conditions.
Apparatus
Electrodepositions and cyclic voltammetry were carried out with an Autolab PGSTAT 30 controlled by the GPES software package, equipped with an Aldrich® glass reference electrode (General purpose, reference, Ag/AgCl, 3 M KCl). Electrochemical impedance spectroscopy was done in the same setup with the Autolab PGSTAT 30 controlled by the FRA software package.The air plasma treatment was carried out in a Femto PCCE low pressure plasma system (Diener Electronic). A 270 W plasma was applied for 2 minutes.
Acknowledgements
We thank Bart van Overbeeke and the ICMS animation studio for providing the images shown in Fig. 4a and 5. This project was carried out within the research programme of BioSolar Cells, co-financed by the Dutch Ministry of Economic Affairs, Agriculture and Innovation. This work is part of the research programme of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organization for Scientific Research (NWO). The research leading to these results has further received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 339031 and the Mujulima project, Grant Agreement No. 604148. The research is part of the Solliance OPV programme and received funding from the Ministry of Education, Culture, and Science (NWO Gravity program 024.001.035).Notes and References
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boetcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogtuan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- G. H. Lin, M. Kapur, R. C. Kainthla and J. O. 'M. Bockris, Appl. Phys. Lett., 1989, 55, 386–387 CrossRef CAS.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, Int. J. Hydrogen Energy, 2001, 26, 653–659 CrossRef CAS.
- G. Peharz, F. Dimroth and U. Wittstadt, Int. J. Hydrogen Energy, 2007, 32, 3248–3252 CrossRef CAS.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- S.-H. Lia, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef PubMed.
- S. Esiner, H. van Eersel, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2013, 25, 2932–2936 CrossRef CAS PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, C. Laberty-Robert, S. Campidelli, R. de Bettignies, V. Artero, S. Palacin and B. Jousselme, Energy Environ. Sci., 2013, 6, 2706–2713 CAS.
- A. Aoki, M. Naruse and T. Abe, ChemPhysChem, 2013, 14, 2317–2320 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- K. Waki, R. A. Wong, H. S. Oktaviano, T. Fujio, T. Nagai, K. Kimoto and K. Yamada, Energy Environ. Sci., 2014, 7, 1950–1958 CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1775 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- W. Li, A. Furlan, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 5529–5532 CrossRef CAS PubMed.
- D. Galizzioli, F. Tantardini and S. Trasatti, J. Appl. Electrochem., 1974, 4, 57–67 CrossRef CAS.
- J. C. Cruz, V. Baglio, S. Siracusano, V. Antonucci, A. S. Aricò, R. Ornelas, L. Ortiz-Frade, G. Osorio-Monreal, S. M. Durón-Torres and L. G. Arriaga, Int. J. Electrochem. Sci., 2011, 6, 6607–6619 CAS.
- H. Ma, C. Liu, J. Liao, Y. Su, X. Xue and W. Xing, J. Mol. Catal. A: Chem., 2006, 247, 7–13 CrossRef CAS.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- D. Miousse and A. Lasia, J. New Mater. Electrochem. Syst., 1999, 2, 71–78 CAS.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Grätzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef CAS.
- E. Tsuji, A. Imanishi, K. Fukui and Y. Nakato, Electrochim. Acta, 2011, 56, 2009–2016 CrossRef CAS.
- S.-H. Lee, P. Liu, H. M. Cheong, C. E. Tracy and S. K. Deb, Solid State Ionics, 2003, 165, 217–221 CrossRef CAS.
- L. D. Burke and N. S. Naser, J. Appl. Electrochem., 2005, 35, 931–938 CrossRef CAS.
- A. J. Esswein, Y. Surendranath, S. Y. Reece and D. G. Nocera, Energy Environ. Sci., 2011, 4, 499–504 CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. Grzelczak, J. Zhang, J. Pfrommer, J. Hartmann, M. Driess, M. Antonietti and X. Wang, ACS Catal., 2013, 3, 383–388 CrossRef CAS.
- M. A. Domínguez-Crespo, E. Ramírez-Meneses, A. M. Torres-Huerta, V. Garibay-Febles and K. Philippot, Int. J. Hydrogen Energy, 2012, 37, 4798–4811 CrossRef.
- M. Gennero de Chialvo and A. Chialvo, J. Electroanal. Chem., 1998, 448, 87–93 CrossRef CAS.
- S. Baranton and C. Coutanceau, Appl. Catal., B, 2013, 136–137, 1–8 CrossRef CAS.
- F. Crnkovic, S. A. Machado and L. Avaca, Int. J. Hydrogen Energy, 2004, 29, 249–254 CrossRef CAS.
- T. J. Jacobsson, V. Fjallstrom, M. Sahlberg, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2013, 6, 3676–3683 CAS.
- M. J. N. Pourbaix, J. van Muylder and N. de Zoubov, Platinum Met. Rev., 1959, 3, 100–106 Search PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2015 DOI:10.1016/j.cattod.2015.08.014.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–7686 CrossRef CAS PubMed.
- C. R. Cox, J. Z. Lee, D. G. Nocera and T. Buonassisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14057–14061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta07325a |
| This journal is © The Royal Society of Chemistry 2015 |