
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-linked polymer-derived B/N co-doped carbon materials with selective capture of CO2†
Wuxue
Zhao
a,
Sheng
Han
b,
Xiaodong
Zhuang
*a,
Fan
Zhang
*a,
Yiyong
Mai
a and
Xinliang
Feng
ac
aSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: fan-zhang@sjtu.edu.cn; zhuang@sjtu.edu.cn
bSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Haiquan Road 100, 201418, Shanghai, P. R. China
cCenter for Advancing Electronics Dresden & Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 9th October 2015
Abstract
A new series of B, N-containing cross-linked polymers (PPs-BN) were achieved via Sonogashira cross coupling. These polymers exhibit very high carbon yields of around 70–80% even at 800 °C, which allow them to be efficiently converted to B/N co-doped porous carbons after pyrolysis at high temperature under an inert atmosphere. The materials have been fully characterized by Fourier transform infrared spectroscopy, X-ray diffraction, thermogravimetric analyses, nitrogen sorption measurements and X-ray photoelectron spectroscopy, revealing their high contents of boron and nitrogen up to ∼3.21% and ∼5.72%, respectively, as well as porous structures with the largest specific surface area of 291 m2 g−1. Their CO2 capacities reached 3.25 mmol g−1 at 273 K under 1 atm. In addition, the very high selectivity for CO2/CH4 with a ratio of more than 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 298 K renders them applicable for gas separation and purification.
:1 at 298 K renders them applicable for gas separation and purification.
Introduction
It is important to reduce greenhouse gases, predominately CO2, one of the chief contributors responsible for global warming, which has a negative impact on the environment and economy.1 Atmospheric CO2 concentrations have increased significantly in the last century and are continuously increasing at a faster rate; so its capture and storage remains a big challenge to scientists. The current technology being employed for post-combustion CO2 capture is based on an amine solution, such as aqueous mono-ethanolamine (MEA) solution (∼30 wt%), through chemically binding CO2, and thus suffering from undesirable issues, such as chemical decomposition, evaporation and high regeneration costs.2 The development of new materials and technologies with efficient uptake for CO2 is still an important challenge. To date, several promising adsorbents for CO2 capture have been explored covering a wide range of porous materials, such as metal organic frameworks (MOFs),3,4 inorganic mesoporous materials,5,6 COFs7 and activated carbons.8–10 Nevertheless, most of them still have the drawback of low stability under stark conditions, such as acidity, oxidant, humidity, and thus their practical applications are seriously limited.11,12 Porous carbon materials13 exhibit intrinsic advantages including excellent hydrophobicity and high thermal and chemical stabilities. In particular, it is possible to tailor such kinds of materials and improve their textural and chemical properties by pre-and post-synthetic methods, through using a diversity of precursors as carbon sources, covering zeolites, silica oxides, polymers, biomass and so on. However, relatively low conversion efficiency of the precursors, releasing of toxic and corrosive by-products, a time-consuming and costly procedure, etc. are always difficult to be avoided in the process of preparation.14 Recently, pyrolysis of porous polymer-based precursors represents an efficient approach to fabricate porous carbons with controlled porosities, and hierarchical architectures.15 It is worth noting that conjugated porous polymers (CMPs) possess shape persistence skeletons combined with rich molecular modification and thermal stability, and thus become one kind of promising precursor for the controlled preparation of carbon materials; for example, heteroatom-doped carbons exhibit remarkably enhanced CO2 capture capacity because of the formation of polarized structures arising from the interactions between heteroatoms and their adjacent carbon atoms or direct adsorption at the heteroatom sites.16,17 Typically, both contents and species of the heteroatoms doped in the resulting carbon materials play an important role in achieving good functional performance. Besides using a single dopant (e.g. nitrogen),18 dual or multi-heteroatom doping can dramatically promote the properties of the materials. Very recently, B/N co-doped carbon nanotubes, graphene, and carbon monoliths exhibited excellent activities in energy storage and conversion, probably ascribed to the synergistic effect of the heteroatoms.19,20 Until now, various precursors for B/N co-doping have been reported, for example, NH3 and boronic acid.21,22 However, using B, N-containing polymers as precursors for the preparation of B/N co-doped materials is still very limited.23,24 With this in mind, we made an effort to develop new types of B, N-containing monomers and corresponding polymers, and further explore these polymers as precursors for the controllable preparation of B/N co-doped carbon functional materials.In this paper, we report the design and synthesis of a series of new B, N-containing cross-linked polymers with well-defined chemical structures and high thermal stabilities. Further, they were efficiently converted into B/N co-doped porous carbons in high yields under pyrolysis treatment. Without any extra activation, the as-prepared materials exhibit high contents of boron and nitrogen, moderate surface area and hierarchical porous structures. They show excellent CO2 uptakes with respect to their relatively low surface areas, and high selectivity of CO2/CH4.
Experimental
Materials
All of the starting materials were purchased from Aladdin and Aldrich. N,N-Dimethylformamide (DMF) and triethylamine (Et3N) were refluxed with calcium hydroxide before use. 1,3,5-Triethynylbenzene,25 tris[p-ethynylphenyl]amine and tetrakis(4-ethynylphenyl)methane26 were synthesized according to the reported procedures. The key starting monomer (1) was synthesized according to the reported procedures.27Synthesis of monomer 2
4,4′,5,5′-Tetraamino-1,1′-binaphthyl (500 mg, 1.59 mmol) was dissolved in toluene (70 mL), and then, to this solution, 4-bromo-phenylboronic acid (798 mg, 3.98 mmol) was added. After refluxing for 2 d, the temperature returned to RT, and the residue was filtered and washed with petroleum ether. After vacuum drying for 6 h at 60 °C, the final product was collected as brown powder (750 mg, yield: 73%).Synthesis of polymers
The preparation of porous polymers containing boron and nitrogen (PPs-BN) was carried out under a nitrogen atmosphere at 100 °C for 72 h. A representative experimental procedure for polymer network 1 is given as follows:1,3,5-Triethynylbenzene (63.0 mg, 0.42 mmol) and monomer 2 (180.0 mg, 0.28 mmol) were dissolved in a mixture of anhydrous DMF (5 mL) and Et3N (5 mL). The mixture was freeze–thawed three times. Then, tetrakis(triphenylphosphine)palladium (20.0 mg), and copper(I) iodide (11 mg) were added and the mixture was stirred for 72 h at 100 °C under a nitrogen atmosphere. The mixture was cooled to room temperature, and the precipitate was filtered and washed with chloroform, water, methanol, and acetone several times to remove any un-reacted monomers and catalyst residues. Further purification of the polymer was carried out by Soxhlet extraction with THF for 24 h. After vacuum drying for 24 h at 70 °C, the B/N containing porous polymer product (denoted as PPs-BN-1) was obtained as yellow-green powder (163.0 mg, yield: 64%).
Tris(4-ethynylphenyl)amine (134.0 mg, 0.42 mmol), monomer 2 (180 g, 0.28 mmol), tetrakis(triphenylphosphine)palladium (20 mg), copper(I) iodide (11 mg), DMF (5 mL) and Et3N (5 mL) were used in the same procedure. Polymer PPs-BN-2 was obtained as yellow-green powder (200.0 mg, yield: 72%).
Tetrakis(4-ethynylphenyl)methane (87 mg, 0.21 mol), monomer 2 (180 mg, 0.28 mmol), tetrakis(triphenylphosphine)palladium (20 mg), copper(I) iodide (11 mg), DMF (5 mL) and Et3N (5 mL) were used in the same procedure. Polymer PPs-BN-3 was obtained as yellow-green powder (196.0 mg, yield: 73%).
Pyrolysis PPs-BN-i (i = 1, 2, 3)
The as-prepared boron and nitrogen contained porous polymers PPs-BN-i (i = 1, 2, 3) were directly pyrolyzed at 800 °C with a heating rate of 5 °C min−1 under a nitrogen atmosphere in a quartz boat for 2 h. The obtained B/N co-doped porous carbons are denoted as PPs-BN-i-800 (i = 1, 2, 3).Characterization
Fourier transform infrared (FTIR) spectra were recorded with a Spectrum 100 spectrometer (Perkin Elmer, Spectrum 100). X-ray powder diffraction patterns were recorded in transmission geometry using a Rigaku X-ray diffractometer with Cu-Kα irradiation (λ = 0.15406 nm) at 40 kV, 20 mA over the 2θ range from 5° to 60°. Thermogravimetric analysis (TGA) was carried out by using a TAQ5000IR with a heating rate of 20 °C min−1 under N2 flow. The morphology of the samples was investigated using a scanning electron microscope (SEM, FEI, Sirion-200) and a transmission electron microscope (TEM, JEOL, JEM-2010). N2 sorption isotherm measurements were performed on a Micromeritics ASAP 2010 surface area and pore size analyzer at 77 K. Prior to the measurement, the samples were degassed in a vacuum at 200 °C for 10 h. Ultra-high-purity grade CO2 and CH4 were used for the adsorption measurements. CO2 and CH4 adsorption measurements were done in an ice-water bath (273 K) under 1 bar and at room temperature (298 K) under 1 bar. X-ray photoelectron spectroscopy (XPS) experiments were carried out on an AXIS Ultra DLD system from Kratos with Al Kα radiation as the X-ray source for radiation.Results and discussion
Synthesis and characterization
Compound 2 as the key building block was first synthesized (yield: 73%) by condensation of 4,4′,5,5′-tetraamino-1,1′-binaphthyl 1 with 4-bromo-phenylboronic acid at 120 °C for 48 h. Upon transition metal-catalyzed Sonogashira–Hagihara cross-coupling reactions, compound 2 was further copolymerized with 1,3,5-triethynylbenzene 3, tris(p-ethynylphenyl)amine 4 and tetrakis(4-ethynylphenyl)methane 5, respectively, to form cross-linked polymers PPs-BN-i (i = 1, 2, 3) in good yields (63–73%) (Scheme 1). The as-prepared polymer samples were insoluble in common organic solvents, e.g. toluene, tetrahydrofuran, dimethylformamide, dichloromethane, methanol, acetone, etc. The structures of polymer networks are shown in Fig. 1.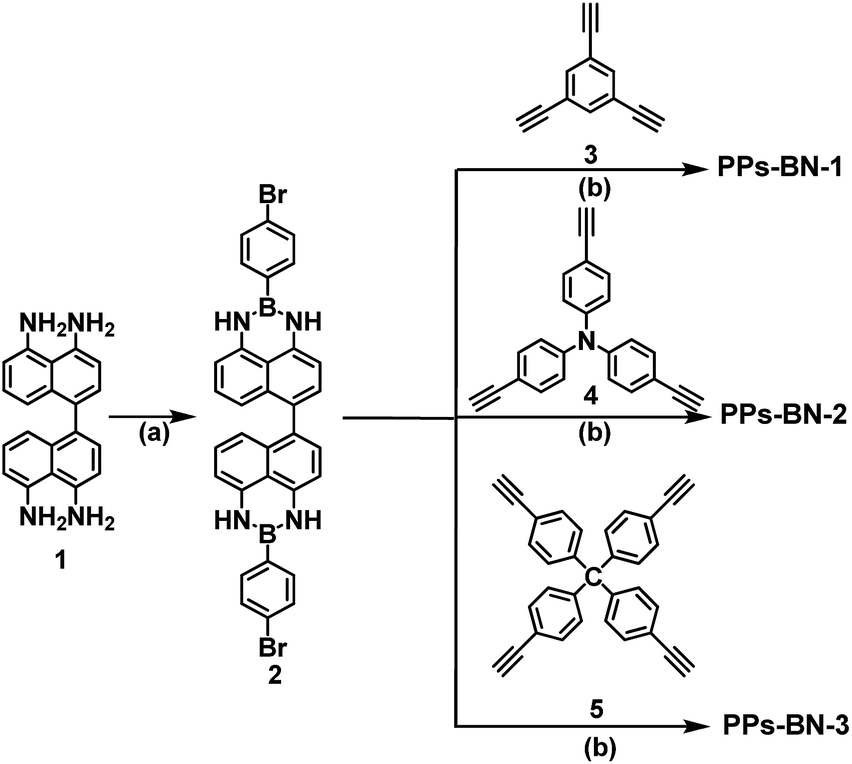 | ||
| Scheme 1 Synthetic procedure for the porous polymers PPs-BN-i (i = 1, 2, 3). (a) 4-Bromophenylboronic acid, 120 °C, 48 h, toluene; (b) Et3N, Pd(PPh3)4, CuI, 100 °C, 72 h, DMF. | ||
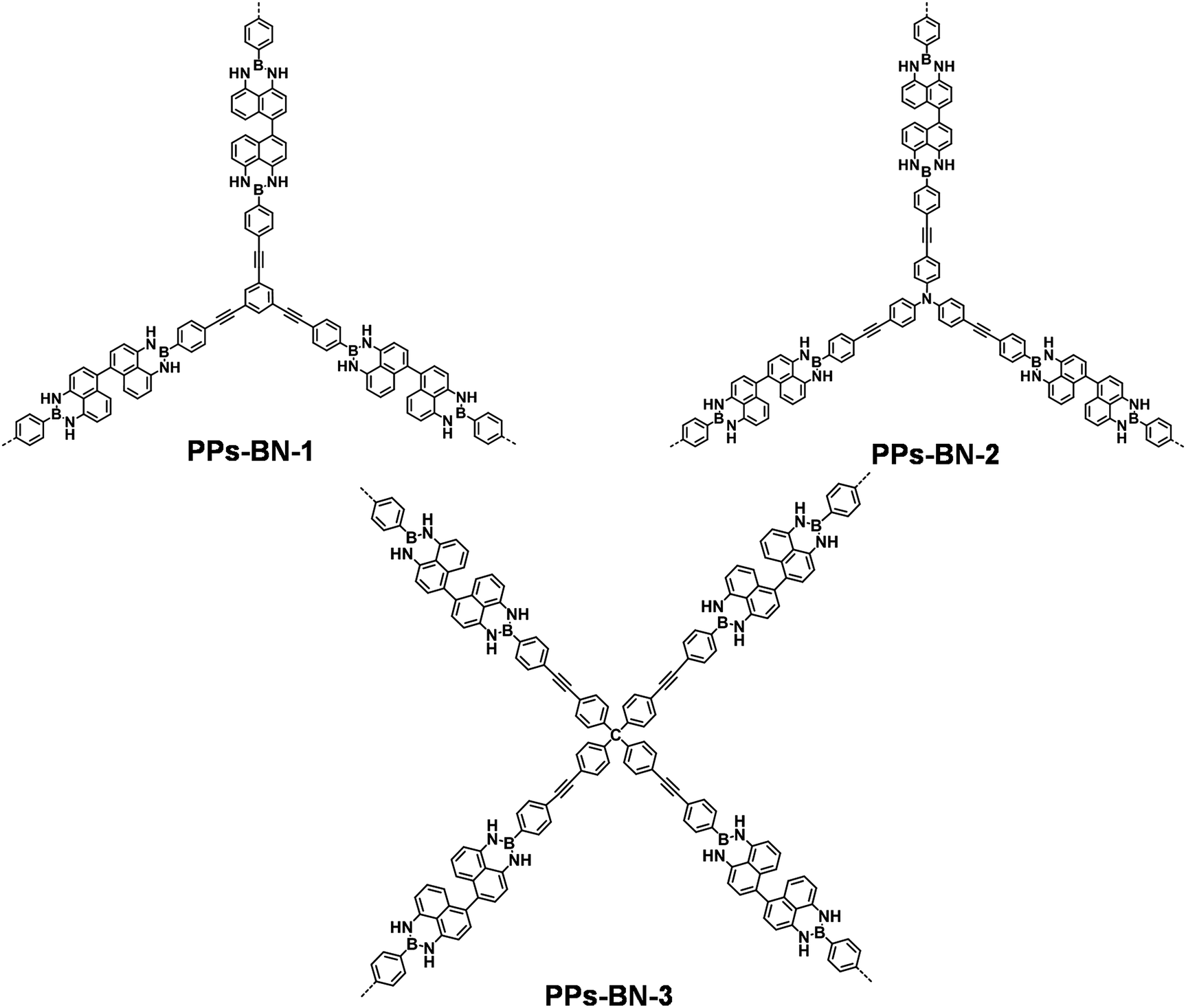 | ||
| Fig. 1 Representative molecular structures of polymer networks PPs-BN-i (i = 1, 2, 3). | ||
The chemical structures of the polymers were confirmed by Fourier transform infrared (FTIR) spectroscopy. The FTIR spectra of PPs-BN-i (Fig. 2a) reveal a peak at 2110 cm−1 with a very low intensity corresponding to –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretching. The peaks at around 3430 cm−1 and 1640 cm−1 are attributed to the stretching and bending vibrations of the N–H (free N–H), respectively. The bands at 2870–3050 cm−1 are attributed to aromatic C–H stretching.28,29 The chemical structures of the polymers were further supported by the solid-state 13C cross-polarization/magic angle spinning (CP/MAS) NMR spectra (Fig. 3); the signal located at 90.0 ppm can be assigned to carbon atoms of the quaternary alkynes (Car–CC–Car). The N-substituted phenyl carbon (N–Car) of PPs-BN-2 is at 147.1 ppm. The quaternary carbon in PPs-BN-3 is observed at 23.0 ppm. The overlapping signals from 121 to 140 ppm belong to the other aromatic carbon in the backbone of the polymer samples. The morphologies of the polymers were observed by using a scanning electron microscope (SEM) and a transmission electron microscope (TEM) (Fig. S1 and S2†). The thermal properties of the as-prepared cross-linked polymers were analyzed by thermogravimetric analyses (TGAs), revealing the very high carbon yields of 71%, 81% and 82% even at 800 °C under a nitrogen atmosphere, for PPs-BN-1, PPs-BN-2 and PPs-BN-3, respectively (Fig. 2b). Such unusual thermal stabilities of these cross-linked polymers could be assigned to their cross-linked aromatic frameworks.
C– stretching. The peaks at around 3430 cm−1 and 1640 cm−1 are attributed to the stretching and bending vibrations of the N–H (free N–H), respectively. The bands at 2870–3050 cm−1 are attributed to aromatic C–H stretching.28,29 The chemical structures of the polymers were further supported by the solid-state 13C cross-polarization/magic angle spinning (CP/MAS) NMR spectra (Fig. 3); the signal located at 90.0 ppm can be assigned to carbon atoms of the quaternary alkynes (Car–CC–Car). The N-substituted phenyl carbon (N–Car) of PPs-BN-2 is at 147.1 ppm. The quaternary carbon in PPs-BN-3 is observed at 23.0 ppm. The overlapping signals from 121 to 140 ppm belong to the other aromatic carbon in the backbone of the polymer samples. The morphologies of the polymers were observed by using a scanning electron microscope (SEM) and a transmission electron microscope (TEM) (Fig. S1 and S2†). The thermal properties of the as-prepared cross-linked polymers were analyzed by thermogravimetric analyses (TGAs), revealing the very high carbon yields of 71%, 81% and 82% even at 800 °C under a nitrogen atmosphere, for PPs-BN-1, PPs-BN-2 and PPs-BN-3, respectively (Fig. 2b). Such unusual thermal stabilities of these cross-linked polymers could be assigned to their cross-linked aromatic frameworks.
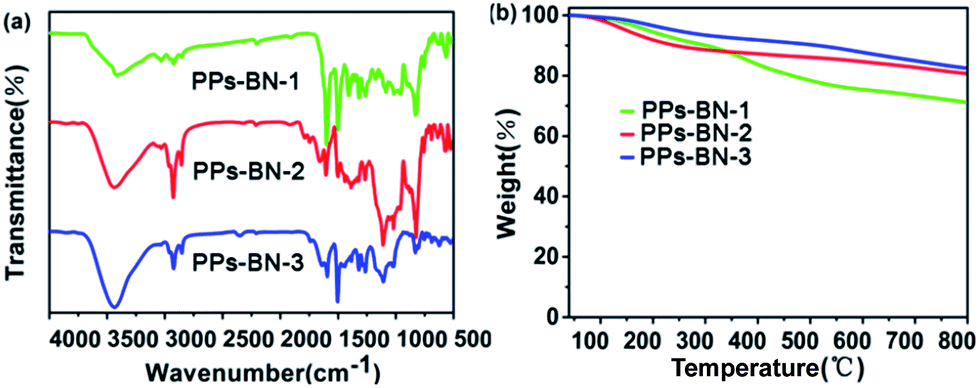 | ||
| Fig. 2 (a) FT-IR spectra of PPs-BN-i. (b) TGA patterns of PPs-BN-i (i = 1, 2, 3). | ||
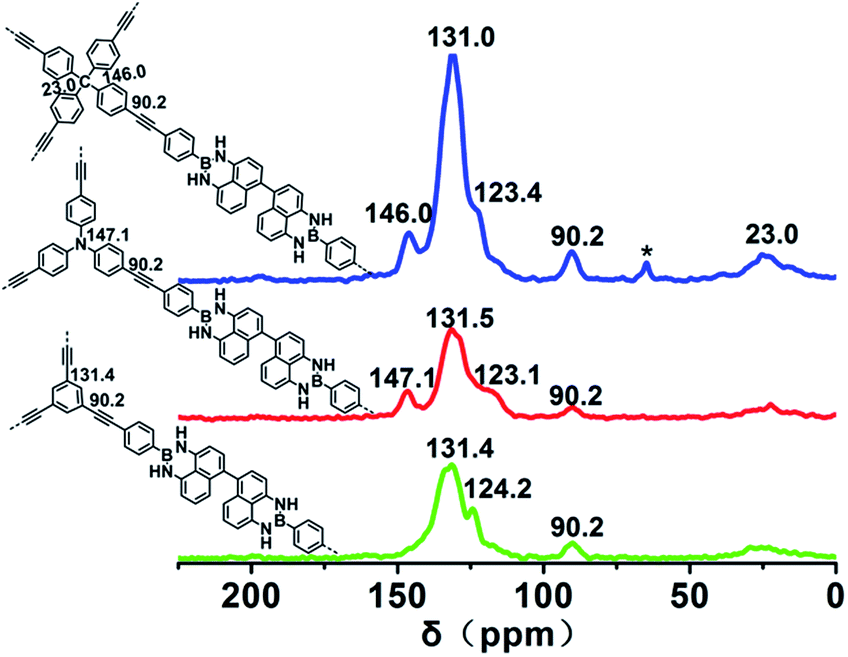 | ||
| Fig. 3 Solid-state 13C CP/MAS NMR spectra of PPs-BN-1 (black), PPs-BN-2 (red) and PPs-BN-3 (blue). | ||
The as-prepared polymers were also characterized by X-ray diffraction (XRD) (Fig. 4a). The two peaks located at 2θ = 23° and 43° are typically attributed to the graphitic (002) and (101) diffractions, respectively.30,31 A series of B/N co-doped porous carbon materials PPs-BN-i-800 (i = 1, 2, 3) were achieved by the direct pyrolysis of the corresponding cross-linked polymers PPs-BN-i (i = 1, 2, 3) at 800 °C for 2 h under a N2 atmosphere. The XRD analyses of the resulting carbon materials showed an obvious broad peak at around 43°, which indicates that the porous carbon materials exhibit a certain degree of graphitization after pyrolysis at 800 °C, suggesting the formation of a large amount of parallel-layered graphite structures32 that are significantly different from those of their cross-linked polymer precursors (Fig. 4b). According to the analysis of Raman spectra of carbon materials (Fig. S3†), two prominent peaks at 1338.4 and 1593.5 cm−1 were observed, which correspond to the D and G bands, respectively. The ID/IG ratio of PPs-BN-i-800 (i = 1, 2, 3) (0.84–0.85) indicates a remarkable degree of graphitization of carbonized samples at 800 °C. The morphology and microstructure of these B/N co-doped porous carbon materials were investigated by transmission electron microscopy (TEM) (Fig. S4†). The alternating areas of light and dark contrast in the TEM images disclosed their disordered porous structures.
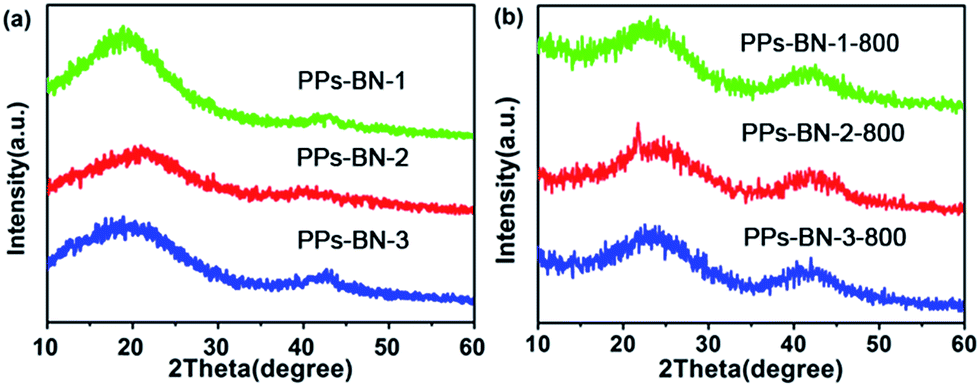 | ||
| Fig. 4 (a) XRD patterns of PPs-BN-i. (b) XRD patterns of PPs-BN-i (i = 1, 2, 3). | ||
Their chemical characteristics were further characterized by X-ray photoelectron spectroscopy (XPS) (Fig. 5). The spectra can be deconvoluted into five peaks at 398.2 eV, ∼398.8 eV, 399.6 eV, ∼400.8 eV and ∼401.6 eV, which were attributed to the presence of N–B bonds, pyridinic-N, N–C bonds, pyrrolic-N and graphitic-N,33,34 respectively. The B 1s spectra can be deconvoluted into three peaks with the binding energies of ∼190.2 eV, ∼191.5 eV and ∼192.6 eV, arising from the B–C bonds, B–N bonds and BC2O or BCO2, respectively,35 indicating that B and N atoms have been efficiently co-doped in the materials.
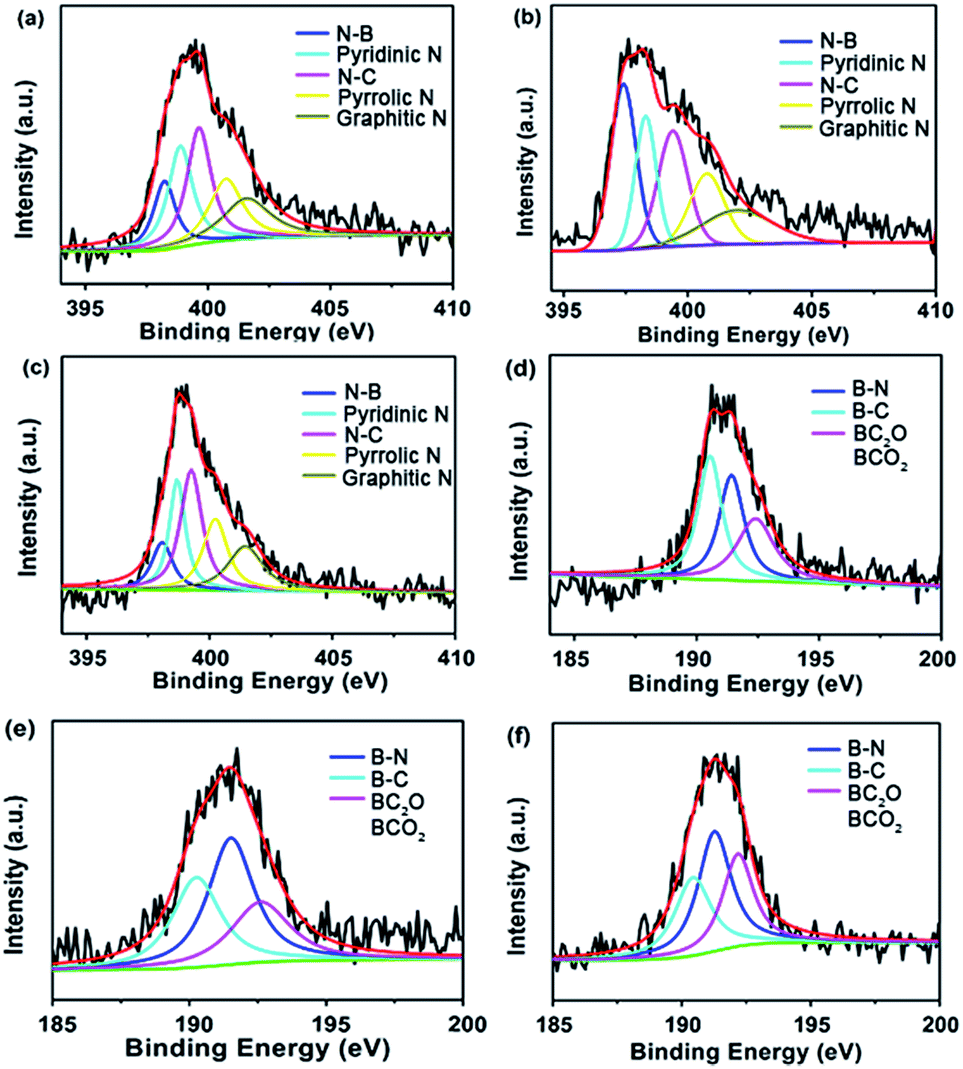 | ||
| Fig. 5 Typical XPS spectra of (a) N 1s and (d) B 1s for PPs-BN-1-800. (b) N 1s and (e) B 1s for PPs-BN-2-800. (c) N 1s and (f) B 1s for PPs-BN-3-800. | ||
The initial porous structures of the PPs-BN-1, PPs-BN-2 and PPs-BN-3 were analysed by using the nitrogen sorption measurements at 77 K, which showed low porosities of 16 m2 g−1, 32 m2 g−1 and 51 m2 g−1 for PPs-BN-1, PPs-BN-2 and PPs-BN-3, respectively (Fig. 6a), probably due to the lack of rigidity of the frameworks with the free rotation of the naphthyl moieties around the carbon–carbon bond. The pore size distribution curves of PPs-BN-i (i = 1, 2, 3) are shown in Fig. 6b. Interestingly, PPs-BN-3 showed a broad pore size distribution covering micropores (with the peak value at about 1.5 nm) and mesopores from 2 nm to ∼30 nm. PPs-BN-2 offered relatively narrow pore size distribution, mainly appearing at 1.2 nm in the micropore region and 2.5 nm and 14 nm in the mesopore region. For PPs-BN-1, only micropores (less than 2 nm) and mesopores with a peak at 2 nm can be observed. These results verified that the porous structures of these cross-linked polymers are highly dependent on their frameworks, which can be tuned by using different building blocks.
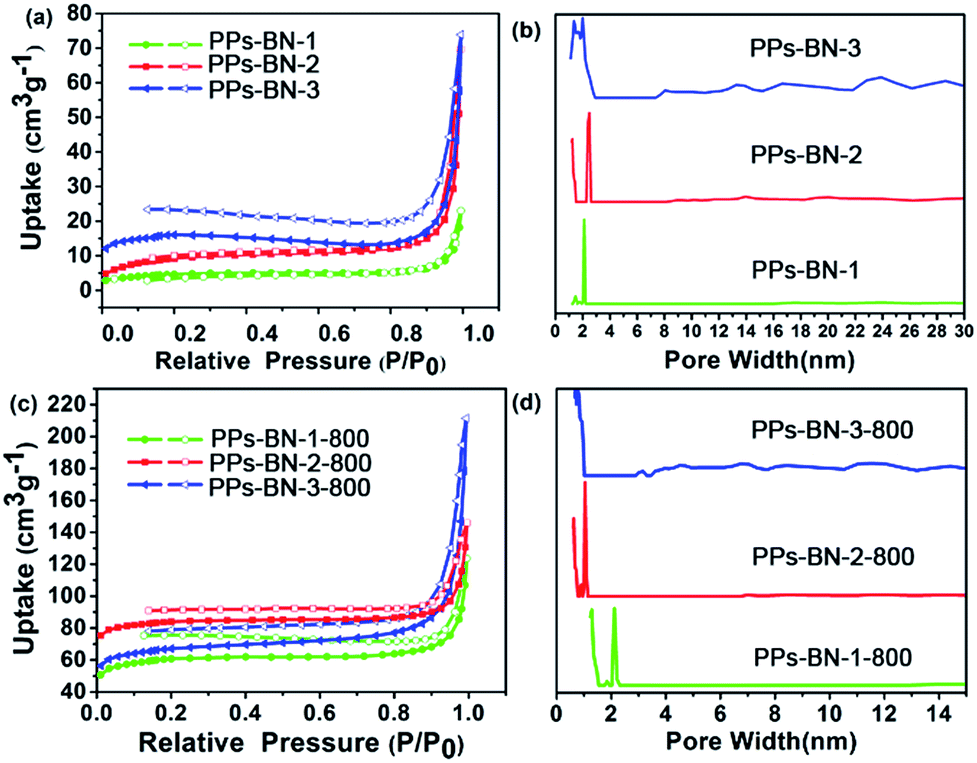 | ||
| Fig. 6 Nitrogen adsorption (filled) and desorption (empty) isotherms of (a) PPs-BN-i and (c) PPs-BN-i-800 measured at 77 K. Pore size distribution curves of (b) PPs-BN-i and (d) PPs-BN-i-800 as calculated by NL-DFT. | ||
After pyrolysis treatment, the porous structures of porous carbon materials PPs-BN-i-800 (i = 1, 2, 3) were further investigated by nitrogen sorption measurements at 77 K (Fig. 6c). The textural parameters of the PPs-BN-i-800 (i = 1, 2, 3) are listed in Table 1. The Brunauer–Emmett–Teller (BET) surface area of 215, 291 and 268 m2 g−1 for PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800, respectively is significantly increased as compared with those of their polymer precursors PPs-BN-i (i = 1, 2, 3) (from 16 to 51 m2 g−1). The PPs-BN-2-800 shows the largest total pore volumes of 0.33 cm3 g−1 followed by PPs-BN-1-800 (0.26 cm3 g−1) and PPs-BN-3-800 (0.23 cm3 g−1). The pore size distribution curves of PPs-BN-i-800 (i = 1, 2, 3) are shown in Fig. 6d, calculated using non-local density functional theory (NL-DFT). Notably, their corresponding carbonized materials PPs-BN-i-800 (i = 1, 2, 3) showed almost sustained pore size distributions. A broad distribution from the micropores with a peak at 1.2 nm to the mesopores at about 2 nm for PPs-BN-3-800 can be observed. And for PPs-BN-2-800, the pore size distribution appears at 1.2 nm in the micropore region and 2.1 nm in the mesopore region. Exceptionally, besides a mesopore peak at ∼2.0 nm, a remarkably intensive micropore peak at around 1.30 nm for PPs-BN-1-800 can be observed, which is different from that of PPs-BN-1. These results suggested that the scaffold of such kinds of cross-linked polymer precursors could provide dominant contribution to the formation of the structures of their corresponding carbon materials in the process of pyrolysis treatment. Furthermore, the use of the CO2 isotherms at 273 K proved to be very beneficial in analyzing micropore distribution of carbon materials more accurately (Fig. S5†). The pore size distribution was mainly centered at 1.18 nm for PPs-BN-1-800, 1.15 nm for PPs-BN-2-800 and 1.16 nm for PPs-BN-3-800. In the case of PPs-BN-2-800 and PPs-BN-3-800, the pore size distributions are similar to those of their corresponding polymer precursors, probably due to the high aromatic contents in the scaffolds of these precursors, and highly in line with their excellent thermal stabilities. Compared with the corresponding cross-linked polymer precursors, the remarkably increased surface areas in the carbonized materials are likely to be attributed to the fact that the high temperature would be helpful to remove the organic small molecules or oligomers, formed in the polymerization.36 The carbon samples do not show classic type I isotherms, and instead offer isomers with steady linear increases at high relative pressure and distinct hysteresis at low pressure between the adsorption and desorption cycles. This is a common phenomenon, arising from an unexpected gate-opening effect at 77 K during the N2 adsorption of porous polymers, which can be explained as the kinetically limited diffusion of N2 molecules through the dynamically open-and-shut gates connecting free volume elements.37 Such a gate-opening phenomenon indicates that polymeric frameworks are not fully condensed into carbons.38 The BET specific surface area values of the as-prepared carbon materials calculated with the relative pressure (P/P0) ranging from 0.01 to 0.1 are consistent with those achieved by T-plot methods (Fig. S6†).
| Sample | S BET [m2 g−1] | V tot [cm3 g−1] | D av [nm] | Bd [wt%] | Nd [wt%] | CO2 uptake [mmol g−1] | CH4 uptake [mmol g−1] | Selectivitye | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 273 K | 298 K | 273 K | 298 K | CO2/CH4 | ||||||
| a Surface area calculated from the N2 adsorption isotherm using the BET method. b Total pore volume at p/p0 = 0.99. c The pore size was derived using the non-local density functional theory. d Derived from XPS analysis. e At 298 K, 1 bar. | ||||||||||
| PPs-BN-1-800 | 215 | 0.26 | 1.81 | 3.34 | 5.63 | 3.23 | 2.31 | 0.98 | 0.46 | 5.1 |
| PPs-BN-2-800 | 291 | 0.33 | 1.84 | 3.21 | 5.72 | 3.25 | 2.40 | 1.10 | 0.59 | 4.1 |
| PPs-BN-3-800 | 268 | 0.23 | 1.72 | 2.91 | 4.89 | 3.11 | 2.34 | 1.15 | 0.54 | 4.3 |
We further studied the capability of the as-made porous carbons for the adsorption of CO2. The CO2 adsorption isotherms showed that PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800 enable capturing 3.23 mmol g−1 (71.8 cm3 g−1), 3.25 mmol g−1 (72 cm3 g−1) and 3.11 mmol g−1 (69 cm3 g−1) at 273 K, 1 bar, respectively (Fig. 8). At a higher temperature of 298 K, the CO2 uptakes of these materials are somehow declined to 2.31 mmol g−1, 2.40 mmol g−1 and 2.34 mmol g−1 for PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800, respectively. No hysteresis on adsorption and desorption isotherms was observed which demonstrated the reversible process of CO2 storage. At 273 K, PPs-BN-2-800 shows a slightly higher CO2 uptake than that of PPs-BN-1-800 (Fig. 7). The nitrogen and boron contents of the two materials are almost similar, but the former material has a larger surface area (268 m2 g−1) than that of 215 m2 g−1 for the latter one (Table 1), and thus positively contributed to the CO2 capturing. While PPs-BN-1-800 with a lower surface area (215 m2 g−1) exhibited a better CO2 uptake than that of PPs-BN-3-800 (268 m2 g−1) under the same conditions, which was probably caused by higher B and N contents in the former case (N: 5.63% and B: 3.34% vs. N: 4.89% and B: 2.91% for PPs-BN-3-800) (Table 1). Eventually, the specific surface area and heteroatom doping would synergistically affect the CO2 adsorption of such kinds of carbon materials. As a further indication (Table S1†), the specific BET surface area (291 m2 g−1) of PPs-BN-2-800 is much lower than that of 490 m2 g−1 for porous carbon MFB-600 derived from melamine, whereas the CO2 loading capacity of the former material (2.4 mmol g−1) is slightly higher than that (2.25 mmol g−1) of the latter one at 1 bar and 298 K.39 The reason may be due to the higher nitrogen content of PPs-BN-2-800 (5.72%) as compared with that of MFB-600 (1.74%). On the other hand, the boron doping can also play a positive role in the CO2 uptake of PPs-BN-3-800, which likely allows these materials even with a low surface area of 268 m2 g−1 to offer a higher CO2 uptake of 3.11 mmol g−1 than that of 2.92 mmol g−1 for the porous carbon FCDTPA-700 (BET = 417 m2 g−1) under the same conditions (1.0 bar and 273 K).40 In addition, the type of nitrogen site can also affect the CO2 adsorption. It has been reported previously that pyrrolic nitrogen may serve as an anchor for CO2 capture, based on the acid–base interactions.41,42 The amount of pyrrolic nitrogen is 23.2, 20.1 and 22.5% for PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800 calculated based on XPS (Table S2†). Notably, these CO2 adsorption isotherms are gradually increasing along with increasing pressure, suggesting further high adsorption capacity for CO2 at elevated pressure. Additionally, the isosteric heats of PPs-BN-i-800 for CO2 adsorption (Fig. 7d) were estimated based on adsorption data collected at different temperatures (273 and 298 K), which are among the highest values for the carbon materials used in CO2 uptake.43 These isosteric heat values increase in the sequence of PPs-BN-2-800 (39.0 kJ mol−1) > PPs-BN-1-800 (37.5 kJ mol−1) > PPs-BN-3-800 (34.8 kJ mol−1), consistent with their capabilities for capturing CO2. Thanks to the relatively low surface areas, the loading amount of CO2 by the as-prepared B/N co-doped porous materials can be taken into account at a higher level, comparable to a lot of heteroatom-doped porous materials with much higher surface areas.
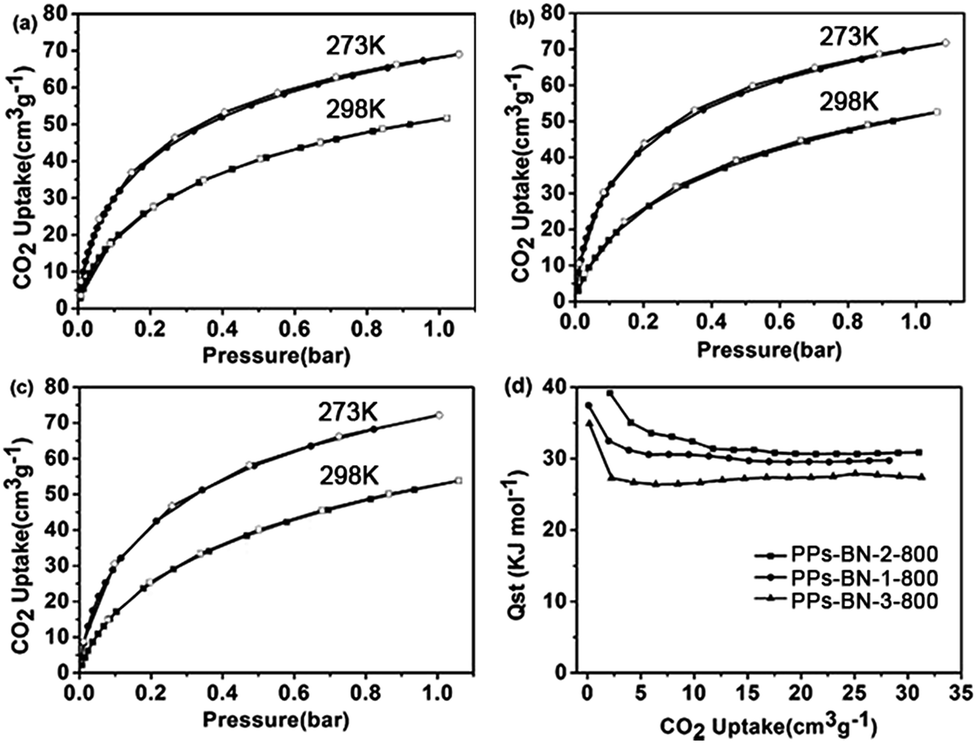 | ||
| Fig. 7 CO2 adsorption isotherms of (a) PPs-BN-1-800, (b) PPs-BN-2-800 and (c) PPs-BN-3-800 at 273 K (red) and at 298 K (blue). (d) Isosteric heat of PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800. | ||
Such a striking phenomenon could be attributed to the rich incorporation of B and N elements into the materials. The different electronegativities among the three components B, N and C in the scaffolds enable the formation of dipole moments in the scaffold of the materials, which are beneficial to absorbing quadrupolar CO2 through the dipole–dipole interactions.44,45 On the other hand, the B/N co-doping into the materials may form defect sites after carbonization, which would enhance the interaction with the CO2 molecule.46 The coverage-dependent adsorption enthalpies of PPs-BN-i-800 for CO2 were calculated based on the virial method, which is a well-established and reliable methodology fitted from their adsorption isotherms at 273 K and 298 K (Fig. S8–S10†).
In addition to the gas absorption, gas selectivity is also an important property of the porous materials applicable for practice. In particular, the separation of CO2/CH4 is one of the very crucial processes in natural gas upgrading since the contamination of CH4 with CO2 will reduce the energy content of natural gas, and cause equipment corrosion.47,48 The adsorption isotherms of PPs-BN-1-800, PPs-BN-2-800 and PPs-BN-3-800 disclosed their CH4 uptakes of 0.98 mmol g−1, 1.10 mmol g−1 and 1.15 mmol g−1 at 273 K, respectively, and somehow decreased to 0.46 mmol g−1, 0.59 mmol g−1, and 0.54 mmol g−1 at 298 K, respectively (Fig. 8). Obviously, the CO2 sorption capacities of the as-prepared materials are much higher than those of CH4. Among them, the PPs-BN-1-800 showed the highest selectivity of CO2/CH4 in a ratio of 5.1/1 at 298 K at 1 bar, which was comparable to those of the reported porous materials with the best performance in this aspect, and much higher than those of activated carbon materials, such as PAF-1-450 (ref. 49) and C168.50 Moreover, some previous N-doped porous carbon materials, for example, reported by Wang et al.,51–53 exhibit much higher surface areas under KOH activation, thus improving CO2 uptake with high selectivity. These results undoubtedly provided a valuable strategy for further optimizing the as-prepared materials in the next work.
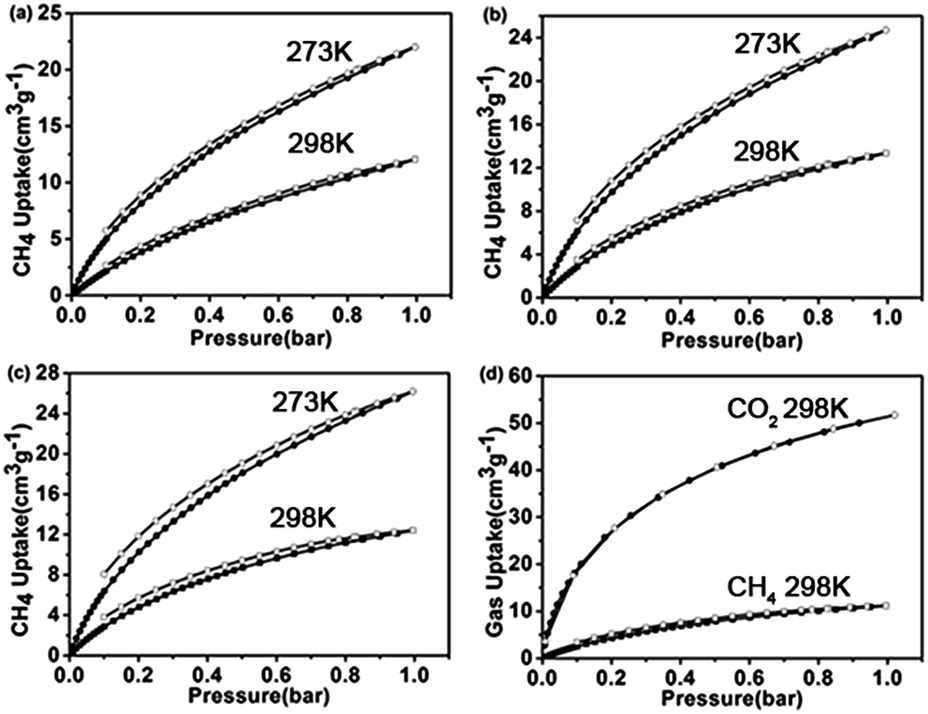 | ||
| Fig. 8 CH4 adsorption isotherms of (a) PPs-BN-1-800, (b) PPs-BN-2-800 and (c) PPs-BN-3-800 at 273 K (red) and at 298 K (blue). (d) Gas selectivity isotherms of CH4 and CO2 at 298 K for PPs-BN-1-800. | ||
Conclusion
In conclusion, we have synthesized a series of new B, N-containing cross-linked polymers (PPs-BN) through transition metal-catalyzed cross-coupling on the basis of a new B, N-containing monomer as the key building block. These cross-linked polymers show outstanding thermal stabilities, allowing them to be efficiently converted into corresponding carbonized PPs-BN-i-800 under pyrolysis treatment. These resulting porous carbon materials with high nitrogen and boron contents possess a moderate specific surface area, and exhibit good capacity for CO2 uptake up to 3.25 mmol g−1 at 273 K under 1 bar, much higher than those of the porous materials with similar specific surface areas, likely due to the B/N co-doping effect. These materials showed a good selectivity for CO2/CH4 with a ratio up to 5.1/1 at 298 K, thus rendering such kinds of materials potentially applicable for the purification of the gases. Improvement of the intrinsic structural characteristics of such kinds of porous materials, including their surface areas, B, N co-doping contents, etc., is in progress. Meanwhile, their use as electrode materials for energy conversion and storage devices, such as lithium batteries, supercapacitors and fuel cells, is also being explored.Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 Program: 2013CBA01602 and 2012CB933404), the Natural Science Foundation of China (21174083, 51403126 and 21102091), and the Shanghai Committee of Science and Technology (15JC1490500). We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for providing some measurements.Notes and references
- C. Song, Catal. Today, 2006, 115, 2 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS PubMed.
- R. V. Siriwardane, M.-S. Shen and E. P. Fisher, Energy Fuels, 2003, 17, 571 CrossRef CAS.
- J. Zhou, W. Li, Z. Zhang, W. Xing and S. Zhuo, RSC Adv., 2012, 2, 161 RSC.
- Z. Xiang, X. Zhou, C. Zhou, S. Zhong, X. He, C. Qin and D. Cao, J. Mater. Chem., 2012, 22, 22663 RSC.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853 CrossRef CAS PubMed.
- G.-P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schüth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781 CrossRef CAS PubMed.
- T. K. Maji, G. Mostafa, H.-C. Chang and S. Kitagawa, Chem. Commun., 2005, 2436 RSC.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS PubMed.
- R. Chatti, A. K. Bansiwal, J. A. Thote, V. Kumar, P. Jadhav, S. K. Lokhande, R. B. Biniwale, N. K. Labhsetwar and S. S. Rayalu, Microporous Mesoporous Mater., 2009, 121, 84 CrossRef CAS PubMed.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574 CAS.
- D. Ko, H. A. Patel and C. T. Yavuz, Chem. Commun., 2015, 51, 2915 RSC.
- Y. Xia, Y. Zhu and Y. Tang, Carbon, 2012, 50, 5543 CrossRef CAS PubMed.
- J. Wang, I. Senkovska, M. Oschatz, M. R. Lohe, L. Borchardt, A. Heerwig, Q. Liu and S. Kaskel, J. Mater. Chem. A, 2013, 1, 10951 CAS.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110 CrossRef CAS PubMed.
- J. Jin, F. Pan, L. Jiang, X. Fu, A. Liang, Z. Wei, J. Zhang and G. Sun, ACS Nano, 2014, 8, 3313 CrossRef CAS PubMed.
- Y. Xue, D. Yu, L. Dai, R. Wang, D. Li, A. Roy, F. Lu, H. Chen, Y. Liu and J. Qu, Phys. Chem. Chem. Phys., 2013, 15, 12220 RSC.
- X. Q. Wang and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5106 CrossRef PubMed.
- P. F. Fulvio, J. S. Lee, R. T. Mayes, X. Q. Wang, S. M. Mahurin and S. Dai, Phys. Chem. Chem. Phys., 2011, 13, 13486 RSC.
- S. H. Lim, Y. Su and S. M. Cohen, Angew. Chem., Int. Ed., 2012, 51, 5106 CrossRef CAS PubMed.
- J.-X. Jiang, A. Trewin, F. Su, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658 CrossRef CAS.
- S. Wiesner, A. Ziesak, M. Reinmuth, P. Walter, E. Kaifer, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2013, 163 CrossRef CAS PubMed.
- W. Zhao, X. Zhuang, D. Wu, F. Zhang, D. Gehrig, F. Laquai and X. Feng, J. Mater. Chem. A, 2013, 1, 13878 CAS.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511 CrossRef CAS.
- Y. Xia and R. Mokaya, Chem. Mater., 2005, 17, 1553 CrossRef CAS.
- P. Pachfule, V. M. Dhavale, S. Kandambeth, S. Kurungot and R. Banerjee, Chem.–Eur. J., 2013, 19, 974 CrossRef CAS PubMed.
- C. N. R. Rao, K. Biswas, K. S. Subrahmanyam and A. Govindaraj, J. Mater. Chem., 2009, 19, 2457 RSC.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS PubMed.
- C.-K. Chang, S. Kataria, C.-C. Kuo, A. Ganguly, B.-Y. Wang, J.-Y. Hwang, K.-J. Huang, W.-H. Yang, S.-B. Wang, C.-H. Chuang, M. Chen, C.-I. Huang, W.-F. Pong, K.-J. Song, S.-J. Chang, J.-H. Guo, Y. Tai, M. Tsujimoto, S. Isoda, C.-W. Chen, L.-C. Chen and K.-H. Chen, ACS Nano, 2013, 7, 1333 CrossRef CAS PubMed.
- X. Zhang, C. Li, Y. Hu, R. Liu, L. He and S. Fang, Polym. Int., 2014, 63, 2030 CrossRef CAS PubMed.
- J. Weber, N. Du and M. D. Guiver, Macromolecules, 2011, 44, 1763 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169 CrossRef CAS.
- C. Pevida, T. C. Drage and C. E. Snape, Carbon, 2008, 46, 1464 CrossRef CAS PubMed.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 15139 CAS.
- X. Zhu, P. C. Hillesheim, S. M. Mahurin, C. Wang, C. Tian, S. Brown, H. Luo, G. M. Veith, K. S. Han, E. W. Hagaman, H. Liu and S. Dai, ChemSusChem, 2012, 5, 1912 CrossRef CAS PubMed.
- X. Zhu, S. Chai, C. Tian, P. F. Fulvio, K. S. Han, E. W. Hagaman, G. M. Veith, S. M. Mahurin, S. Brown, H. Liu and S. Dai, Macromol. Rapid Commun., 2013, 34, 452 CrossRef CAS PubMed.
- J. Cai, J. Qi, C. Yang and X. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 3703 CAS.
- H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2010, 132, 12200 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef PubMed.
- H. Choi, Y. C. Park, Y. H. Kim and Y. S. Lee, J. Am. Chem. Soc., 2011, 133, 2084 CrossRef CAS PubMed.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393 CrossRef CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch and T. Muller, et al. , Chem. Mater., 2010, 22, 5964 CrossRef CAS.
- H. Zhao, Z. Jin, H. Su, J. Zhang, X. Yao and H. Zhao, Chem. Commun., 2013, 49, 2780 RSC.
- R. Babarao, Z. Hu, J. Jiang, S. Chempath and S. Sandler, Langmuir, 2007, 23, 659 CrossRef CAS PubMed.
- J. C. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911 RSC.
- J. C. Wang and Q. Liu, Nanoscale, 2014, 6, 4148 RSC.
- J. C. Wang, I. Senkovska, M. Oschatz, M. R. Lohe, L. Borchardt, A. Heerwig, Q. Liu and S. Kaskel, J. Mater. Chem. A, 2013, 1, 10951 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06702b |
| This journal is © The Royal Society of Chemistry 2015 |