
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodiated carbon: a reversible anode for sodium–oxygen batteries and route for the chemical synthesis of sodium superoxide (NaO2)†
Conrad L.
Bender
a,
Birte
Jache
a,
Philipp
Adelhelm
*b and
Jürgen
Janek
*a
aPhysikalisch-Chemisches Institut, Justus-Liebig-Universität Gießen, Heinrich-Buff-Ring 17, 35392 Gießen, Germany. E-mail: juergen.janek@phys.chemie.uni-giessen.de
bInstitute for Technical Chemistry and Environmental Chemistry, Friedrich-Schiller-University Jena, Center for Energy and Environmental Chemistry Jena (CEEC Jena), Philosophenweg 7a, 07743 Jena, Germany. E-mail: philipp.adelhelm@uni-jena.de
First published on 28th August 2015
Abstract
The common cell design of sodium/oxygen batteries is based on an alkali metal as the negative electrode and a carbon gas diffusion layer (GDL) as the positive (oxygen) electrode. The use of sodium metal anodes maximizes the energy capacity, but on the other hand induces undesired and often unpredictable side reactions that complicate investigations on the oxygen electrode and the overall cell chemistry. Therefore we demonstrate the function of a sodium-ion/oxygen battery by replacing sodium metal with a sodiated carbon electrode that is able to reversibly store up to q = 125 mA h g−1. We use a symmetric “all GDL” arrangement, i.e. the same carbon gas diffusion layer is used as the positive and negative electrode. Overall, this approach increases the cycle life by a factor of 5 and further decreases the sum of the charge and discharge overpotentials (η = 150 mV @ j = 200 μA cm−2), proving that current limitations of the sodium–oxygen battery are mainly determined by the metal anode rather than by the oxygen cathode. We find that sodium storage in the GDL proceeds by at least two different mechanisms which can be distinguished by their different chemical stabilities against oxygen and water. Another important finding is that NaO2 can be also synthesized chemically (rather than electrochemically) under ambient conditions from sodiated carbon and gaseous oxygen – which is interesting with respect to the competition between NaO2 and Na2O2 as discharge products.
1. Introduction
Rechargeable alkali metal–air batteries (or more precisely metal–oxygen batteries) are considered as one ultimate goal for potential future electrochemical energy storage devices mainly because of their unrivaled theoretical energy densities, but fail to fulfill optimistic expectations as practical energy densities are much lower and cycling stability is poor. Recently, sodium-based cells are increasingly studied as they also offer a high (theoretical) specific energy of wth(Na2O2) = 1605 W h kg−1 with a cell potential of E° = 2.33 V for the formation of sodium peroxide (qth(Na2O2) = 689 mA h g−1) from the elements under standard conditions.1 Large overpotentials and poor cycle life are commonly reported,2–9 which is well in line with observations made for the analogue lithium–oxygen cells.1,10 Much lower overpotentials are found for Na/O2 cells in which sodium superoxide (NaO2) forms as a solid discharge product in ether based electrolytes enabling a much more reversible cell reaction.11 The low overpotentials are due to the cell reaction| Na + O2 → NaO2 | (1) |
Until now research on Na/O2 (and also on Li/O2) batteries has been mainly directed towards better understanding and improvement of the oxygen cathode, as the redox chemistry of oxygen at room temperature is considered to be kinetically complex and rate-limiting.3,9,21 Thus, the side reactions and large overpotentials of the oxygen cathode are usually regarded as the major hurdles on the way to well-functioning alkali–oxygen batteries.22,23 Sodium (and lithium) dendrite formation at the anode is usually also considered to be an inherent problem, and it is well known that side reactions with the liquid electrolyte or dendrite formation take place at the metal anode surface. In particular, deep cycling causes severe degradation problems at the alkali metal anode (not only in alkali/O2 cells but also in alkali/sulfur cells) and will lead to drying-out due to continuous consumption of the electrolyte.24–27 In addition, dissolved oxygen diffuses to the anode and may partially passivate the metal surface, leading to increased cell impedance and to the loss of the active material (sodium) which must be compensated by metal excess in the cell. The use of a metal anode is of course motivated by the aim of a maximized cell voltage and energy density. However, superior energy densities (compared to lithium-ion technology) appear currently to be out of reach, and it is questionable whether alkali metal/oxygen cells can be competitive for EV applications in the end.28
In the present study, we replace the sodium metal electrode with a sodiated carbon electrode, without employing any kind of membrane beyond the usual separator. Disturbing effects related to the sodium metal anode (side reactions, shuttle effects, and dendrite formation) should be therefore minimized or avoided. By doing so, the characteristics of the oxygen electrode reaction (OER, ORR) can be studied more accurately without interfering effects and the role of the metal anode can be better judged. Using a three-electrode setup with sodium as the reference electrode, it is then also possible to study the behavior of both the carbon anode and cathode simultaneously and independently.
In essence, the construction of an “all-carbon cell” is primarily motivated by the quest for a better understanding of the individual roles of both electrodes on the cell performance and their “cross-talk” by any kind of soluble reaction or decomposition product. The study is not aiming at maximizing the specific capacity – which indeed is lowered by the use of a carbon anode instead of a sodium metal anode. The replacement of the metal anode has not yet been reported for Na/O2 cells, though dendrite formation appears to be even more severe for sodium than for lithium – the reason for this surprising difference is not yet clear. Several groups reported on dendrite formation, resulting in cell failure and limited cycle life of cells containing a metal anode.12,22,23 Only a few publications address the replacement of lithium metal in a Li/O2 cell by other electrode materials (lithiated silicon–carbon composite, graphite, and lithium titanate (LTO)).23,29
Dendrite formation during metal deposition is the result of an inherent morphological instability of the moving metal surface under the influence of an electric field,30,31 but is often also caused by an inhomogenous current distribution due to nonuniform SEI formation, for example. In any case it poses a severe safety issue in practical cells.22 Short circuiting and cell failure may occur as soon as growing dendrites reach the counter electrode. A number of measures were proposed in order to inhibit or minimize dendrite growth, although to date there is no viable solution that warrants the safe use of alkali metal electrodes in rechargeable cells with liquid electrolytes.32 Solid membranes might be an option and cells with sodium beta alumina and Nafion® membranes were recently presented.12,33,34 Besides, impermeable membranes (for other species than Na+ ions) prevent corrosion of the metal electrode by protecting the metal surface from oxygen access. Bi et al. showed a tenfold increase in cycle life by using a Nafion® membrane.34 In view of practical cells, however, a thick and heavy membrane would drastically reduce the achievable energy density and may also reduce the power density. Whether ion-conducting thin films or layers can provide a durable solution for the dendrite issue has yet to be proven, but it has become a common strategy to reduce metal anode stability issues by pre-forming extended SEI, e.g. by pre-treating lithium metal electrodes in a propylene carbonate solution for several days.35
In LIBs, the issue of dendrite formation is tackled by using graphite (or other carbon materials) as a negative electrode. As carbon materials insert lithium ions at potentials slightly less negative than the Li/Li+ redox couple, metal deposition can be prevented. Today graphite is the standard material for lithium storage at low potentials in the form of a binary intercalation compound LiC6 with a theoretical storage capacity of qth(C) = 372 mA h g−1. Similar binary sodium–graphite intercalation compounds have not yet been reported in the literature, and it appears that graphite cannot intercalate sodium to a sufficient amount.36 On the other hand, ether-based electrolytes might enable the use of graphite in sodium-ion batteries based on a co-intercalation phenomenon and the formation of ternary graphite intercalation compounds, although the capacity is then limited to about q = 100 mA h g−1.37 Higher capacities may be achieved in the case of soft and hard carbons.38–40 For example, q values of around 300 mA h g−1 to 600 mA h g−1 were reported for a specific hard carbon.41 In our cells we employed the gas diffusion layer (GDL) H2315 by Freudenberg which we also use as the oxygen electrode. Though there are carbons with higher storage capacities, the GDL is ideal for our experiments as no binder is used for its preparation and hence another potential source for side reactions is excluded. Furthermore, the GDL can be directly used without an additional current collector as a free-standing electrode.
Employing carbon as the anode material for Na/O2 cells, we performed a series of experiments, which are presented and discussed below: firstly, sodiation/desodiation of the carbon GDL during prolonged cycling is characterized in half cells against a sodium metal counter electrode. Secondly, the beneficial impact of the carbon anode on the cycle life (deep cycling), the combined charge/discharge overpotentials and dendrite formation are presented and compared to conventional cells with a sodium metal anode. Using a carbon anode we exclude the “cross-talk” of both electrodes and study the cathode reactions without disturbance by dendrite formation. We further show that simultaneous recording of the positive and negative electrode potentials is a useful tool to determine the separate contributions of both electrodes on the overall cell voltage. Finally, we report a surprising observation which may shed more light on the mechanism of the one-electron transfer reaction forming NaO2. Sodium superoxide forms on sodiated carbon in contact with dissolved oxygen in a purely chemical reaction, i.e. outside an electrochemical cell. This represents a new chemical approach to synthesize sodium superoxide in an ambient environment, and we suggest the underlying reaction mechanism to be a result of a catalytic effect of carbon.
2. Results and discussion
2.1 Preparation of carbon based anodes (sodiated carbon, NaCX)
Electrochemical sodiation/desodiation of the carbon gas diffusion layer (H2315, Freudenberg) was conducted in a coin cell, i.e. by discharging/charging against a sodium metal counter electrode (the cell setup is depicted in Fig. 2a). The voltage-characteristics of the first two consecutive cycles are shown in Fig. 1. The insertion of sodium into the carbon host is characterized by two sloping regions (note: we use the term insertion instead of intercalation as the non-graphitic nature42 of the carbon GDL does not represent a 3D-ordered host lattice). The first region lies between E = 0.5 V and E = 0.1 V and the second region extends to potentials as low as E = 0.01 V.38,43 Due to decomposition reactions of the electrolyte an irreversible capacity of q ≈ 50 mA h g−1 is observed in the first cycle. The following cycles show very reversible and stable capacity values of up to q = 125 mA h g−1 (see Fig. S1†). The excellent reversibility of the electrode reaction is an important prerequisite for the experiments in “full cells” with both the carbon anode and cathode in 2.3. The potential profiles during prolonged cycling remain stable, and the Coulomb efficiency is higher than 99%. The inset of Fig. 1 shows SEM images of the carbon fiber before and after the first sodiation step. The diameter of the fibers expands significantly due to sodium uptake from about d = 12.5 μm to d = 14.0 μm, and the volume increases by roughly 25%. Some expansion is expected due to SEI formation and Na+-insertion; however, such a large increase is also likely due to solvent co-intercalation effects.37 Along with the volume expansion the surface of the GDL becomes rougher, and many small particles form on the surface. In addition to this electrochemical preparation, we also sodiated the GDL by a solution based chemical method using naphthalene, sodium and diglyme (2G).44 The proof for sodiation is provided by electrochemical measurements (Fig. S2†). In summary, the carbon GDL reversibly stores sodium and the overall results are well competitive to other materials.38 The GDL, however, has the additional advantage of being commercially available and free-standing (i.e. binder-free) and is therefore well suited for replacing metallic sodium in Na/O2 cells for a range of fundamental studies. A more detailed analysis of the electrode reaction will be part of a future study.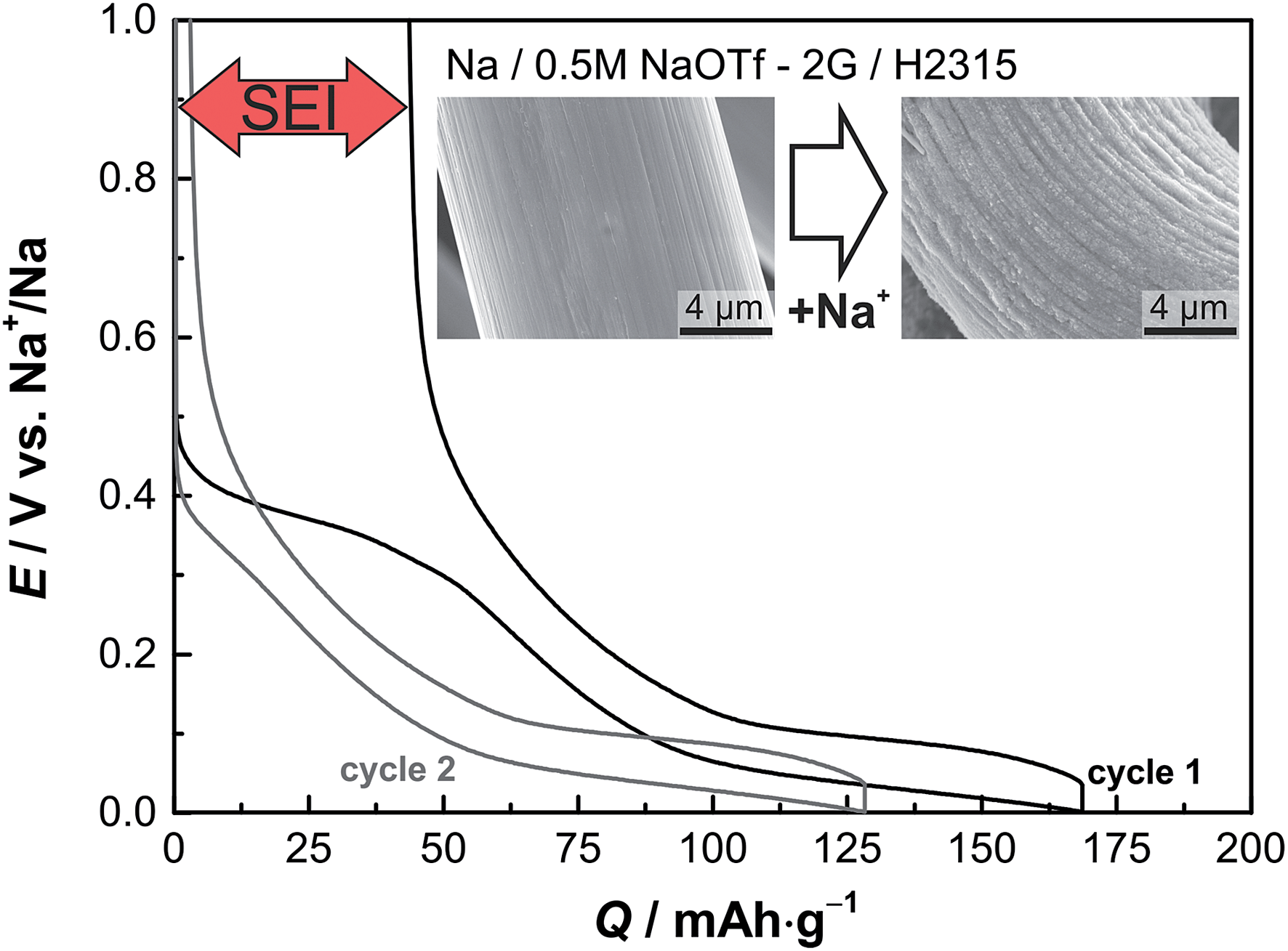 | ||
| Fig. 1 Electrochemical storage of sodium ions in the carbon gas diffusion layer type H2315 in the first and second cycle. The current density was j = 37.2 mA g−1 with a potential-limit set to Elimit = 0.01 V. The reversible specific capacity of the system is about q = 125 mA h g−1. The inset shows two SEM pictures of the GDL before (left) and after (right) the uptake of sodium (see Fig. S1† for more detailed data). | ||
2.2 Chemical stability of NaCX – room temperature NaO2 synthesis
The chemical stability of the resulting NaCX insertion material was tested in two simple experiments: the sodiated GDL electrode was placed in an oxygen cell with sodium as the counter electrode. The experimental setup is depicted in Fig. 2b. The cell was always kept in open-circuit mode, so the net electrochemical reactions driven by electron flow via the external circuit are virtually prohibited. Any change in the open circuit potential (EOCP) therefore indicates chemical reactions taking place at the electrodes. An open circuit potential of typically EOCP = 0.015 V was measured before oxygen or water was purged through the cell. Upon purging the cell with oxygen the potential of the sodiated GDL electrode increases to a potential within the range of E = 30 mV to E = 60 mV vs. Na+/Na (Fig. 2f). Under the remaining oxygen atmosphere this elevated potential is stable for more than 75 hours (Fig. S3†). X-ray diffraction of the sodiated GDL electrode after disassembling the cell showed reflections of sodium superoxide (Fig. 2g). Remembering that no net current has passed through the cell, the sodiated carbon GDL must have reacted chemically with dissolved oxygen to spontaneously form NaO2. The formation of NaO2 on the surface of the GDL extracts sodium and increases the potential vs. the Na+/Na reference. To the best of our knowledge, this is the first proof for chemical (rather than electrochemical) formation of NaO2 under ambient conditions. We emphasize that NaO2 also formed from sodiated carbon and O2 in an experiment without any electrical connections, which is a surprise in view of the putative metastable character of NaO2. Harsh chemical conditions (elevated temperature and high pressure) were so far necessary to chemically form NaO2 from Na2O2 and O2 in an autoclave or in liquid ammonia.45,46 We also never observed that NaO2 forms when bringing metallic Na and dissolved oxygen into direct contact. So one can conclude that NaO2 formation is selectively catalyzed by using sodiated carbon as a sodium source (see visualization of the reaction in the TOC graph).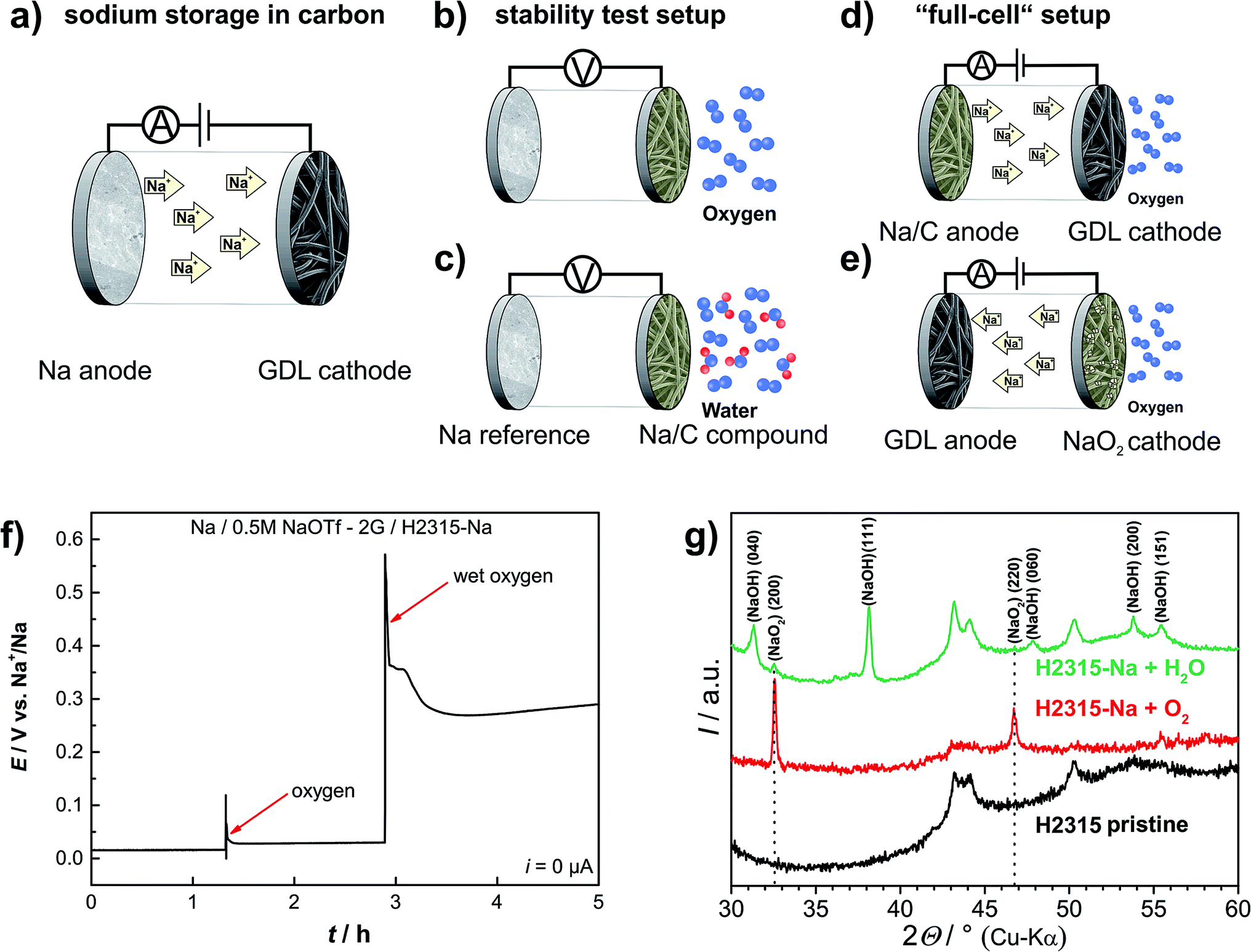 | ||
| Fig. 2 Electrode combinations of the different chemical/electrochemical experiments. Gray disc: sodium metal, black fiber structure: GDL, yellow fiber structure: sodiated GDL. (a) Cell arrangement for the electrochemical sodiation of the GDL. (b) and (c) Cell arrangement for the chemical stability tests of the sodiated GDL under OCP conditions. Sodium metal was used as the reference to record the open circuit potential of the working electrode. j = 0 μA. (d) and (e) Full sodium–oxygen cell with the sodiated GDL as the anode. j = 200 μA cm−2 (in most cases). (f) Results of the measurements of the potential of a sodiated GDL electrode while purging with pure oxygen and oxygen bubbled through water. (g) Corresponding X-ray diffraction patterns of sodiated GDL electrodes after contact with oxygen and water, respectively. All potentials were measured against pure sodium metal. | ||
Using the data shown in Fig. 1, the increase in OCV during the chemical formation of NaO2 can be used to roughly estimate the total amount of NaO2 formed from the sodiated GDL and O2. The contribution to the mixed potential caused by the newly formed NaO2 phase is assumed to be negligible. By doing so the amount of sodium stored in the GDL below 0.06 V can be calculated to be q = 0.67 mA h (q = 0.61 mA h g−1 and q = 0.59 mA h cm−2). Around 35% of the total amount of sodium stored in the GDL therefore must have reacted, corresponding to an absolute amount of NaO2 of about 1.4 mg. One can conclude that sodium stored below 0.06 V is highly reactive and leads to the formation of NaO2 (red area in Fig. S4†). Sodium inserted at potentials above 0.06 V is stable against reaction with oxygen (green area in Fig. S4†). In a consecutive experiment, the oxygen flushed GDL electrode successfully operated as the anode in a sodium–oxygen cell. Thus, if we neglect the loss in capacity, oxygen diffusion from the cathode towards the anode will not obstruct the basic functionality of the battery as long as the potential remains above 0.06 V.
When carrying out the same experiment with water-saturated oxygen (bubbling oxygen through water, see Fig. 2c) a more distinct potential increase of about 350 mV is observed (Fig. 2f). The resulting potential of 0.4 V vs. Na+/Na is significantly higher than that for the reaction with pure oxygen. The X-ray diffraction pattern easily explains this finding as both sodium superoxide and sodium hydroxide (NaOH) could be detected on the carbon GDL. Obviously, water leads to a complete decomposition of the sodiated GDL. As expected we observed no activity of the electrode when using it in a sodium–oxygen cell.
Taking these results together allows us to differentiate between at least two different storage mechanisms of sodium in the present carbon structure (GDL). Sodium stored at low potentials (E < 0.06 V vs. Na+/Na) is unstable against O2 and H2O and will therefore chemically react with these molecules in oxygen cells. Sodium stored above E = 0.06 V vs. Na+/Na is stable against oxygen but remains reactive against water. We note that the underlying principles of sodium storage in non-graphitic carbons are, just as for lithium, very complex and very much depend on the exact type of carbon used (hard or soft carbons), the hydrogen content, the degree of structural disorder and so forth.47,48 By distinguishing between two storage mechanisms, we simply group the different storage principles in the ones that are reactive towards oxygen and water and the ones that are reactive towards water only.
2.3 “Full cells” with sodiated GDL as the anode
This section discusses the use of the sodiated GDL as a replacement for metallic sodium in full cells. In the following experiments a sodiated GDL serves as the anode and a conventional GDL of the same type serves as the oxygen cathode. This “all-carbon cell” is shown in Fig. 2d. In a two-electrode setup we determined the galvanostatic rate capability. Current densities of j = 50, 200, 400 and 800 μA cm−2 were applied. The cut-off potentials for discharge were set to Elimit = 1.4 V and to Elimit = 2.6 V for charge, respectively. Fig. 3a depicts the results of the four different applied currents. The potential during discharge and charge shows a sloping trend in contrast to cells with a sodium metal anode. Using a metal anode, the potential is characterized by a constant potential plateau at about E = 2.1 V with a sharp increase at the end (Fig. 3b). Evaluating the sum of overpotentials at 50% depth of discharge (DOD) the carbon based anode is superior to the sodium metal one. In Fig. 3c the sum of the overpotentials at 50% DOD is plotted vs. the current density. The black hollow squares show the data for the cell with a carbon anode; the green squares show data for the corresponding cell with a sodium metal anode taken from ref. 11. Especially for high current densities the low overpotential of the carbon anode is remarkable. The sum of overpotentials for the measurement at the highest current density of j = 0.8 mA cm−2 is only about η = 350 mV, which is lower than the value for a smaller current density j = 0.3 mA cm−2 employing a sodium metal anode (green squares; η = 480 mV).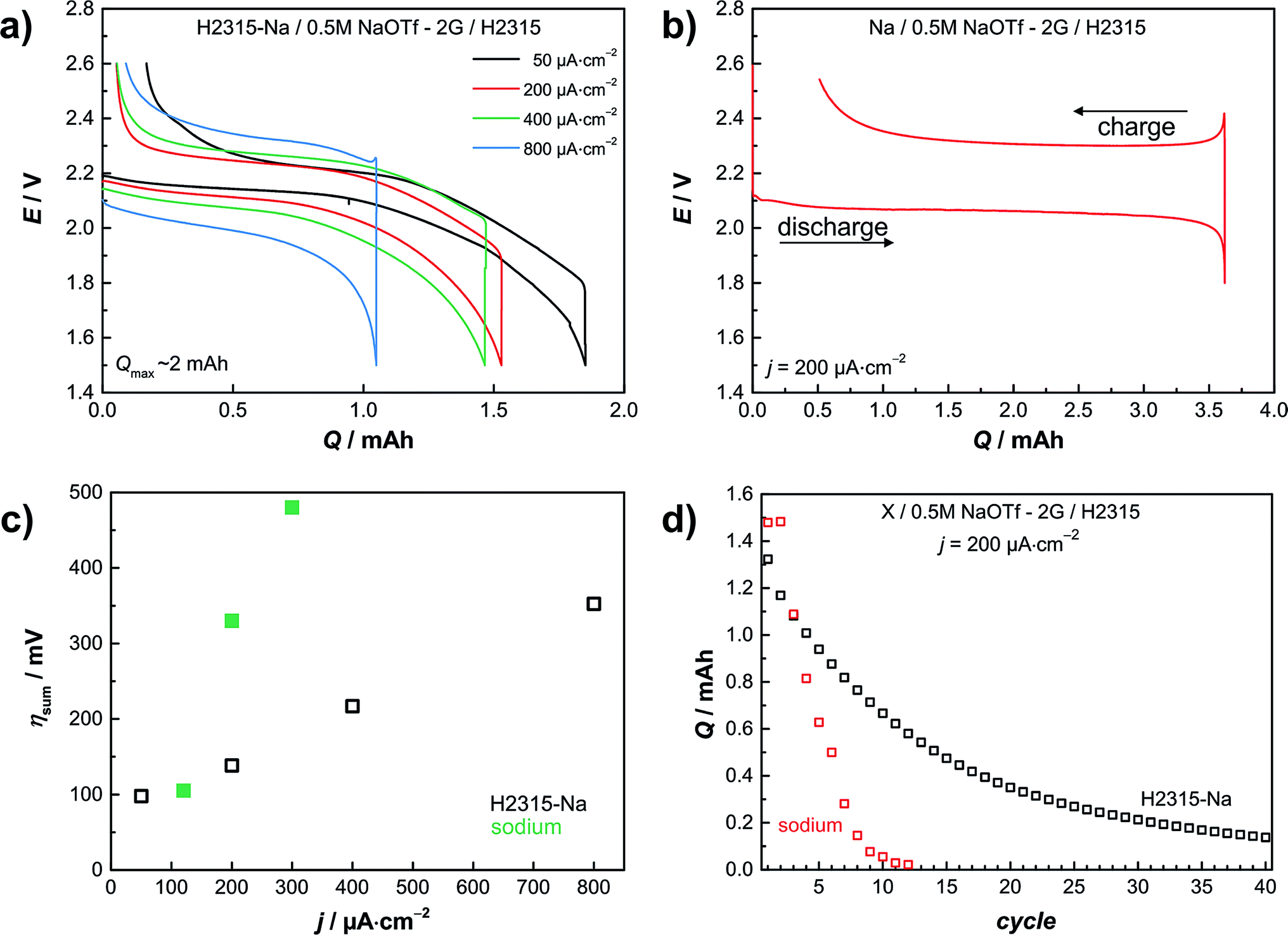 | ||
| Fig. 3 Results from electrochemical measurements with a sodiated carbon anode in a sodium–oxygen cell. (a) Rate capability with different current densities measured in a two-electrode setup and with sodiated carbon as anode. (b) Hysteresis of a Na/O2 cell with a sodium metal anode. (c) Comparison of overpotentials at 50% depth of discharge (DOD) black: sodiated GDL; green: sodium anode by Hartmann et al.11 (d) Cycle lifetime of a Na/O2 cell with a carbon anode measured with a current density of j = 200 μA cm−2 compared to the results of Hartmann et al.11 with a metal anode. The graph compares the discharge capacities of the different cells. See Fig. S10† for details on the cycling protocol. | ||
Fig. 3d shows the cycle life of a Na/O2 cell with a carbon anode in comparison with a sodium metal anode. Both cells were cycled galvanostatically with a current density of j = 200 μA cm−2 in a deep cycling mode. Most of the electrochemical tests in the literature are performed as shallow cycling with a limited discharge capacity. This procedure leads to a better cycle life, but does not use the full capacity of the cell. Clearly the goal of any high energy battery must be to establish a long-life cell in deep cycling mode – or at least in a mode using as much as possible of the theoretical capacity (in fact, deep cycling is not even used in LIBs). Shallow cycling might be necessary to avoid side reactions, but should then still lead to sufficient energy density. As shown, using a carbon anode the cycle life is extended, as after 40 cycles still a capacity higher than q = 0.2 mA h is achieved. To maximize the specific capacity in a full cell (symmetrical, two GDL electrodes) charge balancing of the electrodes becomes important, just as in LIB technology. If one electrode is oversized this electrode will operate in a shallow cycling mode.
The cell with the metal anode fails after 8 cycles, mainly due to the formation of sodium dendrites during deep cycling. Fig. S5 and S6† depict a cell with dendrites grown in the first cycle. In cells with the carbon anode the dendrite problem cannot arise, and indeed we never faced dendrite formation in cells with the carbon anode.
Practical batteries operate in a two-electrode configuration, meaning that only the difference between both electrode potentials is measured, i.e. the cell voltage. Peculiar features of the voltage profile can therefore not be easily assigned to one of the electrodes. We measured the individual potentials of both the anode and cathode in a three-electrode configuration in order to assign features of the cell voltage profile to either the anode or the cathode. Fig. 4a illustrates a typical three-electrode measurement with sodium metal employed as the reference electrode. The black line corresponds to the potential of the oxygen electrode (GDL, cathode). The red line represents the anode potential (sodiated GDL) and the green line depicts the subtracted potentials of the cathode and anode, being identical to the potential difference that is measured in a two-electrode setup. Discharging the cell always causes a decrease in the cathode potential and an increase in the anode potential. For the charging process it is vice versa. The combination of the cathode and anode potential yields the typical sloping profile for two-electrode measurements (green curve, see Fig. 3a). Using a reference electrode it is possible to set individual cut-off potentials for each electrode. Thus the anode was cycled between Elimit = 0.01 V vs. Na+/Na and Elimit = 3.0 V vs. Na+/Na; the cathode limits were set to Elimit = 1.8 V vs. Na+/Na and Elimit = 2.6 V vs. Na+/Na, respectively, and sodium plating is excluded. The two-electrode potential can also be limited and was set to Elimit = 1.2 V vs. Na+/Na for discharge. By controlling the different potentials, the battery always operates in its desired operational window and the issue of exact electrode balancing is circumvented. Sodium metal deposition on the carbon anode is prevented by keeping the anode potential above E = 0 V vs. Na+/Na at all times.
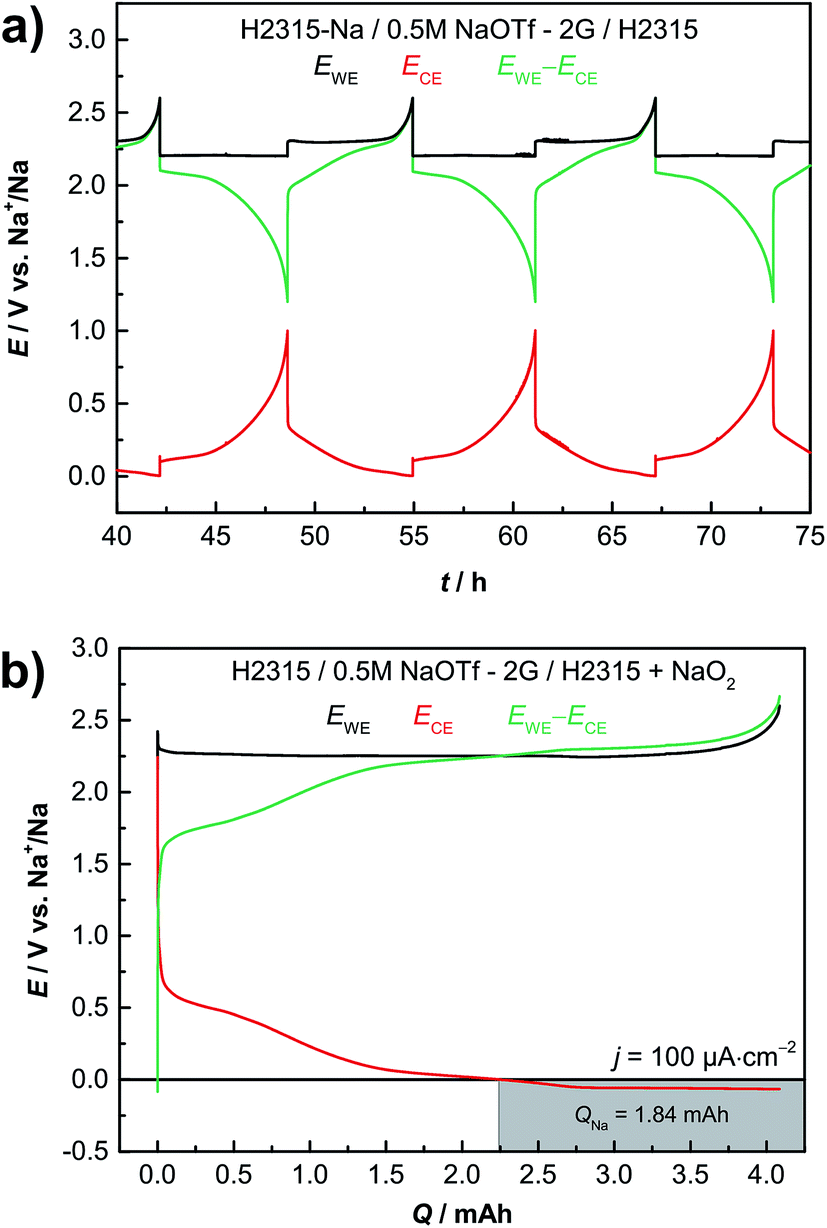 | ||
| Fig. 4 (a) Potential-capacity profiles for discharge and charge of a Na/O2 cell with a carbon anode in a three-electrode setup. The current density was j = 200 μA cm−2. (b) Loading of the carbon GDL with sodium by charging the cathode prefilled with sodium superoxide. The grey area with a potential below 0.0 V depicts sodium plating on the carbon anode. Black curve: working electrode, cathode GDL H2315; red curve: counter electrode, anode GDL H2315; green curve: two-electrode potential of the full cell. | ||
Some expected properties and particular features of the cell reaction become obvious: (1) the cathode alone is responsible for the known potential plateau of Na/O2 cells. (2) Using a carbon anode the cathode is not the limiting factor of the cell capacity, but the limited amount of sodium stored inside the anode. The cathode is cycled in a shallow cycling mode as the potential shows no drop at the end of discharge. As in LIBs balancing becomes important. (3) The potential increase at the end of the charging process is caused by the disappearance of sodium superoxide (cathode side) and not by the anode. (4) The peak at the beginning of the charge process can be assigned to the cathode during deep cycling and is not caused by the anode (see Fig. S7†). Obviously, cycling the cell with different electrodes and/or defining separate cut-off potentials for each of the electrodes and the cell provides a way of improved analysis of the cell reaction, especially considering points (3) and (4).
In another experiment, a “standard” Na/O2 cell with sodium as the anode and GDL as the cathode was discharged down to E = 0.01 V. (see Fig. S8†). During discharging, NaO2 forms on the GDL at potentials around E = 2.2 V vs. Na/Na+. A sudden drop in potential characterizes the end of the oxygen reduction reaction (ORR). As oxygen is available in excess, this “sudden death” of the ORR is usually associated with passivation of the GDL surface. However, when lowering the potential even further, sodiation of the GDL takes place. From this one can conclude that the GDL surface was not fully passivated and hence the sudden end of the ORR must be also related to another phenomenon.
2.4 Full cells with pre-discharged cathode
For safety reasons it is of advantage to assemble batteries in the discharged state. As a proof of principle, we show that this can be easily achieved with the “all-carbon” Na/O2 cell. Here a NaO2 containing GDL is used as the positive electrode and a pristine non-sodiated GDL is used as the negative electrode. (see Fig. 2e for cell geometry). Fig. 4b displays the results of the corresponding experiment including the potential of both electrodes vs. a Na/Na+ reference. The first step is to charge the cell covered with NaO2 particles (about q = 4 mA h; m(NaO2) = 8.2 mg) to form the sodiated GDL.The superoxide decomposes and releases oxygen to the gas phase, electrons and sodium ions. At the anode the sodium ions are inserted into the carbon host of the gas diffusion layer as already shown in Section 2.1 (Fig. 1). The potential of the GDL anode (red curve) falls as sodium is inserted. At about q = 2.25 mA h the potential crosses the x-axis and sodium metal is plated on the carbon fibers as the capacity of the GDL for sodium insertion is exceeded. The potential was intentionally not limited and the plating was intended in this experiment. The end of the charge process is characterized by a sharp increase of the cathode potential (black line) when the NaO2 was almost fully decomposed. The subsequent cycles of discharge and charge are depicted in ESI Fig. S9.† At the beginning of the following discharge step the deposited sodium is oxidized and the potential of the anode increases. As the potential increases above E = 0 V no metallic sodium is left in the cell. For the subsequent cycles, the potentials of both electrodes were independently measured against the sodium metal reference electrode. In addition, the potential limits were fixed in order to prevent recurring sodium plating on the carbon anode during charging. Overall, these results demonstrate that a sodium–oxygen battery can also be assembled in the discharged state.
3. Conclusion
Sodium metal was successfully replaced by sodiated carbon electrodes based on a gas diffusion layer (GDL) in Na/O2 cells. The main aim was to construct a model system that enables studies on the cell chemistry without having interfering effects originating from the use of metal sodium electrodes. Sodiation and desodiation of the GDL were done chemically and electrochemically. The reversible specific capacity of the gas diffusion layer was about q = 120 mA h g−1 for more than 80 cycles. Na/O2 cells with an aprotic electrolyte and the carbon anode showed sodium superoxide (NaO2) as discharge product. The cycle life in deep cycling mode could be improved compared to cells with a metal anode. The overpotentials of Na/O2 cells with sodium superoxide as a product were further reduced to only η = 150 mV @ j = 200 μA cm−2. We also showed that using sodiated carbon instead of sodium metal effectively prevents dendrite formation and hence short circuiting of the cell.An interesting observation is linked to the identification of two different storage mechanisms for sodium. Sodium stored at rather low potentials (high activities) under E = 0.06 V reacts with oxygen chemically to form NaO2 which may open up a new route to the synthesis of NaO2 in an ambient environment. This chemical path might be taken as an indicator for the selective catalytic role of carbon in one-electron transfer and superoxide formation.
4. Experimental section
4.1 Materials and cell assembly
Sodium filled (“sodiated”) carbon anodes were prepared by using a coin cell setup (experiment corresponds to Fig. 2a). The negative electrode consisted of pure sodium metal (BASF SE, Ludwigshafen, Germany) rolled out and punched into electrodes of 10 mm in diameter. For the carbon electrode we used the commercially available gas diffusion layer H2315 (Freudenberg, Weinheim, Germany) punched out in a diameter of 12 mm. Three glass microfiber filters (GF/A, Whatman, Ø 12 mm) were applied to separate the electrodes and soaked with the electrolyte consisting of diglyme (anhydrous, 99.5% Sigma Aldrich) as a solvent and sodium triflate (NaSO3CF3, 98%, Aldrich) as the conducting salt. The concentration of the conducting salt was 0.5 mol L−1. Diglyme was dried over molecular sieves (3 Å, for 1 week), sodium triflate under vacuum at 140 °C for 24 h. The final water content of the electrolyte was determined with an 831KF Karl Fischer coulometer (Metrohm, Filderstadt, Germany) to be less than 10 ppm. About 120 μL of electrolyte were used in one coin cell.The discharged coin cells were opened and the sodiated carbon electrode was then used as the negative electrode for further electrochemical tests in an oxygen cell using the “Giessen cell”.17 The positive electrode consisted as well of the gas diffusion layer H2315 but with a diameter of 10 mm. One glass microfiber filter (GF/A, Whatman, Ø 12 mm) was applied with 45 μL of electrolyte. For three-electrode measurements additional 10 μL were used for connection of the reference electrode made of pure sodium. All cell assembly was done in an argon-filled glove-box (GST4, Glovebox Systemtechnik, Malsch, Germany) with water and oxygen contents in the argon atmosphere below 5 ppm. For a shorter notation we will use the term “full cell” for all cells constructed from two GDL electrodes (one sodiated anode and one oxygen electrode).
4.2 Electrochemical cell testing
The coin cells were discharged by applying a current of 37.2 mA g−1 based on the weight of 11 mg of the GDL (Ø 12 mm). The cut-off potential was set to 0.01 V. All oxygen-cells were cycled galvanostatically (constant current) in a climate chamber at 25 °C using a VMP3 from BioLogic. Coin cells were cycled using a battery cycling system 4300 from Maccor. The gas reservoir of the cells with a volume of about 9 cm3 was flushed with oxygen (purity 5.0, Praxair, Danbury, CT) for 10 s at 105 Pa just before the measurement. Every cell testing included a 30 min holding step with recording of the open-circuit potential. The cut-off limits for the cycling procedure were chosen depending on the cell configuration. For two electrode measurements the limits were usually set to 1.5 V and 2.6 V, respectively. In cells with a reference electrode both electrodes (working and counter) were controlled. The limits for the working electrode were set to 1.8 V vs. Na+/Na and 2.6 V vs. Na+/Na. The counter electrode limits were 0.01 V vs. Na+/Na and 3.0 V vs. Na+/Na, respectively.The following set of experiments was performed (as shown schematically in Fig. 2):
• The GDL was sodiated by discharging against a sodium metal anode (Fig. 2a).
• The stability of the sodiated GDL was tested in an electrochemical cell by measuring the open circuit potential while flushing the cell with oxygen (Fig. 2b) or water (wet oxygen, Fig. 2b).
• In a full cell with two GDL electrodes the performance of sodiated carbon anodes was tested in terms of capacity, overpotential and cycle life (Fig. 2d).
• Another approach to store sodium in a GDL was to charge an electrode with NaO2 deposited against an empty GDL anode (Fig. 2e).
4.3 Structural characterization
Scanning electron microscopy was carried out on discharged positive electrodes on a Merlin high-resolution Schottky field-emission electron microscope (Zeiss SMT) equipped with an X-Max EDS detector (Oxford Instruments). X-ray diffraction was performed on an Empyrean powder X-ray diffractometer (Cu-Kα source, 40 kV, 40 mA) from PANalytical. The electrodes were analyzed using a self-made gas-tight sample holder with a kapton tape as protection against moisture.Acknowledgements
C. L. B. thanks Fonds der Chemischen Industrie (FCI) for a Ph.D. scholarship. The project was supported by the BASF International Scientific Network for Electrochemistry and Batteries.References
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky, H. Mazor, M. Goor and S. Avshalomov, J. Power Sources, 2011, 196, 6835–6840 CrossRef CAS PubMed.
- Y. Li, H. Yadegari, X. Li, M. N. Banis, R. Li and X. Sun, Chem. Commun., 2013, 49, 11731–11733 RSC.
- W.-M. Liu, W.-W. Yin, F. Ding, L. Sang and Z.-W. Fu, Electrochem. Commun., 2014, 45, 87–90 CrossRef CAS PubMed.
- W. Liu, Q. Sun, Y. Yang, J. Y. Xie and Z. W. Fu, Chem. Commun., 2013, 49, 1951–1953 RSC.
- Q. Sun, Y. Yang and Z.-W. Fu, Electrochem. Commun., 2012, 16, 22–25 CrossRef CAS PubMed.
- J. Kim, H. D. Lim, H. Gwon and K. Kang, Phys. Chem. Chem. Phys., 2013, 15, 3623–3629 RSC.
- Z. Jian, Y. Chen, F. Li, T. Zhang, C. Liu and H. Zhou, J. Power Sources, 2014, 251, 466–469 CrossRef CAS PubMed.
- H. Yadegari, Y. Li, M. Norouzi Banis, X. Li, B. Wang, Q. Sun, R. Li, T.-K. Sham, X. Cui and X. Sun, Energy Environ. Sci., 2014, 7, 3747–3757 CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, J. Sann, A. K. Durr, M. Jansen, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 11661–11672 RSC.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- O. Arcelus, C. Li, T. Rojo and J. Carrasco, J. Phys. Chem. Lett., 2015, 6, 2027–2031 CrossRef CAS PubMed.
- B. Lee, D.-H. Seo, H.-D. Lim, I. Park, K.-Y. Park, J. Kim and K. Kang, Chem. Mater., 2014, 26, 1048–1055 CrossRef CAS.
- S. Kang, Y. Mo, S. P. Ong and G. Ceder, Nano Lett., 2014, 14, 1016–1020 CrossRef CAS PubMed.
- C. L. Bender, P. Hartmann, M. Vračar, P. Adelhelm and J. Janek, Adv. Energy Mater., 2014, 4, 1301863 Search PubMed.
- B. D. McCloskey, J. M. Garcia and A. C. Luntz, J. Phys. Chem. Lett., 2014, 5, 1230–1235 CrossRef CAS PubMed.
- C. L. Bender, W. Bartuli, M. G. Schwab, P. Adelhelm and J. Janek, Energy Technol., 2015, 3, 242–248 CrossRef CAS PubMed.
- N. Zhao, C. Li and X. Guo, Phys. Chem. Chem. Phys., 2014, 16, 15646–15652 RSC.
- Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J.-G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- J. Hassoun, H. G. Jung, D. J. Lee, J. B. Park, K. Amine, Y. K. Sun and B. Scrosati, Nano Lett., 2012, 12, 5775–5779 CrossRef CAS PubMed.
- I. C. Jang, Y. Hidaka and T. Ishihara, J. Power Sources, 2013, 244, 606–609 CrossRef CAS PubMed.
- Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS PubMed.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- X. Fang and H. Peng, Small, 2015, 11, 1488–1511 CrossRef CAS PubMed.
- K. G. Gallagher, S. Goebel, T. Greszler, M. Mathias, W. Oelerich, D. Eroglu and V. Srinivasan, Energy Environ. Sci., 2014, 7, 1555 CAS.
- J. Chun, H. Kim, C. Jo, E. Lim, J. Lee and Y. Kim, ChemPlusChem, 2015, 80, 349–353 CrossRef CAS PubMed.
- K. Peppler, M. Pölleth, S. Meiss, M. Rohnke and J. Janek, Z. Phys. Chem., 2006, 220, 1507–1527 CrossRef CAS.
- M. D. Pritzker and T. Z. Fahidy, Electrochim. Acta, 1992, 37, 103–112 CrossRef CAS.
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- S. Wenzel, H. Metelmann, C. Raiß, A. K. Dürr, J. Janek and P. Adelhelm, J. Power Sources, 2013, 243, 758–765 CrossRef CAS PubMed.
- X. Bi, X. Ren, Z. Huang, M. Yu, E. Kreidler and Y. Wu, Chem. Commun., 2015, 51, 7665–7668 RSC.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- R. C. Asher, J. Inorg. Nucl. Chem., 1959, 10, 238–249 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007–15028 RSC.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342–3345 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS PubMed.
- J. Zhao, L. Zhao, K. Chihara, S. Okada, J.-I. Yamaki, S. Matsumoto, S. Kuze and K. Nakane, J. Power Sources, 2013, 244, 752–757 CrossRef CAS PubMed.
- http://goldbook.iupac.org/N04193.html .
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS PubMed.
- Y. Mizutani, T. Abe, M. Inaba and Z. Ogumi, Synth. Met., 2001, 125, 153–159 CrossRef.
- S. E. Stephanou, E. J. Seyb, J. Kleinberg, R. H. Shakely and W. H. Schechter, 1953, Sodium Superoxide, in Inorganic Syntheses, Volume 4 (ed. J. C. Bailar), John Wiley & Sons, Inc., Hoboken, NJ, USA. DOI:10.1002/9780470132357.ch29.
- W. H. Schechter, H. H. Sisler and J. Kleinberg, J. Am. Chem. Soc., 1948, 70, 267–269 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 4428 CrossRef CAS PubMed.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06640a |
| This journal is © The Royal Society of Chemistry 2015 |