
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped carbon-encapsulated SnO2–SnS/graphene sheets with improved anodic performance in lithium ion batteries†
Jieqiong
Shan‡
a,
Yuxin
Liu‡
a,
Ping
Liu
b,
Yanshan
Huang
b,
Yuezeng
Su
*c,
Dongqing
Wu
*b and
Xinliang
Feng
bd
aSchool of Aeronautics and Astronautics, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai, 200240, P. R. China
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai, 200240, P. R. China. E-mail: wudongqing@sjtu.edu.cn
cSchool of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai, 200240, P. R. China. E-mail: yzsu@sjtu.edu.cn
dCenter for Advancing Electronics Dresden (cfaed) & Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, Dresden, 01062, Germany
First published on 28th October 2015
Abstract
A dual-doping approach for nitrogen-doped carbon-coated SnO2–SnS/graphene nanosheets (N–C@SnO2–SnS/GN) has been developed, using hydrothermal carbonization of sucrose with SnO2-decorated graphene and ammonium thiocyanate and a subsequent thermal treatment. The resulting hybrid manifests a typical two-dimensional core–shell architecture, with a N-doped carbon coating over the SnS- and SnO2-decorated graphene nanosheets. Used as the anode material in lithium ion batteries (LIBs), N–C@SnO2–SnS/GN delivers a high specific capacity of 1236 mA h g−1 at a current density of 0.1 A g−1 after 110 cycles, which outperforms most state-of-the-art tin-based LIB anodes with core–shell structures.
Introduction
Lithium ion batteries (LIBs) are being considered as a prominent means of electrochemical energy storage for applications in electric vehicles, portable electronic devices, and smart grids.1–4 A major challenge in developing LIBs is exploring high-performance and cost-effective electrode materials that are capable of delivering greater energy and power densities than those of conventional electrode materials. With high theoretical capacities, tin-based anodes such as tin metal (Sn),5–9 tin oxides (SnO or SnO2),10–16 and tin sulfides (SnS or SnS2)17–23 are regarded as promising electrode materials for next-generation LIBs. However, their practical applications in LIBs have been plagued for a long time by severe capacity fading that arises from enormous volume variation during lithium insertion/extraction.1,5–7,12–21,24,25Current state-of-the-art nanotechnology allows encapsulation of tin-based anodes in a carbon shell, which can provide both effective protection against pulverization of the electrode materials and rapid pathways for the transportation of charge carriers. Nevertheless, introduction of the carbon shell inevitably decreases the mass ratio of the tin components in the core, often leading to a reduction in performance concerning the weight or volume of the whole electrode. An ideal solution is to dope the carbon shell with heteroatoms such as nitrogen (N) and boron (B), which can significantly enhance conductivity and wettability in an electrolyte.6,26–28 Moreover, heteroatoms in carbon have been demonstrated to create extra active sites for accommodating lithium atoms, which is beneficial for further elevating the capacity of tin-based anodes.6,27–30 On the other hand, combining two different tin-containing anode materials can lead to a synergetic effect on the electrochemical properties of the whole electrode material. The varied phase behavior of different tin species such as SnO2 and SnS provides buffers against volumetric changes of electrodes during the charge/discharge process, thereby leading to stabilized cycling performance.20,21,31,32 Diffusion of lithium can also be improved along grain boundaries of different tin-based anode materials.33 Moreover, the rational design and construction of graphene-based carbon composites as highly efficient electrodes for various electrochemical applications has attracted great interest.34–37
Inspired by these results, an unprecedented dual-doping strategy for the fabrication of nitrogen-doped carbon encapsulated SnO2–SnS/graphene nanosheets (N–C@SnO2–SnS/GNs) is developed in this work, through the hydrothermal carbonization of sucrose in the presence of SnO2 nanoparticle-decorated graphene nanosheets (SnO2/GNs) and ammonium thiocyanate (NH4SCN), along with a subsequent thermal treatment of the resultant hybrid under a flow of nitrogen. During the hydrothermal treatment, decomposition of NH4SCN can release multiple gaseous fragments, including carbon disulfide (CS2), hydrogen sulfide (H2S) and ammonia (NH3), which can serve as dopants for SnO2/GNs.38 Due to different reaction energies, carbon and SnO2 are doped with nitrogen and sulfur atoms, respectively, leading to the formation of dual-doped N–C@SnO2–SnS/GN. As the anode material in LIB, N–C@SnO2–SnS/GN can keep a high capacity of 1236 mA h g−1 at a current density of 0.1 A g−1 for over 110 cycles. Even at a fast charge rate of 3.2 A g−1, a good capacity of 350 mA h g−1 can still be obtained by N–C@SnO2–SnS/GN. Such an outstanding electrochemical performance is superior to most state-of-the-art tin-based LIB anodes with core–shell structures.10,39–41
Experimental section
Preparation of SnO2/GN
Graphene oxide (GO) was prepared from natural graphite flakes using the modified Hummers method. An aqueous dispersion of GO (2 mg mL−1, 20 mL) was ultrasonicated with a mixed solvent of ethylene glycol and water (volEG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :volH2O = 9:1, 100 mL) for 1 h to form a homogeneous suspension. Subsequently, SnCl2·2H2O (1 g, dissolved in 20 mL EG) was slowly added to the suspension and stirred for 0.5 h, and then was refluxed at 120 °C for 2 h. After cooling to room temperature, the resulting black suspension was centrifuged and washed three times with distilled water to obtain SnO2 NPs-decorated graphene nanosheets (SnO2/GN); this was then dispersed in distilled water (40 mL) to form an aqueous suspension of SnO2/GN for further use.
:volH2O = 9:1, 100 mL) for 1 h to form a homogeneous suspension. Subsequently, SnCl2·2H2O (1 g, dissolved in 20 mL EG) was slowly added to the suspension and stirred for 0.5 h, and then was refluxed at 120 °C for 2 h. After cooling to room temperature, the resulting black suspension was centrifuged and washed three times with distilled water to obtain SnO2 NPs-decorated graphene nanosheets (SnO2/GN); this was then dispersed in distilled water (40 mL) to form an aqueous suspension of SnO2/GN for further use.
Preparation of N–C@SnO2–SnS/GN, C@SnO2/GN and N–C@SnO2/GN
The suspension of SnO2/GN (40 mL) was mixed with an aqueous solution (20 mL) containing both sucrose (3 mmol) and ammonium thiocyanate (1.65 mmol), followed by a hydrothermal treatment at 180 °C for 12 h and a further thermal treatment at 400 °C for 2 h under a flow of N2. The final product was obtained as a black powder, designated as N–C@SnO2–SnS/GN. In a control experiment, carbon-coated SnO2/GN was fabricated by similar procedures without the addition of ammonium thiocyanate, with the resulting product being denoted as C@SnO2/GN. We also prepared nitrogen-doped carbon-coated SnO2/GN by replacing ammonium thiocyanate with ammonium chloride, and the obtained sample was denoted as N–C@SnO2/GN.Characterization
Structures of the obtained samples were characterized by scanning electron microscopy (SEM, Sirion 200, 25 kV) and transmission electron microscopy (TEM, JEOL JEM-2010, 200 kV). Atomic force microscopy (AFM) characterization was conducted with a multimode nanoscope scanner (DI Co., USA) operated in the tapping mode. Samples were dispersed in water and then dropped onto a silicon wafer for SEM or AFM measurements and a Cu grid covered with a thin amorphous carbon film for TEM measurements. X-ray diffraction (XRD) measurements were carried out on a D/max-2200/PC (Rigaku Corporation, Japan) using Cu (40 kV, 30 mA) radiation. Raman spectra were recorded on a SENTERRA with excitation from the 532 nm line of an Ar-ion laser with a power of about 5 mW. N2 adsorption–desorption isotherms at 77 K were determined by using a Micromeritics ASAP 2010 instrument. XPS experiments were carried out on AXIS Ultra DLD system from Kratos with Al Kα radiation as the X-ray source for radiation. Fourier transform infrared spectra (FT-IR) were measured on a Spectrum 100 (Perkin Elmer, Inc., USA) spectrometer with a scan range of 4000 to 400 cm−1. The samples were ground with KBr and then pressed into disks.Electrochemical measurements
The obtained samples were mixed with carbon black (Super-P), and polyvinylidene fluoride (PVDF) binder in a 8:1:1 ratio in 1-methyl-2-pyrrolidone (NMP) solvent (Aldrich, 99.5%) until homogeneous. This slurry was dried in a vacuum oven at 60 °C for 12 h after casting onto Cu foil. A CR2016-type coin cell was assembled in an argon-filled glovebox (M. Barun glovebox) with the test electrode, a Li metal counter electrode, a polymer membrane separator (Celgard 2400), and an electrolyte of 1 M LiPF6 in a 1:1 (v/v) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (Techno Semichem). Electrochemical experiments were performed using a battery cycler (LAND-CT2001A) with current rates from 0.1 to 3.2 A g−1 at a voltage range of 0.01–3 V. Cyclic voltammograms were obtained over the potential range of 0.01–3 V at a scanning rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements of the electrodes were carried out on an electrochemical workstation (PARSTAT 2273). Impedance spectra were recorded by applying a sine wave with an amplitude of 5.0 mV over a frequency range from 100 kHz to 0.01 Hz. Fitting of the impedance spectra to the proposed equivalent circuit was performed by the code Zview.
Results and discussion
The synthesis strategy of N–C@SnO2–SnS/GN is depicted in Fig. 1. SnO2/GNs were first fabricated by the hydrolysis of tin chloride on graphene oxide (GO) in a mixture of ethylene glycol (EG) and water. Subsequently, N–C@SnO2–SnS/GN was produced via the hydrothermal treatment of SnO2/GN at 180 °C for 12 h in the presence of sucrose and NH4SCN and the following thermal treatment of the resultant hybrid at 400 °C for 2 h in N2 flow. In this step, sulfur-rich gases such as CS2 and H2S generated from the thermal decomposition of NH4SCN can be used as the sulfurizing agent for the sulfurization of SnO2, while nitrogen containing molecules like NH3 can provide a nitrogen source for the doping of the sucrose-derived carbon shells and graphene.38 In a control experiment, carbon-coated SnO2/GN was also fabricated by a similar procedure without the addition of NH4SCN, and the resulting product was denoted as C@SnO2/GN.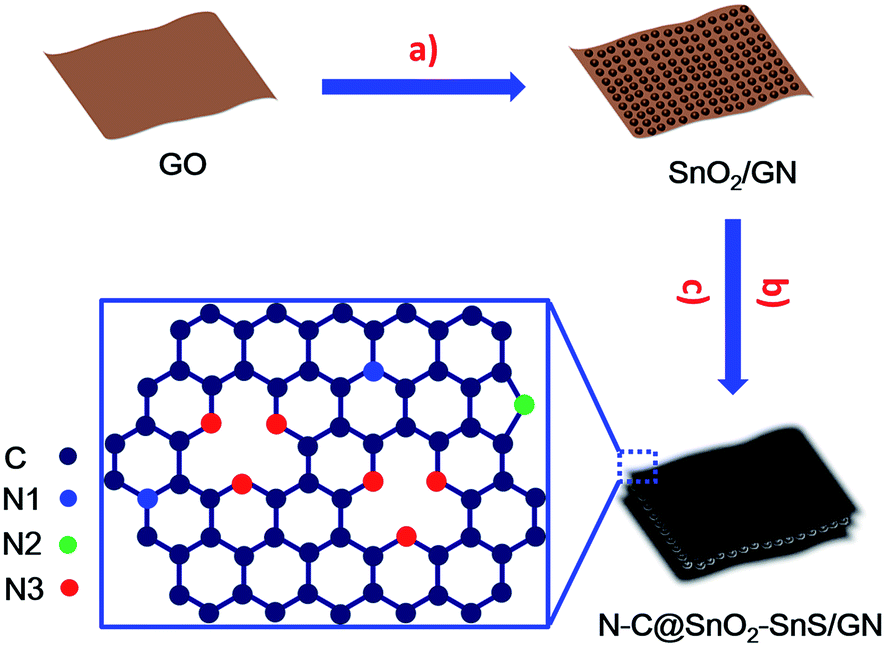 | ||
| Fig. 1 Schematic illustration of the fabrication procedure of N–C@SnO2–SnS/GN. (a) In situ growth of SnO2 NPs on GO in EG/water. (b) Hydrothermal treatment of SnO2/GNs in the presence of sucrose and NH4SCN. (c) Thermal treatment of the hybrid produced from the hydrothermal treatment. | ||
The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of N–C@SnO2–SnS/GN and C@SnO2/GN (Fig. 2a and b, S1a, S1c and S2a†) indicate that both samples have sheet-like architectures with uniform decoration of NPs, which are inherited from the 2D morphology of SnO2/GN (Fig. S1b and S2b†). The smooth strips without SnO2 NPs can be found on the edges of N–C@SnO2–SnS/GN and C@SnO2/GN (Fig. 2c and S2c†), suggesting that the SnO2 NPs are enwrapped in the carbon layers. High-resolution TEM (HRTEM) images (Fig. 2d and e) display the existence of two distinct particle morphologies in N–C@SnO2–SnS/GN. The lattice spacings of the two domains in Fig. 2f are 0.33 and 0.29 nm, corresponding to the (110) plane of SnO2 and the (101) plane of SnS, respectively.19,42 Strongly distinct from N–C@SnO2–SnS/GN, C@SnO2/GN only exhibits the lattice spacing for SnO2 (Fig. S2d†). The atomic force microscopy (AFM) images (Fig. 2f and S3†) suggest that the thickness of SnO2/GN and N–C@SnO2–SnS/GN are about 12 and 26 nm, respectively. These results unambiguously confirm that an additional carbon layer with a uniform thickness of about 7 nm has been successfully introduced on both sides of SnO2/GN, in agreement with the TEM results (Fig. 2c).
 | ||
| Fig. 2 (a) SEM image of N–C@SnO2–SnS/GN; (b) and (c) TEM images of N–C@SnO2–SnS/GN; (d) HRTEM image of N–C@SnO2–SnS/GN; (e) the enlarged image of the square area in (d); and (f) AFM image of N–C@SnO2–SnS/GN. | ||
In the X-ray diffraction (XRD) patterns of C@SnO2/GN (Fig. 3a) and N–C@SnO2/GN (Fig. S4†), five diffraction peaks with 2θ value as 26.61°, 33.89°, 37.94°, 51.78°, and 65.94 °can be assigned to the (110), (101), (211), (200), and (301) faces of rutile SnO2 (JCPDS no. 41-1445).42 In contrast, the additional narrow peaks in the XRD patterns of N–C@SnO2–SnS/GN are attributed to the SnS phase (JCPDS no. 65-3766),19 which suggests the existence of both crystallized SnO2 and SnS NPs in N–C@SnO2–SnS/GN. According to the thermogravimetric analysis (TGA) results (Fig. 3b), the weight content of carbon shells in N–C@SnO2–SnS/GN is ∼34 wt%. As indicated by the Raman spectra of C@SnO2/GN and N–C@SnO2–SnS/GN (Fig. 3c), both samples exhibit two evident peaks at ∼1344 and ∼1584 cm−1, which can be attributed to D (disordered) and G (ordered) bands of carbon, respectively.43 The intensity ratios of D and G bands (ID/IG) of C@SnO2/GN and N–C@SnO2–SnS/GN are 0.70 and 0.83, respectively. The higher ID/IG value of N–C@SnO2–SnS/GN could be ascribed to defects introduced in the carbon shell by nitrogen doping.44 As indicated by their nitrogen adsorption and desorption isotherms (Fig. 3d), the Brunauer–Emmett–Teller (BET) specific surface area of N–C@SnO2–SnS/GN is 129 m2 g−1, which is much higher than that of C@SnO2/GN (52 m2 g−1), presumably owing to the pores and structural defects generated during the thermal decomposition of NH4SCN.38
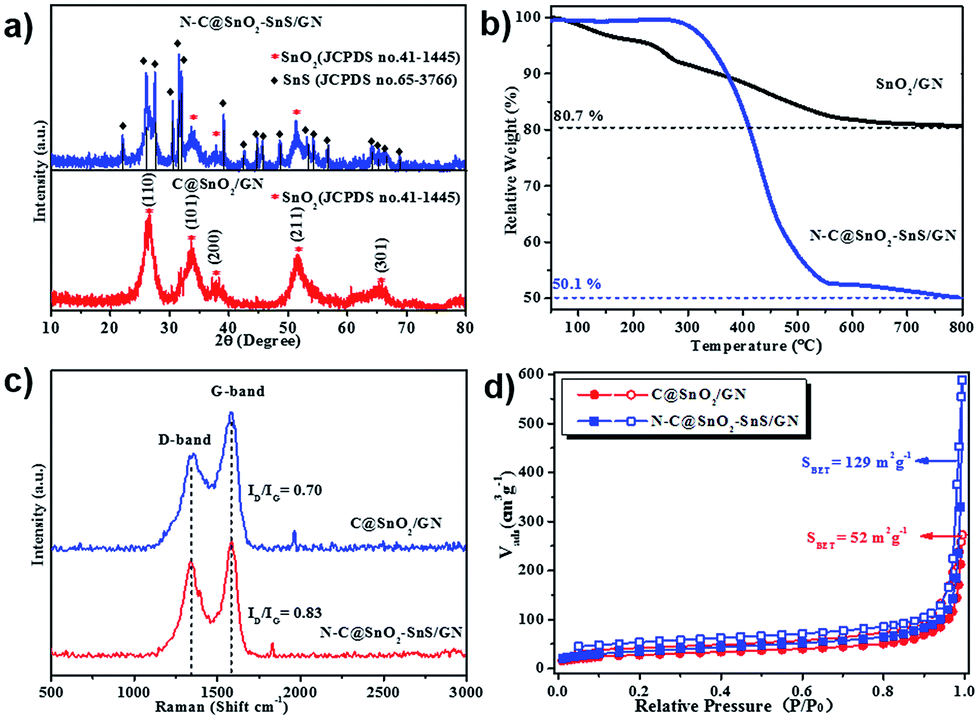 | ||
| Fig. 3 (a) XRD patterns of C@SnO2/GN and N–C@SnO2–SnS/GN; (b) TGA curves of SnO2/GN and N–C@SnO2–SnS/GN; (c) Raman spectra of C@SnO2/GN and N–C@SnO2–SnS/GN; (d) nitrogen adsorption and desorption isotherms of C@SnO2/GN and N–C@SnO2–SnS/GN (obtained at 77.3 K). | ||
The chemical compositions of C@SnO2/GN, N–C@SnO2/GN and N–C@SnO2–SnS/GN were further probed by X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of N–C@SnO2–SnS/GN indicate the presence of C, N, O, S, and Sn elements (Fig. S5a†). The C 1s spectra of N–C@SnO2–SnS/GN (Fig. 4a) display four fitted peaks at 284.7, 285.5, 286.7 and 288.1 eV. The high intensity of the peak at 284.7 eV suggests that the carbon atoms of N–C@SnO2–SnS/GN predominantly exist in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C configuration. Additionally, the strong peaks at 285.5 and 286.7 eV reveal that nitrogen has been doped in the carbon shell and bonded together with carbon as sp2 C–N or sp3 C–N. The peak at 288.1 eV indicates the presence of C–O configuration, confirming the existence of oxygenated functional groups of GO; nevertheless, the low intensity of this peak testifies that most GO has been reduced into graphene.12,45 The N 1s spectra of N–C@SnO2–SnS/GN (Fig. 4b) can be assigned to pyridine N (398.3 eV, 43%), pyrrole N (400.1 eV, 45%), and quaternary N (401.1 eV, 12%), respectively.29,45–47 The S 2p spectra of N–C@SnO2–SnS/GN (Fig. 4c) only show S 2p3/2 peak at 162.1 eV, which can be ascribed to the presence of SnS.48–50 Based on the results from XPS analysis, the contents of N and S in the N–C@SnO2–SnS/GN are calculated to be 1.6 at% and 1.8 at%, respectively. Similarly, the amount of N in N–C@SnO2/GN is also determined as 1.6 at% by its XPS results (Fig. S5b†). In the XPS spectra of C@SnO2/GN, the binding energy (BE) peaks of Sn 3d3/2 (496.0 eV) and Sn 3d5/2 (487.6 eV) should be derived from SnO2 NPs.49 In contrast, an obvious shift of the BE peaks of Sn 3d3/2 and Sn 3d5/2 can be observed in the XPS spectra of N–C@SnO2–SnS/GN, confirming the existence of both Sn4+ and Sn2+ in the hybrid (Fig. 4d).47–51
C configuration. Additionally, the strong peaks at 285.5 and 286.7 eV reveal that nitrogen has been doped in the carbon shell and bonded together with carbon as sp2 C–N or sp3 C–N. The peak at 288.1 eV indicates the presence of C–O configuration, confirming the existence of oxygenated functional groups of GO; nevertheless, the low intensity of this peak testifies that most GO has been reduced into graphene.12,45 The N 1s spectra of N–C@SnO2–SnS/GN (Fig. 4b) can be assigned to pyridine N (398.3 eV, 43%), pyrrole N (400.1 eV, 45%), and quaternary N (401.1 eV, 12%), respectively.29,45–47 The S 2p spectra of N–C@SnO2–SnS/GN (Fig. 4c) only show S 2p3/2 peak at 162.1 eV, which can be ascribed to the presence of SnS.48–50 Based on the results from XPS analysis, the contents of N and S in the N–C@SnO2–SnS/GN are calculated to be 1.6 at% and 1.8 at%, respectively. Similarly, the amount of N in N–C@SnO2/GN is also determined as 1.6 at% by its XPS results (Fig. S5b†). In the XPS spectra of C@SnO2/GN, the binding energy (BE) peaks of Sn 3d3/2 (496.0 eV) and Sn 3d5/2 (487.6 eV) should be derived from SnO2 NPs.49 In contrast, an obvious shift of the BE peaks of Sn 3d3/2 and Sn 3d5/2 can be observed in the XPS spectra of N–C@SnO2–SnS/GN, confirming the existence of both Sn4+ and Sn2+ in the hybrid (Fig. 4d).47–51
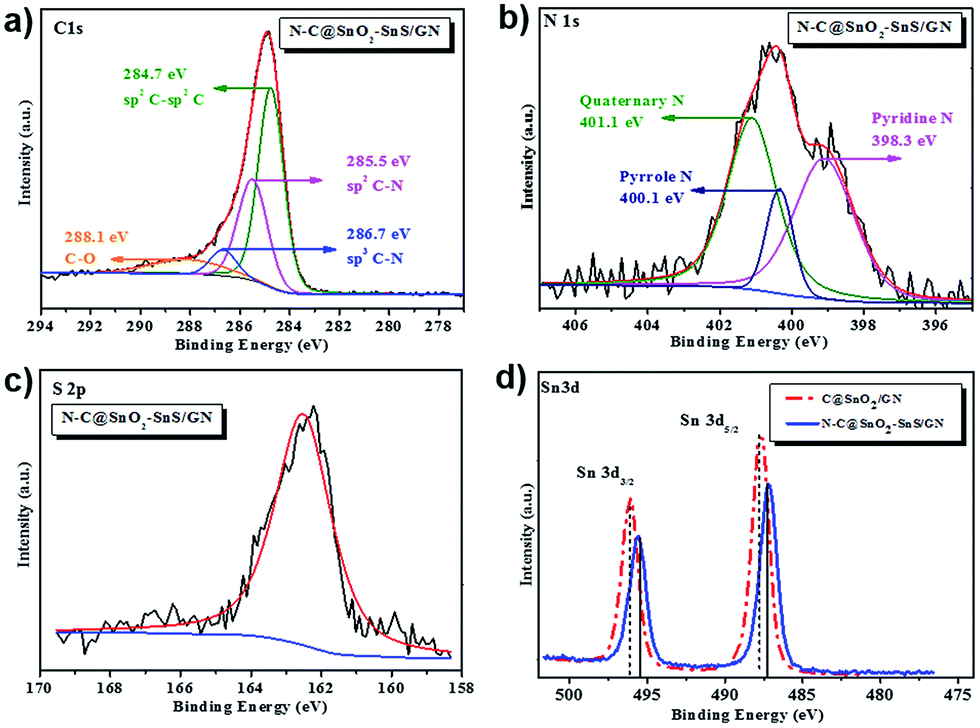 | ||
| Fig. 4 (a) C 1s XPS spectra of N–C@SnO2–SnS/GN; (b) N 1s XPS spectra of N–C@SnO2–SnS/GN; (c) S 2p XPS spectra of N–C@SnO2–SnS/GN; (d) Sn 3d XPS spectra of C@SnO2/GN and N–C@SnO2–SnS/GN. | ||
As shown in the FTIR spectra of N–C@SnO2–SnS/GN (Fig. S6†), the evident peaks at 1220 and 1380 cm−1 can be assigned to the stretching vibrations of C–N and N–CH3,52 respectively. The bands around 1600 cm−1 indicates the existence of CC stretching vibration.53 The absorbance at ca. 650 cm−1 is attributed to the out-of-plane N–H deformation vibration.54 Additionally, the N–H symmetric stretching vibration appears at ca. 3418 cm−1.55 The FTIR analysis clearly demonstrates the existence of C–N, N–H, and C–O56 groups in the N-doped carbon, which agrees well with the XPS results.
The core–shell structure and high heteroatom content of N–C@SnO2–SnS/GN make it an appealing candidate for the anode material of LIBs. To determine the lithiation/delithiation mechanism of N–C@SnO2–SnS/GN, it was assembled as coin cells and tested by a cyclic voltammetry (CV) experiment at a scanning rate of 0.1 mV s−1 in the voltage range of 0.01–3 V vs. Li+/Li (Fig. 5a). During the initial cathodic scan, the first small peak at 1.1 V can be ascribed to the irreversible decomposition of SnS to Sn and Li2S [eqn (1)].17,19–21 It should be note that the inert phase of Li2S can effectively buffer the volume expansion of LixSn (0 < x ≤ 4.4) alloys during the lithiation/delithiation process, which thus is expected to endow N–C@SnO2–SnS/GN with good cycling stability.20,21,31,32 The other three cathodic peaks at 0.7, 0.12, and 0.01 V can be associated with the formation of Li2O [eqn (2)],49–51 LixSn alloys [eqn (3)] and lithium-inserted carbonaceous materials LixC [eqn (4)],57–59 respectively. Accordingly, in the anodic processes, oxidation peaks at 0.12, 0.52, and 1.3 V can be attributed to the extraction of lithium from carbonaceous materials, reversible de-alloying of LixSn, and partial transformation of Li2O to SnO2,57–59 respectively. Furthermore, the small anodic peak at 1.9 V in Fig. 5a is produced by transformation from Li2S to polysulfides.60,61 The shape of the CV profiles from N–C@SnO2–SnS/GN in the following cycles does not change significantly, which implies the completion of the irreversible reactions and the formation of stable SEI layers on the surface of the electrode.
| SnS + 2Li+ + 2e− → Sn + Li2S | (1) |
| SnO2 + 4Li+ + 4e− → Sn + 2Li2O | (2) |
| Sn + xLi+ + xe− ↔ LixSn (0 < x ≤ 4.4) | (3) |
| C + xLi+ + xe− ↔ LixC | (4) |
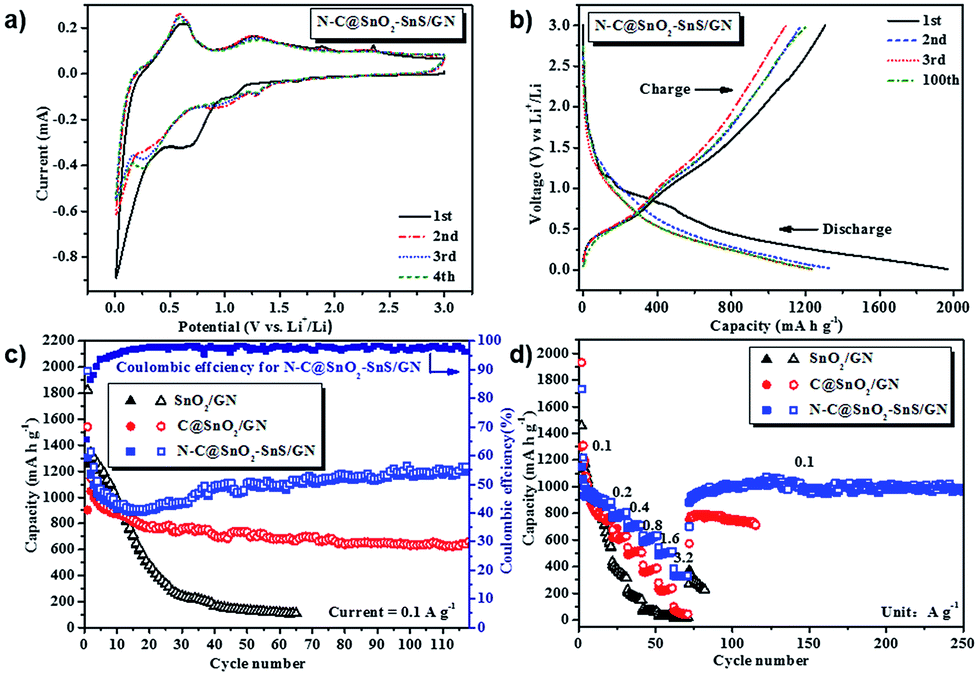 | ||
| Fig. 5 Electrochemical characterizations of the samples: (a) cyclic voltammogram (CV) curves of the N–C@SnO2–SnS/GN electrode at a scanning rate of 0.1 mV s−1 in the voltage range of 0.01–3 V vs. Li+/Li; (b) galvanostatic charge–discharge profile of the N–C@SnO2–SnS/GN electrode at a current rate of 0.1 A g−1 in the voltage range of 0.01–3 V vs. Li+/Li; (c) cycle performances of N–C@SnO2–SnS/GN, C@SnO2/GN, and SnO2/GN electrodes at a current rate of 0.1 A g−1 in the voltage range of 0.01–3 V vs. Li+/Li; (d) rate performances of N–C@SnO2–SnS/GN, C@SnO2/GN, and SnO2/GN electrodes in the voltage range of 0.01–3 V vs. Li+/Li. | ||
Correspondingly, the profile of N–C@SnO2/GN (Fig. S7†) strongly resembles that of N–C@SnO2–SnS/GN, except for the absence of a cathodic peak at 1.1 V and an anodic peak at 1.9 V, which indicates the lack of a SnS phase in N–C@SnO2/GN.
The electrochemical behavior of N–C@SnO2–SnS/GN was further investigated by galvanostatic discharge–charge measurements at a current density of 0.1 A g−1 within a voltage window of 0.01–3 V (Fig. 5b). Initial discharge and charge capacities of the N–C@SnO2–SnS/GN electrode are 1970 and 1305 mA h g−1, respectively, corresponding to a columbic efficiency (CE) of 66.3%. By contrast, N–C@SnO2/GN (Fig. S8†) and C@SnO2/GN (Fig. 5c) offer much lower discharge and charge capacities (1670 and 1029 mA h g−1 for N–C@SnO2/GN and 1543 and 903 mA h g−1 for C@SnO2/GN) in the first cycle, with CEs of 61.6% and 58.5%, respectively. As for SnO2/GN, it delivers initial discharge and charge capacities of 1823 and 1257 mA h g−1, with a CE of 68.9%. The capacity loss in the first cycle can be attributed to the formation of SEI film.17,19–21 It is worth noting that the capacities of the three electrodes are even higher than the theoretical capacities of SnO2 and SnS. In fact, the theoretical capacity of SnO2 (782 mA h g−1) is based on the conventional alloying mechanism, in which the reduction reaction of SnO2 [eqn (2)] is assumed to be irreversible.10,51 In this work, the peak located at 1.0–0.4 V in the CV curve of N–C@SnO2–SnS/GN (Fig. 5a) indicates that this reduction process is highly reversible. This implies that all the Sn in the alloy can be oxidized to Sn4+ during delithiation, which will effectively improve the capacity of the SnO2-based anodes over 1236 mA h g−1.27,62 In addition, the coexistence of two different tin-based phases could help to provide buffers against the volumetric changes of electrodes during cycling63 and improve the diffusion of lithium along grain boundaries of the two phases.33 It is also supposed that an organic polymeric/gel like film can be formed on the interface of the core/shell structured electrodes, which can also enhance their capacity via a “pseudo-capacitance”.64–67 Moreover, the 2D carbon shell of N–C@SnO2–SnS/GN could store lithium at the same time and serve as a mini current collector, facilitating electron transfer during lithiation and delithiation process.68 Besides, the nitrogen atoms doping in the carbon shell may provide accessible lithium active sites, as well as enhanced electrical conductivity.69 The combination of the above advantages thus enables N–C@SnO2–SnS/GN electrode to deliver excellent gravimetric capacity and good volumetric capacity (Fig. S9†).
During the cycling performance tests at a current density of 0.1 A g−1, the N–C@SnO2–SnS/GN electrode displayed an ever-rising capacity from the 15th cycle onwards. The increased capacity should be mainly ascribed to slow activation of electrode materials, promoted by gradual penetration of electrolyte through the nitrogen-doped porous carbon shells.65,70,71 After 110 cycles, a high capacity of 1236 mA h g−1 can still be delivered by N–C@SnO2–SnS/GN (Fig. 5c). Under the same conditions, the C@SnO2/GN-based electrode only gives a reversible capacity of 647 mA h g−1 after 110 cycles. In contrast, the capacity of N–C@SnO2/GN falls to 828 mA h g−1 after 12 cycles (Fig. S8†), while the capacity of SnO2/GN drops drastically to 250 mA h g−1 after 30 cycles, highlighting the crucial role of the heteroatom doping and carbon shell on the cycling stability of the electrode materials.
The rate capability of N–C@SnO2–SnS/GN, C@SnO2/GN and SnO2/GN were further evaluated at progressively increased current densities ranging from 0.1 to 3.2 A g−1 (Fig. 5d). Remarkably, the reversible capacities of N–C@SnO2–SnS/GN are about 800, 700, 650, 500, and 350 mA h g−1 at 0.2, 0.4, 0.8, 1.6, and 3.2 A g−1, respectively. Upon resetting the current density to 0.1 A g−1, a stable reversible capacity of 1000 mA h g−1 can be retained by the N–C@SnO2–SnS/GN electrode. In contrast, the capacities of C@SnO2/GN and SnO2/GN at 3.2 A g−1 are only maintained as 50 and 17 mA h g−1, respectively.
The excellent cycling stability and rate capability of N–C@SnO2–SnS/GN can be attributed to both 2D core–shell architecture and dual-doped heteroatoms. The effective combination of N-doped carbon shells and graphene-supported SnO2–SnS NPs in a 2D sheet-like manner greatly facilitates accommodation of the volume variation of the electrode and the rapid transport of electrons and lithium ions. The SEM with elemental mapping images of the N–C@SnO2–SnS/GN electrode after cycling (Fig. S10 and S11†) demonstrate that the nanosheet morphology was preserved.
Additionally, in their electrochemical impedance spectra (EIS), N–C@SnO2–SnS/GN manifests a more depressed semicircle at high frequencies than C@SnO2/GN (Fig. S12a†). According to the equivalent circuit (Fig. S12b†), the contact resistance (Rf) and charge transfer resistance (Rct) of N–C@SnO2–SnS/GN are fitted to be 17.69 and 70.34 Ω, which are significantly lower than those of C@SnO2/GN (44.6 and 229 Ω) (Table S1†). The small resistances explain the outstanding rate capability of N–C@SnO2–SnS/GN. Obviously, N-doped carbon shells in N–C@SnO2–SnS/GN significantly enhance electrical conductivity and structural stability of the composites. Moreover, the incorporation of nitrogen atoms in the carbon frameworks can facilitate the wetting process of an electrolyte and provide extra active sites for lithium storage.6,26–30,46,47,72
Conclusions
With SnO2/GN as precursor, nitrogen and sulfur dual-doped N–C@SnO2–SnS/GN is constructed in this work by employing NH4SCN as a doping agent for the carbon components and SnO2 NPs. The 2D hybrid contains both N-doped carbon coating layers and a dual-phase component of SnO2–SnS, which efficiently improve the electrochemical reactivity of the hybrid as the anode in LIB. Such dual-doping strategy is expected to be further applied to construct other high performance electrode materials with potential applications in sodium ion batteries, magnesium ion batteries, or lithium sulfur batteries.Acknowledgements
This work was financially supported by 973 Program of China (2012CB933404 and 2014CB239701), Natural Science Foundation of China (21320102006, 61235007, 61575121, 21572132 and 21372155), Professor of Special Appointment at Shanghai Institutions of Higher Learning, MPI-SJTU Partner Group Project for Polymer Chemistry of Graphene Nanoribbons. X. F acknowledges the funding support from ERC grant on 2DMATER and EC under Graphene Flagship (No. CNECT-ICT-604391). We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for the characterization of materials.Notes and references
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- N. F. Sciortino, K. R. Scherl-Gruenwald, G. Chastanet, G. J. Halder, K. W. Chapman, J.-F. Létard and C. J. Kepert, Angew. Chem., Int. Ed., 2012, 51, 9944 CrossRef CAS.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Small, 2013, 9, 1173–1187 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS.
- Z. Zhu, S. Wang, J. Du, Q. Jin, T. Zhang, F. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed.
- B. Luo, B. Wang, X. Li, Y. Jia, M. Liang and L. Zhi, Adv. Mater., 2012, 24, 3538–3543 CrossRef CAS PubMed.
- C. Botas, D. Carriazo, G. Singh and T. Rojo, J. Mater. Chem. A, 2015, 3, 13402–13410 CAS.
- D.-H. Nam, J. W. Kim, J.-H. Lee, S.-Y. Lee, H.-A. S. Shin, S.-H. Lee and Y.-C. Joo, J. Mater. Chem. A, 2015, 3, 11021–11030 CAS.
- X. Zhou, W. Liu, X. Yu, Y. Liu, Y. Fang, S. Klankowski, Y. Yang, J. E. Brown and J. Li, ACS Appl. Mater. Interfaces, 2014, 6, 7434–7443 CAS.
- R. Hu, H. Zhang, J. Liu, D. Chen, L. Yang, M. Zhu and M. Liu, J. Mater. Chem. A, 2015, 3, 15097–15107 CAS.
- X.-T. Chen, K.-X. Wang, Y.-B. Zhai, H.-J. Zhang, X.-Y. Wu, X. Wei and J.-S. Chen, Dalton Trans., 2014, 43, 3137–3143 RSC.
- D. Aurbach, A. Nimberger, B. Markovsky, E. Levi, E. Sominski and A. Gedanken, Chem. Mater., 2002, 14, 4155–4163 CrossRef CAS.
- Y. Huang, D. Wu, J. Wang, S. Han, L. Lv, F. Zhang and X. Feng, Small, 2014, 10, 2226–2232 CrossRef CAS PubMed.
- Y. Su, S. Li, D. Wu, F. Zhang, H. Liang, P. Gao, C. Cheng and X. Feng, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS.
- S. H. Choi and Y. C. Kang, Small, 2014, 10, 474–478 CrossRef CAS PubMed.
- B. Luo, Y. Fang, B. Wang, J. Zhou, H. Song and L. Zhi, Energy Environ. Sci., 2012, 5, 5226–5230 CAS.
- J. Cai, Z. Li and P. K. Shen, ACS Appl. Mater. Interfaces, 2012, 4, 4093–4098 CAS.
- D. D. Vaughn, O. D. Hentz, S. Chen, D. Wang and R. E. Schaak, Chem. Commun., 2012, 48, 5608–5610 RSC.
- Y. Li, J. P. Tu, X. H. Huang, H. M. Wu and Y. F. Yuan, Electrochim. Acta, 2006, 52, 1383–1389 CrossRef CAS.
- J. Liu, Y. Wen, P. A. van Aken, J. Maier and Y. Yu, J. Mater. Chem. A, 2015, 3, 5259–5265 CAS.
- G. Wang, J. Peng, L. Zhang, J. Zhang, B. Dai, M. Zhu, L. Xia and F. Yu, J. Mater. Chem. A, 2015, 3, 3659–3666 CAS.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- X. Meng, Y. Zhong, Y. Sun, M. N. Banis, R. Li and X. Sun, Carbon, 2011, 49, 1133–1144 CrossRef CAS.
- J. Liang, Z. Cai, Y. Tian, L. Li, J. Geng and L. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 12148–12155 CAS.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D.-M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682–2690 CrossRef CAS.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 CAS.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- J.-G. Kang, J.-G. Park and D.-W. Kim, Electrochem. Commun., 2010, 12, 307–310 CrossRef CAS.
- Y. Lee, S.-R. Bae, B. J. Park, D. W. Shin, W. J. Chung and Y.-M. Kang, J. Am. Ceram. Soc., 2012, 95, 2272–2276 CrossRef CAS.
- A. C. Bose, D. Kalpana, P. Thangadurai and S. Ramasamy, J. Power Sources, 2002, 107, 138–141 CrossRef CAS.
- J. Zhu, D. Lei, G. Zhang, Q. Li, B. Lu and T. Wang, Nanoscale, 2013, 5, 5499–5505 RSC.
- D. Zhang, X. Wen, L. Shi, T. Yan and J. Zhang, Nanoscale, 2012, 4, 5440–5446 RSC.
- X. Wen, D. Zhang, T. Yan, J. Zhang and L. Shi, J. Mater. Chem. A, 2013, 1, 12334–12344 CAS.
- Y. Yang, K. Wu, R. Pang, X. Zhou, Y. Zhang, X. Wu, C. Wu, H. Wu and S. Guo, RSC Adv., 2013, 3, 14016–14020 RSC.
- M. Ohta, S. Hirai, H. Kato, V. V. Sokolov and V. V. Bakovets, Mater. Trans., 2009, 50, 1885–1889 CrossRef CAS.
- M. Zhou, Y. Liu, J. Chen and X. Yang, J. Mater. Chem. A, 2015, 3, 1068–1076 CAS.
- R. Liu, D. Li, C. Wang, N. Li, Q. Li, X. Lü, J. S. Spendelow and G. Wu, Nano Energy, 2014, 6, 73–81 CrossRef CAS.
- K.-C. Hsu, C.-E. Liu, P.-C. Chen, C.-Y. Lee and H.-T. Chiu, J. Mater. Chem., 2012, 22, 21533–21539 RSC.
- Y. Wei, C. Gao, F.-L. Meng, H.-H. Li, L. Wang, J.-H. Liu and X.-J. Huang, J. Phys. Chem. C, 2012, 116, 1034–1041 CAS.
- L.-F. Chen, Z.-H. Huang, H.-W. Liang, W.-T. Yao, Z.-Y. Yu and S.-H. Yu, Energy Environ. Sci., 2013, 6, 3331–3338 CAS.
- H. B. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- P. Chen, T.-Y. Xiao, Y.-H. Qian, S.-S. Li and S.-H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- L.-L. Tian, X.-Y. Wei, Q.-C. Zhuang, C.-H. Jiang, C. Wu, G.-Y. Ma, X. Zhao, Z.-M. Zong and S.-G. Sun, Nanoscale, 2014, 6, 6075–6083 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed.
- S. Cheng and G. Conibeer, Thin Solid Films, 2011, 520, 837–841 CrossRef CAS.
- D. Chen, G. Shen, K. Tang, S. Lei, H. Zheng and Y. Qian, J. Cryst. Growth, 2004, 260, 469–474 CrossRef CAS.
- Z. Zainal, M. Z. Hussein and A. Ghazali, Sol. Energy Mater. Sol. Cells, 1996, 40, 347–357 CrossRef CAS.
- Y. Tang, D. Wu, S. Chen, F. Zhang, J. Jia and X. Feng, Energy Environ. Sci., 2013, 6, 2447–2451 CAS.
- H. C. Choi, S. Y. Bae and J. Park, Appl. Phys. Lett., 2004, 85, 5742–5744 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358–364 CrossRef CAS.
- J. H. Kaufman and S. Metin, American Physical Society, 1998, 39, 13053–13060 Search PubMed.
- G.-Q. Yu, et al. , Diamond Relat. Mater., 2002, 11, 1633–1637 CrossRef CAS.
- Z. Wang, M. A. Fierke and A. Stein, J. Electrochem. Soc., 2008, 155, A658–A663 CrossRef CAS.
- M.-Y. Li, C.-L. Liu, Y. Wang and W.-S. Dong, J. Electrochem. Soc., 2011, 158, A296–A301 CrossRef CAS.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868–2874 CrossRef CAS.
- Y. Li, J. P. Tu, X. H. Huang, H. M. Wu and Y. F. Yuan, Electrochem. Commun., 2007, 9, 49–53 CrossRef CAS.
- M. Zhang, D. Lei, X. Yu, L. Chen, Q. Li, Y. Wang, T. Wang and G. Cao, J. Mater. Chem., 2012, 22, 23091–23097 RSC.
- R. Wang, C. Xu, J. Sun, L. Gao and H. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3427–3436 CAS.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2013, 1, 7181–7184 CAS.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252–5256 CAS.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812–1816 CrossRef CAS PubMed.
- Y. Jiang, Z.-J. Jiang, L. Yang, S. Cheng and M. Liu, J. Mater. Chem. A, 2015, 3, 11847–11856 CAS.
- J. Ding, Z. Li, H. L. Wang, K. Cui, A. Kohandehghan, X. H. Tan, D. Karpuzov and D. Mitlin, J. Mater. Chem. A, 2015, 3, 7100–7111 CAS.
- Y. Q. Zhu, H. Z. Guo, H. Z. Zhai and C. B. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 2745–2753 CAS.
- Y. P. Tang, D. Q. Wu, Y. Y. Mai, H. Pan, J. Cao, C. Q. Yang, F. Zhang and X. L. Feng, Nanoscale, 2014, 6, 14679–14685 RSC.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS PubMed.
- X. Li, Y. Ma, L. Qin, Z. Zhang, Z. Zhang, Y.-Z. Zheng and Y. Qu, J. Mater. Chem. A, 2015, 3, 2158–2165 CAS.
- X. Wang, Q. Weng, X. Liu, X. Wang, D.-M. Tang, W. Tian, C. Zhang, W. Yi, D. Liu, Y. Bando and D. Golberg, Nano Lett., 2014, 14, 1164–1171 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06617d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |