
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of efficient photocatalysts for water oxidation and dye degradation reactions using CoCl2 eutectics†
Christian
Fettkenhauer
a,
Xinchen
Wang
b,
Kamalakannan
Kailasam‡
c,
Markus
Antonietti
a and
Dariya
Dontsova
*a
aMax-Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, 14424 Potsdam, Germany. E-mail: dariya.dontsova@mpikg.mpg.de
bState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350002, People's Republic of China
cInstitut für Chemie, Sekr. TC 2, Technische Universität Berlin, Englische Str. 20, 10587 Berlin, Germany
First published on 11th September 2015
Abstract
A simple one-step procedure for the preparation of cobalt oxide–carbon nitride composites by carrying out the thermal condensation reaction of carbon nitride precursors in cobalt(II) chloride-containing salt melts is developed. This method enables us to control the structure of the polymer constituent by the proper selection of the second eutectic component. In this respect, alkali metal chlorides and zinc chloride give rise to poly(triazine imides), while tin(II) chloride delivers melon-based polymers. The crystallinity of the carbon nitride polymer phase can be influenced by varying the concentration of the precursor in the melt. On the other hand, cobalt oxide loading in the final products can be increased by decreasing the precursor concentration in the melt. The products are highly active photocatalysts for Rhodamine B degradation as exemplified by SnCl2/CoCl2-derived solids, and the water oxidation reaction (WOR). The activity of the products in the WOR is comparable with that of the bench composite photocatalyst prepared by using the three-step procedure that includes the synthesis of Co3O4 nanoparticles as a separate step, thus illustrating the advantages of the developed salt melt assisted approach.
Introduction
Photocatalytic water splitting into hydrogen and oxygen over a semiconductor photocatalyst using solar energy is a promising way of converting light energy into chemical energy of hydrogen1 that is inspired by natural photosynthesis. Water oxidation is a crucial step of the overall process since it requires 4-electron and 4-proton transfers as well as O–O bond formation, and is regarded as a critical bottleneck for creating technical photosynthetic devices, since this reaction completes the solar fuel cycle.2,3Graphitic carbon nitride materials (CN-polymers) with the stoichiometry of CxNyHz (x = 3, y ≈ 4.2–5.2, z ≈ 1–1.5) are processable4,5 organic polymeric semiconductors absorbing the high energy fraction of the visible light and having suitable band positions to perform water splitting6 which makes them promising candidates for full artificial photosynthesis. These polymers additionally offer the advantages of high thermal and chemical stability against oxidation.7 However, some carbon nitrides are known to undergo photocorrosion due to the limited mobility of the photogenerated holes and resulting self-oxidation while releasing nitrogen gas. The photocorrosion of the polymers can be minimized by the use of an efficient co-catalyst.6,8
Metal oxides as water oxidation catalysts are attractive because of their robustness, and oxides of noble metals, in particular Ir9–11 and Ru,12–14 are among the most efficient known oxygen evolving catalytic materials. However, their scarcity and the resulting high cost caused the need for exploring robust oxides of earth-abundant metals as efficient multi-electron catalysts for water oxidation, in particular first row transition metal oxides. Among these, cobalt oxides offer a suitable balance between reactivity, structural stability and resistance in the highly oxidative chemical environment of the oxygen evolution reaction.15
Recently, cobalt oxide and cobalt(II) hydroxide containing carbon nitride composites were reported to be promising for water oxidation.8,16 However, so far they were mostly prepared by using multistep procedures. A single-step synthesis procedure by mixing CoCl2 with dicyandiamide (DCDA) resulted in only a slight increase of the photocatalytic activity compared to the bulk CN-polymer.17 Thus, there is still a need for simple preparation methods of highly active composite photocatalysts comprising carbon nitride and catalytically active cobalt species.
Salt melt assisted synthesis of carbon nitrides can be used for the preparation of functional CN-polymers by using one-step procedures.18–21 Bivalent metals remain in the final composites, partially as oxides resulting from the aqueous work-up of the reaction mixtures or the presence of oxygen during the synthesis.20,21 Making use of this observation, we here present a one-step procedure for the preparation of cobalt-containing carbon nitride-based photocatalysts by using the condensation reaction of dicyandiamide in CoCl2-containing eutectic salt mixtures.
Experimental section
Materials
Sodium chloride (99.5%), potassium chloride (99.0%), Rhodamine B (95.0%) and trithiocyanuric acid (95.0) were purchased from Sigma-Aldrich. Zinc chloride (98.0%), tin(II) chloride (98.0%), cobalt(II) chloride (99.0%), and silver nitrate (99.9%) were purchased from Alfa Aesar, and dicyandiamide (DCDA, 98%) from Merck. Lanthanum(III) oxide (99.9%) was purchased from Fluka analytics. All the chemicals were used without further purification.Synthesis procedure
Salts and dicyandiamide were ground together in a glovebox (mBraun Unilab, O2 < 0.1 ppm, H2O < 0.1 ppm) under an argon atmosphere according to eutectic compositions (Table S1†). Reaction mixtures (∼5–10 g) were transferred into porcelain crucibles and covered with lids. Crucibles were placed in the oven under a constant nitrogen flow (15 L min−1) and heated with a ramp of 10 K min−1 up to 550 °C. The temperature was maintained for 6 hours, and then the samples were cooled down. The crude products were washed first with deionized water (50–100 mL) and then with 1 M HCl solution (50–100 mL) in order to remove the salts and the products of their hydrolysis. Each step was carried out for 24 hours. The final products were isolated by filtration, then thoroughly washed with deionized water (500 mL) and dried in a vacuum oven (10 mbar) at 50 °C for 15 h. Reference CN was prepared by heating DCDA with the ramp of 2.3 °C min−1 up to 550 °C and subsequently maintaining at this temperature for 4 h under a constant nitrogen flow (15 mL min−1) in a covered crucible. CNS/3% Co3O4 was prepared according to the described procedure.8Rhodamine B degradation experiments
5 mg of the photocatalyst were dispersed in 5 mL of RhB solution in deionized water (10 mg L−1). The suspension was agitated in the dark for 0.5–2 h in order to reach a dark-adsorption equilibrium and then illuminated with an LED module emitting white light (λ > 420 nm, 50 W, Bridgelux) from the fixed distance of 5 cm. The RhB degradation was monitored as the decrease of the dye concentration over irradiation time. At specific time intervals (t), 300 µL aliquots of the reaction mixture were taken, diluted with 1.7 mL of deionized water, maintained for 30 minutes in the dark to enable catalyst precipitation, and then analyzed spectrophotometrically (T70 UV/VIS spectrophotometer of PG instruments Ltd). The reaction rate constants k were calculated as the slopes of the linear fits of [−ln(A/A0)] dependences on the irradiation time. Here, A0 and A are absorbance of the dye in the solution after dark equilibration and after irradiation time t respectively, measured at 554 nm, which are proportional to the dye concentrations in the solution (C0 and C).Photocatalytic oxygen evolution experiments
Characterization
Powder X-ray diffraction patterns were measured on a Bruker D8 Advance diffractometer equipped with a scintillation counter detector with CuKα radiation (λ = 0.15418 nm) by applying a 2θ step size of 0.05° and a counting time of 3 s per step. FT-IR spectra were recorded on a Varian 1000 FT-IR spectrometer equipped with an attenuated total reflection unit applying a resolution of 4 cm−1. Nitrogen adsorption/desorption measurements were performed after degassing the samples at 150 °C for 20 hours using a Quantachrome Quadrasorb SI-MP porosimeter at 77.4 K. The specific surface areas were calculated by applying the Brunauer–Emmett–Teller (BET) model to adsorption isotherms for 0.05 < p/p0 < 0.3 using the QuadraWin 5.05 software package. Elemental analysis (EA) was accomplished as a combustion analysis using a Vario Micro device. Scanning electron microscopy (SEM) images were obtained by using a LEO 1550-Gemini microscope. Optical absorbance spectra of powders were measured on a Shimadzu UV 2600 equipped with an integrating sphere. The absorption spectra of RhB solutions were recorded on a T70 UV/VIS spectrophotometer (PG instruments Ltd). The emission spectra were recorded using LS-50B, a Perkin-Elmer instrument. The excitation wavelength was 350 nm. Energy-dispersive X-ray spectroscopy (EDS) investigations were conducted on a JEOL JSM-7500F microscope using a Link ISIS-300 system (Oxford Microanalysis Group) equipped with a Si(Li) detector with an energy resolution of 133 eV, at 5–12 keV. X-ray photoelectron spectroscopy (XPS) was performed on a Multilab 2000 (Thermo) spectrometer equipped with the Al Kα anode (hν = 1486.6 eV). All spectra were referenced to the C 1s peak of adventitious carbon at 285.0 eV. For quantification purposes, survey spectra at pass energy of 50 eV and high-resolution spectra at pass energy of 20 eV were recorded and analyzed by using XPS Peak 4.1 software (by Raymond Kwok). The spectra were decomposed assuming line shapes as the sum of Gaussian (80%) and Lorentzian (20%) functions. Raw areas determined after subtraction of a Shirley background23 were corrected according to the following sensitivity factors:24 C1s – 0.25; N1s – 0.42; O1s – 0.66; Cl2p – 0.73, Co2p – 3.8. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed on a Perkin-Elmer ICP OES 8000.Results and discussion
First, the concentration of the carbon nitride precursor, dicyandiamide, was maintained constant by using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 DCDA to salts weight ratio, while the nature of the second component of eutectics was varied (see Table S1†). The composition of the resulting products is shown in Table S2,† as determined by elemental analysis, energy-dispersive X-ray spectroscopy (EDS) and inductively coupled plasma (ICP) studies. The obtained solids are mainly composed of carbon and nitrogen and are characterized by the C/N weight ratios of 0.58 to 0.63. These numbers are higher than the C/N weight ratio in the reference material prepared by the bulk condensation of DCDA (0.58), referred to as ref.-CN, but still slightly lower than the theoretical value for the perfect C3N4 (0.64). The CoCl2-derived solids contain significant amounts of oxygen that comes with the increased hydrogen content if compared to the ref.-CN; both are due to the presence of surface-adsorbed water and surface hydroxyl-groups as corroborated by the X-ray photoelectron spectroscopy (XPS) investigations.
:5 DCDA to salts weight ratio, while the nature of the second component of eutectics was varied (see Table S1†). The composition of the resulting products is shown in Table S2,† as determined by elemental analysis, energy-dispersive X-ray spectroscopy (EDS) and inductively coupled plasma (ICP) studies. The obtained solids are mainly composed of carbon and nitrogen and are characterized by the C/N weight ratios of 0.58 to 0.63. These numbers are higher than the C/N weight ratio in the reference material prepared by the bulk condensation of DCDA (0.58), referred to as ref.-CN, but still slightly lower than the theoretical value for the perfect C3N4 (0.64). The CoCl2-derived solids contain significant amounts of oxygen that comes with the increased hydrogen content if compared to the ref.-CN; both are due to the presence of surface-adsorbed water and surface hydroxyl-groups as corroborated by the X-ray photoelectron spectroscopy (XPS) investigations.
The XPS studies of a sample product obtained from the KCl/CoCl2 salt melt showed that the carbon nitride part of the composite is based on poly(triazine imide) (PTI). Namely, the C1s signal (Fig. 1a) contains four contributions: the major one at 288.5 eV assigned to CN3 bonds,25 and the minor ones at 286.9 eV corresponding to the surface C–OH groups and at 283.1 eV due to some carbide impurities, together with the peak of adventitious carbon at 285.0 eV. The N1s signal (Fig. 1b) can be deconvoluted into three peaks at 400.3 eV due to NHx groups (x = 1, 2), at 398.8 eV assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C bonds and minor contribution of the deprotonated nitrogen atoms at 397.1 eV. The absence of the fourth N1s peak of tertiary nitrogen and the high content of NHx groups expressed as a high peak area suggest that the obtained CN-polymer is based on s-triazine but not tri-s-triazine repeating units. This conclusion is further supported by the previous reports on the simulated26 and measured XPS spectra of poly(triazine imides).20
N–C bonds and minor contribution of the deprotonated nitrogen atoms at 397.1 eV. The absence of the fourth N1s peak of tertiary nitrogen and the high content of NHx groups expressed as a high peak area suggest that the obtained CN-polymer is based on s-triazine but not tri-s-triazine repeating units. This conclusion is further supported by the previous reports on the simulated26 and measured XPS spectra of poly(triazine imides).20
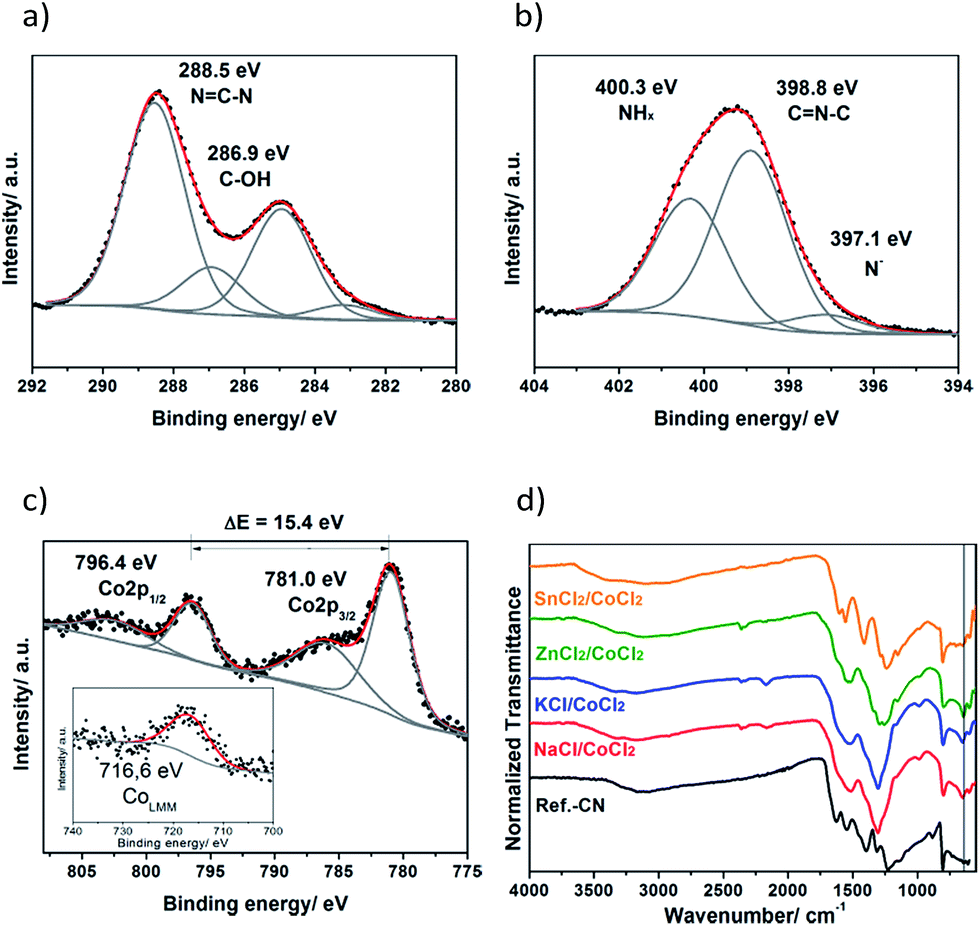 | ||
| Fig. 1 C1s (a), N1s (b) XPS spectra, Co2p spectrum (c) and CoLMM Auger line (inset in c) of the product prepared in KCl/CoCl2; FTIR spectra of the CoCl2-derived products and the reference CN-polymer (d). The vertical line in (d) indicates the 650 cm−1 position. | ||
Inductively coupled plasma (ICP) and energy-dispersive X-ray spectroscopy (EDS) investigations showed that the cobalt content in the composites is 3–6 wt% (Table S2†). Cobalt is present in both Co(II) and Co(III) oxidation states as follows from the results of the XPS studies (Fig. 1c). The Co2p spectrum (Fig. 1c) consists of the Co2p3/2 contribution at 781.0 eV, its corresponding satellite peak at 786.0 eV and the Co2p1/2 contribution at 796.4 eV along with the satellite peak at 802.4 eV. The Co2p1/2–Co2p3/2 spin–orbit splitting makes up 15.4 eV which is in between 15 eV observed for the pure Co(III) state and 16 eV typical for the pure Co(II) state,27 and points to the coexistence of Co(II) and Co(III) species in the products.
Further evidence of the presence of both valence states of cobalt is provided by the position of the CoLMM Auger line (Fig. 1c, inset). The peak is located at 716.6 eV that corresponds to a kinetic energy of 770 eV. The modified Auger parameter, α, can be calculated according to the following equation:28
| α = 1486.6 eV + KE (CoLMM) − KE (Co2p3/2) = 1551.0 eV |
This value of α is in between the reported values for CoO (α = 1549.8 eV) and Co3O4 (α = 1552.9 eV) thus indicating the coexistence of Co(II) and Co(III).
The charge of cobalt is balanced by oxygen and chlorine suggesting that Co is present as cobalt (II, III) oxide and, partially, as entrapped chloride containing complexes, likely, of cobalt(III), since they are known to be kinetically inert and could easily withstand the aqueous workup of the crude products. The O1s signal (Fig. S1a†) consists of three contributions with the binding energies at 533.1 eV due to the surface adsorbed water, at 531.5 eV assigned to the hydroxyl groups and the peak of cobalt (II, III) oxides at 529.9 eV.29,30 The Cl2p spectrum shown in Fig. S1b† reveals a single contribution at 197.6 eV assigned to Co–Cl bonds.31
SEM-EDS investigations revealed that the distribution of Co in the composites is fairly homogeneous, as illustrated by the corresponding elemental maps of KCl/CoCl2 and SnCl2/CoCl2-derived products in Fig. S2.†
The results of the FTIR studies shown in Fig. 1d further corroborate the XPS findings. Poly(triazine imides) are formed in NaCl-, KCl- and ZnCl2/CoCl2 salt melts as can be recognized by the presence of the distinct characteristic peak of the s-triazine ring at 650 cm−1.32 This peak is absent in the cases of the bulk carbon nitride and SnCl2/CoCl2 products suggesting that their structure is based on tri-s-triazine units. PXRD studies further corroborate this assumption (see below). The broad absorption band at 2400–3650 cm−1 suggests the presence of the surface hydroxyl groups, surface adsorbed water and terminal amino-groups in the obtained materials, while the main absorption band at 1100–1650 cm−1 is due to CN stretching and NH bending in aromatic heterocycles33 and is typical for carbon nitride polymers.18,34,35
Therefore, we conclude that the nature of the second component of the eutectic determines the chemical structure of the carbon nitride part of the composite. Here, alkali metal salts and ZnCl2 give rise to poly(triazine imides), while SnCl2 leads to the formation of melon-based products. This can be explained by the strength and reversibility of the binding of mono- (Na, K) and divalent (Zn, Sn) metal ions to the condensation intermediates such as melamine and melam during synthesis. Strong binding, typical for alkali metal ions and for ZnCl2 known to be a strong Lewis acid, stabilizes these s-triazine-based intermediates and prohibits their transformation to tri-s-triazine species, that leads to the s-triazine-based polymer, poly(triazine imide). Conversely, weak and reversible binding of SnCl2, a milder Lewis acid compared to ZnCl2, enables the conversion of the s-triazine ring to the tri-s-triazine one, but stabilizes 1D melon chains preventing their further condensation to form graphitic carbon nitride.21
The products are mainly short-range ordered as suggested by the appearance of the corresponding PXRD patterns (Fig. 2a) due to both poor solubilizing properties of CoCl2 being a weak Lewis acid and relatively high melting points of some eutectics (Table S1†). This results in an early phase demixing of the melt and polymerization intermediates preventing crystallization of the polymers from the melt. The two main peaks characterizing the interlayer stacking at ∼27° 2θ and the in-layer repeating motif at ∼13° 2θ are broadened in the obtained solids compared to the reference bulk CN-polymer. In the pattern of the SnCl2/CoCl2-derived product, the characteristic peak of the in-layer order is shifted to lower angles (2θ = 9.8°) suggesting the formation of a chain-like polymer, presumably melon, as previously reported for the SnCl2-derived products.21
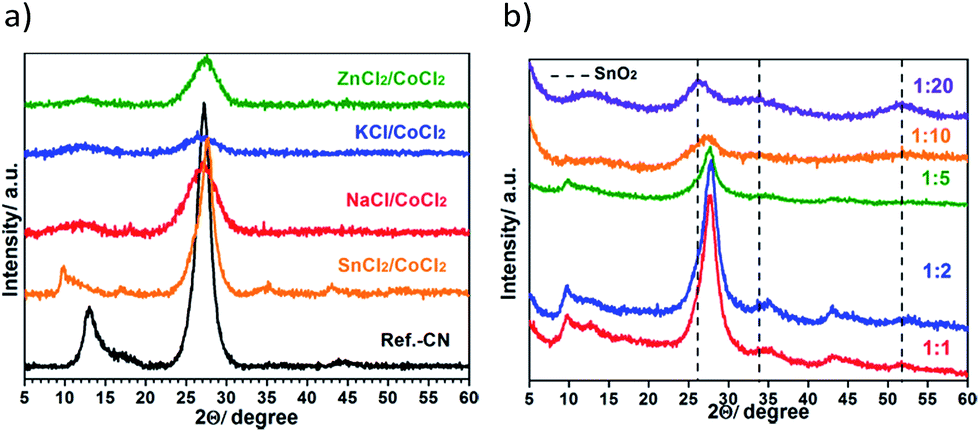 | ||
| Fig. 2 PXRD patterns of the products prepared in NaCl/CoCl2, KCl/CoCl2, ZnCl2/CoCl2 and SnCl2/CoCl2 eutectics at 1:5 precursor to salt weight ratio (a), in the SnCl2/CoCl2 eutectic using different precursor to salt weight ratios (b). | ||
However, the crystallinity of the resulting composites can be tuned by adjusting the precursor concentration in the melt. Fig. 2b shows PXRD patterns of the products prepared in the SnCl2/CoCl2 eutectics using different weight ratios of dicyandiamide to the salts, namely 1:1, 1:2, 1:5, 1:10 and 1:20. High precursor concentrations (1:1 and 1:2) favor the formation of polymers featuring a high structural order, while the decrease of DCDA concentration leads to the less ordered solids. Finally, at 1:20 ratio, mainly SnO2 NP reflections are observed in the corresponding PXRD pattern. The decrease of the crystallinity in the final products upon decrease of the precursor concentration is due to the termination of the crystal growth by the deposition of some metal species from the melt onto the crystal facets, as illustrated by the increase of the metal content in the isolated products (Table S3†). On the other hand, no crystalline cobalt-containing species could be detected under any synthesis conditions used.
The suggested preparation method delivers non-porous composites with the surface areas between 6 and 51 m2 g−1 (Fig. S3†). These values are much higher than provided by bulk synthesis (1 m2 g−1). The selected SEM images of products are shown in Fig. 3. Two different morphologies are typical for the CoCl2-derived products: spherical particles are formed in alkali metal chloride and ZnCl2-containing eutectics, while plate-like particles are obtained from the SnCl2/CoCl2 salt melts. The latter carbon nitride particle morphology along with its chemical structure are typical for CN-polymers prepared in SnCl2-based eutectics, in agreement with the previous report.21
 | ||
| Fig. 3 SEM images of the composites prepared in KCl/CoCl2 (a) and SnCl2/CoCl2 (b) salt melts. | ||
In Fig. 4a, the optical absorption properties of CoCl2-derived materials are compared with those of the composite photocatalyst consisting of tri-thiocyanuric acid derived carbon nitride and 5 nm Co3O4 NPs that was prepared in 3 steps according to the reported procedure8 (Fig. S4†) and used as a reference catalyst in the photocatalytic water oxidation reaction (see below). The products of the salt melt synthesis are characterized by a significantly improved absorption in the visible light range than the reference material. Unlike CNS/3wt%-Co3O4 composites exhibiting a distinct absorption at ∼700 nm due to the O2− → Co3+ electron transition,8,36–38 the smooth appearance of the absorption spectra of CoCl2-derived solids suggests the formation of the dyadic system with a shared electron density, where cobalt species are integrated into the carbon nitride electronic structure enabling a continuum of new electron transitions upon excitation with visible light. The phenomenon was previously reported for carbon@TiO2 composites39 and CN-polymer composites prepared in ZnCl2-containing salt melts.20
 | ||
| Fig. 4 Optical absorption (a) and emission (b) spectra of the products prepared in NaCl/CoCl2, KCl/CoCl2, ZnCl2/CoCl2 and SnCl2/CoCl2 eutectics and the reference CNS/3 wt% Co3O4 composite prepared according to the reported procedure.8 | ||
All CoCl2-derived solids are characterized by almost negligible PL signals (Fig. 4b) that can only partially be explained by the decreased X-ray structural order, but mainly suggest the improved charge separation of the photo-excited charge carriers which is responsible for the high photocatalytic performance of the composites as illustrated below.
Photocatalytic studies
We found that CoCl2-derived composites are active catalysts for the photocatalytic water oxidation reaction. The composites were tested in two different set-ups, whose detailed description is given in the experimental part, using AgNO3 as a sacrificial electron scavenger and La2O3 as a pH buffering agent upon irradiation with a 300 W Xenon lamp (λ > 300 nm). The amounts of the produced oxygen were quantified either by gas chromatography (set up 1) or using an optical oxygen probe (set up 2). Additionally, gas chromatography was used in order to determine the O2/N2 selectivity characterizing the stability of carbon nitride polymers during the photocatalytic reaction.The O2 evolution rates provided by using the CoCl2-derived products are comparable with those achieved by using the benchmark composite, CNS/3wt%-Co3O4 (Table S4†). Moreover, the products of the salt melt synthesis show high O2/N2 selectivity, in most of cases even higher than that of the reference material, suggesting that carbon nitrides are stable during the reaction. Thus, we conclude that cobalt present as cobalt (II, III) oxide clusters and even amorphous chloride-containing cobalt(III) complexes, indeed, successfully serves as an efficient co-catalyst for water oxidation, which is in agreement with the recent reports.15,40,41
Since the crystallinity of a photocatalyst is known to play an important role in its photocatalytic performance, we investigated the influence of the structural order of the CN-polymer component of the SnCl2/CoCl2-derived products on the reaction rates in Rhodamine B degradation and water oxidation (Fig. 5). The products with a high structural order (prepared at 1:1 and 1:2 precursor to salt ratios) are found to be highly active photocatalysts for dye degradation upon irradiation with white light (λ > 420 nm), providing the rate constants (0.226 and 0.291 min−1, respectively) that are 45–58 times higher compared to the ground bulk CN-polymer (k = 0.005 min−1). Upon decrease of the structural order, the reaction constant decreases gradually to k = 0.007 min−1 (Fig. 5a). Such photocatalytic behavior is explained by the lower energy losses via recombination or phonon release and the resulting higher reaction rates in the case of defect-free or more crystalline products.
 | ||
| Fig. 5 The decrease of the relative Rhodamine B concentration and calculated rate constants in min−1 (a) and increase of the O2 concentration (b) as a function of the irradiation time using SnCl2/CoCl2-derived photocatalysts prepared using different precursor to salt ratios. Reaction conditions are given in the Experimental section. | ||
Conversely, the products prepared using low precursor concentrations in the melt (1:20, 1:10 ratios) show superior performance for the water oxidation reaction compared to those prepared at low concentrations (Fig. 5b). This is explained by higher amounts of cobalt species deposited on the surface of the former products (for comparison of cobalt content, see Table S3†), and proves that these serve as active sites for O2 evolution.
Conclusion
The thermal condensation of dicyandiamide in CoCl2-containing eutectics yields cobalt-containing carbon nitride-based photocatalysts. Cobalt is incorporated in the final products as cobalt (II, III) oxide clusters and amorphous chloride-containing cobalt(III) complexes that successfully play the role of co-catalysts in the photocatalytic water oxidation reaction. The composites obtained are characterized by the improved optical absorption in the visible light range due to the formation of the dyadic system between carbon nitride polymer and cobalt species, and almost negligible photoluminescence due to the improved charge separation. The high crystallinity of the carbon nitride phase provides high Rhodamine B degradation rates, while oxygen evolution rates are mainly dependent on the cobalt co-catalyst loading. Both parameters can be successfully tuned by varying the synthesis conditions.References
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- Z. Zhou, J. Wang, J. Yu, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 2179 CrossRef CAS PubMed.
- J. Wang, C. Zhang, Y. Shen, Z. Zhou, J. Yu, Y. Li, W. Wei, S. Liu and Y. Zhang, J. Mater. Chem. A, 2015, 3, 5126 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- J. R. Holst and E. G. Gillan, J. Am. Chem. Soc., 2008, 130, 7373 CrossRef CAS PubMed.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443 RSC.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS PubMed.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578 CrossRef CAS PubMed.
- P. G. Hoertz, Y. I. Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111, 6845 CrossRef CAS PubMed.
- J. Sato, N. Saito, Y. Yamada, K. Maeda, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi, K. Domen and Y. Inoue, J. Am. Chem. Soc., 2005, 127, 4150 CrossRef CAS PubMed.
- P. Baltzer, R. S. Davidson, A. C. Tseung, M. Grätzel and J. Kiwi, J. Am. Chem. Soc., 1984, 106, 1504 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 CAS.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941 CrossRef CAS.
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215 CrossRef CAS PubMed.
- E. Wirnhier, M. Döblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Chem.–Eur. J., 2011, 17, 3213 CrossRef CAS PubMed.
- S. Y. Chong, J. T. A. Jones, Y. Z. Khimyak, A. I. Cooper, A. Thomas, M. Antonietti and M. J. Bojdys, J. Mater. Chem. A, 2013, 1, 1102 CAS.
- C. Fettkenhauer, J. Weber, M. Antonietti and D. Dontsova, RSC Adv., 2014, 4, 40803 RSC.
- C. Fettkenhauer, G. Clavel, K. Kailasam, M. Antonietti and D. Dontsova, Green Chem., 2015, 17, 3350 RSC.
- X. Yang, H. Tang, J. Xu, M. Antonietti and M. Shalom, ChemSusChem, 2015, 8, 1350 CrossRef CAS PubMed.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709 CrossRef.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC.
- E. J. McDermott, E. Wirnhier, W. Schnick, K. S. Virdi, C. Scheu, Y. Kauffmann, W. D. Kaplan, E. Z. Kurmaev and A. Moewes, J. Phys. Chem. C, 2013, 117, 8806 CAS.
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413 CrossRef CAS.
- R. Moreno-Tost, J. Santamaria-Gonzalez, P. Maireles-Torres, E. Rodriguez-Castellon and A. Jimenez-Lopez, Appl. Catal., B, 2002, 38, 51 CrossRef CAS.
- B. W. Lee, J. A. Taylor and J. W. Rabalais, Solid State Commun., 1980, 33, 1205 CrossRef CAS.
- C. R. Brundle, T. J. Chuang and D. W. Rice, Surf. Sci., 1976, 60, 286 CrossRef CAS.
- Insight into the exchange coupling between magnetic molecules and the supporting surface: spectromicroscopy correlation including X-ray magnetic circular dichroism, D. J. Chylarecka, Universität Basel, 2011 Search PubMed.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press limited, 1991 Search PubMed.
- J. Coates, Interpretation of Infrared Spectra, a Practical Approach, John Wiley & Sons Ltd, Chichester, 2000 Search PubMed.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178 CAS.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177 CrossRef CAS PubMed.
- J. D. Cook and M. P. van der Meer, Thin Solid Films, 1986, 144, 165 CrossRef CAS.
- I. D. Belova, Y. E. Roginskaya, R. R. Shifrina, S. G. Gagarin, Y. V. Plekhanov and Y. N. Venevtsev, Solid State Commun., 1983, 47, 577 CrossRef CAS.
- M. Ando, T. Kobayashi and M. Haruta, Sens. Actuators, B, 1996, 32, 157 CrossRef CAS.
- L. Zhao, X. Chen, X. Wang, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 3317 CrossRef CAS PubMed.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301 CrossRef CAS PubMed.
- K. Kwapien, S. Piccinin and S. Fabris, J. Phys. Chem. Lett., 2013, 4, 4223 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Information about the eutectics used, O1s and Cl2p XPS spectra of KCl/CoCl2 derived products, EA and surface area data of products synthesized in different melts, N2 sorption isotherms, summary of activity in photocatalytic water oxidation, and the TEM image of Co3O4 nanoparticles. See DOI: 10.1039/c5ta06304c |
| ‡ Current address: Institute of Nano Science and Technology (INST), Habitat Centre, Phase – 10, Sector – 64, Mohali, Punjab – 160062, India. |
| This journal is © The Royal Society of Chemistry 2015 |