
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The golden gate to photocatalytic hydrogen production†
Philip
Kalisman
a,
Lothar
Houben‡
b,
Eran
Aronovitch
c,
Yaron
Kauffmann
d,
Maya
Bar-Sadan
 c and
Lilac
Amirav
*a
c and
Lilac
Amirav
*a
aSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa 32000, Israel. E-mail: lilac@technion.ac.il
bPeter Grünberg Institut 5 and Ernst Ruska-Centre for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
cDepartment of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel
dDepartment of Materials Science & Engineering, Technion-Israel Institute of Technology, Haifa 32000, Israel
First published on 28th August 2015
Abstract
We demonstrate improved efficiency for the photocatalytic water splitting reduction half reaction by employing Au–Pt bimetallic cocatalysts. We employed nanoparticle-based photocatalysts consisting of CdSe@CdS rods tipped with Au, Pt, Au–Pt core–shell or Au decorated with Pt islands. By tailoring the composition and morphology of the Au–Pt bimetallic catalysts, we achieved more than a fourfold increase in activity for hydrogen production compared to pure Pt.
Solar-driven photocatalytic splitting of water into hydrogen and oxygen is a potential source of clean and renewable fuels. Semiconductor photocatalysts are often loaded with metallic cocatalysts, which promote charge separation of photogenerated electrons and holes and also act as sites for the reduction reaction.1,2 These cocatalysts play an essential role in reactions such as water splitting, as they offer lower activation potentials for hydrogen evolution, thereby greatly enhancing the photocatalytic activity.3–7 The metals demonstrating the greatest catalytic performance do not necessarily form an ideal interface with the semiconductor they are grown on, resulting in less than optimal charge transfer (or accumulation). Utilization of a bimetallic catalyst may enable separate optimization of the different interfaces within such a heterostructure photocatalyst system (i.e. semiconductor–metal interface and metal–liquid junction).
Employing a bimetallic catalyst also extends the range of tunable properties for the metallic compounds, allowing for improved rational design of heterogeneous catalysts.8–17 The reactivity may be tailored by changing the composition, and the exterior electronic and geometric structures of the bimetallic surfaces.18–24 Hence, bimetallic materials are considered highly promising candidates for heterogeneous catalysis. In particular, alloys where the solid solute concentration near the surface differs from the bulk have proven quite successful.25,26 Here we examine a combination of gold and platinum as an improved cocatalyst for the photocatalytic water splitting reduction half reaction.
Two factors differentiate bimetallic catalysts from pure metal surfaces and improve their catalytic performance: (1) the strain effect from the size-mismatch of the component metal atoms, and (2) heterometallic bonding interactions, termed the “ligand effect”, between the surface atoms and the substrate. Both of these effects change the width and subsequently the energetic position of the surface d band, thereby changing the surface chemical and catalytic properties.27 Of relevance is the dissociative adsorption energy of hydrogen, which is controlled by changes induced in the average energy of the d band.
In a semiconductor–metal photocatalytic system, the semiconductor substrate upon which the nanoscale metallic components are grown is likely to affect their electronic and geometric characteristics as well. In addition, modifications to the electronic properties of the metallic components might also affect interfacial charge-transfer processes.
We set out to examine the opportunities for improving the photocatalytic hydrogen production by coupling Pt, a known active catalyst, with Au, a metal that demonstrated the ability to accumulate charges even at the nanoscale.28 We employed a well-controlled nanoparticle-based system for catalyzing the reduction half reaction.29 The light absorption and excitation unit consists of a cadmium selenide (CdSe) quantum dot embedded asymmetrically within a cadmium sulfide (CdS) quantum rod.30–32 This structure has been widely studied optically and photocatalytically and is well characterized.33–40 The pure metals, Au and Pt, and different combinations thereof, were grown onto nanorods forming a single catalytic reduction site, as can be seen in Fig. 1.
 | ||
| Fig. 1 TEM (A and B) and HAADF (C and D) micrographs of CdSe@CdS nanorod photocatalysts with Au–Pt bimetallic tips acting as cocatalyst reduction sites. | ||
Growth of the metal tips was attained with photodeposition, using procedures loosely based on previous protocols for deposition of Pt41 and Au.42 Photodeposition was preferred over colloidal growth43,44 as it allows for metal deposition at room temperature. It is presumed that such moderate growth conditions are necessary for the formation of well-controlled structures. Noteworthy is the fact that despite the use of a seeded rod as the semiconductor substrate, the metal catalyst was deposited almost exclusively at the tip, rather than along the side of the rod, adjacent to the seed, as was previously observed for similar structures.41,42,45
In addition to pure metals that were examined for comparison, sets of different compositions of Au–Pt bimetallic tips were made. This included Pt deposition on premade Au tips (Au@Pt), and codeposition of Au and Pt (varying the relative precursor amounts and growth time). Each set was grown on the same sample of CdSe@CdS rods, and great care was devoted to insuring that the composition of the metal tip was the only altered parameter.
We examined the activity of these sets of rods towards hydrogen production. Solutions of rods suspended in water with IPA (10% by volume) acting as a hole scavenger were placed in a home made gas-tight reaction cell purged with argon. The samples were then illuminated with a 455 nm LED adjusted to 50 mW (a photon flux of 1.15 × 1017 photons per second). The evolved hydrogen was analyzed using an online gas chromatograph equipped with a thermal conductivity detector. The apparent quantum efficiency of the sample, which is defined as QE = 2NH2/Nhν was determined by quantifying the amount of evolved hydrogen at a given photon flux. The results for a representative set are presented in Fig. 2.
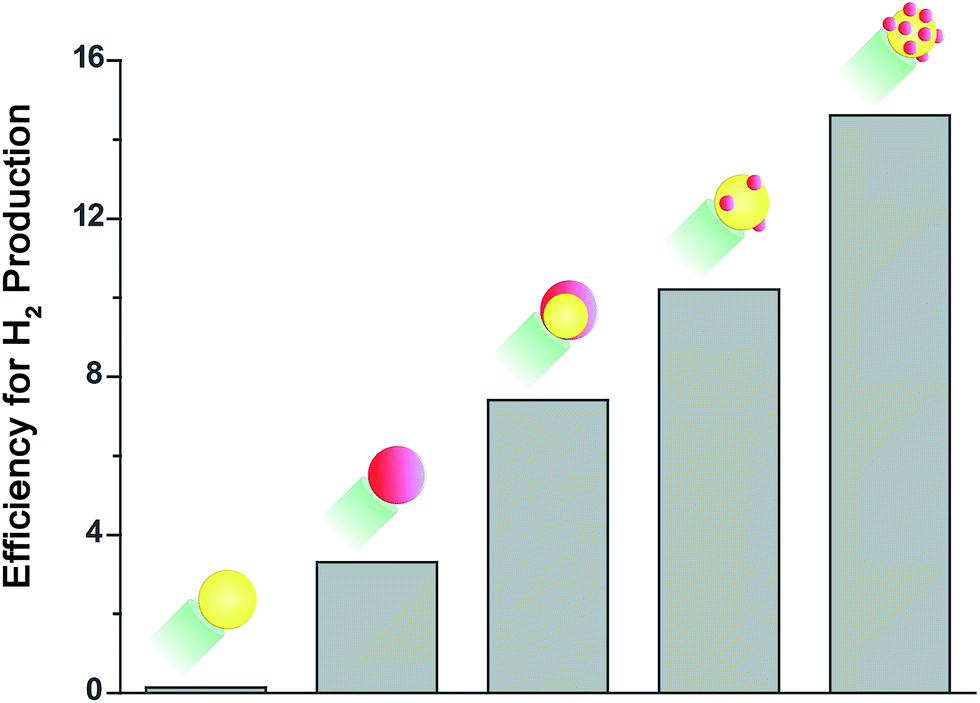 | ||
| Fig. 2 Relative efficiency for the photocatalytic hydrogen production half reaction utilizing CdSe@CdS nanorods decorated with different metal cocatalysts serving as reduction sites: Au (yellow), Pt (red), Au@Pt core–shell and Au–Pt. All measurements were done under identical solution conditions, and at equivalent concentrations. | ||
It is important to note that the absolute activity of the sample is strongly related to the rod morphology.29 Here we worked with 30–40 nm long rods that do not realize the system's full potential for hydrogen production, but allow for improved elemental analysis of the tip. Hence, only relative evaluation of the activity is given significance for these sets.
As expected, rods decorated with a Au cocatalyst showed very low activity, compared to rods with a Pt cocatalyst. When a preformed Au tip was coated with Pt the activity towards hydrogen production improved further, resulting in a twofold increase compared to pure Pt. This improved activity can be explained by the reasoning described in the introduction. Furthermore, Yu et al. reported recently that the injection of photoinduced electrons from CdSe nanorods into a Au tip is faster than their injection into a Pt tip, but only Pt can completely extract the excited electrons from the semiconductor nanorod.46 Consequently, the combination of the two metals is expected to enable both fast and efficient charge transfer, as required for hydrogen production.
Interestingly, the highest activity for the reduction half reaction, with more than a fourfold increase compared to pure Pt, was obtained from the sample in which the two metals were deposited simultaneously.
In order to better understand the structure-property relationship these hybrid nanostructures were characterized at the atomic level. Given the size of the metal tip (ranging from 2 to 4 nm) and a similar Z contrast of Pt and Au, we focused on elemental mapping with X-ray energy dispersive spectroscopy (EDS), for the analysis of the composition. The results, which are presented in Fig. 3, indicate that for the bimetallic tip that was grown with simultaneous codeposition of Au and Pt, a core of Au decorated with islands of Pt was formed.
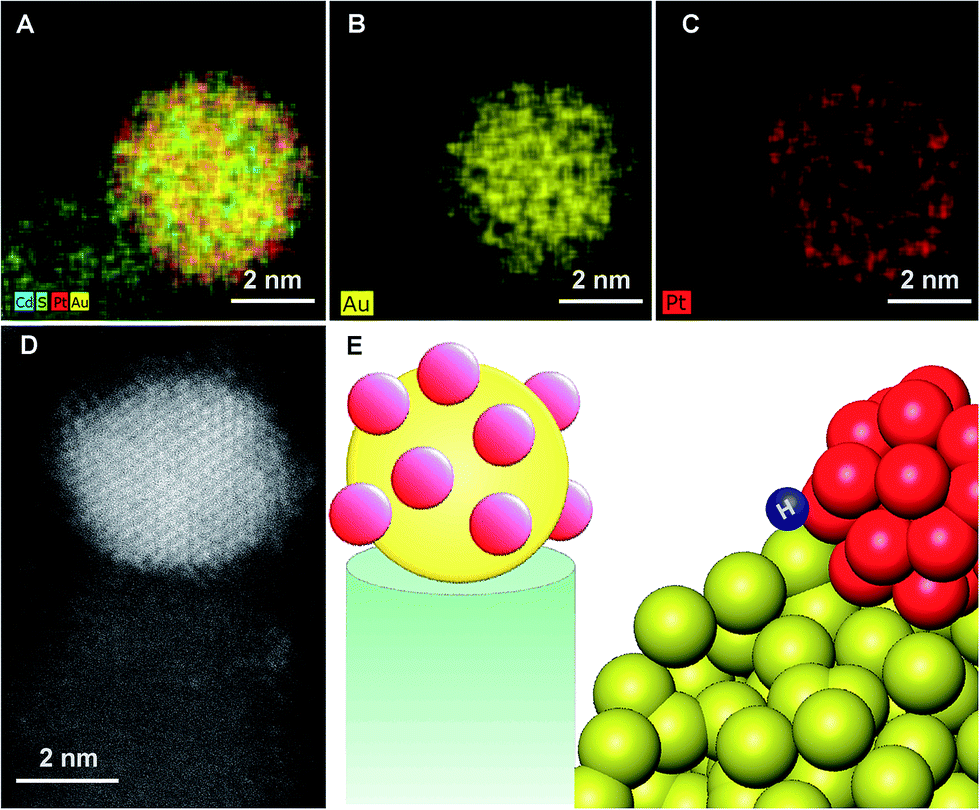 | ||
| Fig. 3 X-ray energy dispersive spectroscopy micrographs with elemental mapping (A–C) and an atomic resolution HAADF image (D) of the bimetallic tip, demonstrating the formation of a Au core that is decorated with islands of Pt (E). The Au–Pt boundary is suggested to be the active catalytic site. | ||
The bulk phase diagram of Au/Pt shows limited miscibility of the two metals.47 The degree of segregation and atomic ordering in nanoscale alloys depends on multiple factors, mainly bond strengths, surface energetics, and atomic sizes.48 Since the Au–Pt bonds are not sufficiently strong compared to Au–Au and Pt–Pt, segregation is favored. Typically, the species forming the strongest homonuclear bonds tends to be at the core of the nanoparticle, while the element with the lowest surface energy tends to segregate to the surface. The observed atomic arrangement for this particular Au–Pt nanoparticle depends critically on the balance of the factors outlined above, as well as on the preparation method and experimental conditions. The bimetallic nanoparticle is supported by the CdS rod on one side and is passivated by ligands on the other. The strength of binding to the support or ligands will also affect the atomic order. The Au core–Pt shell arrangement is consistent with the fact that Au is more noble and hence easier to reduce than Pt, making this the kinetic product, though surface and cohesive energy would favor the reverse configuration.49,50
While bulk Au–Pt alloys exhibit a miscibility gap, Chen et al. report that Au–Pt surface alloys may exhibit a series of ordered striped structures, due to the competition between the energy penalty of Au–Pt bonds at the boundaries and strain relaxation.51 This is consistent with the observed formation of Pt islands, which decorate the Au core.
Of significance is the reported effect of lateral heterogeneity. A strong correlation was found between oxygen binding energy and the local Au/Pt environment of the surface adsorbate.51 DFT calculations indicate that CO binds strongly to both the Pt and Au atoms, with the strongest binding being to Pt atoms that are adjacent to Au.52 This was reflected in high activity of Pt–Au nanoalloys for the electrocatalytic oxidation of CO.53 Hence, we suggest that the unique morphology of the Au core decorated with Pt islands, which introduces numerous exposed interfaces between the two phases, contributes to the improved activity towards hydrogen production.
Privman and co-workers applied a kinetic Monte Carlo model to the shell formation in core–shell particle synthesis. They observed formation of cluster-structured, or smooth epitaxial shells that are formed at a higher temperature.54 We thus suggest that the control over tip morphologies was made possible by the flexibility of photochemical deposition, and the moderate growth conditions it allows. Millstone,55 Khashab,56 Goia57 and their co-workers reported recently on alternative strategies for the formation of organized decoration of Au nanosubstrates with Pt nanoparticles. Here, we demonstrate that this synthetic effort is advantageous also for photocatalysis.
Conclusions
Building a golden gate through which electrons can migrate from the semiconductor photocatalyst to the Pt cocatalyst islands results in increased activity for the water reduction half reaction.Acknowledgements
This research was carried out in the framework of Russell Berrie Nanotechnology Institute (RBNI) and the Nancy and Stephen Grand Technion Energy Program (GTEP). The authors acknowledge the generous support from the I-CORE Program of the Planning and Budgeting Committee, and The Israel Science Foundation (Grant No. 152/11), as well as the Adelis Foundation support in research in renewable energy. We thank the Schulich Faculty of Chemistry and the Technion – Israel Institute of Technology for the renovated laboratory and startup package. P.K. acknowledges the Schulich postdoctoral fellowship for their support. M.B.S. and E.A. gratefully acknowledge ISF grant 475/12. The access to the ER-C and the aberration-corrected instruments was granted through the European Union Seventh Framework Program, under Grant Agreement 312483-ESTEEM2 (Integrated Infrastructure Initiative-I3).Notes and references
- T. Sakata, T. Kawai and K. Hashimoto, Chem. Phys. Lett., 1982, 88, 50–54 CrossRef CAS.
- D. E. Aspnes and A. Heller, J. Phys. Chem., 1983, 87, 4919–4929 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83–86 CrossRef CAS.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- C. H. Christensen and J. K. Norskov, J. Chem. Phys., 2008, 128, 182503 CrossRef PubMed.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS PubMed.
- C. A. Menning and J. G. Chen, J. Chem. Phys., 2008, 128, 164703 CrossRef PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717–4125 CrossRef CAS PubMed.
- Y. G. Ma and P. B. Balbuena, J. Phys. Chem. C, 2008, 112, 14520 CAS.
- O. M. Lovvik and S. M. Opalka, Surf. Sci., 2008, 602, 2840–2844 CrossRef CAS PubMed.
- J. R. Kitchin, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075437 CrossRef.
- R. Callejas-Tovar and P. B. Balbuena, Surf. Sci., 2008, 602, 3531–3539 CrossRef CAS PubMed.
- V. Soto-Verdugo and H. Metiu, Surf. Sci., 2007, 601, 5332–5339 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- W. Chen, W. F. Schneider and C. Wolverton, J. Phys. Chem. C, 2014, 118, 8342–8349 CAS.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- J. Greeley, J. K. Norskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Catal. Today, 2005, 105, 66–73 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS PubMed.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497 CrossRef CAS PubMed.
- A. U. Nilekar and M. Mavrikakis, Surf. Sci., 2008, 602, L89–L94 CrossRef CAS PubMed.
- K. Hartl, K. J. J. Mayrhofer, M. Lopez, D. Goia and M. Arenz, Electrochem. Commun., 2010, 12, 1487–1489 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS.
- R. Costi, A. E. Saunders, E. Elmalem, A. Salant and U. Banin, Nano Lett., 2008, 8, 637–641 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- D. V. Talapin, R. Koeppe, S. Gotzinger, A. Kornowski, J. M. Lupton, A. L. Rogach, O. Benson, J. Feldmann and H. Weller, Nano Lett., 2003, 3, 1677–1681 CrossRef CAS.
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. de Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- C. She, A. Demortiere, E. V. Shevchenko and M. Pelton, J. Phys. Chem. Lett., 2011, 2, 1469–1475 CrossRef CAS.
- K. Wu, W. E. Rodrıguez-Cordoba, Z. Liu, H. Zhu and T. Lian, ACS Nano, 2013, 7, 7173–7185 CrossRef CAS PubMed.
- H. Zhu, N. Song, H. Lv, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2012, 134, 11701–11708 CrossRef CAS PubMed.
- K. Tarafder, Y. Surendranath, J. H. Olshansky, A. P. Alivisatos and L. W. Wang, J. Am. Chem. Soc., 2014, 136, 5121–5131 CrossRef CAS PubMed.
- K. Wu, Z. Chen, H. Lv, H. Zhu, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2014, 136, 7708–7716 CrossRef CAS PubMed.
- A. Demortiere, R. D. Schaller, T. Li, S. Chattopadhyay, G. Krylova, T. Shibata, P. C. dos Santos Claro, C. E. Rowland, J. T. Miller, R. Cook, B. Lee and E. V. Shevchenko, J. Am. Chem. Soc., 2014, 136, 2342–2350 CrossRef CAS PubMed.
- K. Wu, H. Zhu and T. Lian, Acc. Chem. Res., 2015, 48, 851–859 CrossRef CAS PubMed.
- E. Khon, K. Lambright, R. S. Khnayzer, P. Moroz, D. Perera, E. Butaeva, S. Lambright, F. N. Castellano and M. Zamkov, Nano Lett., 2013, 13, 2016–2023 CrossRef CAS PubMed.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS PubMed.
- G. Menagen, J. E. Macdonald, Y. Shemesh, I. Popov and U. Banin, J. Am. Chem. Soc., 2009, 131, 17406–17411 CrossRef CAS PubMed.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed.
- S. E. Habas, P. D. Yang and T. Mokari, J. Am. Chem. Soc., 2008, 130, 3294–3295 CrossRef CAS PubMed.
- M. G. Alemseghed, T. P. A. Ruberu and J. Vela, Chem. Mater., 2011, 23, 3571–3579 CrossRef CAS.
- P. Yu, X. Wen, Y. C. Lee, W. C. Lee, C. C. Kang and J. Tang, J. Phys. Chem. Lett., 2013, 4, 3596–3601 CrossRef CAS.
- H. Okamoto and T. B. Massalski, Bull. Phase Alloy Diagr., 1985, 6, 46–56 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- L. M. Liz-Marzán and A. P. Philipse, J. Phys. Chem., 1995, 99, 15120 CrossRef.
- M. J. Bradley, C. G. Read and R. E. Schaa, J. Phys. Chem. C, 2015, 119, 8952–8959 CAS.
- W. Chen, D. Schmidt, W. F. Schneider and C. Wolverton, J. Phys. Chem. C, 2011, 115, 17915–17924 CAS.
- Q. Ge, C. Song and L. Wang, Comput. Mater. Sci., 2006, 35, 247–253 CrossRef CAS PubMed.
- M. M. Maye, Y. B. Lou and C. J. Zhong, Langmuir, 2000, 16, 7520–7523 CrossRef CAS.
- V. Gorshkov, V. Kuzmenko and V. Privman, J. Phys. Chem. C, 2014, 118, 24959–24966 CAS.
- P. J. Straney, L. E. Marbella, C. M. Andolina, N. T. Nuhfer and J. E. Millstone, J. Am. Chem. Soc., 2014, 136, 7873–7876 CrossRef CAS PubMed.
- H. M. Song, D. H. Anjum, R. Sougrat, M. N. Hedhili and N. M. Khashab, J. Mater. Chem., 2012, 22, 25003–25010 RSC.
- R. K. Roy, J. I. Njagi, B. Farrell, I. Halaciuga, M. Lopez and D. V. Goia, J. Colloid Interface Sci., 2012, 369, 91–95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Includes detailed description of the typical preparation of the different metal decorated CdSe@CdS nanorods, and techniques used in characterization of these particles. See DOI: 10.1039/c5ta05784a |
| ‡ Current address: Chemical Research Support, Weizmann Institute of Science, Rehovot, Israel. |
| This journal is © The Royal Society of Chemistry 2015 |