
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced capacitance of nitrogen-doped hierarchically porous carbide-derived carbon in matched ionic liquids†
J.-K.
Ewert
a,
D.
Weingarth
b,
C.
Denner
a,
M.
Friedrich
a,
M.
Zeiger
bc,
A.
Schreiber
b,
N.
Jäckel
bc,
V.
Presser
*bc and
R.
Kempe
*a
aInorganic Chemistry II, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany. E-mail: kempe@uni-bayreuth.de; volker.presser@leibniz-inm.de
bINM-Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
cDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
First published on 5th August 2015
Abstract
Supercapacitors combine efficient electrical energy storage and performance stability based on fast electrosorption of electrolyte ions at charged interfaces. They are a central element of existing and emerging energy concepts. A better understanding of capacitance enhancement options is essential to exploit the full potential of supercapacitors. Here, we report a novel hierarchically structured N-doped carbon material and a significant capacitance enhancement for a specific ionic liquid. Our studies indicate that matching of the electrode material and the ionic liquid specifically leads to a constant normalized resistance of the electrode material (voltage window up to ±1 V vs. carbon) and a significant enhancement of the specific capacitance. Such effects are not seen for standard organic electrolytes, non-matched ionic liquids, or non-N-doped carbons. A higher N-doping of the electrode material improves the symmetric full cell capacitance of the match and considerably increases its long-term stability at +3 V cell voltage. This novel observance of enhanced specific capacitance for N-doped carbons with matched ionic liquid may enable a new platform for developing supercapacitors with enhanced energy storage capacity.
Introduction
Electrochemical capacitors, also known as supercapacitors or ultracapacitors, capitalize on the high efficiency and performance stability of fast electrosorption of electrolyte ions at the charged interface with nanoporous carbon.1,2 While derived from abundantly available biomass, commonly used high surface area carbons show only a moderate electrical conductivity, which presents a limitation to effective charge screening, leading to limited energy storage capacity.3 At a device level, this shortcoming is usually compensated by the admixing of conductive additives;4 yet, this approach is not an intrinsic solution for the inability to accommodate a high amount of electric charge within the carbon nanopores.The beneficial impact of N-doping on the electrical conductivity and, more generally, the electrochemical performance of supercapacitors has first been investigated by Lota et al. in polymer-derived carbons.5 By now, a series of carbon materials with N-doping has been explored, including carbon nanotubes,6,7 mesoporous carbon spheres,8 biomass-derived porous carbon,9 metal–organic frameworks,10 and graphene-like carbon.11 Nitrogen can be introduced into carbon, generally, top-down or bottom-up. The latter involves the synthesis of a carbon network and subsequent N-doping via annealing, for example in ammonia.12 Alternatively, N-doping can be accomplished very effectively by using nitrogen-containing precursor materials, including bio-materials like prawn shells or yogurt9,13 or ionic liquids.14 Depending on the synthesis procedure and the maximum synthesis temperature, the maximum amount of N-doping in carbon may reach around 21 mass%.15 In general, we find lower nitrogen content when employing higher synthesis temperatures15 and a significant improvement of the electrochemical performance has been reported already for rather low amounts of nitrogen, such as 1–2 mass%.5
Besides improving the electrical conductivity and increasing the charge screening ability of carbon,7 N-sites at the carbon surface may facilitate charge transfer across the electrode/electrolyte interface. Such redox-sites may contribute significantly to the energy storage mechanism by enabling access to reversible faradaic reactions and possibly pseudocapacitance;11,16 yet, this is often accomplished at the cost of sacrificing power handling and longevity to some degree.2
Considering the high ion mobility and the possible benefit of redox-related charge storage, most of the investigations of nitrogen-doped carbons have been carried out in aqueous electrolytes, foremost H2SO4 (ref. 5, 6 and 17) and KOH.5 In such systems, an enhancement of the electrochemical performance is accomplished by the introduction of fast surface redox-reactions of quaternary nitrogen and other N-groups with the protic electrolyte. Only a small number of studies, so far, have been carried out on organic electrolytes,5 including ionic liquids.18 The latter are a highly promising group of electrolytes that are able to push the voltage limit of supercapacitors to at least 3.5 V even for long-term operation,19 while benefitting from a non-volatile and temperature-stable nature at the cost of low ion mobility.20 Since the stored amount of energy scales with the square of the cell voltage, ionic liquids are attractive candidates for high-energy supercapacitor devices.21 While the results for many of the N-doped carbons are promising, one aspect has somewhat been overlooked: the intricate correlation between ions and the carbon surface. Matching the ionic liquid to an optimized voltage window has recently been demonstrated for pure carbon materials,22 but it is still unclear what selection strategy should be applied for matching an ionic liquid to nitrogen-doped carbon.
Herein, we report a novel hierarchically porous N-doped carbon material obtained from meso-structured polymer derived silicon carbonitride and the electrochemical performances in different ionic liquids. We observe a significantly improved capacitance for one specific ionic liquid and an increased N-doping improves the symmetric full cell capacitance as well as the long-time stability of such cells at high voltage.
Results and discussion
Preparation of the hierarchically porous N-doped carbon material
Since the template free synthesis of polymer-derived silicon carbon nitride (SiCN) materials leads neither to micropores nor to mesopores,23 we decided to synthesize a polystyrene-polysilazane nanocomposite regarding meso-structuring.24 Therefore, positively charged polystyrene (PS) spheres with a diameter of about 50 nm (PS50) were synthesized as structuring templates via emulsion polymerization (Fig. 1; see also ESI, Fig. S1†). The particles were mixed in toluene with commercially available polysilazane HTT-1800. Divinylbenzene (DVB) was used as a cross-linker stabilizing the particles in toluene. Thus, an enhanced yield of residual carbon can be obtained. Evaporation of the solvent gave rise to the nano-composite green body. The green body was pyrolyzed at 900 °C in order to remove the PS50 template and led to the thermally stable meso-structured SiCN material PS50SiCN900. (Fig. 1; see also ESI, Fig. S2†). Chlorine treatment, commonly used for the synthesis of carbide-derived carbons (CDC),25 was employed at 800 °C (PS50SiCN900Cl2-800°C) and 1000 °C (PS50SiCN900Cl2-1000°C) to volatize residual silicon and partially mobilize silicon nitride.26 | ||
| Fig. 1 Schematic process of the synthesis of the N-doped hierarchically porous carbon materials and transmission electron microscopy (TEM) images of PS50SiCN900 (left, top) PS50SiCN900Cl2-800°C (left, bottom) following a three step procedure. (1) Mixing 50 nm polystyrene (PS50) particle template, the commercially available SiCN precursor HTT-1800, and dicumylperoxide (DCP, crosslinker) in toluene as well as structuring by removal of the solvent. (2) Pyrolysis at 900 °C obtaining the mesoporous SiCN material PS50SiCN900. (3) Chlorination at 800 °C or 1000 °C leading to the hierarchically porous N-doped carbon material PS50SiCN900Cl2-800°C and PS50SiCN900Cl2-1000°C, respectively. | ||
Material characterization
Transmission electron microscopy (TEM) images of PS50SiCN900Cl2-800°C before and after chlorine treatment are shown in Fig. 1. For scanning electron microscopy (SEM) and TEM images of all materials, see ESI, Fig. S1–S3.† As common for CDC,27 our materials remained conformal after chlorine gas treatment, preserving the structure of the mesoporous SiCN material and adding additional pores so that a very high pore volume (up to 1.67 cm3 g−1) was reached.The SiCN material PS50SiCN exhibits a characteristic nitrogen sorption isotherm typical for a purely mesoporous material (Fig. 2A).28 The nitrogen gas sorption isotherms of the material after chlorine gas treatment were a combination of the IUPAC Type I and Type IV isotherm with a pronounced Type H2 hysteresis in reflectance of the mixed micro- and mesoporous pore structure (Fig. 2B).28 Compared to the mesoporous SiCN material (107 m2 g−1), the BET surface area strongly increases as a result of chlorine etching at 800 °C to 1745 m2 g−1 and at 1000 °C to 1817 m2 g−1 (Table 1). PS50SiCN900Cl2-800°C is dominated by mesopores, 71% of the pore volume (Table 1). In contrast, PS50SiCN900Cl2-1000°C shows approximately a one-to-one distribution of micro- and mesopores at a lower total pore volume (1.19 cm3 g−1 instead of 1.67 cm3 g−1). The smaller pore volume may result from an enhanced carbon sintering and pore coalescing at 1000 °C.29 The hierarchical pore size distributions observed for both N-doped carbon materials facilitates access of ionic liquid ions to the pores (Table 1). The diameter of the largest mesopores is significantly smaller than 50 nm (diameter of the used PS templates) due to incomplete cracking of the PS template during pyrolysis (Fig. 2B).
 | ||
| Fig. 2 Characterization of the hierarchically porous nitrogen-doped carbon materials (A) nitrogen sorption isotherms and (B) calculated pore size distribution of the ceramic template and the chlorinated materials. (C) Raman spectra of the samples PS50SiCN900-800°C and PS50SiCN900-1000°C, (D) XPS survey spectra of the samples PS50SiCN900-800°C and PS50SiCN900-1000°C, (E) N 1s high resolution spectrum of PS50SiCN900-800°C and PS50SiCN900-1000°C, and (F) O 1s high resolution spectrum of PS50SiCN900-800°C and PS50SiCN900-1000°C. | ||
| PS50SiCN900 | PS50SiCN900Cl2-800°C | PS50SiCN900Cl2-1000°C | |
|---|---|---|---|
| BET SSA (m2 g−1) | 106 | 1745 | 1817 |
| DFT SSA (m2 g−1) | 117 | 1536 | 1516 |
| V total (cm3 g−1) | 0.27 | 1.67 | 1.19 |
| V micropores (cm3 g−1) | 0 | 0.47 | 0.59 |
| V mesopores (cm3 g−1) | 0.27 | 1.19 | 0.60 |
| Pore size average (nm) | 4.4 | 3.3 | 2.0 |
| N (mass%)a | Not measured | 5.5 | 1.6 |
| N (mass%)b | Not measured | 4.6 | 1.1 |
The materials after chlorine gas treatment showed Raman spectra with pronounced D- and G-peaks around 1350 cm−1 and 1590 cm−1, respectively, as typical for incompletely graphitized carbons (Fig. 2C).30 With increased chlorination temperature, the degree of carbon ordering increases indicated by the small decrease of the integral ID/IG signal ratio from 2.3 to 2.1.
The insertion of nitrogen into the carbon network was confirmed by X-ray photoelectron emission spectroscopy (XPS) (Fig. 2D–F and Table 1) and elemental analysis (EA) (Table 1). The chlorination temperature had a strong effect on N-doping. The decrease of the temperature during the chlorination process caused a lower nitrogen removal and a higher amount of nitrogen in the final carbon material (Table 1).15
Electrochemical measurements
Next, we investigated the electrochemical behavior of the N-doped carbon materials. As seen from the cyclic voltammograms (CV) of PS50SiCN900Cl2-800°C (Fig. 3A) and PS50SiCN900Cl2-1000°C (Fig. 3B), stable electrochemical performance is obtained in the studied voltage window (up to ±1 V vs. carbon, equivalent to 2 V cell voltage).31 As shown for galvanostatic charge/discharge data (ESI, Fig. S4A and S4B†), a maximum specific capacitance of 151 F g−1 at −1 V vs. carbon is seen for PS50SiCN900Cl2-800°C in combination with 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4), reflecting the behavior of the CV curves. In contrast, 1 M tetraethylammonium tetrafluoroborate (TEA-BF4) in acetonitrile (ACN) results in 121 F g−1 and 122 F g−1 for neat 1-ethyl-3-methylimidazolium bis(trifluoromethyl-sulfonyl)imide (EMIM-TFSI). The same behavior is seen for PS50SiCN900Cl2-1000°C, with a maximum capacitance of 149 F g−1 for EMIM-BF4 and only 126 F g−1 for 1 M TEA-BF4 in either propylene carbonate (PC) or ACN. The only difference between the latter two solvents, as seen from the CV in Fig. 3B, is the lower ion mobility during charging and discharging for PC. Neat 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM-BF4) also yields a lower capacitance, namely 134 F g−1. The capacitance values of different N-doped carbon electrodes with aqueous and non-aqueous electrolytes found in the literature are compared in Table 2.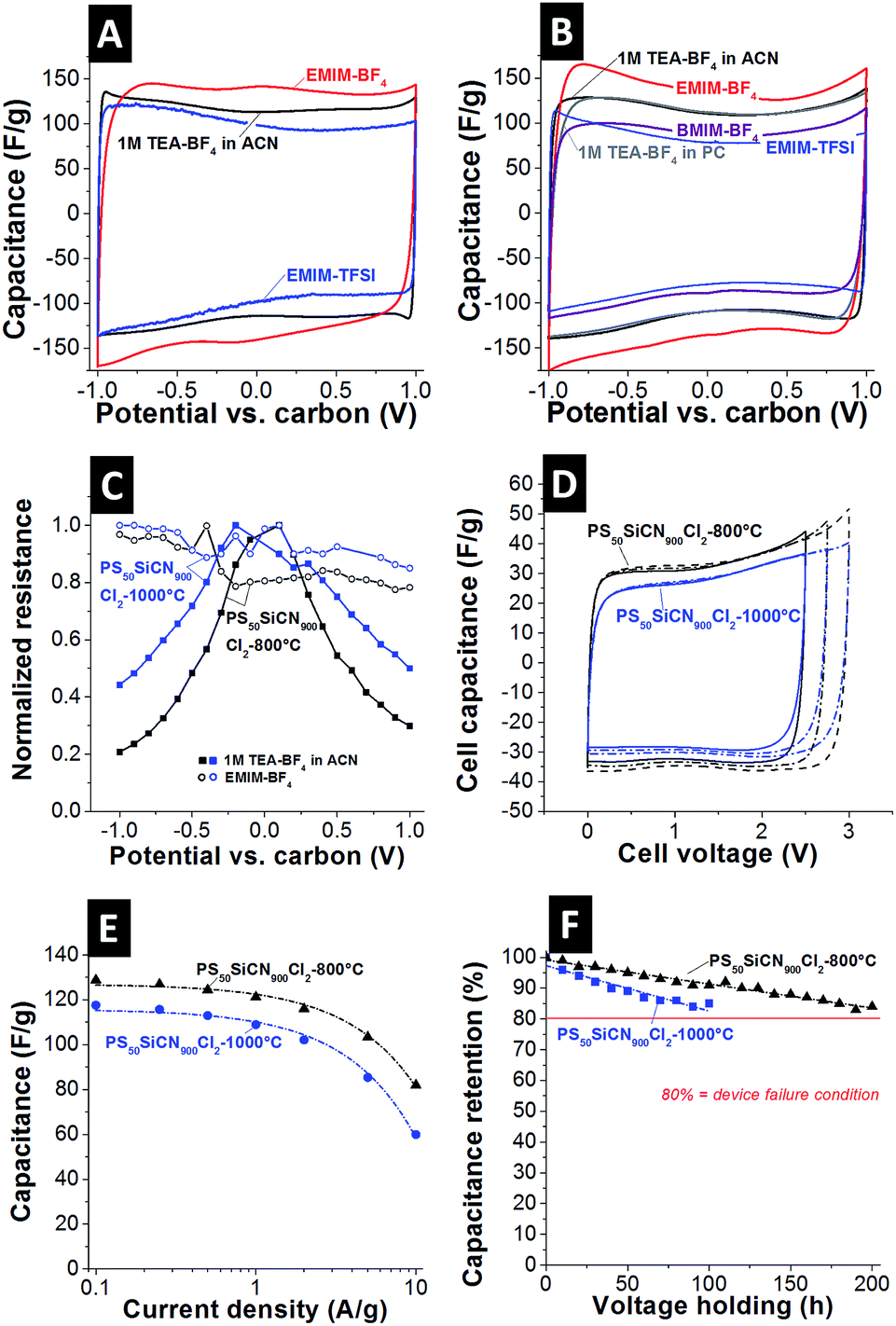 | ||
| Fig. 3 Cyclic voltammograms of PS50SiCN900Cl2-800°C (A) and PS50SiCN900Cl2-1000°C (B) in the range of ±1 V vs. carbon, scan rate: 10 mV s−1. (C) In situ resistivity measurements for PS50SiCN900Cl2-800°C and PS50SiCN900Cl2-1000°C with 1 M TEA-BF4 and EMIM-BF4 as electrolytes. (D) Full cell cyclic voltammograms of PS50SiCN900Cl2-800°C and PS50SiCN900Cl2-1000°C with EMIM-BF4 as the electrolyte, scan rate: 10 mV s−1. (E) Rate handling behavior of PS50SiCN900Cl2-800°C and PS50SiCN900Cl2-1000°C. (F) Long term stability test at 3 V cell voltage of PS50SiCN900Cl2-800°C and PS50SiCN900Cl2-1000°C in EMIM-BF4. | ||
To find a possible explanation for the difference in capacitance, the ohmic resistance of the N-doped carbon materials was measured in the charged state of the electrodes.37,38 This way, it was possible to address the change in electronic properties uninfluenced by the electrolyte. As seen in Fig. 3C, an expected behavior is recorded for the standard electrolyte based on ACN, namely a characteristic bell-shaped curve with a decrease in normalized resistance at increased potential. As shown in Fig. 3B and C the PC-based electrolyte shows comparable capacitance values compared to the ACN based electrolyte, indicating that the resistance behavior is also very similar (cf.ref. 38). For the EMIM-BF4 system, no decrease in normalized resistance is measured (Fig. 3C), indicating interactions of the electrolyte with the electrode material, considering that only the electrolyte was changed. This is additionally confirmed by measuring the in situ resistivity of EMIM-BF4 in a standard activated carbon (YP80F from Kuraray, Fig. S4C†). Here, the typical voltage-dependency of the normalized resistance is seen again for EMIM-BF4. Thus, the unique behavior of a virtually constant profile of the electrical conductivity is only achieved by a suitable match between the electrode material (i.e., N-doped carbon) and the electrolyte. Additionally, the pore hierarchy supports the ion transport. In principle, larger pores (like mesopores) facilitate ion mobility in micrometer-sized particles, while, at the same time, a large amount of mesopores is essential to enable a high specific capacitance (i.e., high ion storage ability).39 Noticeably, the CV shape does not indicate the occurrence of any further processes, such as ion depletion or surface saturation.1b This is further supported when comparing the cyclic voltammograms of SiC-CDC-800°C and PS50SiCN900Cl2-800°C (Fig. S4D†). The performance of the non N-doped material is clearly inferior to that of PS50SiCN900Cl2-800°C in terms of capacitance and rate capability.
To check whether the performance of a single electrode as presented before can be translated to a full cell, symmetric full cells were constructed (Fig. 3D). First, the potential window was opened successively up to 3 V cell voltage with a scan rate of 10 mV s−1. The cell capacitance of PS50SiCN900Cl2-800°C (ca. 34 F g−1) is higher than that of PS50SiCN900Cl2-1000°C (ca. 30 F g−1). Note that the values for the cell capacitance can be transformed to the specific capacitance (for one electrode) by multiplying by a factor of 4. The data from cyclic voltammetry agree well with galvanostatic charge/discharge measurements. At a low current density of 0.1 A g−1, the specific capacitance of PS50SiCN900Cl2-800°C (129 F g−1) is higher than that of PS50SiCN900Cl2-1000°C (118 F g−1). Up to 2 A g−1, both materials display a high rate capability before the capacitance fades at higher current densities.
Finally, the long-term stability at 3 V cell voltage was tested by voltage floating.40 Both cells are characterized by a decay in capacitance over time (Fig. 3F); however, PS50SiCN900Cl2-800°C lasted approximately twice as long as PS50SiCN900Cl2-1000°C at an elevated voltage of 3 V. A drop of 20% in capacitance, the common device failure definition in industry,4 was not seen within 200 h for PS50SiCN900Cl2-1000°C in EMIM-BF4. The cycling stability up to 3 V cell voltage (ESI, Fig. S5†) indicates that after a drop in capacitance at the beginning the cell approaches a stable performance after 1000 cycles.
Conclusions
In summary, we synthesized a hierarchically porous N-doped carbon material with different distributions of meso- and micropores as well as varied N-doping. Both N-doped materials show significant capacitance enhancement for the ionic liquid EMIM-BF4 in comparison to 1 M TEA-BF4 in ACN or EMIM-TFSI. In addition, N-doped carbon in combination with EMIM-BF4 shows a nearly constant normalized resistance from −1 V to +1 V. Distinct differences were observed for both electrode materials in symmetric full cells. The nitrogen-richer carbon material shows a higher cell capacitance and twice as high long-term stability at 3 V cell voltage.For the moment, the mechanisms causing the enhanced specific capacitance for the matched ionic liquid EMIM-BF4 compared to other electrolytes (such as EMIM-TFSI or when using organic solvents) remain unclear. Yet, the electrochemical data show that processes like ion sieving or ion saturation cannot cause the phenomenon. Instead, electrical conductivity shows an anomalous enhancement in electrical conductivity of the N-doped carbon electrode material only when using EMIM-BF4. Noticeably, this is not seen for conventional porous carbon (i.e., without N-doping). The unique solid-state response of N-doped carbon to a specific ionic liquid is an intriguing effect to be unraveled in future work.
Experimental section
Materials
Synthesis was performed in a dry argon atmosphere using standard Schlenk techniques. Halogenated solvents were dried over P2O5 and non-halogenated solvents over sodium benzophenone ketyl. All chemicals were purchased from commercial sources with a purity over 95% and used without further purification unless described detailed below.Synthesis of the PS50 template
The cross-linked polystyrene particles with an average diameter of 50 nm were synthesized by emulsion polymerization. Therefore, a three neck round bottom flask with a KPG stirrer and a reflux condenser was used. Divinylbenzene (DVB, technical grade, 55%, Sigma Aldrich) and styrene (>99% purity, Sigma Aldrich) were purified by destabilization (alumina B column, ICN Biomedicals GmbH). A dispersion of 4.10 g styrene (39.39 mmol) and 0.40 g DVB (3.07 mmol) was stirred in 90 mL ultrapure water. The dispersion was degassed for 30 min. After that, a solution of 0.25 g (1-hexadecyl)trimethyl-ammoniumbromide (0.69 mmol) (CTAB, 98% purity, abcr GmbH) in 5 mL ultrapure water was added under stirring (200 rpm) at 80 °C. After 0.5 h, 0.01 g 2,2′-azobis(2-methylpropionamidine)dihydrochloride (0.04 mmol) (97% purity, Sigma Aldrich) was added initiating the polymerization. Polystyrene particles were dialyzed for purification and freeze dried.Synthesis of PS50SiCN900 material
For several hours the polystyrene template PS50 was evacuated removing residual water. After that, a solution of 0.50 g of KiON HTT1800 (7.77 mmol) (Clariant Advanced Materials GmbH) and 0.05 g dicumylperoxide (1.85 mmol) (97% purity, Sigma Aldrich) in 40 mL toluene was added under stirring. The dispersion was heated to 110 °C for 24 h without stirring. After removal of the solvent the in situ structured green body was treated at 110 °C for 24 h finalizing the cross-linking. The PS50SiCN900 green body was pyrolyzed under a nitrogen atmosphere according to the following procedure: to 300 °C at 1 °C min−1 (held for 3 h), to 400 °C at 1 °C min−1 (held for 3 h), to 500 °C at 0.5 °C min−1 (held for 3 h), to 600 °C at 1 °C min−1 (held for 4 h), to 700 °C at 0.5 °C min−1 (no holding time), and finally to 900 °C at 1 °C min−1 (held for 0.5 h). A Gero furnace was used for this task.Synthesis of N-doped carbon
For the chlorine treatment around 3 g of PS50SiCN900 powder was put into a graphite crucible and placed in a quartz tube furnace (Gero F-A 40-200). The tube was flushed with a constant argon flow of 20 sccm for at least 8 h. During heating, chlorine and hydrogen treatment, the argon background flow was set to 100 sccm. The heating rate was 15 °C min−1 and the oven was held for 6 h at each chlorination temperature (600 °C, 800 °C, and 1000 °C). During the dwell time, the chlorine gas flow was set to 10 sccm. After cooling to 600 °C with 15 °C min−1, the chlorine gas flow was stopped and 10 sccm hydrogen gas was applied for 3 h to remove residual chlorine. At the end, the furnace was cooled down to room temperature with a constant argon flow of 20 sccm. The same procedure was followed for the synthesis of SiC-CDC-800°C (Precursor: Nano SiC, Plasmachem; average particle size ca. 20–30 nm).Materials characterization
Elemental analyses were carried out on a Vario elementar EL III. Thermal gravimetric analysis (TGA) was carried out under a nitrogen atmosphere using a Thermowaage L81 (Linseis, Germany). With a heating rate of 5 °C min−1 the sample was heated to 900 °C. Photon correlation spectroscopy (PCS) was performed using an ALV DLS/SLA-SP 5022F laser goniometer system. The power source was a Ne/Ar ion laser (λ = 632.8 nm at 260 mW). By using an ALV-5000/E multiple tau digital correlator the correlation function was generated. The decalin bath temperature was 20 °C, regulated by using a computer-operated thermostat. 90° fixed angle measurements were carried out. Data were analyzed by the CONTIN analysis. On a Zeiss field emission SEM LEO 1530 GEMINI scanning electron microscopy (SEM) was performed. The acceleration voltage was up to 5 kV and the materials were sputter-coated with a 1.3 nm platinum layer. Fourier transform infrared (FTIR) measurements were carried out using a Perkin-Elmer FTIR Spectrum 100 (from 4400 to 650 cm−1). Raman spectra of the raw materials were recorded with a Renishaw inVia Raman system using an Nd-YAG laser (532 nm) with 0.2 mW power at the sample surface. The peak analysis and peak fitting were performed assuming one Lorentzian peak for both the D-mode and G-mode. Nitrogen gas sorption measurements (−196 °C) of the electrodes (i.e., carbon + binder) were performed with an Autosorb iQ system (Quantachrome, USA). The materials were outgassed at 150 °C for 10 h in a vacuum. The specific surface area was calculated using the ASiQwin-software using the Brunauer–Emmett–Teller (BET) equation in the linear relative pressure range of 0.01–0.2. The SSA and pore size distribution (PSD) were also calculated via quenched-solid density functional theory (QSDFT) with a hybrid model for slit and cylindrical pores and pore size between 0.56 and 37.5 nm. Samples for transmission electron microscopy (TEM) were dispersed and sonicated in chloroform and placed on a copper grid (CF200-Cu-grid, Electron Microscopy Sciences, Hatfield, PA, USA). The TEM images were taken with a Varian LEO 9220 (120 kV, Carl Zeiss) and a JEOL 2100F system at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed using a VG ESCALAB 220iXL spectrometer (Thermo Fisher Scientific) equipped with an Al-Kα mono-source (power: 150 W; spot diameter: 500 μm) and a magnetic lens system. The spectra were recorded in constant analyzer energy mode at a pass energy of 20 eV. The XPS was calibrated using the Ag 3d5/2, the Cu 2p3/2, and the Au 4f7/2 lines as reference signals. The full-width half maximum (FWHM) of the Ag 3d5/2 line was measured to be 0.62 eV at a pass energy of 20 eV. The data were evaluated using the Avantage software provided by Thermo Fisher Scientific. The background subtraction was performed according to Shirley41 and atomic sensitivity factors were used according to Scofield.42Electrode preparation
Electrodes were prepared using sample powder dispersed in ethanol. After homogenization in a mortar, 5–10 mass% of dissolved polytetrafluoroethylene (PTFE, 60 mass% solution in water from Sigma Aldrich) were added as binders. While kneading, the slurry became more viscous and the resulting material was rolled with a rolling machine (MTI HR01, MIT Corp.) to a 200 ± 20 μm thick free standing electrode and dried at 120 °C at 2 kPa for 24 h. We employed a custom-built polyether ether ketone (PEEK) cell with spring loaded titanium pistons as a three electrode system described elsewhere.40 The cells employed electrodes with 12 mm diameter, a glass-fiber separator (GF/A (for full cells) or GF/D (for half cells)) from (Whatman, USA), and carbon-coated aluminum foil current collectors (type Zflo 2653, Coveris Advanced Coatings). PTFE-bound YP-50F was used as the reference electrode.43 The assembled cells were dried at 120 °C for 12 h at 2 kPa in an inert gas glovebox (MBraun Labmaster 130, O2 and H2O < 1 ppm) and, after cooling to room temperature, vacuum-filled with 1 M tetraethylammonium tetrafluoroborate (TEA-BF4) of electrochemical grade (i.e., water content < 20 ppm), acetonitrile (ACN) or propylene carbonate (PC) purchased from BASF. The used ionic liquids 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-imide (EMIM-TFSI; >99%, Sigma Aldrich) and 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4; >99%, IoLiTec Ionic Liquids Technologies) were degassed using a Schlenk tube in a Si-oil bath heated to 100 °C and a vacuum of 1 Pa was applied for at least 6 h to remove residual gas and water.Electrochemical testing: the electrochemical measurements were carried out using a potentiostat/galvanostat VSP300 from Bio-Logic, with cyclic voltammetry (CV), galvanostatic cycling with potential limitation (GCPL), and electrical impedance spectroscopy (EIS). CVs were recorded in half cell mode at 1, 10, 100 and 1000 mV s−1 in the potential range of 0 to 1 V vs. carbon with activated carbon (YP50, Kuraray chemicals) as the reference electrode.43,44 GCPL in half cell mode was performed to access the maximum available capacitance values from discharge in the range of ±1 V. The cell was charged for 10 min up to the desired potential and then discharged to 0 V. The capacitance was determined in 100 mV steps. Full cells were prepared for further testing in CV and GCPL mode. The CVs were recorded up to 3 V with 10 mV s−1. In GCPL mode, the current density was increased in several steps from 0.1 A g−1 to 10 A g−1 with 10 s resting period between charging/discharging to access information on the IR-drop. The voltage holding experiments were performed at 3 V cell voltage with 10 h holding periods followed by 3 galvanostatic charge/discharge cycles to determine the capacitance. This was repeated for at least 10 times. The galvanostatic cycling experiments (see ESI, Fig. S5†) were performed at 1 A g−1.40 The in situ resistance measurements were conducted with a system described in ref. 37. The working electrode was galvanostatically charged to the favored potential and after cell charging, the working electrode cable was removed and a multimeter was used for measuring the resistance between the other two gold contacts (accuracy: ±1.5%). This two-contact-point electrical conductivity probe for in situ measurements at various states of electrode charge was shown to yield data consistent with a four-point probe setup by Kastening et al. shown in ref. 45.
Acknowledgements
DW, MZ, NJ, and VP thank Prof. Eduard Arzt (INM) for his continuing support and acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. RK thanks the SFB 840 for financial support and JKE the Elitenetzwerk Bayern e. V. for a grant. We thank Annett Rabis (Paul Scherrer Institute, Switzerland) for performing the XPS measurements.Notes and references
- (a) P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed; (b) J. Segalini, E. Iwama, P.-L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Commun., 2012, 1, 63 CrossRef.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219 CrossRef CAS PubMed.
- (a) H. Gerischer, R. McIntyre, D. Scherson and W. Storck, J. Phys. Chem., 1987, 91, 1930 CrossRef CAS; (b) A. A. Kornyshev, N. B. Luque and W. Schmickler, J. Solid State Electrochem., 2014, 18, 1345 CrossRef CAS.
- F. Beguin and E. Frackowiak, Supercapacitors, Wiley, Weinheim, 2013 Search PubMed.
- G. Lota, B. Grzyb, H. Machnikowska, J. Machnikowski and E. Frackowiak, Chem. Phys. Lett., 2005, 404, 53 CrossRef CAS.
- L. G. Bulusheva, E. O. Fedorovskaya, A. G. Kurenya and A. V. Okotrub, Phys. Status Solidi B, 2013, 250, 2586 CrossRef CAS.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2009, 113, 19082 CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588 CAS.
- R. J. White, M. Antonietti and M.-M. Titirici, J. Mater. Chem., 2009, 19, 8645 RSC.
- (a) J. Tang, R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572 CrossRef CAS PubMed; (b) R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288 CrossRef CAS PubMed; (c) N. L. Torad, R. R. Salunkhe, Y. Li, H. Hamoudi, M. Imura, Y. Sakka, C.-C. Hu and Y. Yamauchi, Chem.–Eur. J., 2014, 20, 7895 CrossRef CAS PubMed.
- X. Fan, C. Yu, J. Yang, Z. Ling and J. Qiu, Carbon, 2014, 70, 130 CrossRef CAS.
- J. Zhou, Z. Zhang, W. Xing, J. Yu, G. Han, W. Si and S. Zhuo, Electrochim. Acta, 2015, 153, 68 CrossRef CAS.
- M. Wahid, G. Parte, D. Phase and S. Ogale, J. Mater. Chem. A, 2015, 3, 1208 CAS.
- (a) S. Zhang, K. Dokko and M. Watanabe, Chem. Mater., 2014, 26, 2915–2926 CrossRef CAS; (b) N. Fechler, T.-P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75 CrossRef CAS PubMed.
- S. Zhang, S. Tsuzuki, K. Ueno, K. Dokko and M. Watanabe, Angew. Chem., Int. Ed., 2015, 54, 1302 CrossRef CAS PubMed.
- Y. Hu, H. Liu, Q. Ke and J. Wang, J. Mater. Chem. A, 2014, 2, 11753 CAS.
- (a) D.-Y. Kang and J. H. Moon, Sci. Rep., 2014, 4, 5392 CAS; (b) D.-D. Zhou, W.-Y. Li, X.-L. Dong, Y.-G. Wang, C.-X. Wang and Y.-Y. Xia, J. Mater. Chem. A, 2013, 1, 8488 RSC.
- P. Tamilarasan and S. Ramaprabhu, J. Nanosci. Nanotechnol., 2015, 15, 1154 CrossRef CAS PubMed.
- D. Weingarth, I. Czekaj, Z. Fei, A. Foelske-Schmitz, P. J. Dyson, A. Wokaun and R. Kötz, J. Electrochem. Soc., 2012, 159, 611 CrossRef.
- W.-Y. Tsai, R. Lin, S. Murali, L. Li Zhang, J. K. McDonough, R. S. Ruoff, P.-L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403 CrossRef CAS.
- A. Brandt, S. Pohlmann, A. Varzi, A. Balducci and S. Passerini, MRS Bull., 2013, 38, 554 CrossRef CAS.
- K. L. Van Aken, M. Beidaghi and Y. Gogotsi, J. Mater. Chem. A, 2015, 54, 4806 CAS.
- M. Zaheer, C. D. Keenan, J. Hermannsdörfer, E. Roessler, G. Motz, J. Senker and R. Kempe, Chem. Mater., 2012, 24, 3952–3963 CrossRef CAS.
- (a) M. Kamperman, A. Burns, R. Weissgraeber, N. van Vegten, S. C. Warren, S. M. Gruner, A. Baiker and U. Wiesner, Nano Lett., 2009, 9, 2756 CrossRef CAS PubMed; (b) B. H. Jones and T. P. Lodge, J. Am. Chem. Soc., 2009, 131, 1676 CrossRef CAS PubMed; (c) S. K. Pillai, W. P. Kretschmer, C. Denner, G. Motz, M. Hund, A. Fery, M. Trebbin, S. Förster and R. Kempe, Small, 2013, 9, 984 CrossRef CAS PubMed; (d) J.-K. Ewert, C. Denner, M. Friedrich, G. Motz and R. Kempe, Nanomaterials, 2015, 5, 425 CrossRef CAS.
- (a) M. Rose, Y. Korenblit, E. Kockrick, L. Borchardt, M. Oschatz, S. Kaskel and G. Yushin, Small, 2011, 7, 1108 CrossRef CAS PubMed; (b) V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810 CrossRef CAS.
- S.-H. Yeon, P. Reddington, Y. Gogotsi, J. E. Fischer, C. Vakifahmetoglu and P. Colombo, Carbon, 2010, 48, 201 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. V. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- R. Dash, J. Chmiola, G. Yushin, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer and S. Kucheyev, Carbon, 2006, 44, 2489 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 Search PubMed.
- N. D. Kim, W. Kim, J. B. Joo, S. Oh, P. Kim, Y. Kim and J. Yi, J. Power Sources, 2008, 180, 671 CrossRef CAS.
- W. Kim, J. B. Joo, N. Kim, S. Oh, P. Kim and J. Yi, Carbon, 2009, 47, 1407 CrossRef CAS.
- W. Li, D. Chen, Z. Li, Y. Shi, Y. Wan, G. Wang, Z. Jiang and D. Zhao, Carbon, 2007, 45, 1757 CrossRef CAS.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092 CrossRef CAS PubMed.
- L. L. Zhang, X. Zhao, H. Ji, M. D. Stoller, L. Lai, S. Murali, S. Mcdonnell, B. Cleveger, R. M. Wallace and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 9618–9625 CAS.
- P. W. Ruch, R. Kötz and A. Wokaun, Electrochim. Acta, 2009, 54, 4451 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 Search PubMed.
- (a) X. Wen, D. Zhang, L. Shi, T. Yan, H. Wang and J. Zhang, J. Mater. Chem., 2012, 22, 23835 RSC; (b) Z. Peng, D. Zhang, T. Yan, J. Zhang and L. Shi, Appl. Surf. Sci., 2013, 282, 965 CrossRef CAS; (c) H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41 RSC; (d) X. Wen, D. Zhang, T. Yan, J. Zhang and L. Shi, J. Mater. Chem. A, 2013, 1, 12334 RSC; (e) H. Wang, L. Shi, T. Yan, J. Zhang, Q. Zhong and D. Zhang, J. Mater. Chem. A, 2014, 2, 4739 RSC; (f) Q. Wang, J. Yan, Y. Wang, T. Wei, M. Zhang, X. Jing and Z. Fan, Carbon, 2014, 67, 119 CrossRef CAS.
- D. Weingarth, A. Foelske-Schmitz and R. Kötz, J. Power Sources, 2013, 225, 84 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 5, 4709 CrossRef.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129 CrossRef CAS.
- D. Weingarth, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochem. Commun., 2012, 18, 116 CrossRef CAS.
- P. W. Ruch, D. Cericola, M. Hahn, R. Kötz and A. Wokaun, J. Electroanal. Chem., 2009, 636, 128 CrossRef CAS.
- B. Kastening, M. Hahn and J. Kremeskötter, J. Electroanal. Chem., 1994, 374, 159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta04773k |
| This journal is © The Royal Society of Chemistry 2015 |