
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon supported Ru clusters prepared by pyrolysis of Ru precursor-impregnated biopolymer fibers†
Andreas
Kalytta-Mewes
,
Sebastian
Spirkl
,
Sebastian
Tränkle
,
Manuel
Hambach
and
Dirk
Volkmer
*
Chair of Solid State and Materials Chemistry University of Augsburg, Institute of Physics Universitätstrasse 1, 86159 Augsburg, Germany. E-mail: dirk.volkmer@physik.uni-augsburg.de
First published on 6th August 2015
Abstract
Ru clusters deposited on pyrolyzed bacterial nanocellulose (Ru/p-BNC) were prepared in a single step by controlled pyrolysis at 1250 °C (under Ar gas), starting from bacterial nanocellulose (BNC) fibers impregnated with [RuCl2(DMSO)4], which serves as a Ru precursor. Pyrolyzed BNC fibers, featuring a high specific surface area (>600 m2 g−1), are structurally and chemically robust supports for Ru nanoparticles, as demonstrated by temperature programmed pulse chemisorption experiments. The carbon fibers, as well as the carbon supported Ru clusters, were characterized by a range of complementary analytical techniques including high-resolution transmission electron microscopy (HRTEM), selected area electron diffraction (SAED) and Raman spectroscopy.
Introduction
The deposition of Ru containing particles on carbon materials, either as pure Ru clusters, as RuO2 particles, or as a bimetallic phase (e.g. Pd/Ru particles) has been of scientific interest for decades. These materials are widely used as catalysts in a variety of reactions, employing a broad range of different carbon support materials, i.e. carbon nanofibers (CNFs), carbon nanotubes (CNTs), activated carbon fibers (ACFs), activated carbon cloth (ACC), or just amorphous carbon.1–8However, the production of nanostructured carbon supports often requires a number of preparation steps, such as template impregnation, pyrolysis and/or template removal procedures, including the usage of aggressive chemicals (e.g. hydrogen fluoride)9,10 and thus environmentally benign approaches towards structurally and chemically well-defined nanocarbon materials should represent a sustainable and economically attractive alternative. The use of cellulosic precursors for the production of carbon fibers is long since known and dates back to the late 19th century.11 The graphitization of cellulose from different sources (such as plant and bacterial nanocellulose (BNC)), the resulting carbon morphologies and their potential use as catalyst supports have been studied extensively.12–17
A review of the structural evolution of the resulting carbon fibers and the operational parameters is given in ref. 11. While cellulose commonly is not as easily graphitizable as the more frequently used polyacrylonitrile (PAN), the crystallinity of bacterial cellulose is higher than that of cellulose from other biological resources, which possibly enhances its intrinsic propensity for graphitization.11,17 The small sized microfibrils which cellulose-producing bacteria assemble form bands of nanosized fibrils that feature a width of only ∼500 Å, which renders these a technologically attractive biological source for fibrous carbon materials.18 This microstructure leads to highly accessible surface areas of the pyrolyzed carbon fibers, which in turn is advantageous for use as supports for metal catalysts.19
We here report for the first time a highly efficient one-step synthesis of carbon supported ruthenium clusters by controlled pyrolysis of microbial nanocellulose (Ru/p-BNC) impregnated with a Ru precursor, namely cis-[RuIICl2(DMSO)4]. The morphology and structure of the thus-prepared materials were investigated by a range of complementary analytical techniques and preliminary tests of the catalytic properties of Ru/p-BNC were conducted through hydrogenation (methanation) of carbon monoxide by means of temperature controlled pulse chemisorption experiments.
Results and discussion
p-BNC
Pyrolyzed bacterial nanocellulose (p-BNC) was analyzed by IR, TGA, ESEM, HRTEM, temperature controlled pulse chemisorption experiments and argon sorption measurements. The morphologies of the native and the pyrolyzed fibers were investigated by ESEM. Fig. 1a shows the image of an untreated fiber, while Fig. 1b shows the image of a sample pyrolyzed at 1000 °C under a N2 atmosphere. The adsorption isotherms of argon sorption measurements for two different BNC samples treated at 800 °C and 1500 °C under a N2 atmosphere and another sample treated at 1250 °C under an Ar atmosphere were recorded (see Fig. A in the ESI†). According to the BET-plot, the highest surface area value (624 m2 g−1) was obtained for a sample pyrolyzed at 800 °C while samples treated at 1250 °C (in an Ar atmosphere for better comparison with Ru/p-BNC samples) and 1500 °C resulted in a decrease of the specific surface area (307 m2 g−1 and 287 m2 g−1, respectively). The decrease of specific surface area for cellulose treated at very high temperature is a well-known phenomenon, which was reported earlier by Harris.20 | ||
| Fig. 1 ESEM images of the nanocellulose fiber: (a): untreated; (b): pyrolyzed at 1000 °C under N2. | ||
Fourier-transform infrared measurements of the untreated bacterial nanocellulose and samples pyrolyzed at 1000 °C and 1500 °C under N2 are shown in Fig. 2.
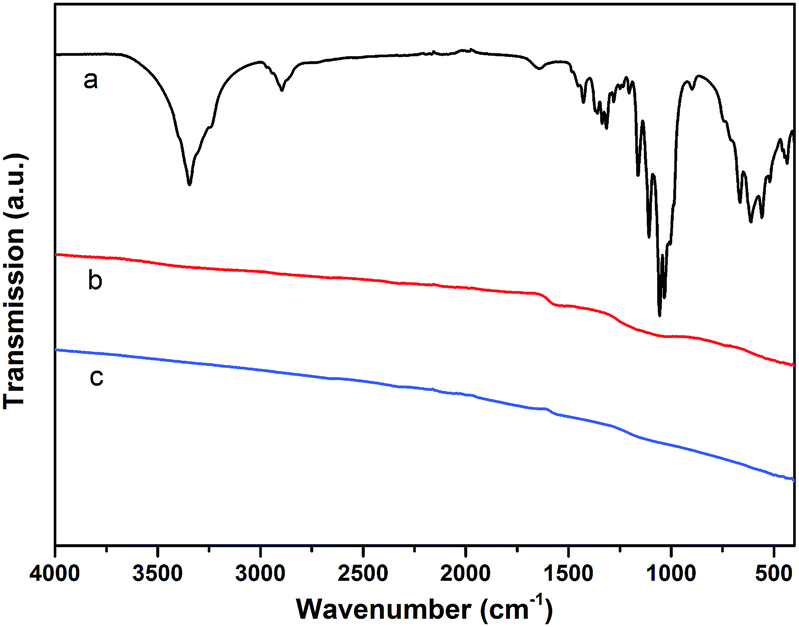 | ||
| Fig. 2 IR-spectra of: (a): untreated polymer fiber; (b): pyrolyzed biopolymer fiber (1000 °C under N2 atmosphere); (c): pyrolyzed biopolymer fiber (1500 °C under N2 atmosphere). | ||
The infrared spectrum of BNC in Fig. 2a shows the typical bands for the cellulose material.21 The bands in the region of 500–700 cm−1 can therefore be assigned to the νO–H vibrations (out of plane bending) of the hydroxyl groups. The bands in the region of 1000–1200 cm−1 are mainly due to the νC–O stretching vibrations of hydroxyl and acetal groups. Bands in the region of 1200–1450 cm−1 can be assigned to the νO–H in-plane bending vibrations of the hydroxyl groups, while bands around 2900 cm−1 are due to the νC–H stretching vibrations of the cellulose structure. Finally, the broad bands in the region from 3000–3600 cm−1 can be assigned to the νO–H stretching modes of hydroxyl groups.21
For the samples heated to 1000 °C under N2 (and 1500 °C respectively, cf.Fig. 2b and c), the characteristic bands for cellulose cannot be observed, which is expected to be due to the carbonization process. The resulting feature-less IR spectra are typical for carbon materials/graphite.22
HRTEM images of BNC fibers pyrolyzed at 1500 °C under N2 in Fig. 3a and b show graphitic domains from which electron density profiles can be extracted (see Fig. 3c). The layer distance was determined to be d002 = ∼0.347 nm, which is close to the d value of turbostratic graphite (random stacking of graphene layers, d = 0.344 nm).20,23 The SAED pattern of the sample in the inset in Fig. 3a shows diffraction rings which can be assigned to the (100) and (110) planes of a graphitic structure. The diffraction ring corresponding to the (002) plane is not clearly visible from the SAED pattern. This is probably due to the small domains of graphitic layers and the resulting very broad ring, which is overlaid by the central spot. This could be revealed by using a different camera length which also resulted in weaker overall intensity. The rotational average of this SAED pattern shows a broad peak for (002) planes which is centred at ∼3.60 Å (see Fig. 4). The peaks for (100) and (110) planes are centred at 2.07 Å and 1.21 Å respectively.
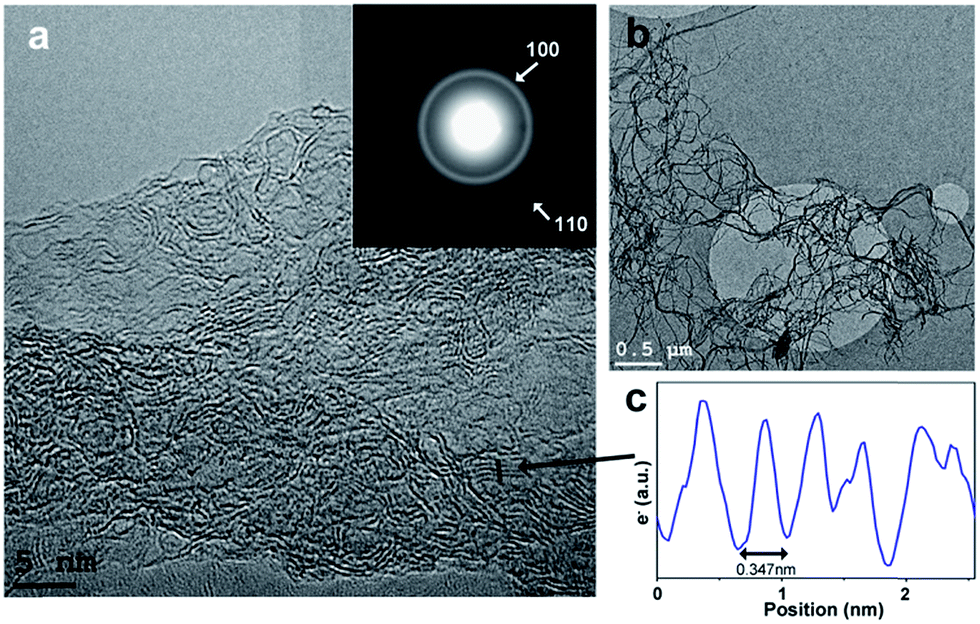 | ||
| Fig. 3 (a and b): TEM images of pyrolyzed biopolymer fibers (p-BPF) heated to 1500 °C under N2 for 3 h; inset in (a): SAED pattern showing diffraction rings associated with a graphitic structure (100 and 110 planes of graphene layers; camera length 60 cm); (c): electron density profile extracted from the path marked in (a) (see arrow) showing layers with a spacing of 0.347 nm. | ||
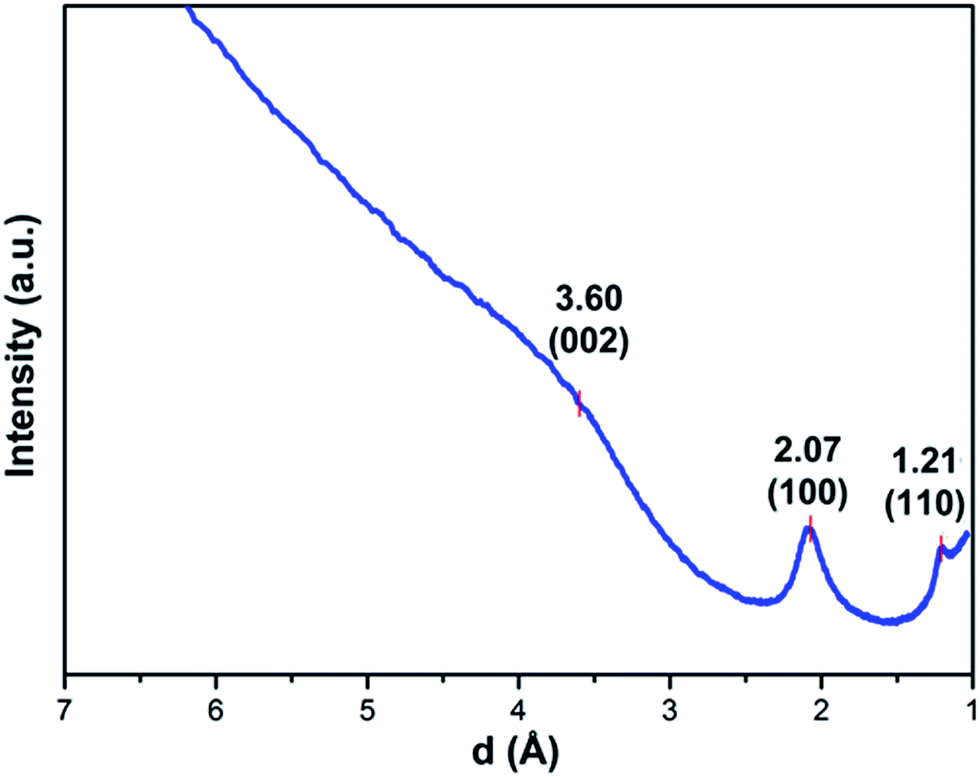 | ||
| Fig. 4 Rotational average of the SAED pattern of BPF with camera length 120 cm. | ||
Thermogravimetric analyses for samples of BNC have been conducted under Ar (purity 99.999%) atmosphere in the temperature range starting from room temperature up to a final temperature of 1100–1400 °C (see Fig. B in the ESI†). The mass losses of four independent samples are almost identical with values ranging from 89.0% loss for the sample heated up to 1100 °C, 93.3% (1200 °C), 91.8% (1300 °C), and 92.7% (1400 °C). The weight loss takes place in one big step with an onset at 311–315 °C for the samples heated to 1200, 1300 and 1400 °C, respectively. For the sample treated at 1100 °C the onset is located at a slightly higher temperature (324 °C). The loss of mass is mainly due to release of water, CO and CO2 (determined by mass spectrometric measurements, see Fig. D in the ESI†).
Ru/p-BNC
HRTEM images of a sample of the Ru precursor impregnated nanocellulose pyrolyzed under Ar at 1400 °C (Ru/p-BNC_28.4%) shows Ru clusters as dark spots confined to the surface of carbon fibers (Fig. 5a and b). The average size of ∼6.5–13 nm (depending on the ruthenium concentration) of the ruthenium clusters was calculated by a method described elsewhere.24 Higher ruthenium-to-cellulose ratios led to larger ruthenium clusters (see Table 1).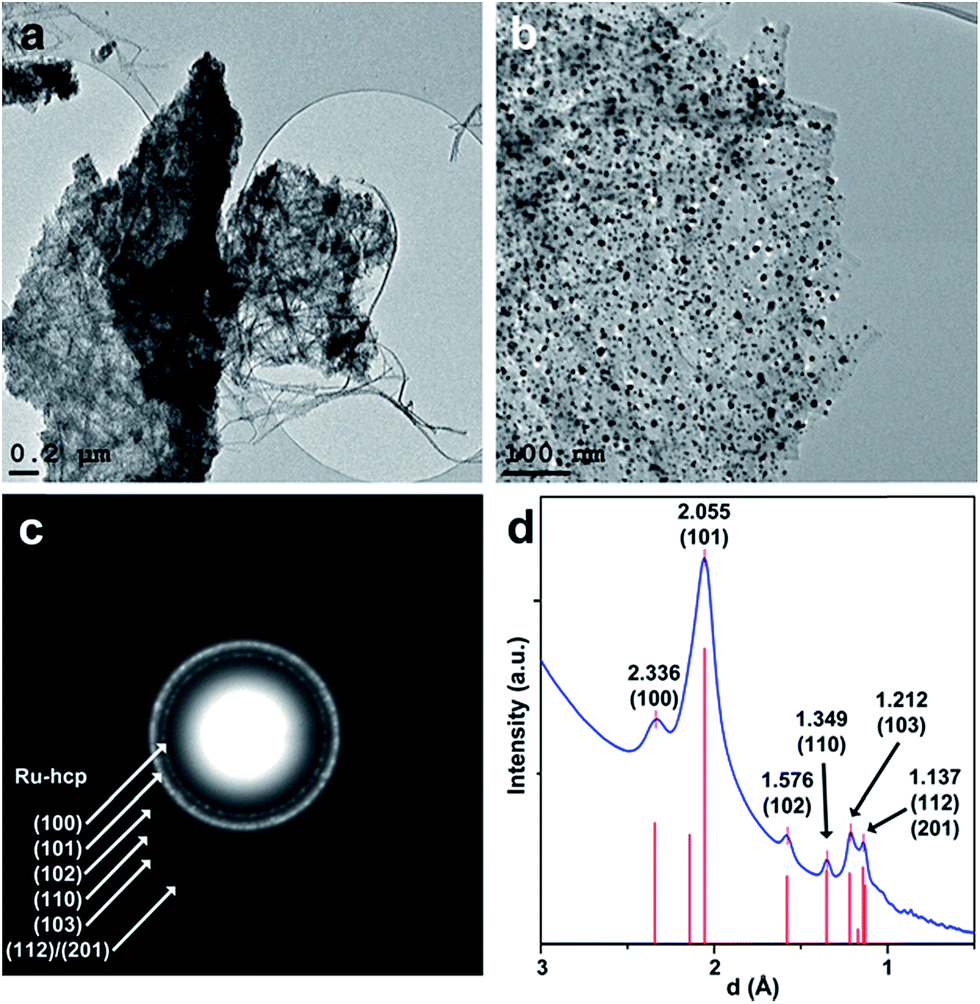 | ||
| Fig. 5 (a and b): TEM images of Ru clusters on pyrolyzed biopolymer fibers (Ru/p-BNC_28.4%); (c): SAED pattern of an area in image (a) showing diffraction patterns of a Ru-hcp structure overlying the diffraction rings of the carbon support; (d): rotational average of the SAED pattern in (c), experimental XRD data-hcp (from ref. 25) are shown as red bars. | ||
| Sample name | Ru content [wt%] | Average cluster size [nm] |
|---|---|---|
| Ru/p-BNC_3.8% | 3.8 | 6.5 ± 2.9 |
| Ru/p-BNC_5.0% | 5.0 | 8.6 ± 4.4 |
| Ru/p-BNC_28.4% | 28.4 | 10 ± 3.7 |
| Ru/p-BNC_64.4% | 64.4 | 13.2 ± 6.4 |
The diffraction rings of the SAED pattern of the area in Fig. 5c can all be assigned to crystalline ruthenium with hexagonal close-packed (hcp) structure. The indexed peaks of the extracted rotational average of the SAED pattern at 2.336 Å, 2.055 Å, 1.576 Å, 1.349 Å and 1.212 Å can be assigned to the (100), (101), (102), (110) and (103) planes of Ru-hcp (Fig. 5d). The peak at 1.137 Å could either be assigned to the (112) or the (201) plane of Ru-hcp with theoretical values of 1.143 Å and 1.129 Å, respectively.25 Broad diffraction rings of the carbon support (compare Fig. 3) can also be observed under the diffraction rings of the Ru-hcp structure at ∼2.05 Å and ∼1.21 Å. These values can be assigned to the (100) and (110) planes of the graphite structure, respectively.26
Thermogravimetric measurements in conjunction with mass spectrometric sampling of the evolved gases of the Ru precursor and the Ru precursor-impregnated nanocellulose were performed under Ar (purity 99.999) up to 1400 °C. The mass ratios of Ru precursor to nanocellulose are listed in Table 2. The decomposition of the Ru precursor (i.e. cis-[RuIICl2(DMSO)4]) takes place in two major mass steps. The temperature onset of the first mass-loss step is ∼187 °C. This first step can be separated into three sub-steps. The first sub-step shows a gas evolution of DMSO (dimethylsulfoxide) and the beginning of a partial decomposition of DMSO in H2O and CO (∼15.2% mass-loss). The second (21.0% mass-loss) and third sub-step (33.5% mass-loss) are much more complicated and not fully understood as yet. As main compounds, gaseous CO and H2O are detected. Apart from these gases chloromethane, DMS (dimethylsulfide) and CH2O (formaldehyde) are also formed. The temperature onset of the second major (7.9% mass-loss) mass-loss step is ∼977 °C; at this temperature SO2 was detected by mass spectrometry (see Fig. E in the ESI†). The residual mass of 21.4% (theoretically calcd: 20.86% for Ru) leads us to conclude that the Ru precursor was not completely reduced to ruthenium upon thermolysis. This assumption was confirmed by an X-ray powder diffraction (XRD) measurement, which, apart from reflections indicative of ruthenium metal, also showed some minor reflections that were assigned to RuO2 (see Fig. I in the ESI†). In agreement with the TGA curves of both pure nanocellulose and bulk cis-[RuIICl2(DMSO)4], the Ru precursor-impregnated nanocellulose as well shows two major decomposition steps. The temperature onset of the first mass-loss step varies between ∼150 °C and ∼240 °C, depending on the loading of the nanocellulose fiber. A loading of the cellulose fibers with the Ru precursor up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio leads to a shift of the decomposition temperature towards a lower onset value. Ratios higher than 1:1 let the decomposition temperature ascend again (see Fig. C in the ESI†). The decomposition of the impregnated nanocellulose also shows two main mass steps. During the first main mass step H2O, CO2, CO, DMSO, DMS, CH2O and chloromethane were detected by mass spectrometry. The temperature onset of the second main mass-loss step is ∼981 °C; at this temperature, SO2 was released. In addition, CO2 was formed due the reduction of RuO2 (see Fig. F in the ESI†).
:1 ratio leads to a shift of the decomposition temperature towards a lower onset value. Ratios higher than 1:1 let the decomposition temperature ascend again (see Fig. C in the ESI†). The decomposition of the impregnated nanocellulose also shows two main mass steps. During the first main mass step H2O, CO2, CO, DMSO, DMS, CH2O and chloromethane were detected by mass spectrometry. The temperature onset of the second main mass-loss step is ∼981 °C; at this temperature, SO2 was released. In addition, CO2 was formed due the reduction of RuO2 (see Fig. F in the ESI†).
| Atmosphere | Temperature [°C] |
|---|---|
| N2 | 800 |
| N2 | 1000 |
| Ar | 1250 |
| N2 | 1500 |
Catalytic activity
Two samples of Ru/p-BNC were used for the preliminary catalytic tests, namely Ru/p-BNC_5.0% and Ru/p-BNC_3.8%. The results obtained with the sample Ru/p-BNC_5.0% (sample mass: 8.0 mg) indicated promising catalytic activity of the material. To verify these results a second independent sample was prepared under the same conditions. The amount of ruthenium in this sample was 3.8 wt%. This sample (45.4 mg) was used to analyze the amount of active sites of the ruthenium clusters by employing carbon monoxide pulse chemisorption. Prior to this, the sample was reduced with 10% hydrogen in argon at an oven temperature of 550 °C (sample: 510 °C). Afterwards, pulse chemisorption measurements were performed at room temperature, injecting pulses of CO/He (5 vol%/95 vol%) into a carrier gas stream of helium. The amount of carbon monoxide adsorbed on the ruthenium particles was 0.24 μmol. Assuming a Ru/CO-ratio of 1:1, ca. 1.3% of the ruthenium atoms are covered with CO. The particle size of this sample was determined to be 6.5 ± 2.9 nm. Thus the amount of titrated surface atoms was calculated to be 6.3% for the average particle size of 6.5 nm.
Ruthenium particles are the most active particles for the Fischer–Tropsch reaction.27 Carbon monoxide is reduced by hydrogen to hydrocarbons and water in the gas phase at higher temperatures and ambient pressure.28
Hence, the carbon supported ruthenium clusters were used in another pulse experiment, applying a temperature ramp up to 400 °C. Hydrogen (10 vol% in Ar) was used as the carrier gas, while carbon monoxide (5 vol% in He) was used as the pulse gas. The reaction products were analyzed by mass spectrometry. At 135 °C the first release of methane was detected by means of mass spectrometry (Fig. 6). Upon reaching 400 °C (sample temp.: 360 °C) the oven temperature was kept constant, showing a constant production of methane (1.18 μL per pulse of carbon monoxide) and water between 250 °C and 360 °C. Another hint for the formation of methane is the delay of the liberation of the pulses with the m/z value of 15, depicted in Fig. H of the ESI,† in comparison to the methane calibration pulses. Typically, the reaction products of a Fischer–Tropsch reaction are higher chains of hydrocarbons and water, which could not be detected during this experiment. Thus the catalytic reaction of Ru/p-BNC can be described as methanation.
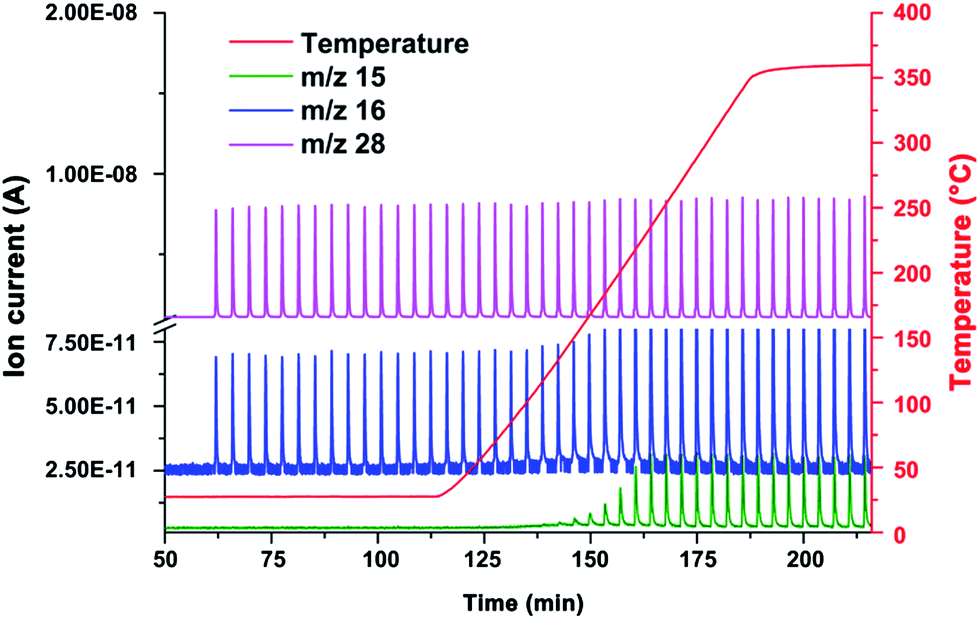 | ||
| Fig. 6 Mass spectrum of CO pulse chemisorption measurements in 10% hydrogen in Ar. A temperature ramp (red) was conducted from room temperature to 400 °C. m/z 28 (purple) represents the carbon monoxide pulses, m/z 16 (blue) represents oxygen from carbon monoxide, as well as the methane formed at temperatures above 135 °C (growth of the peaks). The ratio of m/z 15 (green): m/z 16 is used to prove the formation of methane. | ||
A third pulse experiment was conducted with hydrogen (10 vol% in Ar) and nitrogen (100%) as pulse gas. A temperature ramp up to 800 °C was applied, revealing that the carbon supported ruthenium clusters reduce the support material at temperatures above 525 °C (see Fig. J in the ESI†), as reported elsewhere.29 A comparison of the adsorption isotherms of argon sorption measurements of Ru/p-BNC_5.0% samples showed an increase according to the BET-plot before (288 m2 g−1) and after catalytic tests (384 m2 g−1) including several reduction steps under hydrogen above 550 °C (see Fig. A in the ESI†). The reason for this might be a reaction of the carbon supporting material with hydrogen and the ruthenium clusters at temperatures above ∼525 °C (see Fig. J in the ESI†) or a deposition of carbon according to the Boudouard reaction (2CO ↔ C + CO2). Both effects might lead to the formation of further micropores within the carbon supporting material; hence a higher surface area was generated.
A reference sample of p-BNC (4.0 mg) showed signs of a reaction with hydrogen gas above 450 °C, evolving traces of methane, according to the results of mass spectrometry (m/z 15 and m/z 16).28
Raman spectroscopy
According to the literature the graphitization degree of carbonaceous samples can be determined by different techniques, i.e. X-ray diffraction,30 electron microscopy31 or Raman spectroscopy.32 We chose the latter technique and followed a procedure described by Endo et al. which shows the so-called D- and G-bands of the carbonaceous sample, in which the D-band (centered at approx. 1350 cm−1) is related to disordered graphene layers while the G-band (centered at approx. 1592 cm−1) accounts for the E2g mode at the Γ-point of the graphite crystal lattice.33Thus, determining the ratio between the peak areas (AD-band/AG-band) of a sample provides a means to evaluate its graphitization degree, which can also be employed as a qualitative measure for comparing different samples (sharing similar precursor chemistry and pre-treatment conditions, however).
Raman spectroscopy was used to investigate the graphitization degree of p-BNC and Ru/p-BNC_3.8. For this, different samples were synthesized under Ar- (99.999%) and N2-atmospheres (99.999%) at two different temperatures (1250 °C and 1400 °C, isotherm for 1 h; see Fig. K in the ESI†).
For comparison purposes, the D- (∼1350 cm−1) and G-band (∼1592 cm−1) peak positions, the full width at half maximum (FWHM) and the relative peak area ratio of the two bands (AD-band/AG-band) were fitted using a Gaussian/Lorentzian mixed shape fit function (see Table A in the ESI†).34
BNC pyrolyzed at 1400 °C shows the highest graphitization degree, related to the FWHM from D- and G-band and the relative peak area ratio (AD-band/AG-band). Also the Ru/p-BNC_3.8 samples show an increase in graphitization at higher temperatures. However in direct comparison, the samples including ruthenium have a broader FWHM and a higher relative peak area ratio (AD-band/AG-band) than the p-BNC. This indicates a lower graphitization degree, irrespective of the gas atmosphere and the sample.
Comparing different pyrolysis gases, argon seems to be better suited than nitrogen for the pure BNC, in order to obtain a high graphitization degree of the samples (AD-band/AG-band-ratio and the FWHM of G-bands), which contrasts with the findings of Fey et al. reported in 2002.35 However, the degree of graphitization of Ru/p-BNC_3.8 is higher for samples treated with N2.
Conclusions
By pyrolyzation of bacterial nanocellulose at temperatures ranging from 800–1500 °C it was possible to synthesize a carbon material with a fibrous structure and a moderately high specific surface area of up to 624 m2 g−1. A high surface area represents a crucial feature for carbon materials being used as supports for metal catalysts or as supercapacitors in ionic liquids.19,36,37 The carbon fibers produced from bacterial nanocellulose are partly graphitized, which should give rise to good electrical conductivity, which in turn could also be advantageous for use as supercaps or for electrocatalytic applications.15,17,38A promising method for generating even higher surface area values within the nanocellulose-derived carbon fibers might be a chemical (e.g. KOH) or physical (e.g. CO2) treatment as described elsewhere.17,39
The use of the pyrolyzed bacterial nanocellulose as a catalyst support was tested by in situ preparation of nanosized Ru clusters on p-BNC. The untreated fibers were immersed in [RuCl2(DMSO)4] solution and then heated to 1250 or 1400 °C under an Ar atmosphere. The Ru clusters were identified as ruthenium showing a hexagonal close-packed lattice structure with average metal cluster sizes ranging from ∼6.5 nm to 13 nm, depending on the concentration of the impregnating precursor solution (see Table 1).
Raman measurements show that the graphitization degree of Ru/p-BNC_3.8 is lower than of p-BPF. This is shown for two different pyrolysis temperatures and two different reaction gases. According to the results argon as the inert gas seems to support the graphitization of pure BNC fibers better than nitrogen, whereas nitrogen seems to support the graphitization of Ru/p-BNC_3.8 better than argon.
The efficient generation of carbon nanofiber surface-confined Ru clusters in a single preparation step renders this procedure an economically attractive, sustainable approach toward large-scale production of Ru-based supported catalysts. The fibrous structure of the carbon provides open access for reagents (either in condensed or gas phase) to the catalytically active Ru centers.
As a preliminary test for the catalytic properties of the material the low-pressure (partial pressure of CO: <0.05 bar) reduction of carbon monoxide by hydrogen gas (at temperatures starting at 135 °C) was demonstrated successfully. TEM investigations of a Ru/p-BNC_5.0% sample after several catalytic tests did not show any discernible changes with respect to the carbon fiber structure. The average ruthenium cluster size slightly increased from 8.6 nm to 8.9 nm. These promising results might point to a highly desirable long term stability of the catalyst in gas phase hydrogenation processes.
Experimental section
Preparation of pyrolyzed bacterial nanocellulose fibers (p-BNC)
Bacterial nanocellulose fibers were obtained from Jenpolymer Materials Ltd & Co. KG in the block-shaped form. Pyrolysis was conducted in a Gero HTRH-70-300/16 tube furnace, equipped with an alumina tube 1000 mm in length and 70 mm in diameter (Alsint 99,7 TYP C 799 according to DIN EN 60672), under N2 (purity 99.999%, gas flow 70 mL min−1) atmosphere and under Ar (99.999%, gas flow 70 mL min−1) atmosphere at 1250 °C, respectively. The biopolymer was transferred to an alumina crucible and heated (heating rate 5 K min−1) to the desired temperature (see Table 2). Activated carbon was used in an extra alumina crucible as an oxygen getter and placed near the sample. At the outlet of the tube furnace the oxygen content was monitored during the whole process by means of a solid electrolyte sensor purchased from Zirox® (O2-DF-13.0). After holding the temperature for 3 h the pyrolyzed nanocellulose fibers (p-BNC) were cooled down to room temperature.Preparation of cis-[RuCl2(DMSO)4]
cis-[RuCl2(DMSO)4] was synthesized from RuCl3·nH2O and DMSO as described elsewhere.40,41 RuCl3·nH2O and DMSO were obtained from Merck and Aldrich, respectively, and used without further purification. In a typical synthesis 287 mg RuCl3·nH2O (1.10 mmol assuming n = 3) were dissolved in 25 mL of ethanol in a 100 mL flask and refluxed for 3 h under stirring. After cooling down the undissolved black material was removed by filtration. Ethanol was then removed by rotary evaporation. DMSO (2 mL) was added and the solution was refluxed at 150 °C for 2 h under stirring. The color of the solution changed from green to orange over time. After cooling down to room temperature, a yellow product (cis-[RuCl2(DMSO)4]) was obtained by adding 8 mL of acetone. The slurry was allowed to stand overnight. The precipitate was collected by filtration and washed three times with 5 mL acetone. The product was dried under vacuum for 6 h. Yield: 280 mg (52.5% assuming n = 3).Preparation of carbon supported Ru-clusters (Ru/p-BNC)
The biopolymer fibers were obtained from Jenpolymer Materials Ltd & Co. KG in the block-shaped form. After dissolving a defined amount of [RuCl2(DMSO)4] in demineralised water (see Table 3), the biopolymer fiber-block was immersed in the [RuCl2(DMSO)4] precursor solution. The impregnated biopolymer fiber was then dried in air over night at room temperature. The fiber was transferred to a NETZSCH STA 409 PC Luxx® instrument and heated with a rate of 10 K min−1 to the desired temperature under Ar (purity 99.999%, gas flow 70 mL min−1) atmosphere in an alumina crucible. After holding the temperature for 30 minutes the fiber-block was cooled down to room temperature. The ruthenium content in weight percent (wt%) was calculated from the value of the residual mass from TGA data, under the assumption of a complete conversion of the ruthenium(II)-complex into ruthenium(0) (see Table 3 and Fig. C in the ESI†).| BNC [mg] | [RuCl2(DMSO)4] [mg] | Water [mL] | Temperature [°C] | Ru [wt%] | Sample name |
|---|---|---|---|---|---|
| 5.7 | 2.9 | 0.3 | 1400 | 28.36 | Ru/p-BNC_28.4% |
| 5.6 | 16.2 | 0.3 | 1400 | 64.40 | Ru/p-BNC _64.4% |
| 103.2 | 5.6 | 0.6 | 1250 | 5.01 | Ru/p-BNC _5.0% |
| 203.0 | 10.6 | 1.0 | 1250 | 3.83 | Ru/p-BNC _3.8% |
| 146.9 | 2.82 | 0.8 | 1250 | 2.22 | Ru/p-BNC _2.2% |
| 180 | 1.52 | 0.8 | 1250 | 0.94 | Ru/p-BNC _0.9% |
Characterization
TEM investigations were performed using a JEOL 2100F microscope with a FEG electron source operated at 200 kV. For sample preparation, a small portion of material was dispersed in ∼1 mL of ethanol and sonicated for 2–3 minutes. Holey carbon-coated copper grids were then dipped into the dispersion and dried at RT. The software package DiffTools was used for evaluation of SAED-patterns.42EDX measurements were conducted with an EDAX Genesis detector as part of the JEOL 2100F setup. Data analysis was performed with the corresponding software package.
Crystal phase determinations were performed with a Seifert 3003 TT diffractometer, collecting X-ray diffraction data in the range 10° ≤ 2θ ≥ 100° with a step width of 0.01°. Data collection was carried out by means of a Meteor 1D line detector and took 100 s per data point. (Cu-Kα radiation, Bragg–Brentano geometry). The X-ray tube was operated with 40 kV and 40 mA and a nickel filter was used to suppress Kβ radiation.
Fourier-transform infrared spectra of the samples were recorded using a Bruker Equinox 55 equipped with a Bruker Platinum ATR unit from 4000–400 cm−1 using 4 cm−1 resolution and 32 scans per sample.
Raman spectra were recorded with a DXR™ Raman microscope from Thermo Scientific with a 532 nm laser, a magnification of 10 with an estimated resolution from 5.5 to 8.3 cm−1. The data were collected by applying a Laser power of 0.5 mW and an exposure time of 1 second for each spectrum. Raman spectra of each sample were recorded at seven different positions. Each spectrum was smoothened, base line corrected and fitted with a two peak Gaussian/Lorentzian fit function. For comparison purposes, the peak position, peak area and FHWM of the G- and D-band of the spectra were evaluated (see Table A in the ESI†).
ESEM investigations were performed using a FEI/Philips XL30 FEG ESEM. The samples were sputtered with an Au layer in order to avoid charging effects.
Argon sorption isotherms were measured with a Quantachrome Autosorb-1C instrument at 77 K up to p/p0 = 1. High purity gas was used for the adsorption experiments (argon 99.999%). Prior to measurements, the samples were heated at 300 °C for 2 h in a vacuum.
Thermogravimetric analysis (TGA) was performed with a NETZSCH STA 409 coupled to a mass spectrometer via Skimmer®, and a NETZSCH STA 409 PC Luxx®.
Temperature programmed pulse chemisorption measurements were performed using a BelCat-B catalyst analyzer (Bel Japan, Inc.) equipped with a thermal-conductivity detector and a coupled mass spectrometer (OmniStar GSD 320, Pfeiffer Vacuum). The volume of the pulse loops was 0.920 mL and 0.326 mL, the gas flow rates were set to 30 mL min−1. Two different samples were used for the catalytic tests: Ru/p-BNC_5.0% (8.0 mg) and Ru/p-BNC_3.8% (45.4 mg). Each sample was placed between two plugs of quartz wool in a quartz glass reactor. Prior to hydrogenation experiments the sample was reduced with 10% hydrogen in argon (99.999%) by heating the sample to 550 °C (furnace temperature) for 30 minutes. After this reduction step, 5.01% carbon monoxide in helium was pulsed (pulse loop volume = 0.920 mL) to a helium carrier gas stream (99.999%) to titrate the amount of active sites. To confirm the obtained results of the adsorbed amount of carbon monoxide, the titration was repeated with a smaller pulse loop (0.326 mL). After thermal desorption of the carbon monoxide, 5.01% carbon monoxide in helium was pulsed to a mixture of 10% hydrogen in argon (pulse loop size = 0.920 mL). The furnace temperature was increased from room temperature to 400 °C (ramp rate 10 K min−1) and the reaction products were analyzed by mass spectrometry.
Another experiment was performed with Ru/p-BNC_3.8% and hydrogen (10% in argon) as carrier gas. Nitrogen was applied as the pulse gas (pulse loop size = 0.920 mL), while the furnace temperature was increased from room temperature to 800 °C with a ramp rate of 10 K min−1. The reaction products were analyzed by means of mass spectrometry.
In addition, an experiment with a pure carbon supporting material (p-BNC (1250 °C under Ar), 4.0 mg) was performed to investigate the reduction of the support material under hydrogen (10%, in argon). The reaction temperature for this experiment was increased from room temperature to 950 °C with a ramp rate of 10 K min−1.
Acknowledgements
The authors wish to thank Dana Vieweg for helpful support in the high temperature pyrolysis, Dmytro Denysenko for his valuable help regarding Ar physisorption measurements and Benjamin Baumgärtner for recording ESEM images.References
- Y. Nishi, T. Suzuki and K. Kaneko, J. Phys. Chem. B, 1997, 101, 1938 CrossRef CAS.
- Y. Nishi, T. Suzuki and K. Kaneko, Carbon, 1998, 36, 1870 CrossRef CAS.
- M. L. Toebes, M. K. van der Lee, L. M. Tang, M. H. Huis in 't Veld, J. H. Bitter, A. J. van Dillen and K. P. de Jong, J. Phys. Chem. B, 2004, 108, 11611 CrossRef CAS.
- Y. Zhang, D. Kang, M. Aindow and C. Erkey, J. Phys. Chem. B, 2005, 109, 2617 CrossRef CAS PubMed.
- P. Sivakumar, R. Ishak and V. Tricoli, Electrochim. Acta, 2005, 50, 3312 CrossRef CAS PubMed.
- Y. Motoyama, M. Takasaki, K. Higashi, S.-H. Yoon, I. Mochida and H. Nagashima, Chem. Lett., 2006, 35, 876 CrossRef CAS.
- J. Liu, P. Bai and X. S. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 3758 RSC.
- Z. Li, M. Garedew, C. H. Lam, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2012, 14, 2540 RSC.
- C. Liang, Z. Li and S. Dai, Angew. Chem., 2008, 120, 3754–3776 CrossRef PubMed.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS PubMed.
- A. G. Dumanlı and A. H. Windle, J. Mater. Sci., 2012, 47, 4236 CrossRef.
- B. Azambre, O. Heintz, A. Krzton, J. Zawadzki and J. Weber, J. Anal. Appl. Pyrolysis, 2000, 55, 105 CrossRef CAS.
- D.-Y. Kim, Y. Nishiyama, M. Wada and S. Kuga, Carbon, 2001, 39, 1051 CrossRef CAS.
- K. Ishimaru, T. Hata, P. Bronsveld, D. Meier and Y. Imamura, J. Mater. Sci., 2007, 42, 122 CrossRef CAS.
- Y.-R. Rhim, D. Zhang, D. H. Fairbrother, K. A. Wepasnick, K. J. Livi, R. J. Bodnar and D. C. Nagle, Carbon, 2010, 48, 1012 CrossRef CAS PubMed.
- Y. Kaburagi, M. Ohoyama, Y. Yamaguchi, E. Shindou, A. Yoshida and N. Iwashita, et al. , Carbon, 2012, 50, 4757 CrossRef CAS PubMed.
- K.-Y. Lee, H. Qian, F. H. Tay, J. J. Blaker, S. G. Kazarian and A. Bismarck, J. Mater. Sci., 2013, 48, 367 CrossRef CAS.
- O. M. Astley, E. Chanliaud, A. M. Donald and M. J. Gidley, Int. J. Biol. Macromol., 2001, 29, 193 CrossRef CAS.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259 CrossRef CAS.
- P. J. F. Harris, Int. Mater. Rev., 1997, 42, 206 CrossRef CAS PubMed.
- Y. Maréchal and H. Chanzy, J. Mol. Struct., 2000, 523, 183 CrossRef.
- R. A. Friedel and G. L. Carlson, J. Phys. Chem., 1971, 75, 1149 CrossRef CAS.
- D. D. L. Chung, in Carbon Fiber Composites, Butterworth-Heinemann, Newton, 1994, pp. 55–65 Search PubMed.
- A. Borodzinski and M. Bonarowska, Langmiur, 1997, 13, 5613 CrossRef CAS.
- E. J. Roche, J. Mater. Sci., 1990, 25, 2149 CrossRef CAS.
- H. E. Swanson, R. K. Fuyat and G. M. Ugrinic, in Standard X-ray Diffraction Powder Patterns, National Bureau of Standards, Washington, D.C, 1955, vol. 539, IV, pp. 5–6 Search PubMed.
- J. M. G. Carballo, J. Yang, A. Holmen, S. García-Rodrigue, S. Rojas, M. Ojeda and J. L. G. Fierro, J. Catal., 2011, 284, 102 CrossRef CAS PubMed.
- I. Rodriguez-Ramos, F. Rodriguez-Reinoso, A. Guerrero-Ruiz and J. de Dios Lopez-Gonzalez, J. Chem. Technol. Biotechnol., 1986, 36, 67 CrossRef CAS PubMed.
- F. Rodriguez-Reinoso, Carbon, 1998, 36, 159 CrossRef CAS.
- N. Iwashita and M. Inagaki, Carbon, 1993, 31, 1107 CrossRef CAS.
- H. Daniels, R. Brydson, B. Rand and A. Brown, Philos. Mag., 2007, 87, 4073 CrossRef CAS PubMed.
- M. Endo, K. Nishimura, Y. A. Kim, k. Hakamada, T. Matushita, M. S. Dresselhaus and G. Dresselhaus, J. Mater. Res., 1999, 14, 4474 CrossRef CAS.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2271 CrossRef CAS PubMed.
- Y. Wang, S. Serrano and J. J. Santiago-Avilés, Synth. Met., 2003, 138, 423 CrossRef CAS.
- G. T.-K. Fey and Y.-C. Kao, Mater. Chem. Phys., 2002, 73, 37 CrossRef CAS.
- C. Arbizzani, S. Beninati, M. Lazzari, F. Soavi and M. Mastragostino, J. Power Sources, 2007, 174, 648 CrossRef CAS PubMed.
- A. Balducci, R. Dugas, P. Taberna, P. Simon, D. Plée, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922 CrossRef CAS PubMed.
- L. Deng, R. J. Young, I. A. Kinloch, A. M. Abdelkader, S. M. Holmes, D. A. De Haro-Del Rio and S. J. Eichhorn, ACS Appl. Mater. Interfaces, 2013, 5, 9983 CAS.
- J. A. Maciá-Agulló, B. C. Moore, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1367 CrossRef PubMed.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204 RSC.
- I. Bratsos and E. Alessio, in Inorganic Syntheses, John Wiley & Sons, Hoboken, N.J, 2010, vol. 35, p. 148 Search PubMed.
- D. R. G. Mitchell, Microsc. Res. Tech., 2008, 71, 588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta04253d |
| This journal is © The Royal Society of Chemistry 2015 |