
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urea-containing metal-organic frameworks as heterogeneous organocatalysts†
Alireza Azhdari
Tehrani‡
a,
Sedigheh
Abedi‡
a,
Ali
Morsali
*a,
Jun
Wang
b and
Peter C.
Junk
b
aDepartment of Chemistry, Faculty of Sciences, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran. E-mail: Morsali_a@modares.ac.ir; Fax: +98-21-82884416; Tel: +98-21-82884416
bCollege of Science Technology and Engineering, James Cook University, Townsville, Qld 4811, Australia
First published on 20th August 2015
Abstract
Two novel pillared metal-organic frameworks (MOFs) containing a urea-functional group are introduced. Herein, the urea functional group was incorporated into the MOF backbone by preparing a urea-ditopic ligand. These frameworks (TMU-18 and TMU-19) were fabricated using the synthesized urea-containing ligand, 4,4′-bipyridine (bipy) and 1,2-bis(4-pyridyl)ethane (bpe), and using zinc nitrate as the metal source. Subsequently, TMU-18 and TMU-19 were characterized by X-ray diffraction, IR spectroscopy, elemental analysis, scanning electron microscopy (SEM) and thermogravimetric analysis. Furthermore, their potential efficiency as organocatalysts was evaluated in the regioselective methanolysis of epoxides.
Introduction
Supramolecular organocatalysis is an interdisciplinary research area that includes elements from organic chemistry, supramolecular chemistry and biochemistry.1,2 The design of a supramolecular catalyst is based on using hydrogen bonding and other intermolecular interactions in recognition and activation of substrates for triggering a variety of chemical transformations.3,4 However, supramolecular catalysis often suffers from drawbacks such as the lack of catalyst recycling and low efficiency due to the self-aggregation (self-quenching) of the catalyst.3 The heterogenization of supramolecular organocatalysts may be a logical solution to overcome these obstacles in extending the applicability of these systems.5–7 Recently, metal-organic frameworks (MOFs) were introduced as promising candidates for applications in diverse areas.8–10 Compared to other porous materials, MOFs have given chemists the opportunity to tune the topology, pore size and functionality by rational selection of organic linkers and inorganic metal centers. Owing to this feature, MOFs with uniform and permeable pores and channels have shown to be particularly promising for catalysis.11–14 According to the catalytically active sites, these frameworks may be categorized into four distinct groups, namely metal-organic frameworks with coordinatively unsaturated metal sites (group I), MOFs with metalloligands (group II), MOFs with functional organic sites (group III) and metal nanoparticles embedded in the MOF cavities (group IV). Among these, MOFs with catalytically active functional organic sites have received less attention due to the synthetic complexities in providing guest-accessible functional organic groups in the pore surface of frameworks.15 In this regard, different organic functional groups, such as proline, amide, binol and pyridyl, were successfully incorporated into MOFs.16 Recently, both Cr-MIL-101 and IRMOF-3 are decorated with activated urea and thiourea functional groups, respectively, using a post-synthesis modification method.17,18 The appealing idea of preparing heterogeneous urea-based MOF catalysts was proposed recently by Farha, Hupp and Scheidt et al.6 They have examined the catalytic activity of the urea-based MOF for the Friedel–Crafts reaction between pyrroles and nitroalkenes. In addition, although high conversion was obtained in the case of small substrates, the yield of the addition product was low (39%) under the optimum reaction conditions. They could also successfully synthesize a Zr-based MOF containing a urea functional group used for the Morita–Baylis–Hillman reaction.19 Therefore, designing such a catalytic system based on urea-containing MOFs is in the early stages and needs further work and development. Ring opening of epoxides is one of the most important reactions producing vital intermediates in organic synthesis. There are some reports for this reaction catalysed by MOFs containing Lewis acid sites, especially with Cu, Fe and Hf metal nodes.20–23 In this regard, there is no report of urea-containing MOFs as hydrogen-bond catalysts in this reaction.In this paper, we report an extension of these studies aimed at investigating the organocatalytic role of urea-containing MOFs in the activation of epoxides. Our design is based on two following assumptions: (i) synthesis of a ditopic oxygen-donor ligand containing a urea functional group, which is capable of forming dual hydrogen bonds with organic substrates and (ii) selection of an appropriate pillaring strut able to form MOFs with dinuclear paddle-wheel SBUs, in which five of the six coordination positions of each Zn(II) ion are anticipated to be occupied for network propagation and the sixth is located inside the zinc cluster. Accordingly, two novel urea-containing MOFs were synthesized by combining the ditopic urea “strut”, pillaring struts, and Zn(NO3)2·6H2O using the solvothermal method at 90 °C for 120 h to give suitable X-ray quality crystals of [Zn2(ubl)2(bipy)]·DMF (TMU-18) and [Zn2(ubl)2(bpe)]·DMF (TMU-19), where the ubl (urea-based ligand) is 4,4′-(carbonylbis(azanediyl))dibenzoic acid, and bipy and bpe are 4,4′-bipyridine and 1,2-bis(4-pyridyl)ethane, respectively, Fig. 1.
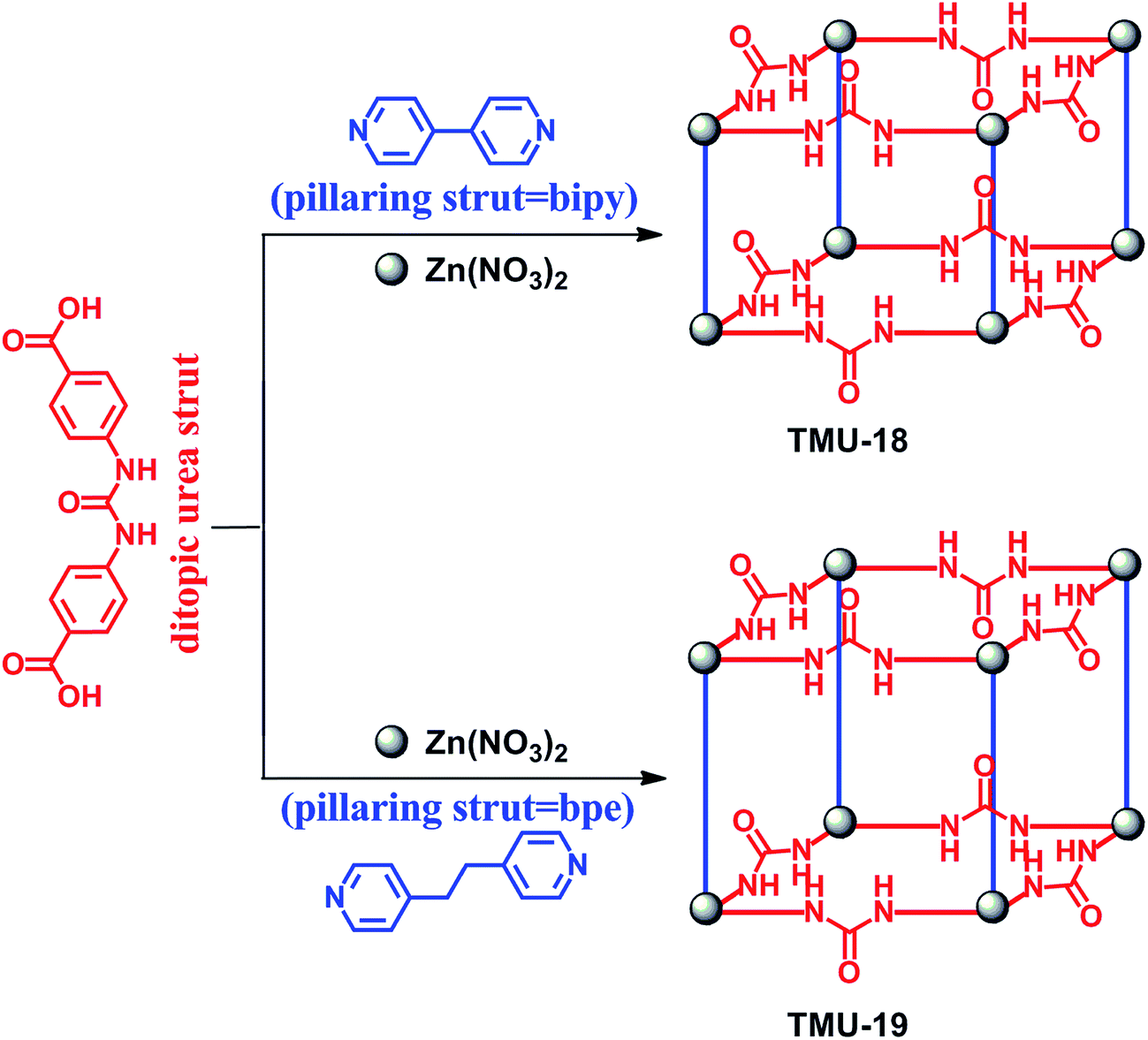 | ||
| Fig. 1 Synthesis of TMU-18 and TMU-19 from the urea-containing dicarboxylate ligand and bipy/bpe as the pillar ligand. | ||
Experimental section
Apparatus and reagents
All starting materials, including 1,1′-carbonyldiimidazole and 4-aminobenzoic acid, were purchased from commercial suppliers (Sigma-Aldrich, Merck) and used as received. The infrared spectra were recorded on a Nicolet Fourier Transform IR, Nicolet 100 spectrometer in the range of 500–4000 cm−1 using the KBr disk technique. Elemental analyses (carbon, hydrogen, and nitrogen) were performed using an ECS 4010 CHN made in Costech, Italy. Melting points were obtained by using a Bamstead Electrothermal type 9200 melting point apparatus and corrected. Thermogravimetric analyses (TGAs) of the compounds were performed on a computer-controlled PL-STA 1500 apparatus. The 1H-NMR spectrum was recorded on a Bruker AC-250 MHz spectrometer at ambient temperature in d6-DMSO and CDCl3. X-ray powder diffraction (XRPD) measurements were performed using a Philips Xpert diffractometer with monochromated Cu-Kα radiation (λ = 1.54056 Å). The samples were also characterized by using a field emission scanning electron microscope (FE-SEM) SIGMA ZEISS and TESCAN MIRA (Czech) with gold coating.Single-crystal diffraction
X-ray crystal structure determination: crystals in viscous paraffin oil were mounted on cryoloops and intensity data were collected on the Australian Synchrotron MX1 beamline at 100 K with wavelength λ = 0.71073 Å. The data were collected using the BlueIce24 GUI and processed with the XDS25 software package. The structures were solved by conventional methods and refined by full-matrix least-squares on all F2 data using SHELX9726 or SHELX2014 in conjunction with the X-Seed27 or Olex228 graphical user interface. Anisotropic thermal parameters were refined for non-hydrogen atoms and hydrogen atoms were calculated and refined with a riding model.Crystallographic data: TMU-18: C83H63N13O21Zn4, M = 1839.94 g mol−1, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 21.179(4) Å, b = 22.560(5) Å, c = 23.138(5) Å, α = 105.30(3)°, β = 114.19(3)°, γ = 102.88(3)°, V = 9013(4) Å3, Z = 2, ρcalc = 0.678 g cm−3, λ = 0.71073, T = 100 K, R1 = 0.0688, wR2 = 0.1817, S = 0.897, CCDC = 1041981†; TMU-19: C21H16N3O5Zn, M = 455.74 g mol−1, orthorhombic, Pnna, a = 21.035(4) Å, b = 16.064(3) Å, c = 29.891(6) Å, V = 10
, a = 21.179(4) Å, b = 22.560(5) Å, c = 23.138(5) Å, α = 105.30(3)°, β = 114.19(3)°, γ = 102.88(3)°, V = 9013(4) Å3, Z = 2, ρcalc = 0.678 g cm−3, λ = 0.71073, T = 100 K, R1 = 0.0688, wR2 = 0.1817, S = 0.897, CCDC = 1041981†; TMU-19: C21H16N3O5Zn, M = 455.74 g mol−1, orthorhombic, Pnna, a = 21.035(4) Å, b = 16.064(3) Å, c = 29.891(6) Å, V = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100(3) Å3, Z = 8, ρcalc = 0.599 g cm−3, λ = 0.71073, T = 100 K, R1 = 0.0786, wR2 = 0.2428, S = 1.103, CCDC = 1041982.†
100(3) Å3, Z = 8, ρcalc = 0.599 g cm−3, λ = 0.71073, T = 100 K, R1 = 0.0786, wR2 = 0.2428, S = 1.103, CCDC = 1041982.†
Synthesis of the urea ditopic ligand
The urea ditopic ligand was synthesized in three steps, starting from commercially available 4-aminobenzoic acid. Detailed synthetic procedures and characterization of the synthesized frameworks are provided in the Experimental section and ESI.†Synthesis and activation of TMU-18
Zn(NO3)2·6H2O (0.297 g, 1 mmol), 4,4′-(carbonylbis(azanediyl))dibenzoic acid (0.300 g, 1 mmol) and 4,4′-bipyridine (0.156 g, 1.0 mmol) were dissolved in 20 mL DMF. The mixture was placed in Teflon-lined stainless steel autoclaves and heated to 90 °C for 72 h and then it was gradually cooled to room temperature over 48 h. The crystals were obtained in 72% yield. The products were characterized by different techniques such as powder X-ray diffraction (PXRD), IR spectroscopy, elemental analysis and SEM microscopy. (Before activation) FT-IR (KBr pellet, cm−1): 3320 (br), 1657 (vs), 1604 (vs), 1530 (vs), 1398 (vs), 1308 (m), 1230 (m), 1171 (m), 856 (w), 779 (m), 626 (w), 500 (w). Anal. calcd for ZnC23H23N4O7: C, 51.84; H, 4.35; N: 10.51, found: C, 49.96; H, 4.79, N: 10.85.The sample was activated by immersing the crystals of TMU-18 in anhydrous chloroform followed by heating at 80 °C in a vacuum for 24 h. The activation was also confirmed by PXRD and FT-IR spectroscopy. (After activation) FT-IR (KBr pellet, cm−1): 3390 (br), 1604 (vs), 1532 (vs), 1400 (vs), 1308 (s), 1228 (m), 1172 (m), 856 (w), 779 (m), 628 (w), 500 (w). Anal. calcd for ZnC20H14N3O5: C, 54.38; H, 3.19; N: 9.51, found: C, 54.12; H, 3.82, N: 9.94.
Synthesis and activation of TMU-19
Zn(NO3)2·6H2O (0.297 g, 1 mmol), 4,4′-(carbonylbis(azanediyl))dibenzoic acid (0.300 g, 1 mmol) and 1,2-bis(4-pyridyl)ethane (0.184 g, 1.0 mmol) were dissolved in 20 mL DMF. The mixture was placed in Teflon-lined stainless steel autoclaves and heated to 90 °C for 72 h and then it was gradually cooled to room temperature over 48 h. The crystals were obtained in 60% yield. The products were characterized by different techniques such as powder X-ray diffraction (PXRD), IR spectroscopy, elemental analysis and SEM microscopy. (Before activation) FT-IR (KBr pellet, cm−1): 3346 (br), 1655 (vs), 1605 (vs), 1532 (s), 1397 (s), 1308 (m), 1231 (m), 1172 (m), 855 (m), 780 (m), 622 (w), 500 (w). Anal. calcd for ZnC24H25N4O7: C, 52.71; H, 4.61; N: 10.25, found: C, 50.99; H, 4.83, N: 11.05.The sample was activated by exchanging the DMF molecules with chloroform and then evacuating at room temperature for 8 h. FT-IR spectroscopy confirmed that some of the DMF molecules are removed from TMU-19, while the rest of them could be necessary to stabilize the MOF framework. (After activation) FT-IR (KBr pellet, cm−1): 3346 (br), 1606 (vs), 1527 (s), 1391 (s), 1306 (m), 1227 (m), 1169 (m), 855 (m), 778 (m), 619 (w), 505 (w). Anal. calcd for ZnC21H16N3O5: C, 55.34; H, 3.54; N: 9.22, found: C, 55.17; H, 3.88, N: 9.96.
Catalysis experiments
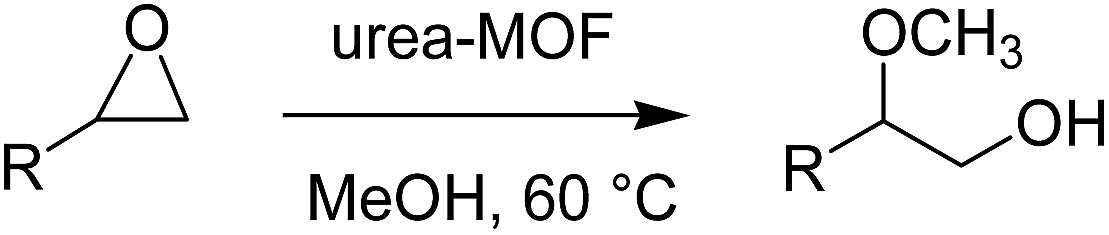
In a typical reaction, the urea-based MOFs (25 mg, ca. 0.05 mmol equiv. of urea species) were added to a CH3OH solution (3 mL) containing the epoxide substrate (0.2 mmol). The reaction mixture was stirred at 60 °C for the indicated times mentioned in Table 1. After the required reaction time, the reaction mixture was cooled to room temperature and analysed by GC analysis using an internal-standard method.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Major product | Time [h] | Conversiona [%] TMU-18 | Conversiona [%] TMU-19 |
| a GC yield using an internal-standard method; conditions: styrene oxide (25 mg, 0.2 mmol), catalyst (25 mg, 0.05 mmol of urea moiety ∼25 mol%), 60 °C, and methanol (3 mL). b Reaction control (without catalyst). c With 40 mol% urea in a homogeneous system. d With recycled catalysts. e With 50 mg (0.1 mmol) of the catalysts. f The data in parentheses are the selectivity calculated for the major product. | |||||
| 1 |
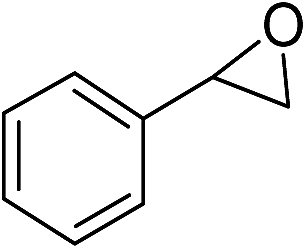
|
|
24b | 14 | |
| 2 | 110b | 19 | |||
| 3 | 24c | 15 | |||
| 4 | 24 | 35 | 31 | ||
| 5 | 40 | 45 (95)f | 41 (95) | ||
| 6 | 110 | 78 (96) | 72 (97) | ||
| 7 | 140 | 100 (98) | 95 (98) | ||
| 8 | 40d | 39 (95) | 29 (94) | ||
| 9 | 110e | 100 (98) | 100 (98) | ||
| 10 |
|
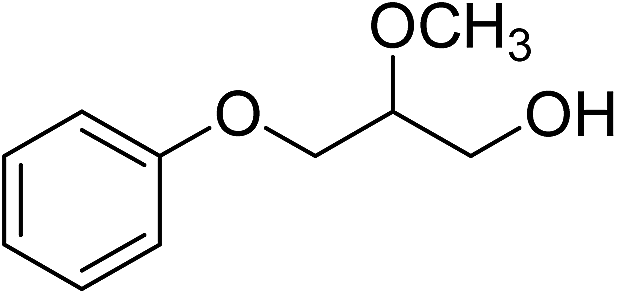
|
110b | 22 | |
| 11 | 55 | 40 | 33 | ||
| 12 | 110e | 78 (96) | 67 (91) | ||
| 13 |
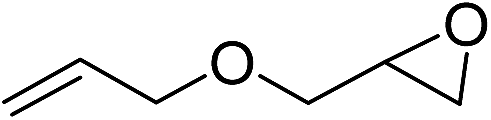
|
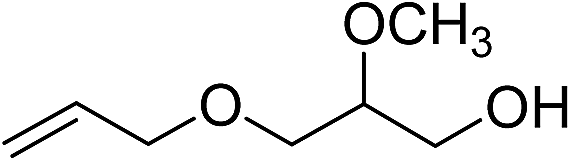
|
110b | 26 | |
| 14 | 55 | 37 | 35 | ||
| 110e | 64 (81) | 53 (77) | |||
| 15 | |||||
| 16 |
|
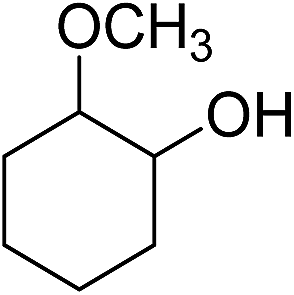
|
110b | 16 | |
| 17 | 55 | 34 | 19 | ||
| 18 | 110e | 51 (78) | 48 (72) | ||
| 19 |
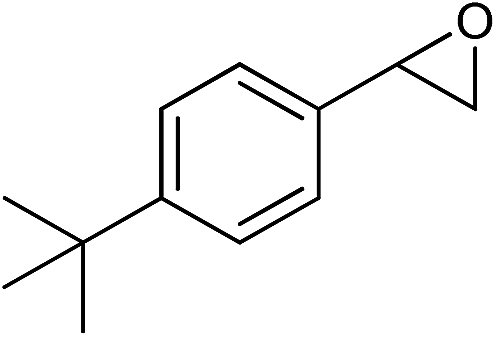
|
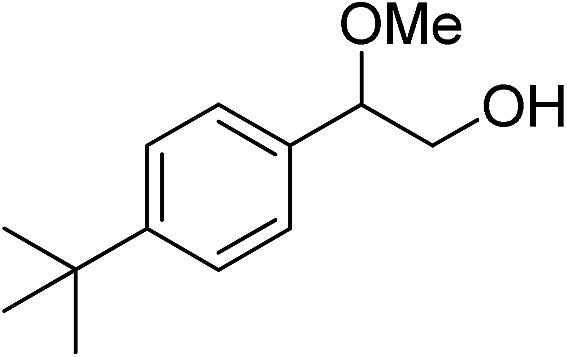
|
40 | <5 | <5 |
| 140 | 9 | 6 | |||
Catalyst recycling
The reusability of TMU-18 and TMU-19 was tested for the methanolysis of styrene oxide. After stirring for 40 h, the heterogeneous mixture was allowed to settle completely followed by decantation of the supernatant liquid. The TMU-18 and TMU-19 catalysts were filtered off after the 40 h reaction, washed with excess MeOH and respectively dried at 80 °C and under vacuum at room temperature. The recovered catalyst was then reused without further purification for the second run with fresh styrene oxide and methanol.Typical procedure for the methanolysis of styrene oxide
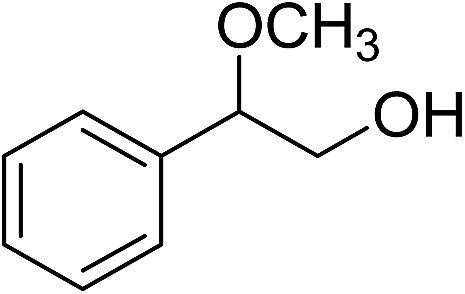
The prepared urea MOF (25 mg, ca. 0.05 mmol) catalysts are suspended in a MeOH (3 mL) solution of styrene oxide (25 mg, 2 mmol) and stirred at 60 °C for 140 h. Then, the solid catalyst was filtered off. The reaction mixture was analyzed using gas chromatography. Then the excess of solvent was removed under reduced pressure to give the corresponding product. The major product (2-methoxy-2-phenylethanol) is determined by NMR. 1HNMR (250 MHz, CDCl3): 7.26–7.39 (m, Ph, 5H), 4.3 (dd, J = 3.74, 8.1 Hz, 1H), 3.58–3.72 (m, CH2OH, 2H), 3.31 (s, OMe, 3H), 2.91 (bs, OH, 1H).Typical procedure for the methanolysis of other epoxides
The prepared urea MOF catalysts are suspended in a MeOH (3 mL) solution of epoxide (25 mg) and stirred at 60 °C for 110 h. Then, the solid catalysts were filtered off. Determination of the major product was performed based on an internal-standard method. All standard samples for determination of the major products in the reaction mixture were prepared using the separate methanolysis reactions of the epoxides in the presence of catalytic amounts of HCl. Under this acidic condition, the major products are formed through the formation of a more stable carbocation. Thus the retention time for GC analysis was obtained in this manner. For this reaction, 2 drops of concentrated HCl were added to the solution of epoxides (2 mmol) and MeOH (5 mL). The reaction solution was stirred at 60 °C for 1 h. The progress of the reaction was monitored using GC analysis.Results and discussion
Structural analysis and characterization
TMU-18 and TMU-19 were synthesized by combining the ditopic urea ligand, pillaring struts, and Zn(NO3)2·6H2O using the solvothermal method at 90 °C for 120 h to give suitable X-ray quality crystals. X-ray crystallography analyses reveal that TMU-18 and TMU-19 crystallize in triclinic P and orthorhombic Pnna, respectively. In these compounds, the coordination geometry around the Zn(II) can be described as distorted octahedra, with four sites occupied by oxygen atoms of four different urea ligand carboxylate groups in an approximately square configuration and the fifth site occupied by a nitrogen atom of the bipy/bpe ligand (Fig. 2a and d). The remaining coordination site of each metal center is located inside the zinc paddle-wheel cluster. The Zn–Zn distances are 2.930(1) and 2.9319(7) Å for TMU-18 and TMU-19, respectively. Both compounds are composed of paddle-wheel dinuclear zinc carboxylate clusters (Zn2(COO)4) bridged by the urea struts to form a two-dimensional square grid. The 2D square grids are further linked to each other by pillaring bipy/bpe forming a 3D framework which can be described as a doubly interpenetrated pcu network, Fig. 2. Both compounds possess large channels (along the bc-plane with an aperture size of 13.5 × 9.9, for TMU-18 and along the b-axis with an aperture size of 12.1 × 10.6 Å for TMU-19, including van der Waals radii of the atoms), Fig. 2(c) and (f). Also, the calculated void space per unit cell for disorder- and guest-free TMU-18 and TMU-19 frameworks is 66.3% (5977.6 Å3) and 69.6% (7034.1 Å3), respectively.29 X-ray crystallography analysis reveals that the N–H groups of TMU-18 are involved in N–H⋯O hydrogen bonding interactions with the oxygen atom of the N,N-dimethylformamide (DMF) molecule. In the case of TMU-19, the DMF molecule cannot be located in the electron-density map, due to severe disorder and therefore was squeezed out with the help of PLATON squeeze.29 However, spectroscopic analyses suggest that TMU-18 and TMU-19 may have similar chemical compositions.
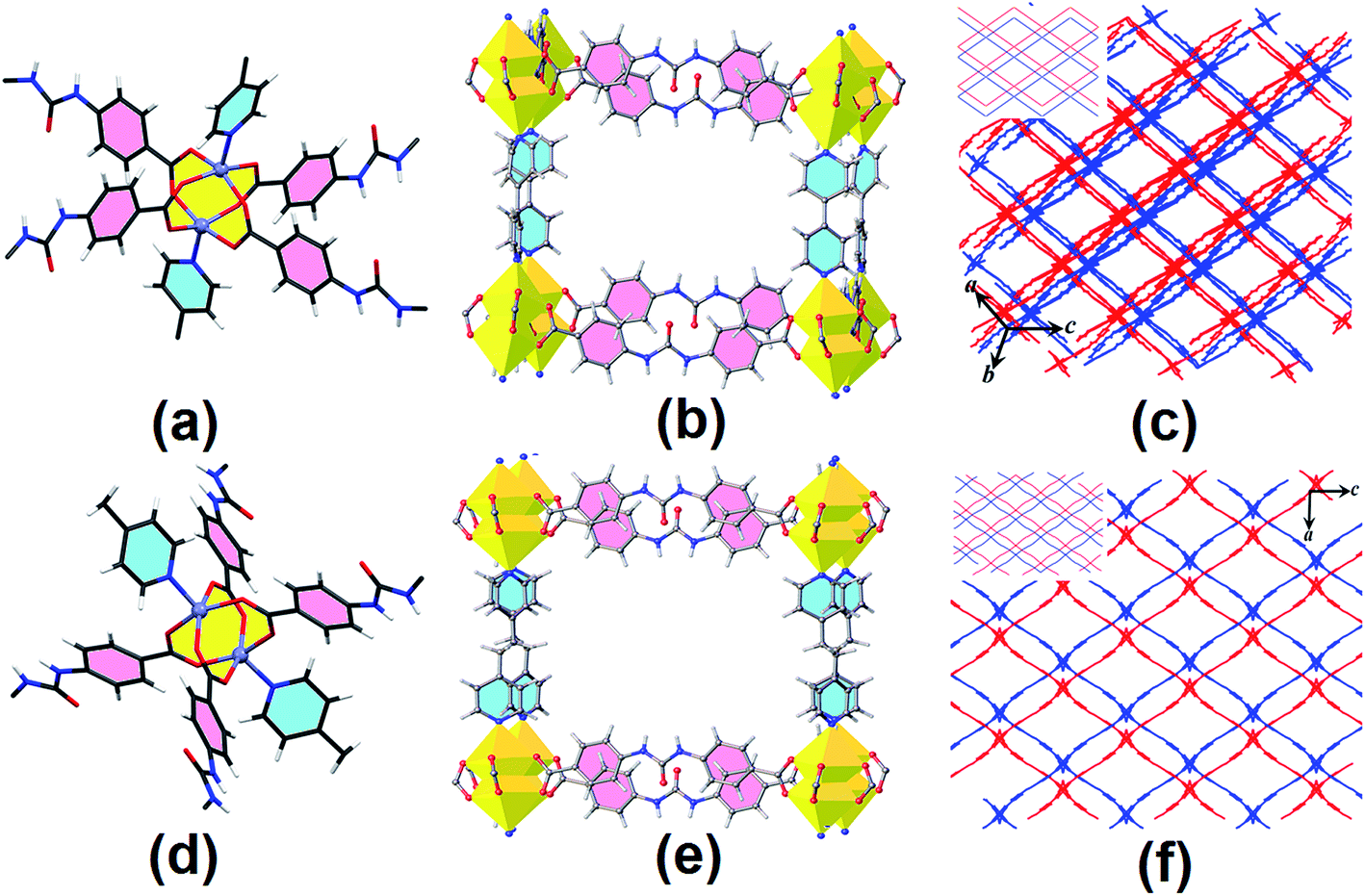 | ||
| Fig. 2 Paddle-wheel dinuclear zinc carboxylate clusters (a) and (d), representation of the cubic structure (b) and (e) and a view along the pore direction (c) and (f). The two interpenetrating frameworks are shown in red and blue. All guest molecules were omitted for clarity. The left top insets illustrate the simplified interpenetration in TMU-18 and TMU-19, respectively. | ||
Thermogravimetric analysis (TGA) indicates that TMU-18 has a much better thermal stability compared to TMU-19. The TGA data of TMU-18 show an initial weight loss (4%, after heating to 120 °C) which is attributed to the loosely bound water molecule. The other weight loss occurred between 120 and 200 °C (13%) corresponding to the removal of DMF. In contrast to TMU-18, TGA analysis of TMU-19 indicates a large mass loss in two steps in the range of 30 to 260 °C, indicating its low thermal stability. Thus, in order to activate the potential catalytic sites of TMU-18, the crystals were immersed in anhydrous chloroform for 72 h, filtered and vacuum-dried at 80 °C for 24 h. The activation was confirmed by FT-IR spectroscopy and PXRD analysis (see the ESI†). An attempt to activate TMU-19 by the same procedure was unsuccessful probably due to the low thermal stability of this compound at elevated temperatures. Accordingly, this compound was activated by exchanging the DMF molecules with chloroform and then evacuating at room temperature for 8 h. FT-IR spectroscopy confirmed that some of the DMF molecules are removed from TMU-19, while the rest of them may be necessary to stabilize the MOF framework.
Catalytic studies of TMU-18 and TMU-19
The ring-opening reaction of epoxides, by alcoholic compounds known as “alcoholysis”, was chosen as a probe to study the catalytic activity of the synthesized urea-containing MOFs. The alcoholysis reaction of epoxides is facile providing 1,2-bifunctional compounds such as 1,2-diols, β-amino alcohols and other interesting compounds from the pharmaceutical and agrochemical points of view.30,31 Actually, the activation of the oxygen atom of the epoxides is catalytically performed using either Lewis or Brønsted acids. Methanolysis of epoxides, which was traditionally attained using corrosive sulfuric acid,32 has been widely investigated in heterogeneous catalytic systems including polymers and silica-based materials.33,34 In recent years, some noteworthy attempts for the methanolysis reaction using MOFs have been reported, in which the epoxide activation is mostly achieved by the function of metal Lewis acidity centers within the MOF structures.20,22,23 Generally, the coordinating ability of solvents such as MeOH which can act as a nucleophile and the requisite vacant metal sites within the MOF catalyst led to the low structural stability of these kinds of catalysts, even those which provided excellent catalytic activity and selectivity.35 Therefore, incorporating the linkers containing Brønsted acids36 or hydrogen-bond donating (HBD) moieties into a MOF structure which involves locked metal centers such as paddle-wheel nodes can hopefully provide a MOF catalytic system that would be durable even for recycling purposes. According to the valuable report presented by Hupp and co-workers, incorporating a urea strut into MOF structures can significantly enhance its HBD ability, preventing the intrinsic unproductive self-quenching behavior of the urea units.6Regarding the above synthesized urea-containing MOFs of TMU-18 and TMU-19, we subsequently examined their potential as organocatalysts for the methanolysis reaction of epoxides. The ring opening of the styrene oxide in MeOH as a probe reaction was selected to explore the reaction conditions. No methanolysis reaction proceeded at room temperature. Moreover, in the presence of mixed solvent systems, including toluene, CH2Cl2, CHCl3, THF and CH3CN in combination with MeOH (1:1 ratio), no additional product was observed during the 48 h reaction with styrene oxide. The alcoholysis reaction in the presence of 25 mg (0.05 mmol) of these urea-based MOFs and net MeOH as the solvent gave 5% conversion as obtained by GC. However, in the absence of a catalyst, when the reaction temperature was increased to 60 °C, during 24 and 110 h, the reaction proceeded with 14 and 19% conversions (Table 1, entries 1 and 2) while within 24 h, in the presence of TMU-18 and TMU-19, 35 and 31% of styrene oxide was converted, respectively (entry 4). This observation clearly revealed the catalytic effect of the prepared urea-containing MOFs. In addition, the same reaction runs were carried out for optimization of the catalyst and also solvent amounts. During the survey of the reaction conditions in the presence of 25 mg of styrene oxide, the best results were obtained by using 25 mg of the catalysts (∼25 mol%, indicated by ICP analysis) and at 60 °C in 3 mL of MeOH. Under the optimized reaction conditions, 45 and 41% of the corresponding products were respectively formed after 40 h (entry 6). It is noteworthy to mention that 15% of the methanolysis reaction took place in the presence of 40 mol% of urea powder as the catalyst, in a homogeneous system (entry 3). It should be noted that using the urea-containing ligand could not be an appropriate choice for the control reaction. In this case, the reaction can proceed via the catalytic role of the two carboxylic acid groups instead of urea species. Considering the paddle-wheel nodes established by X-ray analysis, in these urea-based heterogeneous catalytic systems the self-quenching phenomena raised from aggregation of urea molecules in homogeneous systems has been suppressed through accommodation into MOF structures. Further investigation of the heterogeneous character of the catalytic systems as well as stability of the structures was carried out using a hot filtration test in addition to ICP analysis. After 40 h of the methanolysis reaction of styrene oxide, the reaction mixtures were centrifuged and the catalysts were filtered off. Then, the supernatants of methanolic liquids were left stirring at 60 °C. Interestingly, within 40 h of further reaction time, no distinguishable changes were recognized in the reaction conversion using GC analysis.
Moreover, in other sets of the same reactions, the catalysts were filtered off after 40 h, washed thoroughly with MeOH and subsequently the filtrates were examined by ICP analysis. 0.13 and 0.19% of residual zinc were identified respectively which significantly confirmed that more than 99% of the zinc metal center does not leach into the reaction mixture under the methanolysis conditions. Not only all of these observations confirm the reliable chemical stability of the prepared MOFs, but also they reject the Lewis acid catalytic role of metal species in epoxide activation. Finally, these observations may confirm the HBD character of the urea moieties through the MOF structure.
The productivity of both catalysts was evaluated by determination of the reaction selectivity for the conversion of styrene oxide to 2-methoxy-2-phenylethanol as the major product in our catalytic system. As shown in Table 1, after 40 h reaction time, 95% selectivity was calculated for the above mentioned major product (entry 6). When the reactions prolonged to 140 h, with TMU-18, quantitative conversion of styrene oxide was obtained with 98% selectivity for 2-methoxy-2-phenylethanol that was characterized by GC and NMR (ESI†). During the same time 95% conversion with 98% selectivity was achieved in the presence of the TMU-19 catalyst (entry 7).
The observed catalytic activity as well as regioselectivity achieved in methanolysis of styrene oxide encouraged us to examine other epoxides in the reaction. The reaction of three epoxides including γ-phenoxypropylene oxide, allyl(2,3-epoxypropyl)oxide and cyclohexene oxide was screened with both catalysts under the same optimum reaction conditions (Table 1, entries 10–18). However, the reactivity changed in methanolysis of these less reactive substrates especially cyclohexene oxide and the corresponding products which were formed with relatively moderate yield and regioselectivity, even in the presence of twice the amount of the catalysts (entries 12, 15 and 18). Actually, increasing the amount of the catalyst, in the case of styrene oxide, did not lead to doubling of the catalytic activity (entry 9) and a little improvement was observed (110 h vs. 140 h for completion the reaction). Although more detailed studies are needed to identify the real cause of this issue, this may be explained by engaging or hiding some urea functional groups within the complexities of the bulk matrix of the framework. We also checked the reaction with a bulky substrate, t-butyl styrene oxide, to clarify that the catalysis occurred within the pores of the frameworks. As tabulated in Table 1 (entry 19), the methanolysis of t-butyl styrene oxide proceeded negligibly even after 140 h.
In order to further investigate the comparison of the catalytic performance of these heterogeneous urea catalytic systems, the time-conversion for both catalysts was plotted and compared with the control methanolysis reaction of styrene oxide, Fig. 3. Although both catalyst systems are carried out with significant diversity relative to the control reaction and moderately implement the reaction within 140 h, the TMU-18 catalyst shows somewhat higher activity than the TMU-19 catalyst. The comparison of the PXRD diffraction peaks of the catalysts upon activation, represented in Fig. S1 (ESI†), indicates that little change occurred in the TMU-19 structure. Accompanied by the lower thermal stability of TMU-19 indicated by TG analysis, these results demonstrate higher catalytic performance of the TMU-18 compared to TMU-19 thereafter it was also confirmed through the recycling experiment.
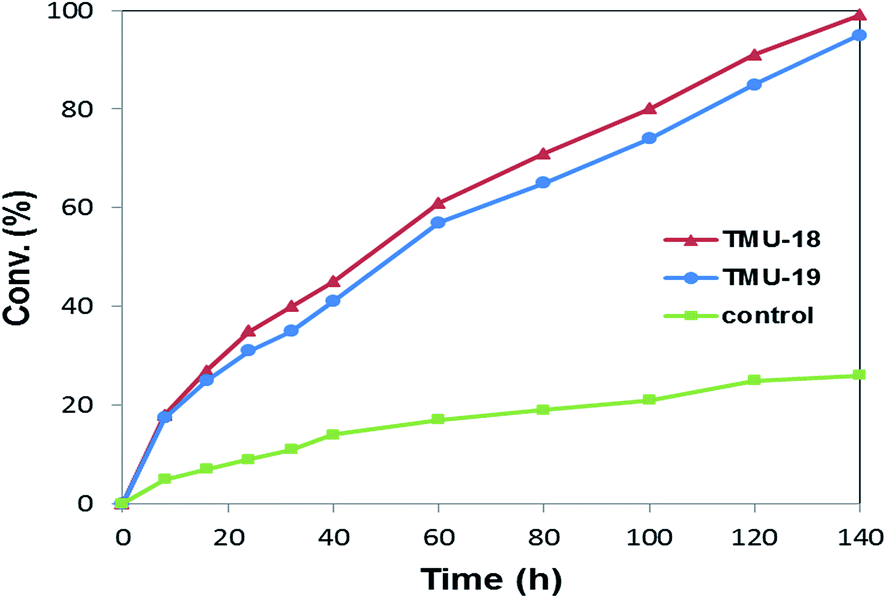 | ||
| Fig. 3 Comparison of the time conversion plot for methanolysis of styrene oxide catalyzed by TMU-18, TMU-19 and the reaction control system. Conditions: styrene oxide (25 mg, 0.2 mmol), catalyst (25 mg, 0.05 mmol of urea moiety), 60 °C, and methanol (3 mL); reaction control (without catalyst). | ||
Moreover, to evaluate the durability and the catalyst recycling ability, TMU-18 and TMU-19 catalysts were filtered off after the 40 h reaction, washed with excess MeOH and dried at 80 °C and under vacuum at room temperature, respectively. The methanolysis reaction of styrene oxide with these recovered catalysts proceeded with more diminished catalytic reactivity in the case of the TMU-19 structure (Table 1, entry 8). In addition, the comparison of the PXRD patterns of these catalysts clearly shows that a decrease in crystallinity occurred in both recovered catalyst structures (ESI†). As shown in Fig. S1(ESI†), the prominent changes of the TMU-19 structure take place after its activation, while the activated TMU-18 structure has more similar PXRD patterns to the simulated pattern. Considering the instability observed after extraction of entire DMF molecules in TMU-19, these data confirm the foundation role of the solvent molecules in preserving the whole skeleton. Although the solvent exchange with the epoxide molecules propels the methanolysis reaction, it seems that the remaining DMF within the pores slightly suppresses the organocatalytic activity of this structure.
The foundation role of the solvent within the pores was additionally proved for both MOFs by immersing them in deionized water. The catalysts were removed after 48 h, washed with CHCl3 and subsequently the aliquots were monitored using GC. Interestingly, no segregated residues of the organic pillars were detected for both samples. Furthermore, preservation of the PXRD patterns of these two water-treated samples along with their PXRD patterns after 140 h of the methanolysis reaction of styrene oxide (Fig. S2†) explicitly reveals the aforementioned effect. In addition, these data may show the genuine heterogeneous character and actual chemical stability in the reaction.
Conclusion
Two new pillared metal-organic frameworks containing urea functional groups were synthesized aiming at their application as heterogeneous organocatalysts. Structural analysis revealed that both TMU-18 and TMU-19 could be described as doubly interpenetrated pcu networks. These frameworks were characterized by different techniques and were further utilized as organocatalysts in the methanolysis of epoxides. Compared with the previous report,6 in which the number of electron withdrawing carboxyl groups per urea unit is doubled, the results of the methanolysis reaction indicate that TMU-18 and TMU-19 have weaker hydrogen bond donating ability, while they are more chemically stable MOF structures. In addition, the elucidation of additional structural features of these organocatalysts to design more active urea-based MOF structures is an ongoing project in our laboratory and needs further work and refinement.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
This work was funded by the Iran National Science Foundation (INSF) (grant number: 92024961), Iran Nano technology Initiative Council (INIC) and Tarbiat Modares University.References
- P. M. Pihko, Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed.
- D. W. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
- A. Zamboulis, N. Moitra, J. J. Moreau, X. Cattoën and M. W. C. Man, J. Mater. Chem., 2010, 20, 9322–9338 RSC.
- J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334–3337 CrossRef CAS PubMed.
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2–27 CrossRef CAS PubMed.
- S. Abedi and A. Morsali, ACS Catal., 2014, 4, 1398–1403 CrossRef CAS.
- L. R. MacGillivray, Metal-organic Frameworks: Design and Application, Wiley, Hoboken, N.J, 2010 Search PubMed.
- S. Abedi and A. Morsali, New J. Chem., 2015, 39, 931–937 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734–777 CrossRef PubMed.
- A. Corma, H. García, F. X. Llabrés and i. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS PubMed.
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- X.-W. Dong, T. Liu, Y.-Z. Hu, X.-Y. Liu and C.-M. Che, Chem. Commun., 2013, 49, 7681–7683 RSC.
- Y. Luan, N. Zheng, Y. Qi, J. Tang and G. Wang, Catal. Sci. Technol., 2014, 4, 925–929 CAS.
- P. W. Siu, Z. J. Brown, O. K. Farha, J. T. Hupp and K. A. Scheidt, Chem. Commun., 2013, 49, 10920–10922 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem.–Eur. J., 2010, 16, 8530–8536 CrossRef CAS PubMed.
- K. Tanaka, K.-i. Otani, T. Murase, S. Nishihote and Z. Urbanczyk-Lipkowska, Bull. Chem. Soc. Jpn., 2012, 85, 709–714 CrossRef CAS.
- D. Jiang, A. Urakawa, M. Yulikov, T. Mallat, G. Jeschke and A. Baiker, Chem.–Eur. J., 2009, 15, 12255–12262 CrossRef CAS PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- T. M. McPhillips, S. E. McPhillips, H.-J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter and R. P. Phizackerley, Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- D. Jiang, T. Mallat, F. Krumeich and A. Baiker, J. Catal., 2008, 257, 390–395 CrossRef CAS PubMed.
- K. Tanaka, S. Oda and M. Shiro, Chem. Commun., 2008, 820–822 RSC.
- W. Reeve and I. Christoffel, J. Am. Chem. Soc., 1950, 72, 1480–1483 CrossRef CAS.
- G. A. Olah, A. P. Fung and D. Meidar, Synthesis, 1981, 1981, 280–282 CrossRef.
- P. Salehi, M. Dabiri, A. Zolfigol and A. B. Fard, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1113–1121 CrossRef CAS PubMed.
- P. Garcia-Garcia, M. Muller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- S. J. Garibay, Z. Wang and S. M. Cohen, Inorg. Chem., 2010, 49, 8086–8091 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: PXRD patterns, TGA, IR spectroscopy, and SEM images. CCDC 1041981 and 1041982. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ta03835a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |