
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjugated microporous polymer nanoparticles with enhanced dispersibility and water compatibility for photocatalytic applications†
Beatriz Chiyin
Ma
a,
Saman
Ghasimi
a,
Katharina
Landfester
a,
Filipe
Vilela
*b and
Kai A. I.
Zhang
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
bSchool of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: f.vilela@hw.ac.uk
First published on 30th June 2015
Abstract
Here we report the design of well-dispersible conjugated microporous polymer nanoparticles as highly stable heterogeneous organic photocatalysts in aqueous medium. This class of materials combines two different heterogeneous catalytic model systems: (i) nanostructured catalysts and (ii) highly porous materials. By incorporating different electron donor and acceptor moieties, the nanoparticle shape can be controlled ranging from nanospheres via nanorods to nanorings. The high photocatalytic activity, stability and reusability of the materials were demonstrated in the reductive activation of molecular oxygen and photooxidation of amines under irradiation of a household energy-saving light bulb.
Introduction
Using sunlight as an inexpensive, non-polluting, abundant and clean energy source is undoubtedly a strategy that will aid in reducing our fossil fuel dependence as well as reducing anthropogenic CO2 emissions. Taking inspiration from Nature, where various chromophores or photocatalysts for converting solar energy into chemical energy have been utilized for millions of years, synthetic chemists have developed a vast number of visible light-driven systems involving photo-redox processes in order to mimic the natural photosynthesis in recent years.1–6 Besides well-developed transition metal-based complexes,7,8 metal-free and organic dyes have also been successfully employed as non-metal photo-redox catalysts in the last two decades.9–11 However, a number of disadvantages associated with these homogeneous catalytic systems still affect the long-term usage. For instance, they are either highly expensive, toxic, and have limited availability in nature; or suffer from solvolytic attack in the reaction medium, which can be troublesome for the recycling ability of the catalyst. It is therefore beneficial to develop a new class of organic heterogeneous photocatalysts with high stability and reusability. For this purpose, two different heterogeneous photocatalytic model systems, (i) nanometer-sized system and (ii) porous materials are herein investigated. Nanoparticles (NPs) offer a huge advantage over conventional bulk catalysts due to their high surface-to-volume ratio.12,13 Porous materials offer an enlarged active interface during the catalytic process due to their large surface area.14 Recently, conjugated microporous polymers (CMPs), which combine the photoactive π-electron backbone and microporous properties, have been employed as stable heterogeneous photocatalysts for organic synthesis under irradiation of visible light.14–19 Recent research activities have demonstrated their ability for singlet oxygen generation,20 high selective oxidation of organic sulfides,21 oxidative C–C bond formation,22 oxidative hydroxylation of arylboronic acids,23 reductive dehalogenation reaction,24 visible light-initiated free radical and cationic polymerization,25,26 and light-induced hydrogen evolution.27 A transformation of bulk CMPs into nanostructured systems (CMP NPs) offers a combination of the aforementioned advantages in heterogeneous catalytic systems.Herein, we report the design and synthesis of well-dispersible nanoparticles of conjugated microporous polymers as highly efficient metal-free visible light photocatalysts. CMP NPs combine high porosity with enhanced solution dispersibility, an important feature which allows for a heterogeneous material to behave closer to a homogeneous system. Therefore, this property also opens the possibility for the use of CMP NPs in solution processable methodologies such as spin-coating. Via the incorporation of different electron donor and acceptor building-blocks, CMP NPs were synthesized in a variety of shapes, ranging from nanospheres, nanorods to nanorings. The possible mechanism of non-spherical formation of CMP NPs was studied by monitoring the polymerization process. The high photocatalytic activity of CMP NPs was demonstrated in the reductive activation of molecular oxygen for degradation of the dye rhodamine B and the photooxidation of N,N,N′,N′-tetramethyl-p-phenylenediamine under irradiation of a household energy-saving light bulb.
The design principle and the synthesis of CMP NPs are illustrated in Fig. 1. CMP NP backbone structures were designed to demonstrate the formation of electron–hole pairs under visible light irradiation. After the light-induced charge separation of the electron–hole pair, the valence band (VB) and the conduction band (CB) should function as oxidative and reductive sites of CMP NPs for further redox reactions (Fig. 1b).
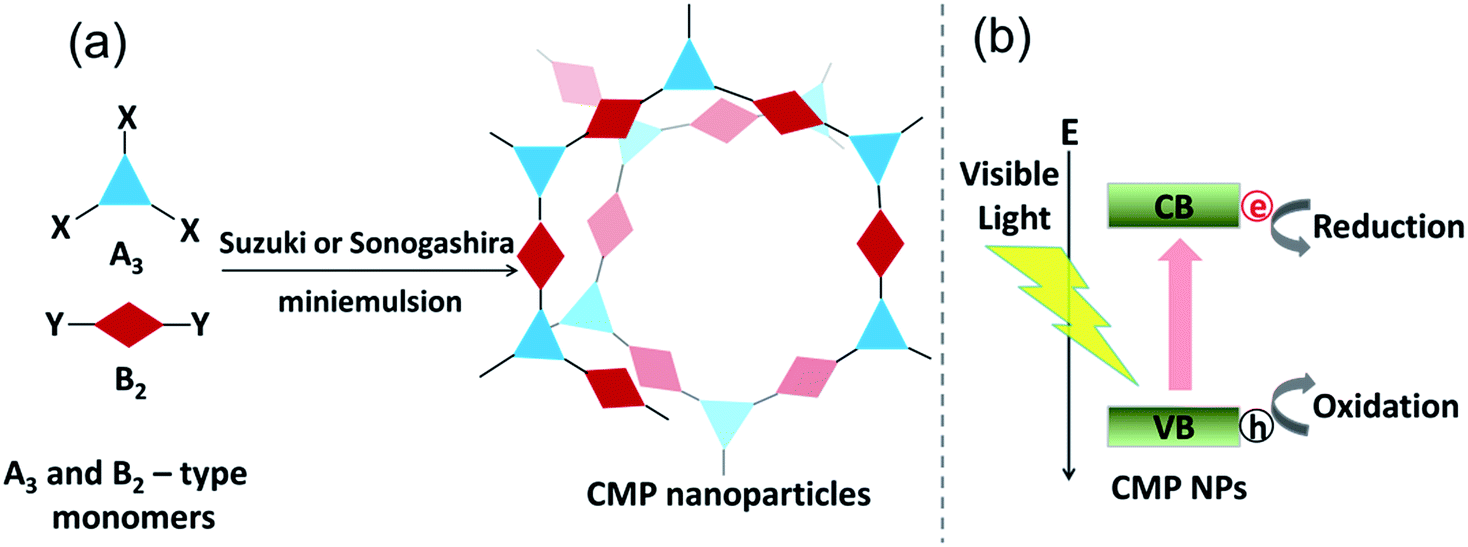 | ||
| Fig. 1 (a) Synthetic route to CMP NPs via (A3 + B2)-type Suzuki–Miyaura or Sonogashira–Hagihara cross-coupling polycondensation reactions in an oil-in-water miniemulsion system. (b) Design principle of CMP NPs for photo-induced reduction and oxidation reactions using the photogenerated electron–hole pair as reductive and oxidative sites. | ||
Results and discussion
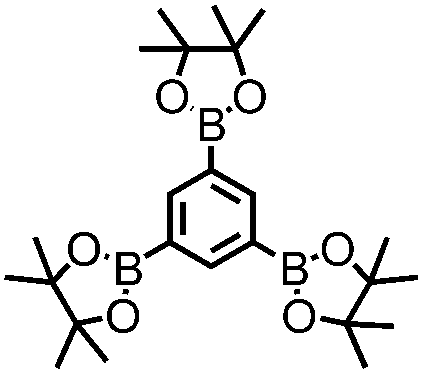
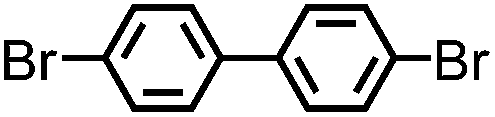
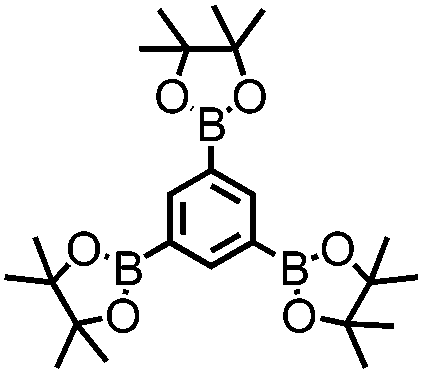
CMP NPs were obtained via palladium-catalysed Suzuki–Miyaura and Sonogashira–Hagihara cross-coupling polycondensation reactions in an oil-in-water miniemulsion (Fig. 1a).28,29 For each polymerization reaction, the same amount of starting compounds and surfactant were used to achieve similar reaction conditions within the miniemulsion droplets. The detailed reaction conditions along with detailed characterization of the polymers are described in the Experimental section and in the ESI.† The building-blocks, i.e. the A3-type cross-linkers and B2-type co-monomers used for different CMP NPs and their physical properties are listed in Table 1. The obtained CMP NPs showed a high stability as dispersion in organic solvents, and despite their hydrophobic nature, also in water (Fig. S1 in the ESI†).| CMP NPs | Cross-linker (A3-type) | Co-monomer (B2-type) | Shape | Surface area [m2 g−1] | Pore volume [cm3 g−1] | Pore diameter [nm] | Band gap/opt. [eV] |
|---|---|---|---|---|---|---|---|
| B-FL3-a |
|

|
Spherical | 18 | 0.058 | 7.5 | 3.04 |
| B-BPh3-a |

|
|
Irregular shape | 281 | 0.264 | 1.5 | 2.52 |
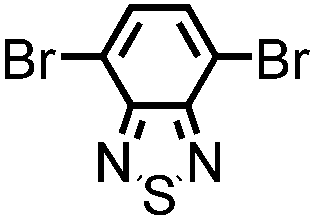
| B-BT3-a |
|
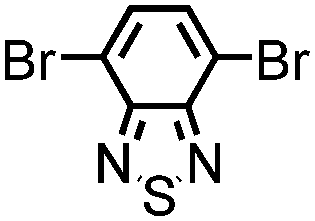
|
Rod-shape | 93 | 0.220 | 1.5 | 1.96 |
| B-FL3-b |
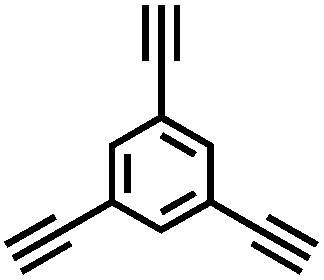
|
|
Spherical | 48 | 0.067 | 3.8 | 2.64 |
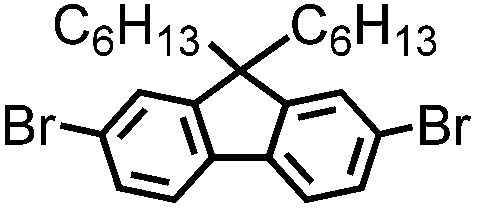
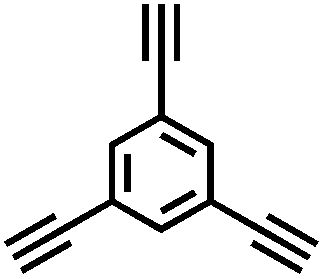
| B-BPh3-b |
|
|
Spherical | 512 | 0.382 | 1.4 | 2.36 |
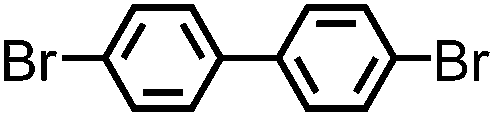
| B-BT3-b |
|
|
Ring-shape | 200 | 0.560 | 1.4 | 1.76 |
Solid state 13C/MAS NMR spectroscopy showed typical chemical shifts between 110 and 150 ppm for all CMP NPs, which can be assigned to aromatic rings in the polymer backbone (Fig. S2–S7 in the ESI†). For B-FL3-a and B-FL3-b, alkyl signals of the dihexylfluorene (FL) unit appeared between 14 and 56 ppm. Signals between 85 and 94 ppm of B-FL3-b, B-BPh3-b and B-BT3-b can be assigned to the triple bonds of the polymer backbone. For B-BT3-a and B-BT3-b, the benzothiadiazole (BT) unit can be characterized by the signals at about 155 ppm, which are assigned to the adjacent carbon next to nitrogen in the BT unit. TGA analysis showed a clear tendency of higher stability of the triple bond-containing CMP NPs B-FL3-b, B-BPh3-b and B-BT3-b (up to 350 °C) in comparison to the other series without triple bonds (up to 200 °C) (Fig. S9†). To note, B-FL3-b and B-BPh3-b showed a high “left weight”, which could indicate the carbonized content after being heated under nitrogen.
The Brunauer–Emmett–Teller (BET) surface area of CMP NPs varied from 18 m2 g−1 to 512 m2 g−1 (Table 1). The low surface area of B-FL3-a and B-FL3-b can be due to the dihexylfluorene unit, which most likely blocked the micropores with the alkyl side chains, leading to almost impossible diffusion of nitrogen gas molecules during the gas sorption analysis. The biphenyl (BPh) containing B-BPh3-b showed the highest surface area of 512 m2 g−1, which is lower than the bulk CMP reported.30 The same effect also applies to B-BT3-b with a surface area of about 200 m2 g−1, which is lower in comparison to the bulk CMP from our previous work.18 The overall trend of the CMP NPs obtained via Sonogashira coupling showed that they have higher surface areas than the ones prepared via Suzuki-coupling. This can be explained because the preparation of the polymers via Sonogashira coupling results in the formation of triple bonds, which act as rigid bridges between the building blocks, therefore, adding an extra space in the polymer backbone resulting in higher surface area. The FL-containing CMP NPs showed broad pore size distribution with size larger than 2 nm. This could be caused by the alkyl side chains of the fluorene unit, which blocked the micropores and required larger space inside the porous polymers as reported before.24 A previous study of Cooper et al. about the formation mechanism of CMPs via Sonogashira coupling showed that the high surface area was mainly obtained at the last phase of the reaction, after the fusion of small porous fragments.31 Here, the lower surface area of CMP NPs could be partially due to the lack of small fragments inside the droplets during the reaction.
In Fig. 2, SEM and TEM images of CMP NPs are displayed. As it can be seen, a variety of nanoparticle shapes was obtained. As expected, the FL-containing B-FL3-a and B-FL3-b appeared as round spheres, exhibiting an average size ranging from 60 nm to 100 nm. B-BPh3-a was obtained as irregular shape particles of ca. 80 nm, whereas B-BPh3-b appeared as spherical NPs with size ranging between 100 and 150 nm, despite its great rigid polymer backbone structure. The overall trend here, similar to the porous properties, is that CMP NPs obtained via Sonogashira-coupling are larger in comparison to the ones obtained via Suzuki-coupling. This can be due to a more rigid and extended network structure of CMP NPs containing triple bonds than the ones without them. Significantly, the BT containing CMP NPs were absolutely non-spherical, and B-BT3-a showed a rod-like shape with a length of ca. 200 nm and a width of ca. 20 nm. Most surprisingly, B-BT3-b appeared as nanometer-sized rings with a diameter of ca. 200 nm and a width of 20 nm (Fig. 2).
 | ||
| Fig. 2 (a) SEM and (b) TEM images of CMP NPs with different shapes. | ||
To investigate the ring formation mechanism of B-BT3-b, the polymerization process was monitored with defined reaction time intervals. As displayed in Fig. 3, the UV/vis spectra of the as-made nanoparticle samples were taken, indicating their development upon the completion of the polymerization process. The graph shows that the main absorption band of B-BT3-b with a maximum peak at ca. 430 nm appeared directly after the miniemulsion preparation using ultrasonication. Fig. 3A–F display the SEM images of B-BT3-b particles after the corresponding reaction time. Directly after the ultrasonication, small particles of ca. 20 nm were already formed (Fig. 3A). These could be the pre-formed oligomers, which subsequently formed the ring-like particles inside the droplets of the miniemulsion within the first minutes (Fig. 3B). After 5 minutes, the ring-shaped particles were clearly formed (Fig. 3C), which did not differentiate largely from the ones obtained after 10, 30 or 60 minutes (Fig. 3D–F) and the final product after the completion of the polymerization (Fig. 2).
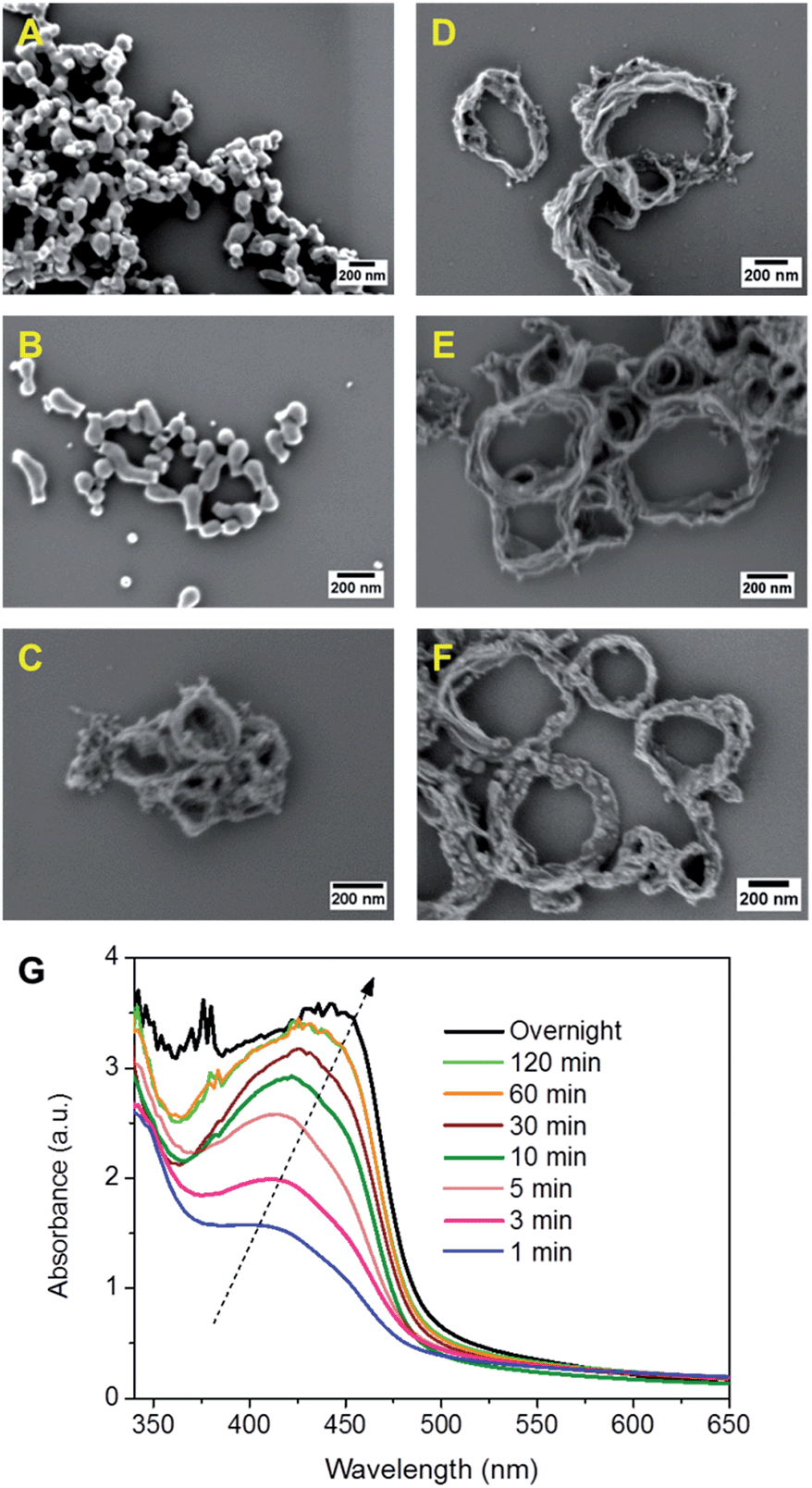 | ||
| Fig. 3 Monitoring experiments of the nanoparticle formation of B-BT3-b. SEM images of nanoparticles formed in different time intervals after ultrasonication and heating at 80 °C. (A) 1 min, (B) 3 min, (C) 5 min, (D) 10 min, (E) 30 min, and (F) 60 min. (G) UV/vis spectra of the NPs formed after defined time intervals. | ||
The monitoring experiment indicates a possible, but still not totally clear, ring formation mechanism of B-BT3-b. An average droplet size of ca. 250 nm determined by dynamic light scattering (DLS) suggests that the final ring formation of B-BT3-b might have first taken place at the interface (Fig. S12 in the ESI†), followed by an aggregation of the oligomers formed within the first minutes after the ultrasonication. According to the very rigid structure caused by the BT-based polymer backbone, a possible contraction of aggregates might have occurred, which led only to the formation of ring-shaped CMP NPs instead of capsule-like particles. After changing from an ionic surfactant (sodium n-dodecylsulfate) to a nonionic one (Lutensol® AT50) in the reaction, ring-shaped CMP NPs were still formed (Fig. S13 in the ESI†). This result suggests that the formation mechanism is not dependent on the type of surfactant used. And no strong evidence led to the assumption that the nanorings could likely be collapsed capsules in a dry state.
The UV/vis absorption spectra of CMP NPs are displayed in Fig. 4 and all of them show a broad absorption band. The donor-based CMP NPs, B-FL3-a and B-FL3-b, B-BPh3-a and B-BPh3-b, absorb mainly in the blue light range. After introducing a strong electron acceptor, BT unit into the polymer backbone, a large bathochromic shift up to 100 nm occurred, indicating a narrower band gap of the polymers. According to our recent studies on CMPs with various donor–acceptor combinations, the introduction of strong acceptors lowers mostly the LUMO level and therefore the redox-potential of the polymer, which could favor catalytic reactions initiated by light.32,33 The UV/vis absorption spectra also show that the triple bond-containing B-FL3-b, B-BPh3-b, and B-BT3-b have clearly broader absorption bands than the ones without it. A narrower optical band gap can be derived from the absorption edges (Table 1), the reason behind it can be due to a more ordered structure obtained via efficient π–π stacking during the CMP synthesis, which led to a better conjugation throughout the CMP networks, and therefore, lowering the energy levels.
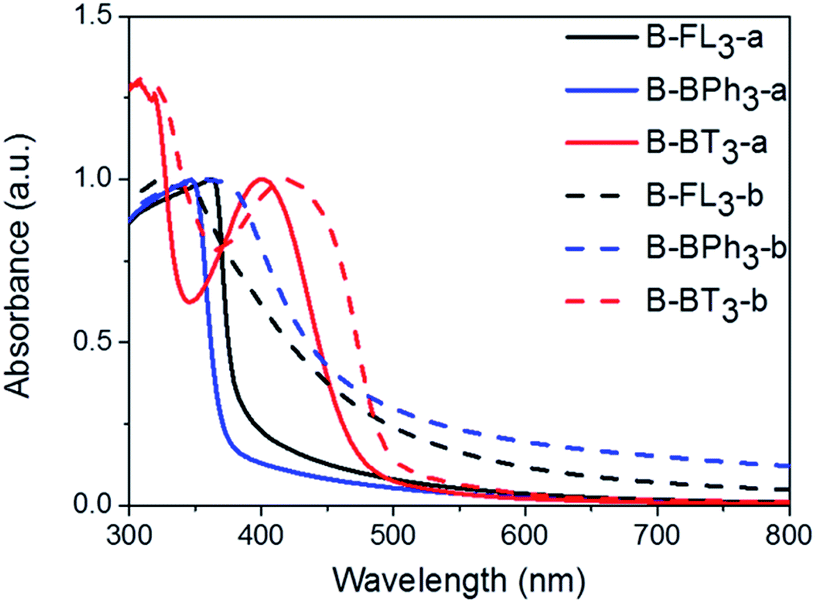 | ||
| Fig. 4 UV/vis absorption spectra of CMP NPs in water dispersion. | ||
Taking advantage of the excellent dispersibility of hydrophobic CMP NPs in water, we first explored the photocatalytic activity of CMP NPs in the reductive activation of molecular oxygen, in which the photon energy was transferred from the photogenerated electron of the electron–hole pair onto oxygen. The obtained activated oxygen species was then subsequently used for the degradation of a water-soluble organic dye, rhodamine B (RhB) (Fig. 5a). It is worth mentioning that a 23 W household energy-saving light bulb was used as the light source for the experiments (Fig. S15 in the ESI†). B-BT3-b showed the highest photodegradation rate of RhB. This result is in accordance with the lowest optical band gap from B-BT3-b with 1.76 eV. After 25 min, over 80% of RhB was already degraded, while its counterpart without triple bond, B-BT3-a (band gap: 1.96 eV), only degraded about 50% of RhB after the same period. Interestingly, B-BPh3-b showed, despite its larger band gap (2.36 eV), also a slightly higher photocatalytic efficiency in the first minutes in comparison to B-BT3-a. This could be attributed to its high BET surface area (512 m2 g−1), which could offer a large reactive interface during the catalytic process. B-BT3-a initially showed a low degradation rate of RhB in the first minutes, however after 20 min it showed an accelerated degradation process of RhB, which might indicate a different reaction order in comparison to its triple-bond containing counterpart B-BT3-b. Other CMP NPs only showed very low photodegradation rates of RhB. At the same time, it is worth mentioning that B-BT3-b also showed higher photocatalytic efficiency in comparison to its bulk form, which indicates the enhanced photocatalytic activity of B-BT3-b in the nanostructured form (Fig. S16 in the ESI†). The efficiency of CMP NPs, especially of B-BT3-b is comparable to that of porous carbon nitride (C3N4), a state-of-art non-metal based visible light photocatalyst under similar reaction conditions.34
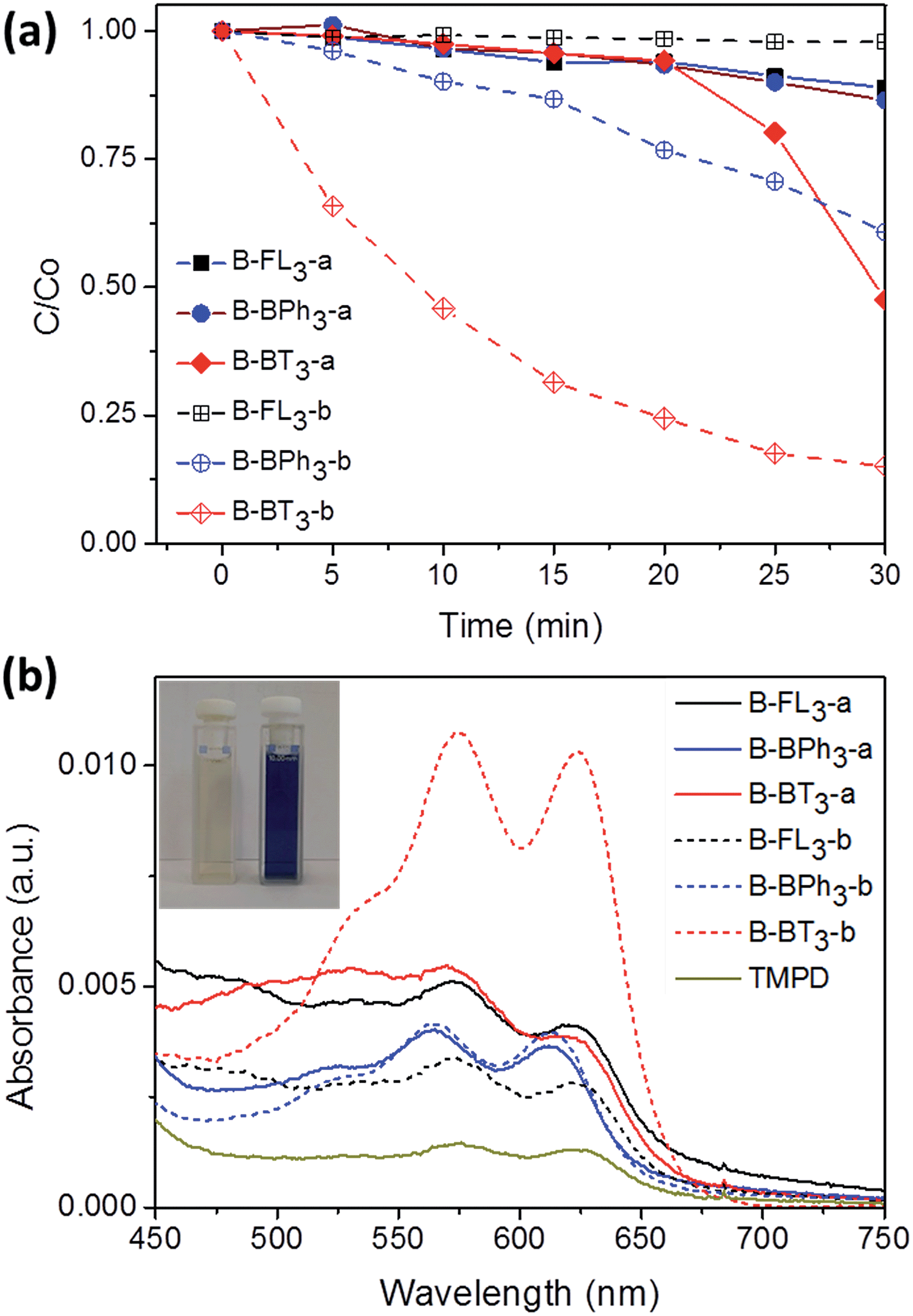 | ||
| Fig. 5 (a) Photocatalytic degradation of RhB in the presence of CMP NPs in water. C is the concentration of RhB after light irradiation for a certain period and C0 is the concentration of RhB after reaching adsorption/desorption equilibrium in the dark. Concentration of CMP NPs: 1 mg mL−1, concentration of RhB: 10 mg L−1. (b) Photooxidation of TMPD using different CMP NPs in acetonitrile. Concentration of CMP NPs: 1 mg mL−1 and concentration of TMPD: 32.85 mg mL−1. The inset image shows: (left) pure TMPD solution after light irradiation and (right) TMPD solution mixed with B-BT3-b after light irradiation. | ||
In addition, CMP NPs showed high stability during the photodegradation reaction. Five extra repeating experiments using B-BT3-b as the photocatalyst were performed. RhB was degraded almost in a quantitative manner after every cycle, indicating the high stability and reusability of CMP NPs in water. As shown in Fig. S17 in the ESI,† the absorption band of B-BT3-b barely changed after the photodegradation reaction, whilst the UV/vis absorption spectrum of RhB almost disappeared completely. It is important to mention that almost no degradation of RhB occurred under light irradiation without using CMP NPs as photocatalysts.
In the photodegradation of organic dyes in water, it is well known that reactive species such as superoxide (˙O2−), singlet oxygen (1O2), ˙OH and the photogenerated hole (h+) play important roles in the photocatalytic process.18,34,39 In order to better understand whether those species are involved in the RhB degradation using CMP NPs, we conducted a series of control experiments (Fig. 6) using B-BT3-b as the catalyst with the following radical scavengers: sodium azide (NaN3) as the 1O2 scavenger, benzoquinone (BQ) as the ˙O2− scavenger, isopropanol (IP) as the ˙OH scavenger and ammonium oxalate (AO) as the h+ scavenger. As shown in Fig. 6, the addition of NaN3 and the absence of oxygen in the reaction led to an expressive decrease in the photodegradation process of RhB. These results suggest that under visible light irradiation the B-BT3-b is able to induce the formation of radical oxygen species, which are responsible for the degradation of RhB, with the main role played by 1O2. In our previous study, it was shown that the bulk-made CMP similar to B-BT3-b was able to generate singlet oxygen species (1O2) under visible light irradiation. Therefore, as an additional control experiment, we further confirmed the role of 1O2 in the photocatalytic system by using the well-studied furoic acid as an 1O2 trap.33,35–39 5-Hydroxy-2(5H)-furanone was obtained as the oxidized product in a quantitative manner using B-BT3-b as the photocatalyst (Fig. S18 in the ESI†).
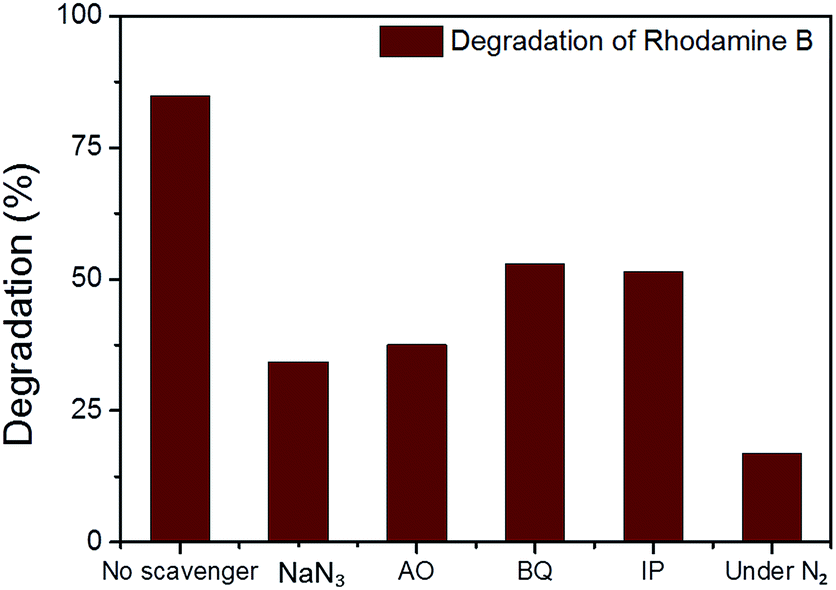 | ||
| Fig. 6 The effect of different scavengers, sodium azide (NaN3), ammonium oxalate (AO), benzoquinone (BQ), isopropanol (IP) and the absence of oxygen (under N2) on the degradation of rhodamine B (RhB) over B-BT3-b under 30 min of visible light irradiation. | ||
A significant decrease in the photocatalytic reaction was also observed by adding AO as the hole scavenger (Fig. 6), indicating the vital role of the photogenerated hole for the direct oxidation of RhB in a photocatalytic system.41 We then further evaluated the electron transfer between the photogenerated hole inside CMP NPs and substrate compounds, i.e. the photooxidation reaction. We examined the transformation of N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) into its cationic radical form (TMPD˙+) under an oxidative process using CMP NPs as photocatalysts. The oxidized form TMPD˙+ can be determined by a color change from colorless to blue, and thus makes the intermolecular oxidative electron transfer reaction from TMPD to CMP NPs visible.40 As displayed in Fig. 5b, after the same reaction time under light irradiation, the absorption spectra of the dispersion containing B-BT3-b and TMPD exhibited the highest intensity. B-BT3-a showed slightly higher photooxidative ability than other CMP NPs, but significantly lower than B-BT3-b. Similar to the photodegradation experiments of RhB, the absence of any color changes in the TMPD solution occurred without using CMP NPs as photoactive oxidizing agents under the same light irradiation conditions. It can be observed that B-BT3-b can generate more and longer-living electron–hole pairs efficiently, which can further catalyze reactions driven either under reductive or oxidative processes.
Conclusions
In conclusion, we report a new platform of well-dispersible CMP NPs as a combination of nanostructures and highly porous and heterogeneous photocatalytic systems. The variety of shapes of the NPs can be controlled by introducing different electron donors and acceptors as building blocks into the polymer backbone, leading to shape variations ranging from nanospheres, nanorods to nanorings. The high photocatalytic activity of CMP NPs was demonstrated in the reductive activation of molecular oxygen for degradation of rhodamine B and the photooxidation of N,N,N′,N′-tetramethyl-p-phenylenediamine under visible light irradiation. It was shown that the transformation of CMPs into nanoparticles could enhance the photocatalytic activity due to the higher dispersity and larger active surface content of the material. Additionally, it is important to note that a household energy-saving light bulb was used as a light source, which provides an economically low-cost solution for the photocatalytic process in broader application fields.Experimental procedure
Materials
1,3,5-Triethynylbenzene (97%), copper(I) iodide (99%), tetrakis(triphenylphosphine)palladium(0) (99%), potassium carbonate (K2CO3) (99%), ammonium oxalate (99%), p-benzoquinone (98%), sodium azide (99%), N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) (97%), triethylamine (99%), rhodamine B (RhB) (99%) and all other solvents were purchased from Sigma-Aldrich (Missouri, USA). Lutensol® AT50 (99%) was purchased from BASF (Ludwigshafen, Germany). Sodium n-dodecylsulfate (99%), 4,4′-dibromobiphenyl (98%), and 9,9-dihexyl-2,7-dibromofluorene (98%) were obtained from Alfa Aesar (Massachusetts, USA). 4,7-Dibromobenzo[c]-1,2,5-thiadiazole (97%) and 1,3,5-phenyltriboronic acid tris(pinacol) ester (97%) were purchased from Combi Blocks (California, USA). All chemicals and solvents were used as received unless otherwise specified.Synthesis of B-FL3-a, B-BPh3-a and B-BT3-a
B-FL3-a, B-BPh3-a and B-BT3-a nanoparticles were synthesized using the Suzuki-Miyaura cross coupling reaction via miniemulsion polymerization. First, 1,3,5-phenyltriboronic acid tris(pinacol) ester (2.5 mg mL−1) and tetrakis(triphenylphosphine)palladium(0) (0.32 mg mL−1) were dissolved in 2 mL of toluene (99%). Then, 9,9-dihexyl-2,7-dibromofluorene (4.06 mg mL−1) was added to the mixture for B-FL3-a, 4,4′-dibromobiphenyl (2.57 mg mL−1) was added for B-BPh3-a and 4,7-dibromobenzo[c]-1,2,5-thiadiazole (2.44 mg mL−1) was added for B-BT3-a. An aqueous solution (18 mL) of sodium n-dodecylsulfate (10 mg mL−1) and K2CO3 (4.56 mg mL−1) was added to the organic phase and vigorously stirred using a Branson W-450D Digital Sonifier operating at 70% of amplitude for 2 min. Later, the homogeneous reaction mixture was stirred and heated at 80 °C overnight. The resulting CMP nanoparticles were then transferred to a dialysis tube (MWCO 14.000 Da) with continuous water substitution to remove the surfactant.Synthesis of B-FL3-b, B-BPh3-b and B-BT3-b
The synthesis of B-FL3-b, B-BPh3-b and B-BT3-b nanoparticles was performed using the Sonogashira–Hagihara cross-coupling reaction via miniemulsion polymerization. 1,3,5-Triethynylbenzene (2.5 mg mL−1), tetrakis(triphenylphosphine)palladium(0) (0.98 mg mL−1) and copper(I) iodide (0.16 mg mL−1) were dissolved in 2 mL of toluene. Then, 9,9-dihexyl-2,7-dibromofluorene (12.29 mg mL−1) was added to the mixture for B-FL3-b, 4,4′-dibromobiphenyl (7.8 mg mL−1) was added for B-BPh3-b and 4,7-dibromobenzo[c]-1,2,5-thiadiazole (7.35 mg mL−1) was added for B-BT3-b. Triethylamine (2 mL) and an aqueous solution (17 mL) of sodium n-dodecylsulfate (10 mg mL−1) were added to the organic phase and vigorously stirred using a Branson W-450D Digital Sonifier operating at 70% of amplitude for 2 min. Later, the homogeneous reaction mixture was stirred and heated at 80 °C overnight. The resulting CMP nanoparticles were transferred to a dialysis tube (MWCO 14.000 Da) with continuous water substitution to remove the surfactant.Characterization
UV/vis absorption spectra were taken on a Perkin-Elmer Lambda 25 UV-vis spectrometer. FT-IR spectra were recorded on a Nicolet 730 FT-IR spectrometer. DLS measurements were carried out using a PSS Nicomp 380 Submicron Particle Sizer. Solid state 13C CP/MAS NMR spectroscopy was performed on a Bruker Avance solid state NMR spectrometer operating at 300 MHz Larmor frequency equipped with a standard 4 mm magic angle spinning (MAS) double resonance probe head. The polymer surface area and pore size distribution were measured by nitrogen adsorption and desorption at 77.3 K using an Autosorb-1 (Quantachrome Instruments). Data were evaluated using QuadraWin software from Quantachrome Instruments. Pore size distributions and pore volumes were derived from the adsorption branches of the isotherms using Quenched Solid Density Functional Theory (QSDFT, N2, assuming carbon adsorbent with slit pores). Samples were degassed at 100 °C for 24 h under vacuum before analysis. TGA measurements were conducted on a Mettler Toledo TGA-851 system. Sample scanning was carried out under nitrogen with a temperature increase from 25 °C to 1000 °C using a heating rate of 10 °C min−1. Electron paramagnetic resonance (EPR) was measured on a Magnettech Miniscope MS200 spectrometer. SEM images were acquired on a LEO Gemini 1530 (Carl Zeiss AG, Germany) using an in lens SE detector. TEM images were recorded on a JEM 1400 (JEOL, USA).Photodegradation of RhB
A mixture of CMP NPs dispersed in water (1 mg mL−1) and RhB solution (10 mg L−1) were stirred in the dark until reaching the adsorption/desorption equilibrium (60 min) between the nanoparticles and the solution. After that, the mixture was illuminated with a 23 W household energy-saving light bulb (Osram) during different time intervals in air. Changes in RhB concentration were analyzed using UV-vis spectroscopy. A control experiment with the reaction mixture under N2, therefore, without oxygen in the reaction, was also performed. For the repeating experiments using B-BT3-b as the photocatalyst, the nanoparticles were separated via centrifugation after each photocatalytic reaction, and added to another RhB solution with the same concentration. The radical scavenger experiments were performed using ammonium oxalate (0.142 g mL−1), p-benzoquinone (0.108 g mL−1), sodium azide (65 mg mL−1) and isopropanol (99%) (60 μL mL−1). Each of the radical scavengers was added to a mixture of B-BT3-b (1 mg mL−1) and RhB solution (10 mg L−1). After reaching the adsorption/desorption equilibrium the mixture was illuminated with a 23 W household energy-saving light bulb (Osram) for 30 min and changes in RhB concentration were analyzed using UV-vis spectroscopy.Photooxidation of TMPD
A 10 mL glass vial containing Ar-saturated acetonitrile solutions (2 mL) of TMPD (32.85 mg mL−1) with CMP NPs (4 mg mL−1) was irradiated by a 23 W household energy-saving light bulb for 5 h. After the irradiation, CMP NPs were completely removed by centrifugation and filtration. The absorbance changes in UV-visible absorption spectra of the resulting solutions before and after irradiation were used to estimate the relative photocatalytic efficiency between different CMP NPs.Acknowledgements
The authors appreciate the financial support from the Max Planck Society. B. C. M. acknowledges the financial support from DAAD, CAPES and CNPq. K. A. I. Z. thanks Fonds der Chemischen Industrie, FCI for funding.Notes and references
- D. Gust and T. A. Moore, Science, 1989, 244, 35–41 CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- M. Neumann, S. Fuldner, B. Konig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- H. J. Liu, W. Feng, C. W. Kee, Y. J. Zhao, D. Leow, Y. H. Pan and C. H. Tan, Green Chem., 2010, 12, 953–956 RSC.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS PubMed.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720–18724 CAS.
- Z. J. Wang, K. Landfester and K. A. I. Zhang, Polym. Chem., 2014, 5, 3559–3562 RSC.
- S. Dadashi-Silab, H. Bildirir, R. Dawson, A. Thomas and Y. Yagci, Macromolecules, 2014, 47, 4607–4614 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- A. Patra, J.-M. Koenen and U. Scherf, Chem. Commun., 2011, 47, 9612–9614 RSC.
- P. Zhang, Z. Weng, J. Guo and C. Wang, Chem. Mater., 2011, 23, 5243–5249 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- A. Laybourn, R. Dawson, R. Clowes, T. Hasell, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2014, 5, 6325–6333 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- H. Li, Y. Liu, X. Gao, C. Fu and X. Wang, ChemSusChem, 2015, 8, 1189–1196 CrossRef CAS PubMed.
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed.
- J. D. White, J. P. Carter and H. S. Kezar, J. Org. Chem., 1982, 47, 929–932 CrossRef CAS.
- M. I. Burguete, R. Gavara, F. Galindo and S. V. Luis, Catal. Commun., 2010, 11, 1081–1084 CrossRef CAS PubMed.
- H. Tokuyama and E. Nakamura, J. Org. Chem., 1994, 59, 1135–1138 CrossRef CAS.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- J. R. Choi, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2010, 26, 10437–10443 CrossRef CAS PubMed.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and characterization data such as 1H NMR and solid state 13C CP/MAS NMR, extra synthetic procedures, TGA, UV/vis, fluorescence spectroscopy and photocatalytic procedures, additional SEM images, N2 sorption isotherms and pore size distribution of the polymers are shown. See DOI: 10.1039/c5ta03820k |
| This journal is © The Royal Society of Chemistry 2015 |