
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural evolution of mixed valent (V3+/V4+) and V4+ sodium vanadium fluorophosphates as cathodes in sodium-ion batteries: comparisons, overcharging and mid-term cycling†
V.
Palomares
*a,
P.
Serras
ab,
H. E. A.
Brand
c,
T.
Rojo
ad and
N.
Sharma
*e
aInorganic Chemistry Department, University of the Basque Country UPV/EHU, P.O. Box 644, 48080, Bilbao, Spain. E-mail: veronica.palomares@ehu.es
bBCMaterials, Building 500–1st. Floor, Bizkaia Science and Technology Park, 48160 Derio, Spain
cAustralian Synchrotron, 800 Blackburn Road, Clayton, Victoria 3168, Australia
dCIC ENERGIGUNE, Parque Tecnológico de Álava, Albert Einstein 48, ED. CIC, 01510, Miñano, Spain
eSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: neeraj.sharma@unsw.edu.au
First published on 28th September 2015
Abstract
Sodium vanadium fluorophosphates belonging to the Na3V2O2x(PO4)2F3−2x family of compounds have recently shown very good electrochemical performance versus Na/Na+ providing high working voltages (3.6 and 4.1 V) and good specific capacity values. In this work the electrochemical behaviour and structural evolution of two compositions, Na3V2O1.6(PO4)2F1.4 (V3.8+) and Na3V2O2(PO4)2F (V4+), are detailed using time-resolved in situ synchrotron X-ray powder diffraction. For the first time in sodium-ion batteries the effects of overcharging and mid-term cycling are analyzed using this technique. Differences in the composition of both materials lead to different combinations of biphasic and single-phase reaction mechanisms while charging up to 4.3 V and overcharging up to 4.8 V. Moreover, the analysis of particle size broadening of both samples reveals the higher stress suffered by the V4+ compared to the more disordered V3.8+ sample. The more “flexible” structure of the V3.8+ sample allows for maximum sodium extraction when overcharging up to 4.8 V while in the case of the V4+ sample no evidence is shown of more sodium extraction between 4.3 V and 4.8 V. Furthermore, the analysis of both materials after 10 cycles shows the appearance of secondary phases due to the degradation of the material or the battery itself (e.g. electrolyte degradation). This study shows examples of the possible degradation mechanisms (and phases) while overcharging and mid-term cycling which is in turn crucial to making better electrodes, either based on these materials or generally in cathodes for sodium-ion batteries.
Introduction
Batteries are not only part of our present lives in the form of portable energy sources for our numerous electronic devices, but also part of our future as the tools that will allow us to move further with the integration of clean renewable energy sources in the electricity grid. This will be achieved by using batteries, storing the excess energy generated from renewable sources and releasing the energy when necessary. However, to achieve widespread integration of batteries into the electricity grid, essentially better, safer, longer lasting and cheaper batteries are required. In this context, research on battery materials has become broader, in the search for new ions that can reversibly insert/extract from a range of hosts, for example the use of sodium ions, instead of lithium ions.1–4The development of Na-ion based batteries demands research into new materials, electrodes and electrolytes, to make these devices a commercial reality. Many compounds have been examined as possible cathodes for more reliable, long-lasting and economic Na-ion batteries.5,6 Among them, sodium vanadium fluorophosphates show high voltage, high capacity and long cycle-life. The performance arises from the two reaction voltages, 3.6 and 4.1 V vs. Na/Na+, the high theoretical specific capacity of 130 mA h g−1 and their 95% capacity retention over 200 cycles.7 In the last two years, it has been demonstrated that these sodium vanadium fluorophosphates belong to the Na3V2O2x(PO4)2F3−2x family, where the substitution of F by O in the same crystallographic site controls the vanadium oxidation state from a V3+ compound when x = 0 (or Na3V2(PO4)2F3), to a V4+ phase when x = 1 (or Na3V2O2(PO4)2F). Mixed valent phases are readily evident for intermediate values of x (ref. 8–10) and Fig. 1 shows the structural model and the F/O substitution sites.
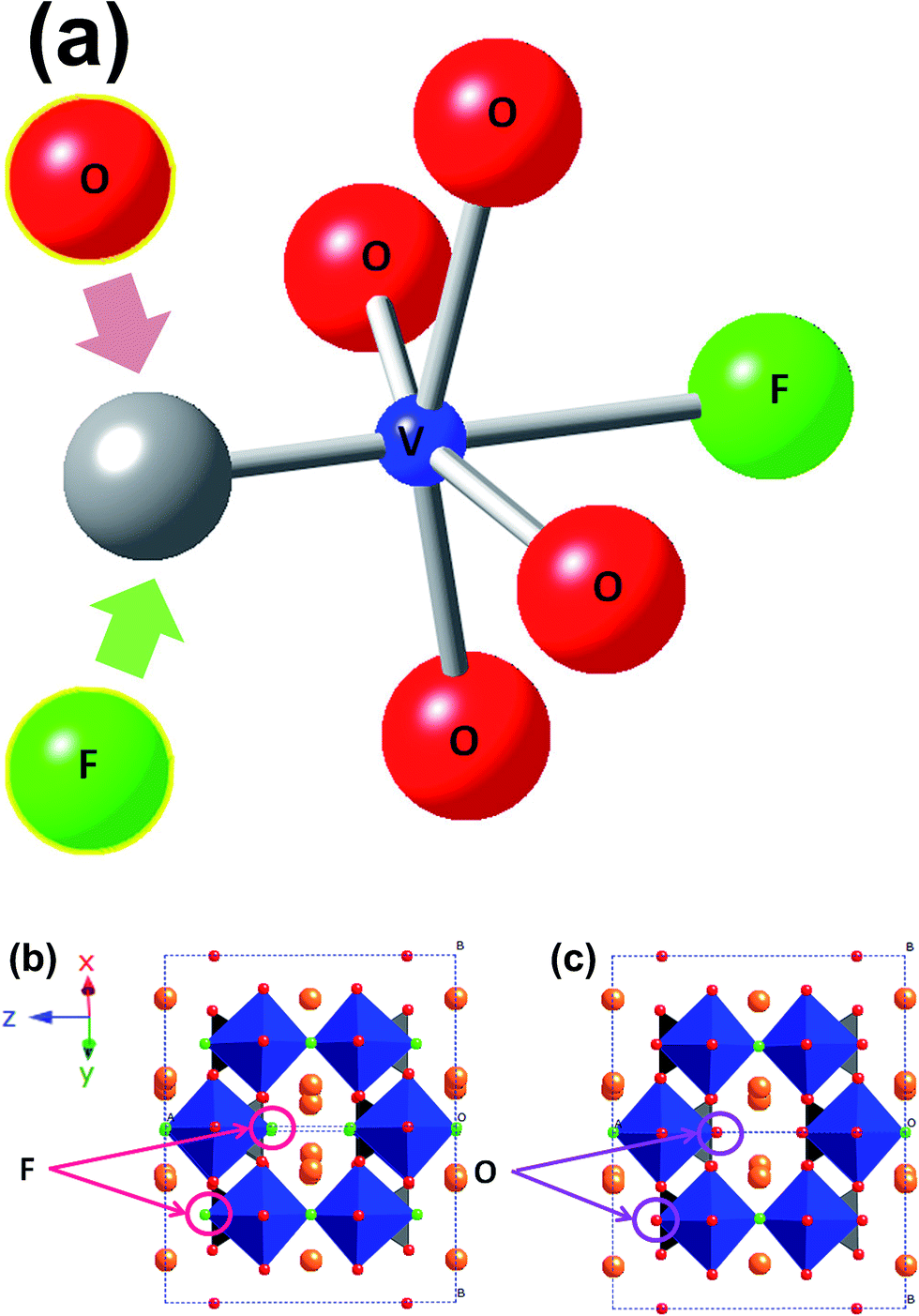 | ||
| Fig. 1 (a) Vanadium octahedra of Na3V2O2x(PO4)2F3−2x (0 < x < 1) compounds. Vanadium is represented in blue, oxygen in red and fluorine in green. The crystallographic structure of (b) Na3V2(PO4)2F3 and (c) Na3V2O2(PO4)2F in the tetragonal P42/mnm space-group. Phosphate tetrahedra are represented in blue. | ||
In spite of belonging to the same family, there are slight differences between the V4+ compound and the thoroughly-studied mixed valent phase of the composition x = 0.8 with V3.8+. First, they both are produced by hydrothermal syntheses but from different precursors. The choice of precursor provides the V3.8+ compound with a carbonaceous coating that the V4+ material lacks. Second, the electrochemical performance of the V3.8+ material has been demonstrated to be better than the V4+ material, especially when cycled at high rates. This can, in part, be related to the carbon coating of the V3.8+ materials, or to the higher intrinsic conductivity of the mixed valent material (with V3+ and V4+) compared to the V4+ materials (with only V4+). It was recently demonstrated that these materials undergo different reaction mechanisms when extracting/inserting Na+ ions from/into the structure.11,12 Recent calculations on the Na3V2O2x(PO4)2F3−2x family have suggested that the third Na can be extracted at 4.9 V for x = 0 and 5.3 V for x = 1;13 however for the x = 1 sample the oxidation state of all V atoms is V5+ when 2 Na atoms are removed so the removal of the third Na would need to involve another oxidation state or mechanism. Additionally, favourable high rate performance has also been shown with this family of compounds when combined with graphene and its derivatives.13,14
A thorough understanding about these materials in terms of electrochemical performance and structure is required before they can be commercially used. In particular, the mechanisms that appear under cell stress or aging conditions, such as overcharging and mid-term cycling are of practical importance to manufacturers. To date, some in situ X-ray diffraction studies of materials in Na-ion batteries have been published, both from laboratory15–17 and synchrotron X-ray sources,18–20 but none of them have focused on the degradation mechanisms of the electrode materials by overcharge or mid-term cycling, until now.
In this study, we detail two possible degradation mechanisms of these cathodic materials, overcharging and mid-term cycling. For this purpose, we have used time-resolved in situ synchrotron X-ray diffraction (XRD) data obtained from coin-cells designed to be analyzed during operation and without removal. In situ XRD provides insight into the reaction mechanism and crystal structure evolution of electrodes in a battery during charge/discharge and this technique has already proved its relevance.11,12,19–22 Such data are distinctly different from providing a snapshot of the equilibrated structure under equilibrium conditions as ex situ (post mortem) or pseudo-equilibrium in situ data provide. With sufficient time-resolution the in situ data show detailed evolution under non-equilibrium and real battery operation conditions. Using this technique, we determined the Na extraction mechanisms of both mixed valent and V4+ phases, when they are overcharged up to 4.8 V vs. Na/Na+. Then, the structures and sodium content of both compounds at 4.8 V are compared, and the effect of a potentiostatic step at high voltages on the mixed valent sample is analyzed. For mid-term cycling analysis, both compounds have been examined using synchrotron XRD data after a sequence of offline cycles. The reaction mechanism, appearance of new secondary phases and the finally discharged cathode structure are illustrated.
Experimental
Na3V2O2(PO4)2F, the V4+ phase,9 was prepared under mild hydrothermal conditions (170 °C) and autogenous pressure, by reacting V2O3 (Sigma-Aldrich, 98% purity), NaH2PO4·H2O (Fluka, 99% purity) and NaF (Sigma-Aldrich, 99% purity) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 molar proportion. The reaction mixture was sealed in a polytetrafluoroethylene (PTFE)-lined steel pressure vessel, which was maintained at 170 °C for 65 hours. The reaction product was washed repeatedly with distilled water and acetone.
:2:1 molar proportion. The reaction mixture was sealed in a polytetrafluoroethylene (PTFE)-lined steel pressure vessel, which was maintained at 170 °C for 65 hours. The reaction product was washed repeatedly with distilled water and acetone.
Na3V2O2x(PO4)2F3−2x where x = 0.8, the V3.8+ phase,8 was synthesized by the hydrothermal method from a ceramic precursor. The synthesis process took place in two steps. First, the VPO4/C composite was synthesized by the ceramic method. V2O5 (Sigma-Aldrich, 99.99% purity) and NH4H2PO4 (Fluka, 99.5% purity) were mixed in an agate mortar in a stoichiometric ratio with a 25% molar excess of Kejten black. This mixture was annealed twice under a nitrogen atmosphere at 300 and 850 °C. Second, the sodium fluorophosphate was prepared under mild hydrothermal conditions (170 °C) and autogenous pressure by reacting NaF (Sigma-Aldrich, 99% purity) and the VPO4/C composite in a 3.3/1 molar proportion. The reaction mixture was sealed in a polytetrafluoroethylene (PTFE)-lined steel pressure vessel, which was maintained at 170 °C for 65 hours.
The positive electrodes were manufactured by mixing 80% wt Na3V2O2x(PO4)2F3−2x active material, 10% wt conductive carbon (Super C65, Timcal) and 10% wt polyvinylidenefluoride binder (PVDF 5130, Solvay). A few mL of N-methylpyrrolidone (NMP, Aldrich) were added and the resulting slurry was stirred for 1 h. This slurry was then coated on an aluminium foil using the “doctor blade” technique. The electrode film was dried at 80 °C in a vacuum oven for 24 h. The electrode sheets were pressed to 100 kN using a flat plate press (MTI corporation) and dried overnight at 100 °C before transfer to the Ar-filled glovebox. Coin cells with 3 mm diameter holes in the casing and 5 mm diameter holes in the stainless spacer were used for the construction of the coin cells for the in situ measurements. The coin cells contained Na metal (∼1 mm thickness), glass fibre separators with 1 M NaPF6 in dimethyl carbonate and diethyl carbonate (1:1 wt%) electrolyte solution.
In situ synchrotron XRD experiments were performed within two days after the construction process for the overcharge investigation. Overcharge studies were performed by charging both starting phases up to 4.8 V at 0.3 mA. The mixed valent sample was held for 75 minutes at 4.8 V, whereas the V4+ phase discharged immediately. Although the experiment was designed to hold for both samples, unfortunately an error in the cell cycler during the experiment meant that for the V4+ sample the hold step finished rapidly. Both materials were subsequently discharged to 2.5 V vs. Na/Na+.
The cells for the mid-term cycling were started after construction and synchrotron XRD data were collected within a day of completion of the cycles. The V4+ material was cycled offline for 10 cycles between 2.5 and 4.3 V at 0.3 mA for the first cycle and 0.1 mA for the following cycles. The open circuit voltage (OCV) was 3.57 V, the cell was cycled in situ at 0.3 mA but for comparative purposes to the mixed valent electrode only the initial state after the offline cycling is considered. The mixed valent sample was only cycled 5 times, and then it failed.
In situ synchrotron XRD data were collected on the Powder Diffraction beamline23 at the Australian Synchrotron with a wavelength (λ) of 0.68816(2) Å, determined using the NIST 660b LaB6 standard reference material. Data were collected continuously in 4 minute acquisitions on the coin cell in transmission geometry throughout the charge/discharge cycles described above. Rietveld refinements were carried out using the GSAS24 software suite with the EXPGUI25 software interface. A similar Rietveld procedure to our previous work was used.11,12 Particle size distributions were determined using the lorentzian profile function as implemented in GSAS and the Scherrer equation.
Results and discussion
First, results related to the overcharge experiments will be described followed by the results of cathode degradation or mid-term cycling for both materials, V3.8+ and V4+. It should be noted that the Na3V2O2x(PO4)2F3−2x family of compounds have been studied recently where the V oxidation state has been allowed to vary. However, in order to achieve this variation the synthetic methods are typically very different, e.g. a one-step hydrothermal method for V4+9, a two-step hydrothermal method using a carbon containing precursor for the V3.8+8 or a ceramic method using a carbon containing precursor for the V3+20. These approaches lead to inherent differences in electrochemical performance, e.g. with or without carbon coating. In addition, for in situ experiments different rates have been used and reported.8,9,20 The phase evolution will in part be related to the current rate applied during charge and discharge, the current modifies the galvanostatic charge/discharge rates and these are manifested in the structural evolution. Bearing this in mind, future work can be targeted to understand the phase evolution as a function of the applied current. This is particularly pertinent due to the high rate performance recently observed with these materials.13,14,26Overcharging of mixed valent and V4+ phases up to 4.8 V
The upper cutoff voltage is often determined by either the electrochemical curve increasing rapidly without the addition of significant amount of further capacity or by the limits of electrolyte stability. The cutoff voltage also corresponds to the maximum amount of Na that can be removed from the cathode. In our attempt to address what is the maximum amount of Na extraction from the Na3V2O2x(PO4)2F3−2x electrodes we charged the cells in situ to 4.8 V. Fig. 2a and b show the 2D diffraction plots of the V4+ and the mixed valent samples focused on 220 and 113 reflections (13.1° to 13.7° 2θ range) corresponding to charge up to 4.8 V.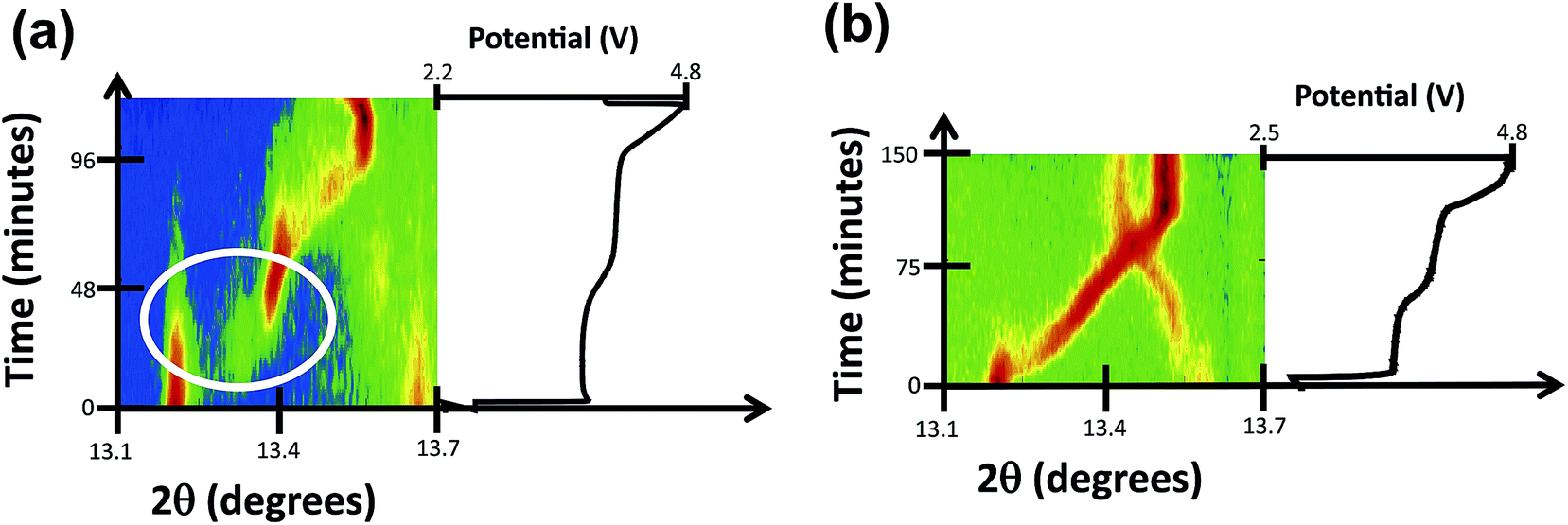 | ||
| Fig. 2 2D diffraction plots of the charge process of (a) V4+ and (b) mixed valent fluorophosphate electrodes focused on the 220 and 113 reflections (from 13.1° to 13.7° 2θ range). | ||
Similar to our previous report,12 charging of the V4+ sample proceeds via a short initial solid solution mechanism followed by two sets of two-phase transitions (P to P′ to P′′12) around 3.6 V linked to the lower potential plateau-like feature in the charge curve (Fig. 2a), then a single phase region for the majority of the higher potential plateau until close to 4.3 V where an abrupt two-phase transition occurs (see Fig. 2a). After this transition and to higher voltages a solid solution mechanism continues until the overcharge cutoff voltage. In the case of the mixed valent sample, again this is similar to the literature as expected, beginning with a solid solution region, followed by a two-phase region and then a solid solution region. The solid solution region continues until the end of overcharge, at 4.8 V. The detailed structural evolution of both these materials when overcharging is parallel to that reported earlier without an overcharge step.11,12
Both materials present a different structural evolution with charge, probably induced by the subtle changes in the vanadium oxidation state (and the F:O ratio) in Na3V2O2x(PO4)2F3−2x (x = 0.8) and Na3(VO)2(PO4)2F phases (and possibly due to the synthetic approach, e.g. with or without carbon coating). The compositional changes slightly modify the structure of the compound. It must be recalled that the mixed valent phase has a crystallographic site occupied by either F or O, so the V3.8+ compound is more disordered than the V4+ one. Thus, structural evolution is strongly affected by this phenomenon and each phase does not evolve in the same way when extracting Na from the structure.
Overcharging: what happens at high voltage charge?
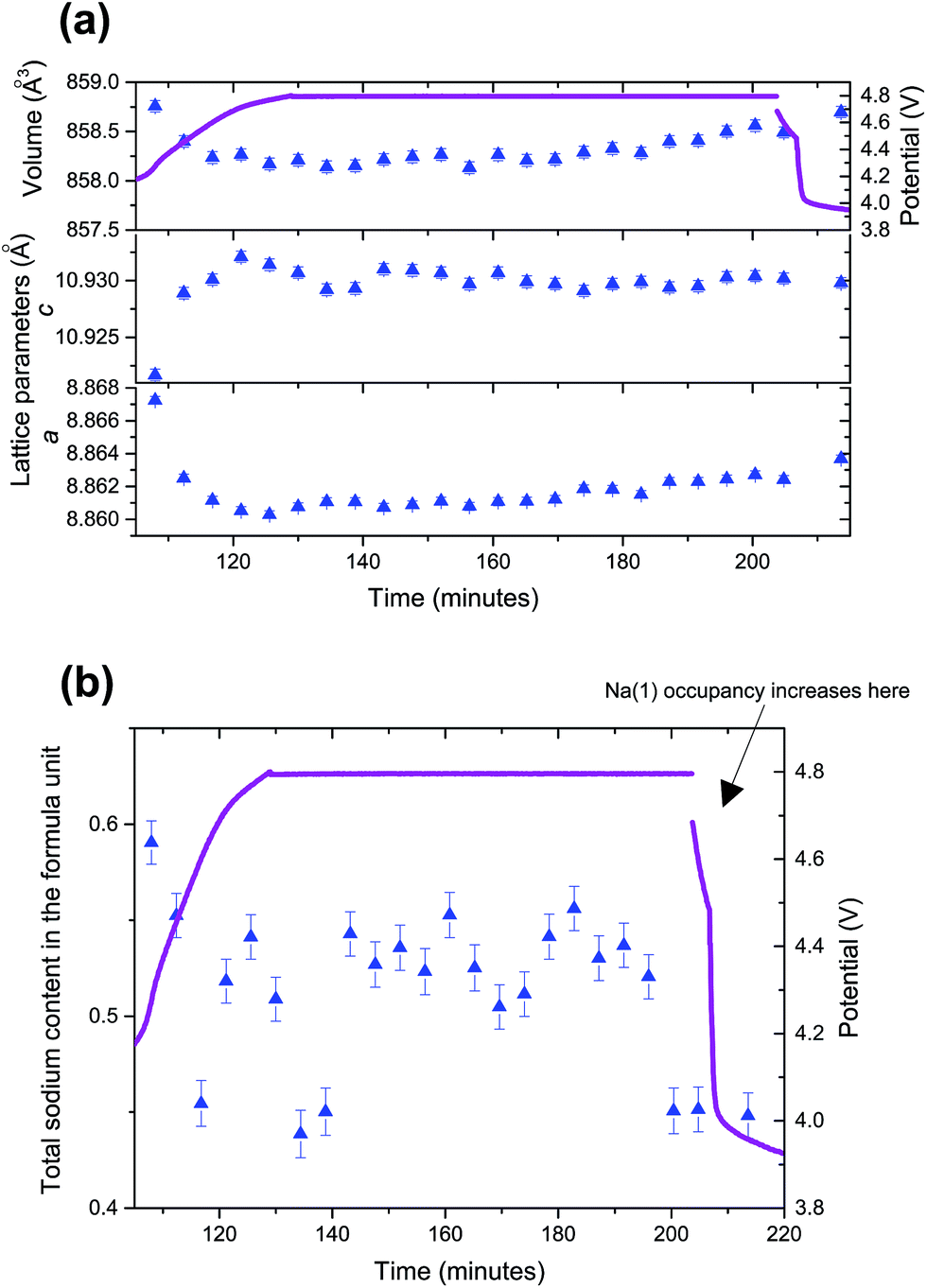
As the structural evolution during charge has been established, the behaviour of the V4+ compound under overcharge conditions, in the 3.8–4.8 V range (104 to 120 minutes) is now discussed. The structural details of the overcharged V4+ phase are shown in the ESI, Table S1.†Fig. 2 shows the lattice and Na site occupation evolution in this high voltage region. Very small fluctuations are detected in the lattice, which is consistent with the small variations in sodium content from 4.3 V onwards. An average total sodium content of 1.21(10) with a clear preference for the Na(1) site was found in the high voltage region, which indicates that the vanadium oxidation state is ∼4.90. This value, sodium content and preferential occupation are very similar to the ones obtained when applying a potentiostatic step at 4.3 V to this phase,12 so it can be concluded that no extra specific capacity is obtained from this phase when going to voltages above 4.3 V. The fact that the phase reaches 4.8 V very fast, with a sharp rise in the voltage curve is also in good agreement with the minimal addition of extra capacity during overcharge.The mixed valent material undergoes subtle structural evolution during overcharge that can be analyzed in terms of diffraction intensity, position and full width at half maximum (FWHM) as shown in Fig. 3. Between 4.3 and 4.8 V the intensity of the 002/111 reflection significantly increases, whereas 220 and 113 intensities slightly decrease and the 222/311 reflection intensity shows a minor increase. In the constant potential region all the reflection intensities tend to stabilize, with the exception of the 220 reflection, which appears to increase at the later stages of the 4.8 V constant potential.
 | ||
| Fig. 3 (a) Lattice parameter and cell volume evolution and, (b) sodium site occupation in the high voltage range (3.8–4.8 V) for the V4+ electrode. Purple lines represent the voltage curves and, in (b), nominal 4.3 V voltage. | ||
In terms of reflection positions, 222/311 and 220 reach their highest position at 4.3 V and stabilize during the constant voltage region. On the other hand, the 113 reflection position decreases continuously up to 4.8 V and 002/111 reflections reach their lowest positions at 4.3 V and slightly increase until 4.8 V and stabilize during the constant potential region. When the FWHMs of these reflections are compared, the 220 and 222/311 reflections remain quite stable, while the 002/111 FWHM decreases between 4.3 and 4.8 V, and then continues to slightly decrease during the potentiostatic step. It must be also remarked that, at the end of the potentiostatic step when a wait step is applied before discharge, from 203 to 207 minutes, the structure changes abruptly. This happens because the structure, which is at a high potential, starts to relax quickly when the potential/current is not applied.
Summarizing, two regions must be distinguished for the mixed valent sample at high voltages; the one that corresponds to the potential rise up to 4.8 V, and the one corresponding to the constant potential step. In the first section, structural changes coming from the charge process are more evident, and these changes are minimized during the potentiostatic step.
Finally, all the changes that are observed in the diffraction intensity turn into modifications of the lattice and sodium occupation in the material (Fig. 4). The lattice (volume) shrinks during the voltage rise up to 4.8 V and then stabilizes with slight fluctuations of a and c lattice parameters. In order to refine sodium occupation, a variety of models were attempted. Finally, it was necessary to fix or sequentially refine most atomic parameters (positions/ADPs) and set Na(1) occupation as 0. The constraints were necessary due to the presence of some correlations when using more refinable parameters. However, it must be noted that the Na(2) occupation results obtained when constraining Na(1) occupation to 0 and when allowed to freely refine were very similar. The structural details of the overcharged mixed valent phase are shown in the ESI, Table S2.†
 | ||
| Fig. 4 (a) Integrated intensity, (b) 2θ position and (c) FWHM of 002, 111, 220, 222, 311 and 112 reflections of the mixed valent electrode between 100 and 200 minutes, which corresponds to the overcharge and potentiostatic step at 4.8 V. | ||
Structural refinements of the mixed valent model with the diffraction data at the end of charge at 4.8 V (before the potentiostatic step) indicate an average total sodium content of ∼0.51. There is a clear preference for the Na(2) site, leaving Na(1) site empty (with or without including this site into the model). This implies that the vanadium oxidation state at the end of the charge process and before the potentiostatic step is V5+, which is the maximum possible oxidation state of this element. Thus the mixed valent material would have reached the maximum possible specific capacity for this phase, since the structure is at the electrochemical V5+ extreme.
The comparison between the two 4.8 V overcharged electrode materials, V4+ and mixed valent, is shown in Fig. 5 and key crystallographic data for sodium in both materials are displayed in Table 1.
 | ||
| Fig. 5 (a) Lattice parameter and unit cell volume evolution of the mixed valent sample, and (b) Na(2) site occupation which is the total sodium content evolution, as Na(1) = 0, during the high voltage range and potentiostatic step (3.8–4.8 V). Purple lines represent the voltage curves. | ||
| Atom | x | y | z | Site occupancy factor |
|---|---|---|---|---|
| V 4+ overcharged material | ||||
| Na(1) | 0.5091 | 0.252 | 0 | 0.60(6) |
| Na(2) | 0.794 | 0.035 | 0 | 0.01(8) |
|
||||
| Mixed valent overcharged material | ||||
| Na(1) | 0.5495 | 0.2155 | 0 | 0 |
| Na(2) | 0.7403 | −0.0353 | 0 | 0.254(12) |
The first feature that must be noted is that only Na(2) sites are represented in the mixed valent overcharged material, since Na(1) site occupation is zero; whereas both Na(1) and Na(2) are partially occupied for the V4+ sample. There is an extremely low but not insignificant Na(2) content in the V4+ compound. Notably, the sodium distribution amongst the crystallographic sites is surprisingly inverse in both oxidized materials, with overcharged V4+ showing preferential Na(1) site occupation and the oxidized mixed valent electrode showing preferential Na(2) site occupation. This exchange can be due to the fact that both sodium sites are located inside the tunnels of the structure and these can be filled/extracted alternatively. There is likely to exist a close relationship between these sites, such that cooperative insertion/extraction of sodium in the channels is possible or speculatively a concerted filling/removing into/from the Na sites. It must be also remarked that, in spite of having a lower overall sodium content, the cell volume of the mixed valent electrode at the oxidized state is greater than the V4+ volume. A smaller volume may influence the probability of preferential occupation on a particular sodium site.
The difference in volume is also found in the raw materials,8,9 possibly due to the greater disorder that exists in the mixed valent sample, with the partial substitution of O by F in the vanadium coordination octahedra and the lower overall vanadium oxidation state. Thus the different sodium distributions between the V4+ and mixed valent electrodes at the overcharged state can be related to the different structural transitions leading to the overcharged state, the ease of transport between the two sodium sites and the influence the cell volume has on the site-specific occupancy.
Electrochemically the performance of V4+ compared to the mixed valent electrode can be related in part to the structural differences observed in these overcharged structures. Table 2 shows the total sodium content and V oxidation state of V4+ and mixed valent electrodes at 4.3 V (normal charge) and 4.8 V (overcharge). It appears that the V4+ electrode approaches its Na extraction limit at 4.3 V and it is not possible to extract more, even going up to 4.8 V, whereas the mixed valent electrode can go beyond the Na extraction at conventional cut-off voltages of 4.3 V. This can partially explain the inferior performance of the V4+ relative to the mixed valent electrode. While V4+ is pushed up to the conventional Na extraction limit in normal charge, the mixed valent 4.3 V charged structure suffers less structural stress because it doesn't reach its limit and can be cycled to a higher potential. This fact can be also considered as a consequence of the initial structural differences in both materials. In addition, calculations suggest that further desodiation might be possible.13
Overcharging – particle size broadening
The changes observed in the FWHM during overcharge, Fig. 3, shows a potential anisotropic change of crystallite size, e.g. preference in the 002 direction, for the mixed valent sample at the overcharged state. This indicates that the crystallite and particle size may play a role in conventional and overcharge regimes.Crystallinity evolution of these fluorophosphates can be evaluated by the fitting of selected reflections or in whole pattern refinements in the discharged materials before and after the overcharge. In the case of the V4+ sample two sets of reflections are depicted in Fig. 6. 111 at 7.7° and 404 at ca. 24.6° were selected for comparison and a SEM image of the pristine material is also shown in Fig. 6. Two phases coexist in the V4+ discharged electrode after 4.8 V overcharge, which is unlike what was observed in our previous investigation of this material when cycled up to 4.3 V, in normal charge conditions where a single phase electrode is found on discharge.12 Thus, whereas the V4+ material comes back to its original (single phase) state when charging and discharging under normal conditions, the structure does not reset when overcharge conditions are used. This can be considered as an adverse influence of the high voltage used. Moreover, diffraction reflection broadening can be observed when the overcharged electrode is discharged, indicating the crystallinity loss. The Scherrer equation was used to semi-quantitatively study the changes in the crystallite size in the material based on whole pattern refinement. The obtained data have been compared to the crystallite size obtained from our previous in situ cycling of this material.12Table 3 displays the obtained particle size broadening of the V4+ material before cycling and in the discharged state after the 4.3 V potentiostatic step, with conventional charge up to 4.3 V and overcharge conditions up to 4.8 V vs. Na/Na+.
 | ||
| Fig. 6 Structures of the overcharged (a) V4+ phase, Na1.21(10)V2O2(PO4)2F and (b) mixed valent phase, Na0.508(12)V2O1.6(PO4)2F1.4. | ||
Note that the particle size analysis is relevant in terms of relative changes, not necessarily in absolute particle size values. This is because calculated sizes are affected by the peak shape function used in all cases. A significant size difference can be observed for the initial V4+ phase in the material analyzed in ref. 12 and the one from this work. This could be due to the fact that the V4+ electrode in ref. 12 had been precycled offline (1 cycle), so this could account for the differences. When comparing the initial particle size to the size of the discharged material after conventional charge/discharge up to 4.3 V, no significant changes are observed, whereas, when subjecting the material to a potentiostatic step at 4.3 V there is a large 52% reduction in particle size distributions. The use of an overcharge potential of 4.8 V leads to an even more massive reduction in particle size. Thus the overcharge conditions induce significant structural changes and stresses on the V4+ phase, as it causes the existence of two phases when discharged, with a concerted reduction in particle size. This can lead to a premature failure of the material, shortening its cycle life.
In the case of the mixed valent sample, the material is monophasic after overcharge and a potentiostatic step at 4.8 V, as it can be seen in Fig. 7. Furthermore, no significant diffraction peak broadening is observed, which indicates that crystallinity is preserved after overcharge, see Table 4. SEM images of the pristine mixed valent electrode are also shown in Fig. 8. As it can be seen in Table 4, changes in the particle size distributions for the mixed valent electrode are of a lower magnitude under the three conditions analysed compared to the V4+ electrode. The three conditions are normal charge up to 4.3 V, after a potentiostatic step at 4.3 V, and after overcharge at 4.8 V. This can be due to the slight structural disorder characteristic of the mixed valent phase that allows the material to adapt better to structural changes. Overcharge at 4.8 V gives the largest change in size (65%) relative to the conventional and potentiostatic charge (∼25%).
 | ||
| Fig. 7 Selected diffraction peaks before and after overcharge of the V4+ material. 7.7° corresponds to 111, 7.9° to 002 and 24.6° to 404 reflections. The inset shows an SEM image of the sample. | ||
 | ||
| Fig. 8 111 and 002 reflections before (top) and after overcharge (bottom) of the mixed valent material. Example of a single peak fit is shown but the whole pattern fits are used to derive the particle size distributions in Tables 3 and 4. The inset shows a SEM image of the pristine electrode material prior to electrochemical cycling. | ||
It should be noted that one sample is carbon coated (V3.8+) while the other is not (V4+) and this may also contribute to the observed changes in the particle size. The coating layer may act to enhance or restrict the dynamics of ion insertion/extraction. However, there is no method at the moment to synthesize both V4+ and V3.8+ phases under the same conditions.
Mid-term cycling
The cathodes in selected batteries were not extracted, they were cycled in in situ cells and data were collected after 10 cycles while the batteries remained in the cell. In situ or ex situ analysis of a battery after ten cycles offline can shed light on the processes that take place in the cathode, that can be related to the degradation of the active material. This knowledge is of great value to evaluate the stability of the cathode in the battery system and, in case it is possible, to propose ways to avoid this degradation.V4+ material: fresh cell vs. start of the 11th cycle
The V4+ electrode structure has been analyzed after 10 charge/discharge cycles. Fig. 9 shows the XRD patterns of the fresh cell and at the start of charge corresponding to the 11th cycle. It is worth noting that the amorphous broad feature that is observed in the starting material lowers in intensity and stabilises at the beginning and over the course of the 11th cycle (which was conducted in situ). Usually this broad background feature at low angles is attributed to the liquid structure factor of the electrolyte,27 thus it is consistent that, after 10 cycles, some electrolyte has been consumed or even lost during cell operation. | ||
| Fig. 9 (a) XRD patterns of the fresh V4+ electrode and the electrode at the start of charge in the 11th cycle. (b) XRD pattern of the material at the start of the charge in the 11th cycle fitted to that of the initial V4+ material. Red arrows mark the broad reflections and blue arrows the sharp ones, showing that two species have been formed. Upper diagram is a zoom of the wider one below. | ||
In terms of diffraction reflections, the main reflections associated with the V4+ phase are maintained, although some new peaks are observed. These new reflections can be assigned to degradation products generated during material cycling. Two kinds of new reflections are marked in Fig. 9b, broad and sharp peaks which could be attributed to a crystalline secondary or by-product phase and to a secondary or by-product with a marked amorphous or nanocrystalline character. In addition, the V4+ electrode component appears to be of two-phases at this state of charge. Thus, we can observe that the electrode contains broad and sharp reflections associated with possibly at least two phases and the V4+ phases.
V4+ material: start and end of the 11th cycle
When comparing the beginning and end points of the 11th cycle it can be seen that they are very similar (Fig. 10). The two-phase mixture at the beginning of the 11th charge mentioned above is not obvious in the discharged state, similar to the monophasic phase when fresh. This suggests that the electrode relaxed after the offline cycling at the beginning of the 11th cycle which is consistent with the lattice parameters being different from the initial fresh electrode found in ref. 12. | ||
| Fig. 10 (a) XRD patterns at the start of charge (red), and the discharged state (black) of the 11th cycle and (b) detail of XRD patterns in the 7–8.6° range with the multiple phases seen at the beginning of the 11th cycle by the split reflection at ∼7.9°. | ||
The analysis of the 7–8.6° range in the XRD patterns provides important clues about the extra peaks found at the start and end of the 11th cycle. It can be seen that there are small differences in the diffraction reflections related to the V4+ phase (at about 7.7 and 7.9° characteristic of two-phase reactions), whereas the 7.4° reflection does not change (even in the in situ data, not shown). This indicates that the phase this peak belongs to is not electroactive and it can be formed by degradation over time or slow crystallization during electrochemical reactions.
Mixed valent material: fresh cell vs. cycled
The mixed valent electrode was cycled offline for 5 times and then failed, so the analysis of the XRD pattern for the failed, discharged material will comprise both secondary phases produced during cycle life and degradation products from the failure. Fig. 11 shows, in the lower part, the XRD pattern of the failed mixed valent cell fitted to the mixed valent structural model. There are a number of extra reflections, which indicates the variety of by-products present in the cathode. The upper part of the figure is an enlargement of the 7 to 14° range, where the arrows indicate some reflections that are observed in the cycled V4+ electrode above. Surprisingly most of these are also observed in the mixed valent failed electrode in addition to some new reflections. | ||
| Fig. 11 Fit of the mixed valent model to the failed cell synchrotron XRD data (red crosses are the data, the black line is the calculated model, the purple line is the difference between the data and model and the vertical black lines are the reflection markers for the mixed valent model). Some reflections found in Fig. 10 are very similar to those found here, thus there are distinct similarities between mixed valent and V4+ cells. Red arrows mark the broad reflections and blue arrows mark the sharp ones, showing that at least two species have been formed. Upper diagram is a zoom of the wider one below. | ||
In order to compare the similarities and differences between the cycled V4+ and failed mixed valent electrode they are plotted together in Fig. 12. The zones highlighted in blue correspond to mixed valent or V4+ phases, black arrows mark new reflections only seen in the failed mixed valent electrode and the purple arrows point out new reflections observed in both cases. It must be remarked that the relative intensity of these latter reflections are higher in the failed mixed valent sample than in the cycled V4+ material.
 | ||
| Fig. 12 Comparison of XRD patterns of the cycled V4+ electrode (in red) and failed mixed valent material (in black). Zones highlighted in blue are those corresponding to mixed valent or V4+ phases, purple arrows point out new reflections observed in both cases and black arrows mark new reflections only seen in the failed mixed valent electrode. | ||
The reflections that are common for both materials can be divided into sharp and broad peaks, so the composition of the V4+ cycled material consists of the V4+ phases and at least two unidentified by-products, whereas the failed mixed valent electrode is made up of the mixed valent phases, at least two unidentified by-products that are related to the aging of these materials with cycle life, common with the V4+ electrode, and at least one other phase produced by the cell failure. This failure can be due to different reasons, such as exposure to air, formation of dendrites, etc. The next step for the analysis of mid-term cycling-related degradation mechanisms will be undertaken on full cells with an anode, so the possibility of dendrites would be eliminated.
Conclusions
In this work, the electrochemical mechanisms involved in overcharge and repeated cycling-related degradation have been illustrated. The structural evolution during cycling is affected by the compositional differences between V4+ and V3.8+ phases, leading to different combinations of biphasic and single-phase stages for both, with normal charge up to 4.3 V and overcharge up to 4.8 V.The charge limits of both compounds have been explored. Whereas the V4+ phase reaches the Na+ extraction limit at the usual cutoff voltage of 4.3 V; the V3.8+ phase is able to reach the maximum theoretical Na+ extraction limit when charging up to 4.8 V. Based on the conditions of our work, e.g., V4+ not carbon-coated and V3.8+ carbon coated, the V4+ sample appears to reach a point where the structure does not easily provide further sodium when charged to 4.3 V while V3.8+ appears to be more flexible at this voltage. It should be noted that the computational calculations suggest that the V4+ can be further desodiated to 5.3 V (ref. 13) but these voltages require specialised electrolytes and are not easily accessible at this stage. It may be possible to try using highly concentrated electrolyte solutions27–29 to achieve these voltages in sodium-ion batteries in order to determine the mechanism at 5.3 V in the future. The other experimental variable to be considered is the use of carbon coating which was applied to the V3.8+ sample but not the V4+ sample. Overall, the mixed valent electrode is not at the extreme limit during the normal 4.3 V operation which positively affects the cycle-life performance of this compound compared to the V4+ phase.
The higher stress suffered by the more ordered V4+ phase is also revealed when analyzing particle size broadening. During conventional 4.3 V charging the particle size distribution is not significantly affected, but there is a large 52% reduction when using a potentiostatic step at 4.3 V and an even larger reduction in particle size distribution when charging up to 4.8 V. On the other hand, the mixed-valent phase exhibits a significantly smaller variation in particle size, even when overcharging. Moreover, the V4+ electrode shows two phases when discharged after overcharge, while the V3.8+ material remains monophasic.
The higher tolerance of the V3.8+ material to stress can be related to the structural disorder characteristic of this phase, where some O are substituted by F atoms in the vanadium coordination octahedra, conferring to this material the “flexibility” to adapt better to the structural changes induced by continuous Na+ extraction up to different cutoff limits analysed in this work.
Examination of medium-term cycling for the V4+ phase indicates that secondary products are already present after 10 cycles, and these phases can be classified into crystalline and more amorphous or nanocrystalline phases. Analysis of the failed cathode of the V3.8+ material showed that the by-products observed in the cycled V4+ phase were also present in this case, together with another set of diffraction peaks, probably linked to the cell exposure to air or the failure mechanism.
Acknowledgements
This work was financially supported by the Ministerio de Economía y Competitividad (MAT2013-41128-R) and the Gobierno Vasco/Eusko Jaurlaritza (IT570-13). Dr Sharma would like to thank AINSE Ltd for providing support through the research fellowship scheme. This research was undertaken on the Powder Diffraction beamline at the Australian Synchrotron, Victoria, Australia. The University of Basque Country is acknowledged for funding under project UFI11/53.References
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS PubMed.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- B. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martinez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- P. Serras, V. Palomares, A. Goni, P. Kubiak and T. Rojo, J. Power Sources, 2013, 241, 56–60 CrossRef CAS PubMed.
- P. Serras, V. Palomares, A. Goni, I. Gil de Muro, P. Kubiak, L. Lezama and T. Rojo, J. Mater. Chem., 2012, 22, 22301–22308 RSC.
- P. Serras, V. Palomares, P. Kubiak, L. Lezama and T. Rojo, Electrochem. Commun., 2013, 34, 344–347 CrossRef CAS PubMed.
- Y. U. Park, D. H. Seo, H. Kim, J. Kim, S. Lee, B. Kim and K. Kang, Adv. Funct. Mater., 2014, 24, 4603–4614 CrossRef CAS PubMed.
- P. Serras, V. Palomares, T. Rojo, H. E. A. Brand and N. Sharma, J. Mater. Chem. A, 2014, 2, 7766–7779 CAS.
- N. Sharma, P. Serras, V. Palomares, H. E. A. Brand, J. Alonso, P. Kubiak, M. L. Fdez-Gubieda and T. Rojo, Chem. Mater., 2014, 26, 3391–3402 CrossRef CAS.
- M. Xu, P. Xiao, S. Stauffer, J. Song, G. Henkelman and J. B. Goodenough, Chem. Mater., 2014, 26, 3089–3097 CrossRef CAS.
- M. Xu, L. Wang, X. Zhao, J. Song, H. Xie, Y. Lu and J. B. Goodenough, Phys. Chem. Chem. Phys., 2013, 15, 13032–13037 RSC.
- M. Guignard, C. Didier, J. Darriet, P. Bordet, E. Elkaim and C. Delmas, Nat. Mater., 2013, 12, 74–80 CrossRef CAS PubMed.
- C. Vidal-Abarca, J. M. Ateba Mba, C. Masquelier, J. L. Tirado and P. Lavela, J. Electrochem. Soc., 2012, 159, A1716–A1721 CrossRef CAS PubMed.
- M. Galceran, D. Saurel, B. Acebedo, V. V. Roddatis, E. Martin, T. Rojo and M. Casas-Cabanas, Phys. Chem. Chem. Phys., 2014, 16, 8837–8842 RSC.
- J. C. Pramudita, R. D. Aughterson, W. M. Dose, S. W. Donne, H. E. A. Brand and N. Sharma, J. Mater. Res., 2015, 30, 381–389 CrossRef CAS.
- J. C. Pramudita, S. Schmid, T. Godfrey, T. Whittle, M. Alam, T. Hanley, H. E. A. Brand and N. Sharma, Phys. Chem. Chem. Phys., 2014, 16, 24178–24187 RSC.
- M. Bianchini, F. Fauth, N. Brisset, F. Weill, E. Suard, C. Masquelier and L. Croguennec, Chem. Mater., 2015, 27, 3009–3020 CrossRef.
- N. Sharma, E. Gonzalo, J. C. Pramudita, M. H. Han, H. E. A. Brand, J. N. Hart, W. K. Pang, Z. Guo and T. Rojo, Adv. Funct. Mater., 2015, 25, 4994–5005 CrossRef CAS PubMed.
- N. Sharma, M. H. Han, J. C. Pramudita, E. Gonzalo, H. E. A. Brand and T. Rojo, J. Mater. Chem. A, 2015 10.1039/C5TA04976H.
- K. S. Wallwork, B. J. Kennedy and D. Wang, AIP Conf. Proc., 2007, 879, 879 CrossRef CAS PubMed.
- A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory Report LAUR 86-748, 1994 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- H. Jin, J. Dong, E. Uchaker, Q. Zhang, X. Zhou, S. Hou, J. Li and G. Cao, J. Mater. Chem. A, 2015, 3, 17563–17568 CAS.
- R. Petibon, J. Li, N. Sharma, W. K. Pang, V. K. Peterson and J. R. Dahn, Electrochim. Acta, 2015, 174, 417–423 CrossRef CAS PubMed.
- Y. Yamada, Y. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic structures and details of the overcharged V4+ and mixed valent phase. See DOI: 10.1039/c5ta03780h |
| This journal is © The Royal Society of Chemistry 2015 |