
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emulsion soft templating of carbide-derived carbon nanospheres with controllable porosity for capacitive electrochemical energy storage†
M.
Oschatz
ab,
M.
Zeiger
c,
N.
Jäckel
c,
P.
Strubel
a,
L.
Borchardt
a,
R.
Reinhold
d,
W.
Nickel
a,
J.
Eckert
d,
V.
Presser
c and
S.
Kaskel
*a
aDresden University of Technology, Department of Inorganic Chemistry, Bergstraße 66, 01069 Dresden, Germany. E-mail: stefan.kaskel@chemie.tu-dresden.de
bUtrecht University, Inorganic Chemistry and Catalysis, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
cINM-Leibniz Institute for New Materials and Saarland University, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
dLeibniz Institute for Solid State and Materials Research (IFW), Institute for Complex Materials, Helmholtzstraße 20, 01069 Dresden, Germany
First published on 25th June 2015
Abstract
A new approach to produce highly porous carbide-derived carbon nanospheres of 20–200 nm diameter based on a novel soft-templating technique is presented. A platinum catalyst is used for the cross-linking of liquid (allylhydrido)polycarbosilane polymer chains with para-divinylbenzene within oil-in-water miniemulsions. Quantitative implementation of the pre-ceramic polymer can be achieved allowing precise control over the resulting materials. After pyrolysis and high-temperature chlorine treatment, the resulting particles offer a spherical shape, very high specific surface area (up to 2347 m2 g−1), and large micro/mesopore volume (up to 1.67 cm3 g−1). The internal pore structure of the nanospheres is controllable by the composition of the oil phase within the miniemulsions. The materials are highly suitable to be used as supercapacitor electrodes with high specific capacitances in aqueous 1 M Na2SO4 solution (110 F g−1) and organic 1 M tetraethylammonium tetrafluoroborate in acetonitrile (130 F g−1).
Introduction
Carbide-derived carbons (CDCs)1 are an emerging class of nanoporous carbon materials which has received tremendous attention in the last decade because of the unique sub-nanometer tunability of the pore size distribution (PSD).2 CDCs are produced by the removal of metal or semi-metal atoms from carbide precursors using thermal/hydrothermal decomposition, electrochemical etching, or, most widely used, high-temperature halogen treatment leading to the formation of well-defined pores.1 The narrow and controllable PSD renders selected CDCs highly attractive candidates in applications such as gas storage and separation,3 catalysis,4 water desalination,5 and electrochemical energy storage.6 CDCs are widely applied as model substances for profound investigation of ion- and gas adsorption phenomena in porous carbons7 and unique hierarchical porous CDC can be obtained from ternary carbide precursors which were found to be promising for biomedical applications.8In addition to the PSD, the morphology of CDCs is precisely controllable as well. As the carbide-to-carbon transformation is fully conformal,2 precursors with various shapes can be subjected to high-temperature chlorine treatment under conservation of the initial morphology. Hence, a plethora of CDC shapes, including monoliths,9 bio-inspired structures,10 thin films,11 fibers,12 aerogels,13 and powders,14 has evolved in recent years. The respective morphological advantages of these structures can be combined with the high purity and the high specific surface area (SSA) of CDCs (more than 2000 m2 g−1)14 which can be further increased by post-synthesis treatment.15 Secondary mesopore systems can be inserted into CDCs by using hard- or sacrificial templating approaches16 and polycarbosilane polymers as silicon carbide precursors to ensure efficient material transport within the interior of the mostly μm- or mm-sized carbon particles. In addition, emulsion techniques are suitable for the implementation of a void-window-type macropore architecture and for the functionalization of CDCs with catalytically active species.17 CDCs obtained from polymeric precursors provide the advantages of higher micropore volumes and specific surface areas in comparison to materials based on crystalline bulk carbides.18
Besides the implementation of a transport pore system, the use of sub-μm-sized precursor particles for CDC synthesis is another approach to enhance the accessibility of the micropore system due to the large external surface area and short transport pathways. CDC nanopowders have been prepared from SiC and TiC precursor powders with particle sizes below 20 and 30 nm.19,20 Compared to materials obtained from bulk carbides, CDC nanoparticles can be prepared at lower temperatures due to the higher reactivity of the precursors and the insertion of nitrogen-containing functional groups is possible.19 Such properties are not achievable by chlorine treatment of μm-sized particles. When used as the electrode material in electrical double-layer capacitors (EDLCs, also known as supercapacitors), CDC nanoparticles provide advanced access to their surface for charge carriers.19 However, as CDC nanoparticles often show distinct graphitization and since they are prepared from nanocrystalline precursors, they suffer from comparably low micropore volume and SSA. Compared with quasi-zero-dimensional CDC nanoparticles, another way of utilizing quasi-one-dimensional CDC nanoforms for electrochemical energy storage with superior power handling is electrospinning. Starting with TiC/carbon hybrid fibers, where ca. 20 nm small carbide nanodomains are engulfed by amorphous carbon,21 facile production of binder-free and free-standing non-woven textile mats of CDC is possible via halogen treatment.22 While the resulting fiber mats of microporous CDC exhibit very high power density, the volumetric performance is very low due to the low electrode density (ca. 0.1 g cm−3, compared to ca. 0.6 g cm−3 for conventional carbon powder electrodes).22
In this study, we present a novel synthesis pathway towards CDC nanospheres (CDC-NS), using miniemulsions. Miniemulsions are dispersions of kinetically stable oil droplets within a size range of 50–500 nm into a continuous aqueous phase.23 The emulsion system is obtained by the introduction of shearing forces (e.g., by ultrasonication or high-pressure homogenizers). The high stability of the droplets is ensured by the combination of a surfactant and a co-stabilizer which is soluble and homogeneously distributed in the droplet phase. The oil phase of the miniemulsions reported here consists of a mixture of an allylhydridopolycarbosilane (trade name SMP-10) and the cross-linker para-divinylbenzene (p-DVB). The droplets act as nano-reactors for the platinum-catalyzed cross-linking of the polycarbosilane chains at a moderate temperature of 80 °C. After the removal of the aqueous phase by evaporation, the obtained cross-linked PCS nanospheres (PCS-NS) are pyrolyzed to silicon carbide and subsequently transformed to CDCs by high-temperature chlorine treatment (Fig. 1). The resulting CDC-NS exhibit an SSA and micropore volume of up to 2350 m2 g−1 and 0.69 cm3 g−1, respectively. Their pore structure can be controlled by the composition of the oil phase. Due to the combination of high SSA and large micropore volume, the novel CDC structures are promising as electrode materials for EDLCs as shown in both organic (tetraethylammonium tetrafluoroborate in acetonitrile) and aqueous (Na2SO4 solution) electrolyte systems providing specific capacities of up to 130 F g−1 and 110 F g−1, respectively.
 | ||
| Fig. 1 Synthesis procedure for carbide-derived carbon nanospheres. | ||
Experimental
Synthesis of CDC-nanospheres
In a 50 mL beaker, 3 g allylhydridopolycarbosilane SMP-10 (Starfire Systems) and 2.74 g para-divinylbenzene or 4.2 g SMP-10 and 1.64 g para-divinylbenzene (80%, Sigma Aldrich) were mixed according to volume ratios of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (CDC-NS-50–50) or 70:30 (CDC-NS-70–30). Then, 250 mg of hexadecane (98%, Fluka) were added dropwise and to this was added a solution of 0.072 g sodium dodecyl sulfate (SDS, 99%, Fluka) in 24 g of deionized water followed by mild stirring for 10 min. The oil-in-water miniemulsion was prepared by ultrasonicating the emulsion for 2 min using an ultrasound finger Labsonic P (Sartorius) with 0.5 cycles at 90% amplitude. During the ultrasonic treatment, the mixture was cooled with an ice bath. The miniemulsion was then filtered over glass wool and 0.1 g of a solution of 0.1 mass% of H2PtCl6 in water was added. This was followed by a treatment at 80 °C in a polypropylene bottle overnight for cross-linking of the organic phase. The miniemulsion was then transferred to a Petri dish and the water phase was gently evaporated at 80 °C. Resulting polycarbosilane nanospheres (PCS-NS) were transferred to an alumina boat and placed in a horizontal tubular furnace equipped with an alumina tube. After purging under flowing argon, the temperature was increased to 800 °C and maintained there for 2 h (60 °C h−1 heating rate). For the high-temperature chlorine treatment, ca. 2 g of the obtained silicon carbide nanospheres (SiC-NS) were transferred to a quartz boat and placed in a horizontal tubular furnace equipped with a quartz tube of 25 mm diameter. After purging under 150 mL min−1 argon flow, the SiC-NS were heated to 700 °C (450 °C h−1 heating rate) and the gas flow was changed to a mixture of 80 mL min−1 chlorine and 70 mL min−1 argon. After 3 h of chlorination, the gas flow was changed to 150 mL min−1 argon and the furnace was cooled to 600 °C. After 1 h of argon purging, the gas flow was changed to 80 mL min−1 hydrogen. This post-reductive treatment for the removal of chlorine and metal chlorides present in the CDC pores after high-temperature chlorine treatment was carried out for 1 h and finally the CDC-NS were cooled to room temperature under flowing argon.
:50 (CDC-NS-50–50) or 70:30 (CDC-NS-70–30). Then, 250 mg of hexadecane (98%, Fluka) were added dropwise and to this was added a solution of 0.072 g sodium dodecyl sulfate (SDS, 99%, Fluka) in 24 g of deionized water followed by mild stirring for 10 min. The oil-in-water miniemulsion was prepared by ultrasonicating the emulsion for 2 min using an ultrasound finger Labsonic P (Sartorius) with 0.5 cycles at 90% amplitude. During the ultrasonic treatment, the mixture was cooled with an ice bath. The miniemulsion was then filtered over glass wool and 0.1 g of a solution of 0.1 mass% of H2PtCl6 in water was added. This was followed by a treatment at 80 °C in a polypropylene bottle overnight for cross-linking of the organic phase. The miniemulsion was then transferred to a Petri dish and the water phase was gently evaporated at 80 °C. Resulting polycarbosilane nanospheres (PCS-NS) were transferred to an alumina boat and placed in a horizontal tubular furnace equipped with an alumina tube. After purging under flowing argon, the temperature was increased to 800 °C and maintained there for 2 h (60 °C h−1 heating rate). For the high-temperature chlorine treatment, ca. 2 g of the obtained silicon carbide nanospheres (SiC-NS) were transferred to a quartz boat and placed in a horizontal tubular furnace equipped with a quartz tube of 25 mm diameter. After purging under 150 mL min−1 argon flow, the SiC-NS were heated to 700 °C (450 °C h−1 heating rate) and the gas flow was changed to a mixture of 80 mL min−1 chlorine and 70 mL min−1 argon. After 3 h of chlorination, the gas flow was changed to 150 mL min−1 argon and the furnace was cooled to 600 °C. After 1 h of argon purging, the gas flow was changed to 80 mL min−1 hydrogen. This post-reductive treatment for the removal of chlorine and metal chlorides present in the CDC pores after high-temperature chlorine treatment was carried out for 1 h and finally the CDC-NS were cooled to room temperature under flowing argon.
Structural characterization
Nitrogen physisorption measurements of the PCS-NS and the SiC-NS were performed at −196 °C using a Quadrasorb apparatus (Quantachrome Instruments). Measurements of the CDC-NS were performed on an Autosorb 1C apparatus (Quantachrome). Specific surface areas were calculated with the multi-point Brunauer–Emmett–Teller equation (0.05 < p/p0 < 0.2). Internal pore volumes (excluding external porosity) and total pore volumes (including external porosity) were calculated at p/p0 = 0.8 and p/p0 = 0.99, respectively. Pore size distributions were calculated with the quenched solid density functional theory (QSDFT) method (based on the adsorption branch) for nitrogen at −196 °C on carbon surfaces with slit/cylindrical pore geometry.24 Micropore volumes are the cumulative pore volumes at a diameter of 2 nm obtained from the QSDFT-PSD.Scanning electron microscopy with a DSM982 microscope (Zeiss, Germany) was performed at a beam voltage of 5 kV. The samples were immobilized on an aluminum grid equipped with a carbon tape. Transmission electron microscopy with a 2100F microscope (JEOL, Japan) was performed at 200 kV. Samples were prepared by dispersing powders in ethanol, followed by tip sonication for 10 s, and drop casting them on a copper grid with a lacey carbon film (Gatan, USA).
Dynamic light scattering (DLS) experiments were carried out at room temperature with an aqueous dispersion of the cross-linked polycarbosilane nanospheres (before solvent evaporation) in a plastic cuvette using a Zetasizer Nano-ZS (Malvern Instruments).
Raman spectra were recorded with a Renishaw inVia Raman Microscope using a Nd–YAG laser with an excitation wavelength of 532 nm. A grating with 2400 lines per mm and a 50× objective (numeric aperture: 0.9) were used with a spectral resolution of ∼1.2 cm−1. The spot size on the sample was in the focal plane of ∼2 μm using an output power of 0.2 mW. The acquisition time was 30 s and 10 accumulations were used to eliminate cosmic rays and to improve the signal-to-noise ratio. Peak fitting was performed assuming one Lorentzian peak for each of the D- and the G-modes and two other peaks for a sufficient approximation of the spectrum between 1200 and 1700 cm−1.
Electrochemical characterization
Film electrodes were prepared by mixing 95 mass% of carbon material (CDC-NS-50–50 and CDC-NS-70–30) with 5 mass% polytetrafluoroethylene (60 mass% PTFE in water, Sigma Aldrich) together with ethanol to make a slurry. The slurries were tip sonicated for 10 min at ∼4 W, and then constantly stirred until a dough-like mass was obtained. Using a twin roller (MTI HR01, MTI Corporation) the PTFE-bound mass was rolled to sheets with a thickness of ∼200 μm and a mass loading of 11–14 mg cm−2. Prior to use, the electrodes were dried at 120 °C in a vacuum at 2 kPa for 24 h. Electrochemical characterization was performed in 1 M tetraethylammonium tetrafluoroborate (TEA-BF4) in acetonitrile (ACN) from BASF (electrochemical grade) using a two-electrode set-up. The electrodes were 12 mm in diameter, ∼200 μm thick and were separated by a glass-fiber separator with a diameter of 13 mm (type GF/A, Whatman). The electrode/separator/electrode arrangement was placed between two carbon-coated aluminum foils (diameter: 12 mm, type Zflo 2653, Exopack technologies) and compressed with spring-loaded titanium pistons. The cells were dried at 120 °C and a vacuum of 2 kPa for 12 h before they were put in an argon-filled glove box (MBraun Labmaster 130; O2, H2O < 1 ppm), where they were vacuum back-filled with a syringe containing 1 M TEA-BF4 in ACN. Using aqueous 1 M Na2SO4 (Sigma Aldrich) as the electrolyte the same set-up as for the organic electrolyte was used except that the current collector was changed to platinum disks with a diameter of 12 mm and a thickness of 100 μm (Carl Schaefer). All electrochemical measurements were carried out using a VSP300 or VMP300 potentiostat/galvanostat from Bio-Logic in cyclic voltammetry (CV) and galvanostatic mode (GCPL). Cyclic voltammograms at 10 mV s−1 for cell voltages of 2.5 V (organic electrolyte) and 0.8 V (aqueous electrolyte) were applied 3-times and the gravimetric capacitance for one electrode dependent on the voltage was calculated using eqn (1).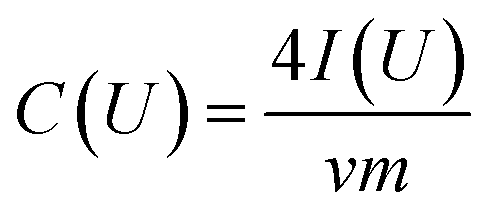 | (1) |
Using GCPL mode, different current densities from 0.1 to 10 A g−1 with 10 s resting time between charging/discharging and a potential limitation of 2.5 V (organic electrolyte) and 0.8 V (aqueous electrolyte) were applied. The resulting gravimetric capacitance for one electrode was calculated from the current and the time for discharge with respect to the IR drop of the cell using eqn (2).
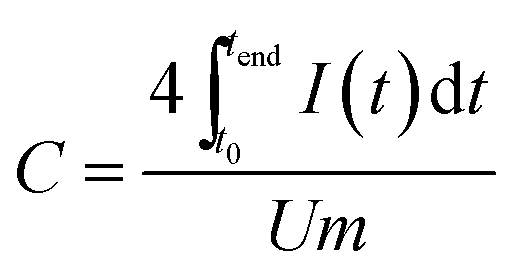 | (2) |
Results and discussion
Structure and porosity control of CDC-NS
CDC-NS are prepared starting from miniemulsions with different compositions of the oil phase. The SMP-10:p-DVB volume ratio is adjusted to 70:30 or 50:50. Thermogravimetric analyses of the materials reveal precise control over the amount of polycarbosilane inserted into the solidified PCS-NS (ESI, Fig. S1†). During heating in air, the mass of the cross-linked polycarbosilane spheres increases starting from a temperature of 150 °C due to the oxidation of the silicon within the PCS. At around 300 °C, volatile species (e.g., hydrogen, methane, and carbon oxides) are formed and the mass decreases. Due to the cross-linking of SMP-10 and p-DVB, the PCS-NS-70–30 shows a higher initial mass gain as well as a higher residual mass as compared to the material with lower silicon content and higher amount of cross-linker.
Dynamic light scattering (DLS) measurements of the cross-linked PCS-NS in aqueous dispersion (ESI, Fig. S2†) show that the hydrodynamic diameters of the particles are 20–400 nm (polydispersity index: 0.117) and 20–600 nm (polydispersity index: 0.197) for SMP-10:p-DVB ratios of 50:50 and 70:30, respectively. The corresponding z-average sizes are 147 nm (50:50) and 172 nm (70:30). The absence of larger particles indicates that the spheres are highly separated and that agglomeration or Ostwald ripening does not take place in the miniemulsion system.
In the nitrogen physisorption isotherms of the PCS nanospheres (Fig. 2A), a large uptake associated with condensation of nitrogen can be observed at relative pressure p/p0 > 0.9 due to the adsorption of nitrogen on the outer surface of the nanospheres and the condensation in the inter-particular pore space. In accordance with the DLS measurements, the condensation occurs at higher relative pressure for the PCS-NS-70–30 due to the ca. 30 nm large diameter of the spheres and hence the larger inter-particular pores. The latter are also responsible for the higher total pore volume (including inter-particular pores) of the particles with higher SMP-10 content. Independent of the elevated SMP-10:p-DVB ratio, the PCS-NS do not contain significant intra-particular pore volume and the SSA mainly arises from external surface of the spheres.
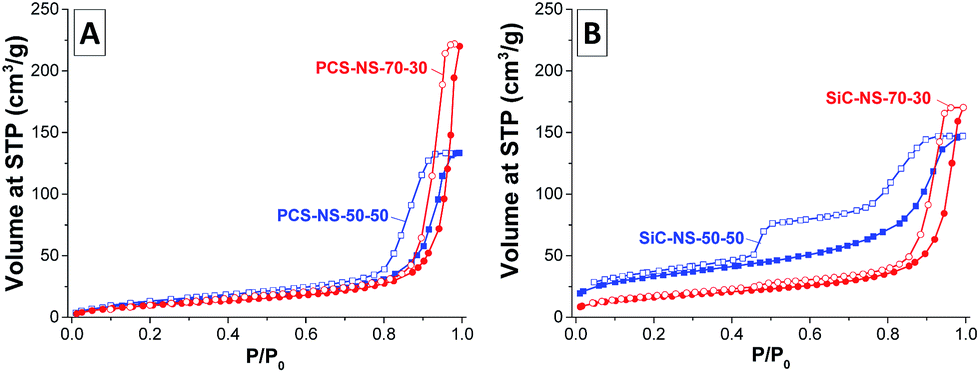 | ||
| Fig. 2 Nitrogen adsorption/desorption (filled symbols/empty symbols) isotherms (measured at −196 °C) of (A) the PCS-NS and (B) the SiC-NS obtained from miniemulsions with SMP-10:p-DVB volume ratios of 70:30 (red) or 50:50 (blue). | ||
After pyrolysis of PCS-NS to silicon carbide, the SSAs increase slightly due to mass loss during decomposition of the polymer chains (Table 1). While the shape of the isotherm of the SiC-NS-70–30 remains comparable to the corresponding PCS-NS, an additional pore system can be observed for SiC-NS-50–50 (Fig. 2B). The larger fraction of gaseous decomposition products during pyrolysis leads to the formation of an irregular mesopore network within the SiC-NS-50–50, as previously reported for other silicon carbide and CDC materials obtained from emulsion-based SMP-10/p-DVB precursor systems.13,17 The large hysteresis and the sharp step at about 0.5 relative pressure in the desorption branch of SiC-NS-50–50 indicates that the mesopores empty by a cavitation mechanism, that is, they are located within the nanospheres and are accessible only via necks of diameters below 5–6 nm.25 Therefore, a larger intra-particular pore volume is present in the silicon carbide material obtained from miniemulsions with higher content of cross-linker. A small step is also observable in the isotherm of SiC-NS-70–30 and the presence of defects within the spheres cannot be ruled out. However, the increase of internal pore volume and SSA is less pronounced for this material and the spheres are rather dense.
| Material | Specific surface area [m2 g−1] | Micropore volume [cm3 g−1] | Total internal pore volume [cm3 g−1] | Total pore volume [cm3 g−1] |
|---|---|---|---|---|
| a Not determined. | ||||
| PCS-NS-50–50 | 44 | n.d.a | 0.05 | 0.21 |
| PCS-NS-70–30 | 39 | n.d.a | 0.04 | 0.34 |
| SiC-NS-50–50 | 120 | 0.02 | 0.11 | 0.23 |
| SiC-NS-70–30 | 60 | 0.004 | 0.05 | 0.26 |
| CDC-NS-50–50 | 2347 | 0.69 | 1.27 | 1.53 |
| CDC-NS-70–30 | 2298 | 0.64 | 1.21 | 1.67 |
Scanning electron microscopy (SEM) images of the SiC-NS (Fig. 3A and B) reveal that the spherical structure of the oil-droplets can be successfully transformed to silicon carbide with a spherical shape. Due to the fully conformal characteristics of the high-temperature chlorine treatment, the spherical shape is perfectly retained in the CDC-NS (Fig. 3C–F). Their diameters of 20–200 nm are slightly below those of the PCS-NS determined by DLS due to minor shrinkage processes during solvent evaporation and pyrolysis. It can be seen that the CDC-NS-50–50 contain a larger number of defects and partially hollow structures compared to the CDC-NS-70–30. This observation is likely related to the formation of more gaseous decomposition products during pyrolysis of the PCS-NS-50–50. In accordance, transmission electron microscopy images of the CDC-NS (Fig. 3G and H) show a less smooth outer sphere surface morphology for the CDC-NS-50–50 due to the formation of defects during decomposition of the polymeric spheres.
 | ||
| Fig. 3 Scanning electron microscopy images of (A) SiC-NS-50–50, (B) SiC-NS-70–30, (C and D) CDC-NS-50–50, and (E and F) CDC-NS-70–30 as well as transmission electron microscopy images of (G) CDC-NS-50–50 and (H) CDC-NS-70–30. | ||
After the high-temperature chlorine treatment, the SSAs and micropore volumes significantly increase to values of 2347 m2 g−1 and 0.69 cm3 g−1, respectively (Table 1). Despite the higher uptake of nitrogen at low relative pressure, the shape of the nitrogen physisorption isotherms of the CDC-NS (Fig. 4A) is comparable to that of the SiC-NS precursors, that is, CDC-NS-50–50 still contains a certain fraction of internal mesopores emptying by a cavitation mechanism. The introduction of high micropore volumes and the mass loss during silicon removal further lead to significant increase of the total pore volume and the total internal pore volume. While the latter is higher for the CDC-NS-50–50 due to the presence of the internal mesopores, the fraction of inter-particular pores is larger in case of the CDC-NS-70–30. The PSDs of the CDC-NS (Fig. 4B) show the presence of micropores of 0.9–1.0 nm size as is typical for polycarbosilane-based CDCs prepared at comparable temperatures.16 In accordance with the shape of the isotherms, CDC-NS-50–50 further contains a minor volume of mesopores between 6 and 11 nm in size. The cavities > 10 nm present in the CDC-NS-70–30 are related to inter-particular porosity.
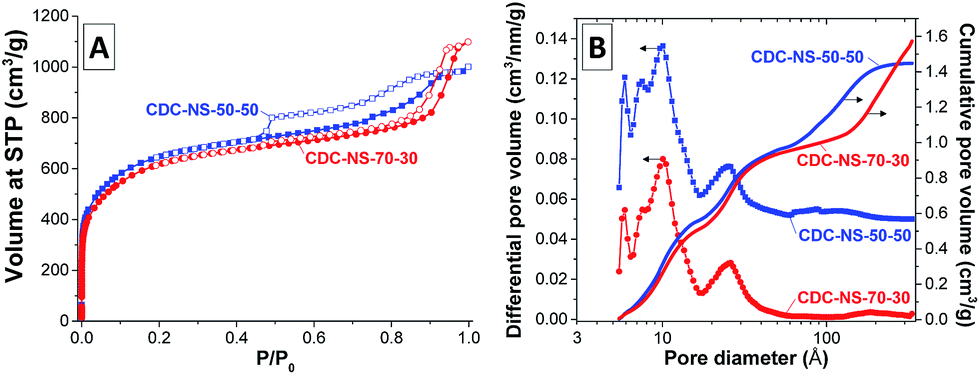 | ||
| Fig. 4 (A) Nitrogen adsorption/desorption (filled symbols/empty symbols) isotherms (measured at −196 °C) and (B) corresponding QSDFT pore size distributions of the CDC-NS-70–30 (red) and CDC-NS-50–50 (blue). Note a +0.05 cm3 nm−1 g−1y-axis offset of the CDC-NS-50–50 material for the differential pore volume plot. | ||
Independent of the SMP-10:p-DVB ratio in the initial oil phase, the CDC-NSs show a highly disordered carbon microstructure with randomly arranged sp2 carbon fringes (Fig. 5). This observation aligns well with Raman spectroscopy (Fig. 6) and the measured spectra indicate a highly disordered carbon microstructure with the characteristic D-mode at ∼1350 cm−1. Furthermore, the G-mode (assigned to graphitic sp2 carbon) and the A-mode (assigned to amorphous sp2 carbon) are observed at ∼1595 cm−1 and ∼1550 cm−1, respectively. The shoulder transpolyacetylene-(TPA)-like mode present in both samples and the absence of sharp second order and combination modes in the range of 2400–3000 cm−1 also indicate a low degree of graphitization in the CDC-NS, that is, no excessive stacking and formation of parallel oriented graphene sheets take place. Due to the equal synthesis temperatures, no significant differences between the CDC-NS are observed as can be seen from the comparable full width at half maximum (FWHM) of the D- and G-modes as well as ID/IG and IG/IA ratios.
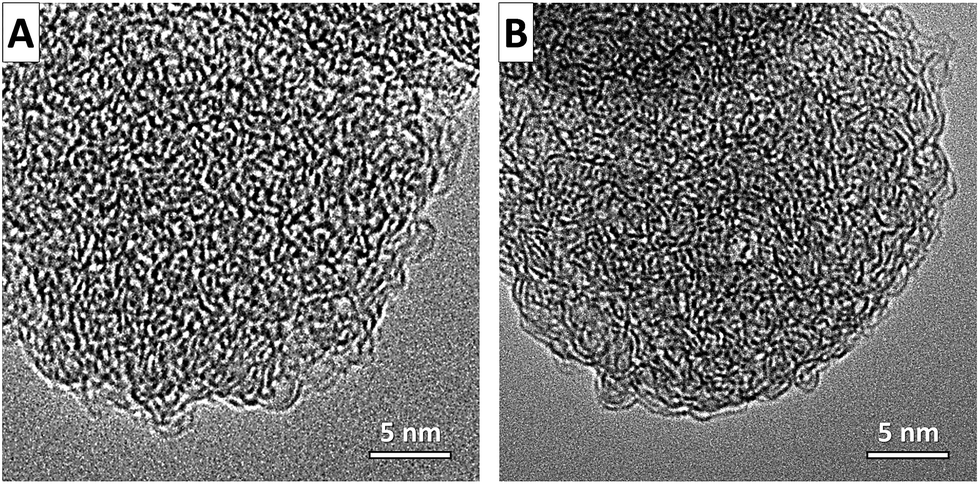 | ||
| Fig. 5 Transmission electron micrographs of (A) CDC-NS-50–50 and (B) CDC-NS-70–30. | ||
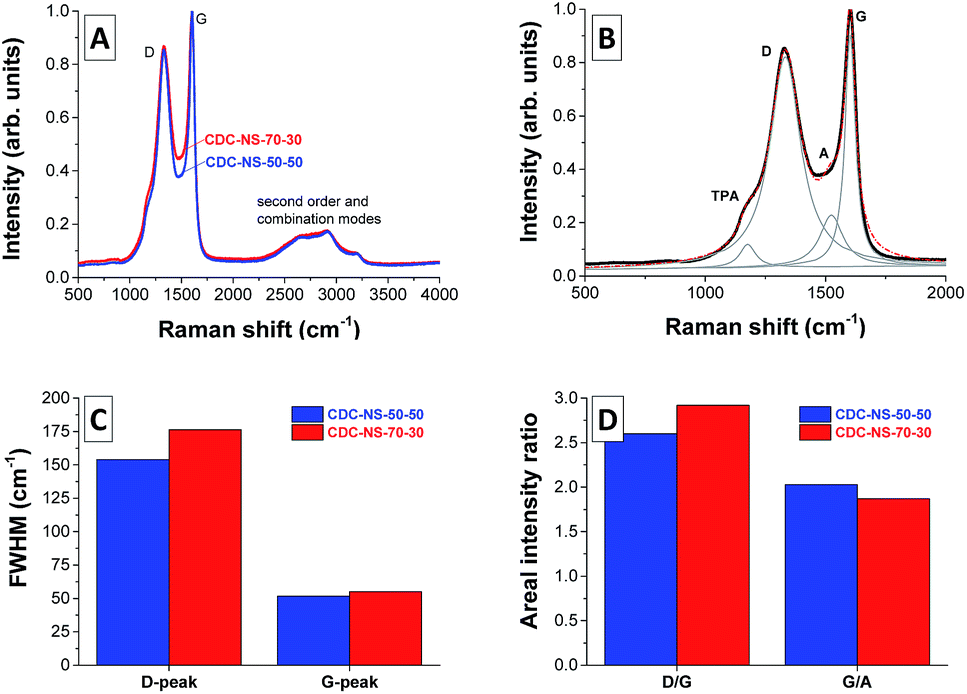 | ||
| Fig. 6 (A) Raman spectra of CDC-NS-70–30 (red) and CDC-NS-50–50 (blue), both normalized to the G-peak. (B) Lorentzian peak fitting of CDC-NS-50–50, and the fitting parameters of both materials in (C) full-width at half maximum and in (D) the areal intensity ratios. | ||
CDC-NS as a supercapacitor electrode material
Due to their high micropore volume and SSA, CDC-NS are highly attractive electrode materials for electrical double-layer capacitors in different electrolyte systems. To exemplify the beneficial electrochemical performance, we chose a typical pH-neutral aqueous system (1 M Na2SO4) and the most commonly used organic system (1 M tetraethylammonium tetrafluoroborate (TEA-BF4) in acetonitrile). Cyclic voltammograms (CVs) of the materials in 1 M aqueous Na2SO4 electrolyte solution are nearly rectangular within the applied potential range of 0–0.8 V (Fig. 7A) indicating the absence of a large number of surface functional groups and the high purity of the CDC-NS resulting in a nearly ideal (purely capacitive) EDLC behavior. Besides CVs, galvanostatic charge–discharge (C–D) measurements are suitable for a quantitative analysis of the electrochemical performance at higher current densities to explore power handling ability (Fig. 7C and 7E). At a current density of 0.1 A g−1, the CDC-NS provide high specific capacities of 110 F g−1 (CDC-NS-50–50) and 103 F g−1 (CDC-NS-70–30). At a high rate of 10 A g−1, 74% (CDC-NS-50–50) and 80% (CDC-NS-70–30) of the initial capacitances are still retained. The values for specific capacitance are very similar considering an experimental error of ±5%, which is reflective of the comparable porosity and surface area of the two materials (Table 1). This effect is likely related to the rapid accessibility of the ion adsorption sites (i.e., the micropores) of the CDC-NS due to the combination of 20–50 nm-sized spherical particles and relatively large 0.9–1.0 nm-sized micropores suitable for rapid diffusion of the electrolyte ions. | ||
| Fig. 7 (A) and (B) Cyclic voltammograms at scan rates of 10 mV s−1, (C) and (D) galvanostatic charge–discharge plots, and (E) and (F) galvanostatic rate handling plots of CDC-NS-70–30 (red) and CDC-NS-50–50 (blue) in (A), (C) and (E) 1 M aqueous Na2SO4 and (B), (D) and (F) 1 M TEA-BF4 in ACN electrolyte. The solid lines in panels (E) and (F) were fitted assuming a linear regression. | ||
While aqueous electrolytes in general provide the advantages of high ionic conductivity and low cost, they suffer from the narrow electrochemical stability window of water (1.23 V). In contrast, organic electrolytes can operate at higher potential (typically up to 2.7 V) and, therefore, the achievable energy density of the organic EDLCs is much higher. In consequence, most of the commercially available devices make use of organic electrolytes. The CVs of the CDC-NS-based electrodes in a 1 M solution of TEA-BF4 in acetonitrile (ACN) show, just like in aqueous media, a nearly rectangular shape as well (Fig. 7B). Also, the C–D curves exhibit a triangular shape typical of an exclusive capacitive charge–discharge mechanism (Fig. 7D). In contrast to the C–D measurements in Na2SO4, the CDC-NS-70–30 now shows a slightly higher specific capacitance of 130 F g−1 at a current density of 0.1 A g−1 (Fig. 7F). This sample further provides high capacity retention of 119 F g−1 (92%) at 10 A g−1. For comparison, CDC-NS-50–50 exhibits at 0.1 A g−1 a capacitance of 120 F g−1 which drops to 79 F g−1 (i.e., 67%). Although CDC-NS-50–50 are slightly smaller, CDC-NC-70–30 show a larger volume of inter-particular mesopores which greatly favors an enhanced performance at high current density in organic electrolytes when the higher viscosity compared to aqueous media is considered.
In summary, the CDC-NSs are highly attractive candidates for the use as supercapacitor electrode materials as they provide high specific capacities in combination with good charge retention capabilities at high current densities. Operating in aqueous media (0.8 V) or organic media (2.5 V), the maximum energy densities were 2.7 W h kg−1 (CDC-NS-50–50) and 28.2 W h kg−1 (CDC-NS-70–30), respectively. The performance of the materials in a given electrolyte is dependent on the pore structure of the CDCs and hence the elevated SMP-10:p-DVB ratio of the initial miniemulsion, that is, CDC-NS are highly versatile for the application in EDLCs based on a specific electrolyte system.
Perspectives of CDC-NS for flow electrode applications
Besides the use of CDC-NS as the electrode material in conventional EDLCs, the application of high surface area, mono-dispersedly sized carbon beads is highly attractive for flow electrode applications,26 such as electrochemical flow capacitors (EFCs) for scalable energy storage,27 flow electrode capacitive deionization (FCDI) for continuous water treatment,28 or flow electrode capacitive mixing energy extraction (Capmix).29 In EFCs, which combine the advantages of rapid charge–discharge and decoupling of energy- and power ratings, a fluidized carbon–electrolyte slurry is employed as the active material. For charging and discharging by the formation of an electrical double-layer it is pumped into a polarized electrochemical cell and for charge storage it is pumped into reservoirs. In most cases, EFCs make use of active materials with spherical geometry/shape to minimize viscous effects and, hence, the energy to pump and transport the suspension electrode.30 Moreover, carbon electrodes with small diameters are expected to show better electrochemical characteristics due to the higher accessible SSA and better flow characteristics. Hence, CDC-NS are highly promising candidates for EFC devices and related flow electrode applications due to their combination of high micropore volumes with well-defined size suitable for ion adsorption and the nearly perfect spherical shape with low diameters in the nm-range.Conclusions
A novel approach to produce carbide-derived carbon nanospheres of 20–200 nm diameter based on a novel soft-templating technique was presented. Platinum catalysis is used for the cross-linking of liquid (allylhydrido)polycarbosilane polymer chains with para-divinylbenzene within oil-in-water miniemulsions. Quantitative implementation of the pre-ceramic polymer can be achieved allowing precise control over the resulting materials. After pyrolysis and high-temperature chlorine treatment, resulting particles offer a spherical shape in combination with very high specific surface area (up to 2347 m2 g−1) and very large micro/mesopore volume (up to 1.67 cm3 g−1). The internal pore structure of the nanospheres is controllable by the composition of the oil phase within the miniemulsions. CDC-NS are versatile materials for supercapacitors with a specific capacitance of up to 130 F g−1 in organic media and excellent power handling at high current density. Their performance in a given electrolyte system is tunable by the composition of the oil phases in the initial miniemulsions.Acknowledgements
MO, SK, LB, PS, and WN gratefully acknowledge financial support by the German Research Foundation (DFG) for the project “Adsorption kinetics in hierarchical carbide-derived porous carbon materials” (KA 1698/18-1). MO acknowledges financial support from the German Academic Exchange Service (DAAD). MZ, NJ, and VP acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework and thank Prof. Eduard Arzt (INM) for his continuing support.Notes and references
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810 CrossRef CAS; M. R. Lukatskaya, J. Halim, B. Dyatkin, M. Naguib, Y. S. Buranova, M. W. Barsoum and Y. Gogotsi, Angew. Chem., Int. Ed., 2014, 53, 4877 CrossRef PubMed.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591 CrossRef CAS PubMed.
- Y. Gogotsi, R. K. Dash, G. Yushin, T. Yildirim, G. Laudisio and J. E. Fischer, J. Am. Chem. Soc., 2005, 127, 16006 CrossRef CAS PubMed; M. Sevilla, R. Foulston and R. Mokaya, Energy Environ. Sci., 2010, 3, 223 Search PubMed; A. Silvestre-Albero, S. Rico-Frances, F. Rodriguez-Reinoso, A. M. Kern, M. Klumpp, B. J. M. Etzold and J. Silvestre-Albero, Carbon, 2013, 59, 221 CrossRef; S. K. Bhatia and T. X. Nguyen, Ind. Eng. Chem. Res., 2011, 50, 10380 CrossRef; V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059 Search PubMed.
- J. J. Niu, V. Presser, C. J. Karwacki and Y. Gogotsi, Mater. Express, 2011, 1, 259 Search PubMed; A. Schlange, A. R. dos Santos, B. Hasse, B. J. M. Etzold, U. Kunz and T. Turek, J. Power Sources, 2012, 199, 22 CrossRef CAS.
- S. Porada, L. Borchardt, M. Oschatz, M. Bryjak, J. S. Atchison, K. J. Keesman, S. Kaskel, P. M. Biesheuvel and V. Presser, Energy Environ. Sci., 2013, 6, 3700 CAS; S. Porada, L. Weinstein, R. Dash, A. van der Wal, M. Bryjak, Y. Gogotsi and P. M. Biesheuvel, ACS Appl. Mater. Interfaces, 2012, 4, 1194 Search PubMed.
- L. Borchardt, M. Oschatz and S. Kaskel, Mater. Horiz., 2014, 1, 157 RSC; J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392 CrossRef CAS PubMed; J. Leis, M. Arulepp, A. Kuura, M. Laett and E. Lust, Carbon, 2006, 44, 2122 CrossRef; M. Arulepp, J. Leis, M. Laett, F. Miller, K. Rumma, E. Lust and A. F. Burke, J. Power Sources, 2006, 162, 1460 CrossRef.
- L. Borchardt, M. Oschatz, S. Paasch, S. Kaskel and E. Brunner, Phys. Chem. Chem. Phys., 2013, 15, 15177 RSC; M. Oschatz, L. Borchardt, S. Rico-Frances, F. Rodriguez-Reinoso, S. Kaskel and J. Silvestre-Albero, Langmuir, 2013, 29, 8133 CrossRef CAS PubMed; M. Oschatz, H. C. Hoffmann, J. Pallmann, J. Schaber, L. Borchardt, W. Nickel, I. Senkovska, S. Rico-Frances, J. Silvestre-Albero, S. Kaskel and E. Brunner, Chem. Mater., 2014, 26, 3280 CrossRef; C. Merlet, B. Rotenberg, P. A. Madden, P.-L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306 CrossRef PubMed; A. C. Forse, J. M. Griffin, H. Wang, N. M. Trease, V. Presser, Y. Gogotsi, P. Simon and C. P. Grey, Phys. Chem. Chem. Phys., 2013, 15, 7722 RSC; S. Kondrat, C. R. Perez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474 Search PubMed.
- G. Yushin, E. N. Hoffman, M. W. Barsoum, Y. Gogotsi, C. A. Howell, S. R. Sandeman, G. J. Phillips, A. W. Lloyd and S. V. Mikhalovsky, Biomaterials, 2006, 27, 5755 CrossRef CAS PubMed.
- S.-H. Yeon, I. Knoke, Y. Gogotsi and J. E. Fischer, Microporous Mesoporous Mater., 2010, 131, 423 CrossRef CAS; T. Fey, B. Zierath, A. M. Kern, P. Greil and B. J. M. Etzold, Carbon, 2014, 70, 30 CrossRef; M. Schmirler, T. Knorr, T. Fey, A. Lynen, P. Greil and B. J. M. Etzold, Carbon, 2011, 49, 4359 CrossRef; W. Nickel, M. Oschatz, M. von der Lehr, M. Leistner, G.-P. Hao, P. Adelhelm, P. Mueller, B. M. Smarsly and S. Kaskel, J. Mater. Chem. A, 2014, 2, 12703 Search PubMed.
- M. Kormann, H. Gerhard and N. Popovska, Carbon, 2009, 47, 242 CrossRef CAS; M. Kormann and N. Popovska, Microporous Mesoporous Mater., 2010, 130, 167 CrossRef; M. Kormann, H. Ghanem, H. Gerhard and N. Popovska, J. Eur. Ceram. Soc., 2008, 28, 1297 CrossRef.
- J. Chmiola, C. Largeot, P.-L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480 CrossRef CAS PubMed; M. Heon, S. Lofland, J. Applegate, R. Nolte, E. Cortes, J. D. Hettinger, P.-L. Taberna, P. Simon, P. Huang, M. Brunet and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 135 Search PubMed.
- V. Presser, L. Zhang, J. J. Niu, J. McDonough, C. Perez, H. Fong and Y. Gogotsi, Adv. Energy Mater., 2011, 1, 423 CrossRef CAS; M. Rose, E. Kockrick, I. Senkovska and S. Kaskel, Carbon, 2010, 48, 403 CrossRef.
- M. Oschatz, W. Nickel, M. Thommes, K. A. Cychosz, M. Leistner, M. Adam, G. Mondin, P. Strubel, L. Borchardt and S. Kaskel, J. Mater. Chem. A, 2014, 2, 18472 CAS.
- R. K. Dash, A. Nikitin and Y. Gogotsi, Microporous Mesoporous Mater., 2004, 72, 203 CrossRef CAS; R. K. Dash, G. Yushin and Y. Gogotsi, Microporous Mesoporous Mater., 2005, 86, 50 CrossRef; G. N. Yushin, E. N. Hoffman, A. Nikitin, H. Ye, M. W. Barsoum and Y. Gogotsi, Carbon, 2005, 43, 2075 CrossRef; J. Leis, A. Perkson, M. Arulepp, M. Kaarik and G. Svensson, Carbon, 2001, 39, 2043 CrossRef; J. Leis, M. Arulepp, M. Kaeaerik and A. Perkson, Carbon, 2010, 48, 4001 CrossRef.
- M. Sevilla and R. Mokaya, J. Mater. Chem., 2011, 21, 4727 RSC; S. Osswald, J. Chmiola and Y. Gogotsi, Carbon, 2012, 50, 4880 CrossRef CAS; S. Osswald, C. Portet, Y. Gogotsi, G. Laudisio, J. P. Singer, J. E. Fischer, V. V. Sokolov, J. A. Kukushkina and A. E. Kravchik, J. Solid State Chem., 2009, 182, 1733 CrossRef; S.-H. Yeon, S. Osswald, Y. Gogotsi, J. P. Singer, J. M. Simmons, J. E. Fischer, M. A. Lillo-Rodenas and A. Linares-Solano, J. Power Sources, 2009, 191, 560 CrossRef; M. Oschatz, L. Borchardt, I. Senkovska, N. Klein, M. Leistner and S. Kaskel, Carbon, 2013, 56, 139 CrossRef.
- M. Oschatz, L. Borchardt, K. Pinkert, S. Thieme, M. R. Lohe, C. Hoffmann, M. Benusch, F. M. Wisser, C. Ziegler, L. Giebeler, M. H. Ruemmeli, J. Eckert, A. Eychmueller and S. Kaskel, Adv. Energy Mater., 2014, 4, 1300645 CAS; M. Oschatz, J. T. Lee, H. Kim, W. Nickel, L. Borchardt, W. I. Cho, C. Ziegler, S. Kaskel and G. Yushin, J. Mater. Chem. A, 2014, 2, 17649 Search PubMed.
- L. Borchardt, F. Hasche, M. R. Lohe, M. Oschatz, F. Schmidt, E. Kockrick, C. Ziegler, T. Lescouet, A. Bachmatiuk, B. Buechner, D. Farrusseng, P. Strasser and S. Kaskel, Carbon, 2012, 50, 1861 CrossRef CAS; M. Oschatz, L. Borchardt, M. Thommes, K. A. Cychosz, I. Senkovska, N. Klein, R. Frind, M. Leistner, V. Presser, Y. Gogotsi and S. Kaskel, Angew. Chem., Int. Ed., 2012, 51, 7577 CrossRef PubMed.
- S.-H. Yeon, P. Reddington, Y. Gogotsi, J. E. Fischer, C. Vakifahmetoglu and P. Colombo, Carbon, 2009, 48, 201 CrossRef CAS; C. Vakifahmetoglu, V. Presser, S.-H. Yeon, P. Colombo and Y. Gogotsi, Microporous Mesoporous Mater., 2011, 144, 105 CrossRef.
- C. R. Perez, S.-H. Yeon, J. Segalini, V. Presser, P.-L. Taberna, P. Simon and Y. Gogotsi, Adv. Funct. Mater., 2013, 23, 1081 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, J. Electrochem. Soc., 2008, 155, A531 CrossRef CAS; B. Rufino, S. Mazerat, M. Couvrat, C. Lorrette, H. Maskrot and R. Pailler, Carbon, 2011, 49, 3073 CrossRef.
- L. Zhang, J. Hu, A. A. Voevodin and H. Fong, Nanoscale, 2010, 2, 1670 RSC.
- Y. Gao, V. Presser, L. Zhang, J. J. Niu, J. K. McDonough, C. R. Perez, H. Lin, H. Fong and Y. Gogotsi, J. Power Sources, 2012, 201, 368 CrossRef CAS.
- L. Borchardt, M. Oschatz, R. Frind, E. Kockrick, M. R. Lohe, C. P. Hauser, C. K. Weiss, K. Landfester, B. Buechner and S. Kaskel, Beilstein J. Nanotechnol., 2011, 2, 638 CrossRef CAS PubMed; K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488 CrossRef PubMed; K. Landfester, N. Bechthold, F. Tiarks and M. Antonietti, Macromolecules, 1999, 32, 5222 CrossRef; H.-D. Koh, M. Changez, J.-P. Lee and J.-S. Lee, Macromol. Rapid Commun., 2009, 30, 1583 CrossRef PubMed.
- G. Y. Gor, M. Thommes, K. A. Cychosz and A. V. Neimark, Carbon, 2012, 50, 1583 CrossRef CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059 CrossRef CAS.
- B. Kastening, Ber. Bunsen-Ges. Phys. Chem., 1988, 92, 1399 CrossRef CAS.
- V. Presser, C. R. Dennison, J. Campos, K. W. Knehr, E. C. Kumbur and Y. Gogotsi, Adv. Energy Mater., 2012, 2, 895 CrossRef CAS.
- S.-I. Jeon, H.-R. Park, J.-G. Yeo, S. C. Yang, C. H. Cho, M. H. Han and D. K. Kim, Energy Environ. Sci., 2013, 6, 1471 CAS.
- S. Porada, D. Weingarth, H. V. M. Hamelers, M. Bryjak, V. Presser and P. M. Biesheuvel, J. Mater. Chem. A, 2014, 2, 9313 CAS.
- B. Kastening, N. Busscher and U. Asskamp, J. Electroanal. Chem. Interfacial Electrochem., 1989, 265, 77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal analyses of the PCS-NS and CDC-NS as well as dynamic light scattering measurements of PCS-NS. See DOI: 10.1039/c5ta03730a |
| This journal is © The Royal Society of Chemistry 2015 |