
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long cycle life of CoMn2O4 lithium ion battery anodes with high crystallinity†
Mirjana
Bijelić
a,
Xiang
Liu
b,
Q.
Sun
b,
Aleksandra B.
Djurišić
*b,
Mao Hai
Xie
b,
Alan M. C.
Ng
cd,
Christian
Suchomski
e,
Igor
Djerdj
f,
Željko
Skoko
a and
Jasminka
Popović
*f
aDepartment of Physics, Faculty of Science, University of Zagreb, Bijenička c. 32, 10000 Zagreb, Croatia
bDepartment of Physics, University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: dalek@hku.hk
cDepartment of Physics, South University of Science and Technology of China, Shenzhen, China
dDepartment of Physics, University of Hong Kong, Pokfulam Road, Hong Kong
eInstitute of Physical Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 58, D-35392 Gießen, Germany
fDivision for Materials Physics, Ruđer Bošković Institute, Bijenička c. 54, 10000 Zagreb, Croatia. E-mail: Jasminka.Popovic@irb.hr
First published on 25th June 2015
Abstract
CoMn2O4 nanomaterials are prepared by a low temperature precipitation route employing metal acetates and NaOH. Structural changes, induced by different annealing temperatures, are comprehensively analyzed by X-ray powder diffraction and Raman spectroscopy. With rising annealing temperature the crystal lattice of CoMn2O4 undergoes changes; AO4 tetrahedra expand due to thermally induced substitution of Co2+ by larger Mn2+ metal ions on the A-site of the spinel structure, while in contrast, BO6 octahedra shrink since the B-site becomes partially occupied by smaller Co3+ metal ions on account of the migrated Mn ions. CoMn2O4 particle sizes are easily fine-tuned by applying different annealing temperatures; the particle size increases with increasing annealing temperature. During the battery operation, pulverization and reduction of particle sizes occurs regardless of the initial size of the particles, but the degree of division of the particles during the operation is dependent on the initial particle properties. Thus, contrary to the common assumption that nanostructuring of the anode material improves the battery performance, samples with the largest particle sizes exhibit excellent performance with a capacity retention of 104% after 1000 cycles (compared to the 2nd cycle).
Introduction
Rechargeable lithium-ion batteries (LIBs) have a wide range of applications in various fields, starting with the telecommunication and entertainment industry all the way to medicine and aeronautics. LIBs are nowadays used in notebook computers, tablets, E-books, smart phones, digital cameras, pacemakers, etc. In addition, LIBs also possess great potential for utilization in high power applications, such as electric vehicles and hybrids, as well as energy power storage.1–4 The performance of LIBs is greatly determined by the chemical composition, structural characteristics and microstructural features of both anode and cathode materials. Different simple micro- and nano-structured metal oxides, such as manganese oxide,4–6 iron oxide,7–13 cobalt oxide,14–17 and tin oxide,18–23 have been recently investigated as alternative anode materials for use in LIBs. Manganese-based oxide electrodes are characterized by lower operating voltages (1.3–1.5 V for lithium extraction) and deliver higher energy density than the commonly used cobalt oxide-based electrodes. Therefore, it is a logical consequence that Mn2O3, Mn3O4, ZnMn2O4, NiMn2O4 and CoMn2O4 have recently attracted significant research interest,24–29 especially if one additionally considers that manganese has many advantages, such as multiple valence states, high abundance in nature, low toxicity and low cost.In recent times, several papers evidenced clearly that CoMn2O4 is a promising candidate for prospective LIB anode materials. For this purpose, different morphologies were prepared and their battery performances were extensively examined.30–34 For example, Courtel et al.30 fabricated CoMn2O4 powders that exhibit capacities of approximately 515 mA h g−1 with 64% retention at a current of 69 mA g−1 after 50 cycles. Also, double-shelled CoMn2O4 hollow microcubes were made by heat treatment of Co0.33Mn0.67CO3 at 600 °C and demonstrate initial discharge capacities of 624 mA h g−1, which drop down to 200 mA g−1 after 50 cycles.31 Moreover, hierarchical CoMn2O4 microspheres were prepared by a two-step procedure which included precipitation of alkoxide salt precursors from ethylene glycol solutions followed by thermal decomposition at 600 °C.32 The obtained hierarchical microspheres have been shown to possess high discharge capacities of 894 mA h g−1 (at a current density of 100 mA g−1) after 65 cycles and, moreover, indicate excellent cycling stability.32 Additionally, electrochemical properties of CoMn2O4 nanorods obtained by a solid state reaction starting from hydrothermally synthesized β-MnO2 serving as the template and Co(OH)2 have also been reported recently.33 The as-made CoMn2O4 nanorods show outstanding electrochemical performances: both excellent rate capability (400 mA h g−1 at a current density of 1000 mA g−1), and cycling performance and high Coulombic efficiency (98% after 100 cycles).33 In addition, quasi-hollow CoMn2O4 microspheres were prepared by thermal decomposition of Mn0.33Co0.67CO3 at 600 °C.34 These quasi-hollow microspheres exhibit reversible capacities of 706 mA h g−1 after 25 cycles at a current density of 200 mA g−1.34
From the literature review, it can be concluded that the long-established solid-state route which involves repeated grinding and firing at elevated temperatures of carbonates and oxides has been almost completely abandoned. Indeed, all previously mentioned reports30–34 are focused on low cost preparation methods which are conducted at low temperatures (usually 600 °C). In regard to finding a simple and straightforward preparation method for manganese spinels, room temperature synthesis of CoMn2O4 by a reduction of amorphous MnO2 precursors in an aqueous solution containing Co2+ ions has been reported.35 Finally, Liu et al.36 also reported a room temperature route for the preparation of AMn2O4 (A = Zn, Co, Cd) from metal acetates. Although the synthetic route shown by Liu et al.36 represents a facile and efficient method for low temperature synthesis of spinel materials, in order to utilize this route for the targeted design of nanomaterials it is necessary to precisely establish the correlations between specific preparation conditions (i.e., concentration of NaOH, aging period, and temperature of additional heat treatments), structure, morphology and electrochemical properties.
In this work we report a systematic study of CoMn2O4 anode materials prepared by a precipitation route using metal acetates and NaOH. Detailed structural investigations employing X-ray powder diffraction (XRPD) and Raman spectroscopy have been carried out in order to understand structural changes along with the accompanying electrochemical properties. The majority of existing work on battery performance appears to focus on the complex morphologies of the synthesized CoMn2O4. On the other hand, the studies on the effect of crystal structures on battery performance are quite rare. We have performed an extensive investigation of the battery performance of different CoMn2O4 samples, and discuss the relationship between the crystallinity and the electrochemical properties of CoMn2O4.
Experimental
Synthetic procedures
Cobalt-dimanganate (CoMn2O4) compounds were prepared by the original synthetic route reported by Liu et. al.36 and, in addition, by the modified synthetic route which we report here. In each case 0.626 g Co(Ac)2·4H2O (Sigma-Aldrich, 99.995%) and 1.238 g Mn(Ac)2·4H2O (Sigma-Aldrich, purum p.a., ≥99.0%) were dissolved in 40 ml of distilled water. In order to induce precipitation, 150 ml of 0.25 M NaOH solution was added, dropwise, to the mixture solution at room temperature under vigorous magnetic stirring. After 45 min of the reaction, the resulting precipitate was collected by centrifugation and repeatedly washed with distilled water until the pH became neutral. Finally, the precipitate was dried at 80 °C for a period of 12 h, and the as-prepared sample is denoted as C1. Alternately, the synthesis was also carried out with the same quantities of starting materials but a higher concentration of NaOH was used for precipitation, in addition to a prolonged drying time at higher temperature. Samples denoted as D1 and S1 were prepared by using 0.4 M and 0.8 M NaOH, respectively, followed by drying at 100 °C for 24 h. Additionally, sample S1 was heat treated at different temperatures T = 300, 400 and 500 °C; the samples are denoted as S2, S3, and S4, respectively.Methods
Raman spectra were collected on a SENTERRA dispersive Raman microscope from Bruker Optics equipped with an objective from Olympus MPlan N 100× (FN 22 and NA 0.9) and a Nd:YAG laser λ = 532 nm, P = 0.2 mW. Structural features of cobalt(II) dimanganates were studied by XRPD at room temperature using a Philips MPD 1880 counter diffractometer with Cu Kα radiation. Samples C1 and D1 were recorded in the 2θ range of 15–100°, step 0.02° with a fixed counting time of 10 s per step. For each sample in the series S, S1–S4, two datasets were collected: (i) XRPD pattern of the sample mixed with 10 wt% of a silicon standard reference material (Koch-Light Lab. Ltd, 99.999% purity), 2θ range: 15–80°, step 0.02°, fixed counting time 6 s per step, for the purpose of precise determination of unit cell parameters; (ii) XRPD pattern of the pure sample, 2θ range: 15–100°, step 0.02°, fixed counting time 10 s per step, for the purpose of Rietveld structure refinement. Determination of unit cell parameters was performed by the whole-powder-pattern decomposition method proposed by Toraya, and implemented in the program WPPF37 by using the split-type pseudo-Voigt profile function and the polynomial background model. Structure refinement was carried out by the Rietveld method within the program X'Pert HighScore Plus, version 2.1 (PANalytical 2004), using a pseudo-Voigt profile function and also the polynomial background model. Isotropic vibration modes were assumed for all atoms. During the refinement a zero shift, scale factor, half-width parameters (U, V, W), asymmetry parameters and peak shape parameters were simultaneously refined. Occupancies of different cations residing on 4a and 8d sites were constrained to 1. Isotropic displacement parameters for cations sharing the same crystallographic site were constrained to change unisonous.The morphologies of electrodes before and after cycling were examined by scanning electron microscopy (SEM) using a JEOL JMS-7001F system. Transmission electron microscopy (TEM) and energy dispersive X-ray (EDX) mapping were measured using a FEI Tecnai G2 20 S-TWIN scanning transmission electron microscope system. The active materials were mixed with conductive carbon additives (carbon black, Super-P@Li, Timcal) and a binder (polyvinylidene fluoride, PVDF, MTI) in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The loading amount of the active material was ∼2 mg cm−2, and the specific capacity was calculated using the actual weight of the active material. Electrochemical measurements were conducted using a coin-cell (CR2032) with lithium metal (15.6 mm in diameter, 0.25 mm in thickness) as a counter-electrode. After coating and drying, electrodes were cut into 14 mm diameter disks. Cells were assembled in an Ar-filled glove box. The electrolyte which consisted of 1 M LiPF6 in a 1:1:1 (in volume) mixture of ethylene carbonate (EC)/dimethyl carbonate (DMC)/diethyl carbonate (DEC) was purchased from MTI Corporation. Cyclic-voltammetry (CV) measurement was conducted at the rate of 0.1 mV s−1 in the range between 0.005 and 3.0 V on a BioLogic VMP3 electrochemical workstation. The galvanostatic charge/discharge cycles were studied with a Neware BTS3000 battery test system at various current densities of 100 mA g−1 to 1 A g−1 between 3.0 and 0.005 V. EIS measurements were performed using a BioLogic VMP3 electrochemical workstation by employing an ac voltage of 5 mV amplitude in the frequency range of 0.01–100 kHz.
:10:10. The loading amount of the active material was ∼2 mg cm−2, and the specific capacity was calculated using the actual weight of the active material. Electrochemical measurements were conducted using a coin-cell (CR2032) with lithium metal (15.6 mm in diameter, 0.25 mm in thickness) as a counter-electrode. After coating and drying, electrodes were cut into 14 mm diameter disks. Cells were assembled in an Ar-filled glove box. The electrolyte which consisted of 1 M LiPF6 in a 1:1:1 (in volume) mixture of ethylene carbonate (EC)/dimethyl carbonate (DMC)/diethyl carbonate (DEC) was purchased from MTI Corporation. Cyclic-voltammetry (CV) measurement was conducted at the rate of 0.1 mV s−1 in the range between 0.005 and 3.0 V on a BioLogic VMP3 electrochemical workstation. The galvanostatic charge/discharge cycles were studied with a Neware BTS3000 battery test system at various current densities of 100 mA g−1 to 1 A g−1 between 3.0 and 0.005 V. EIS measurements were performed using a BioLogic VMP3 electrochemical workstation by employing an ac voltage of 5 mV amplitude in the frequency range of 0.01–100 kHz.
Results and discussion
All prepared CoMn2O4 compounds crystallize in the tetragonal spinel-type structure. However, a pronounced difference in crystallinity was observed for as-prepared samples C1, D1 and S1, as shown in Fig. 1. The exceptionally high crystallinity of sample S1, observed from well-defined X-ray powder diffraction (XRPD) maxima, was achieved by increasing the concentration of NaOH solution from 0.25 M, 0.4 M to 0.8 M followed by a longer annealing period of 24 h at 100 °C. Interestingly, Liu et al.36 also reported an increase in crystallinity with increase in NaOH concentration. However, a further increase above 0.25 M resulted in the appearance of an impurity phase. On the other hand, samples S1–S4 obtained by our modified synthetic route where a high concentration of NaOH was used as well as longer annealing period did not show any unwanted additional phases as nicely seen from the XRPD patterns and additionally confirmed by Raman spectroscopy. In addition to qualitative XRPD analysis, a model-free whole pattern decomposition method and Rietveld refinement on XRPD data collected on samples S1–S4 at RT were carried out in order to track the fine structural changes in the CoMn2O4 lattice induced by changes in the heat treatment temperature. Even though occupancy parameters for 4a and 8d sites have been refined and therefore a cation distribution parameter has been quantified, we prioritized changes in the unit cell parameters and interatomic distances as a more reliable indicator for tracking fine structural changes within the spinel lattice. The changes in unit cell parameters a and c as obtained by WPPF program are given in Fig. 2. A decrease in both parameters has been observed. However, the change in the c-direction is significantly more pronounced. The observed decrease in unit cell parameters is inconsistent with a simple spinel-type exchange of Co2+ by Mn3+ on the tetrahedral site, and vice versa, Mn3+ by Co2+ on the octahedral site. Considering the ionic radii, substitution of the smaller Mn3+ cation by the larger Co2+ at the octahedral site would cause an increment of the octahedral bond lengths, i.e. an observable increment of lattice parameters should have been noticed since octahedral bonds, being parallel to crystallographic axes a and c, directly influence the unit cell parameters.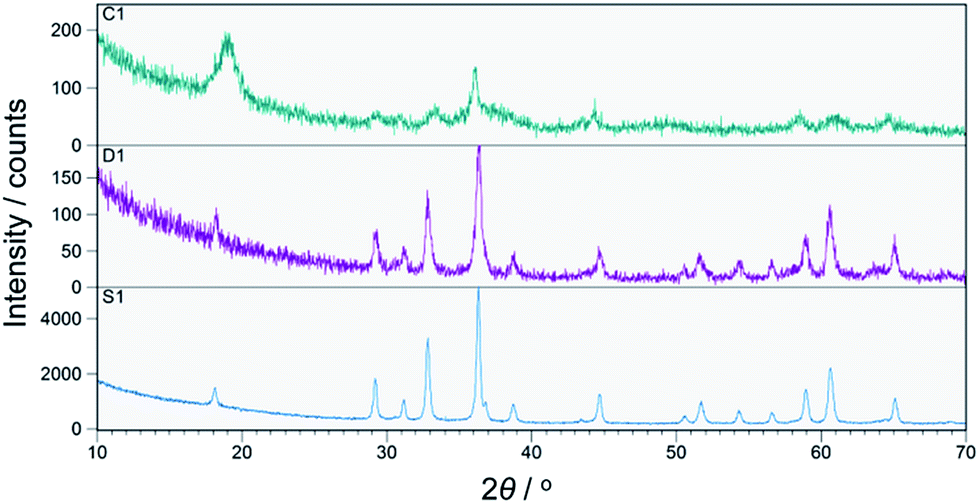 | ||
| Fig. 1 XRPD patterns of as-prepared samples C1, D1 and S1. | ||
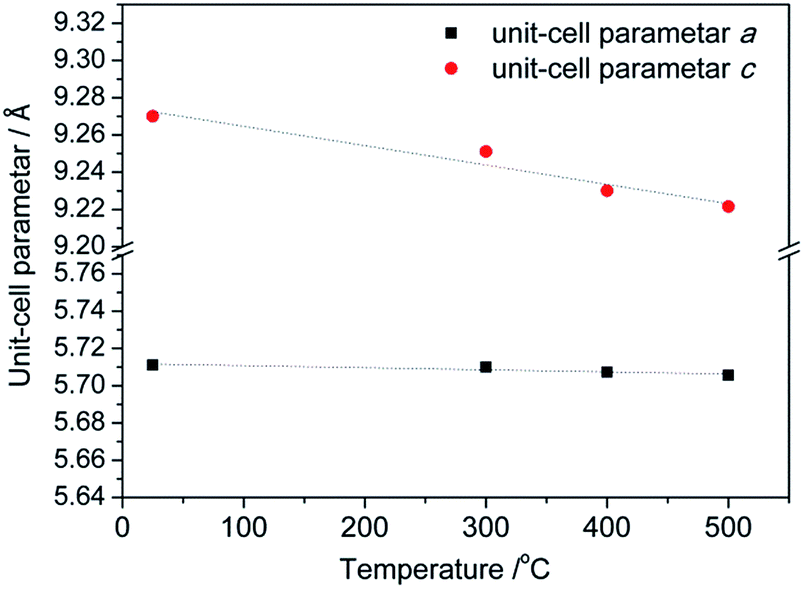 | ||
| Fig. 2 Changes in the unit cell parameters a and c as a function of the temperature of thermal treatment. | ||
Since the first indications about cation distribution and their oxidation states at octahedral and tetrahedral sites, based on the results from a precise determination of the unit cell parameters, pointed out that structure refinement should not be performed assuming a typical spinel composition with non-zero inversion [Co1−x2+Mnx3+]tetra[Cox2+Mn2−x3+]octaO4, further attempts at establishing a suitable model were made by means of Raman spectroscopy. Although results of Rietveld refinement on XRPD data are generally unaffected by different oxidation states used in the structural model (due to small differences between X-ray scattering factors), still only an exact model offers meaningful interpretation of the observed changes in structural parameters. It should be noted that additional information on the oxidation states could possibly be obtained using X-ray photoelectron spectroscopy.38,39 Based on the results of Raman spectroscopy (see below) a plausible model described by the formula [Co1−x2+Mnx3+]tetra[Cox2+Mn2−x3+]octaO4 has been proposed and tested upon structural data. Table S1† contains refined structural data for samples S1–S4 while a graphical result is given for sample S3 in Fig. S1 (ESI†). Crystallographic Information Files have been deposited at ICSD – Inorganic Crystal Structure Database, FIZ Karlsruhe, under No. 429387, 429388, 429389 and 429390 for samples S1 (as prepared), S2, S3 and S4 (treated at 500 °C), respectively. From the refined atomic coordinates and unit cell parameters a list of octahedral and tetrahedral bond lengths for samples S1–S4 have been derived and are shown in Fig. 3.
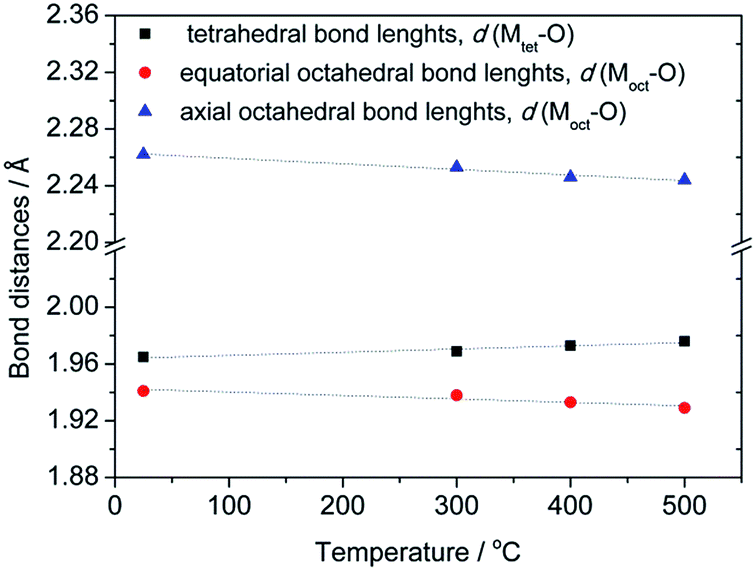 | ||
| Fig. 3 Temperature evolution of octahedral and tetrahedral bond lengths for as-synthesized compounds annealed at different temperatures. | ||
An observed increase of metal–oxygen interatomic distances within the tetrahedra, d(Mtet-O), from 1.965(1) Å (for sample S1) to 1.976(1) Å (for sample S4), resulted from the thermally enhanced substitution of Co2+ by larger Mn2+ cations at the A-site of the spinel structure. Consequently, the octahedral B-site becomes partially occupied by Co3+ on account of the transferred Mn cations. Indeed, the substitution of Mn3+ cations by smaller Co3+ led to an observed decrease of interatomic distances within the octahedra from dax(Moct-O) = 1.941(2) and deq(Moct-O) = 2.262(1) Å for sample S1 to dax(Moct-O) = 1.929(3) and deq(Moct-O) = 2.244(2) Å for sample S4.
Because vibrational spectroscopy is a much more sensitive method of observing minor amounts of impurity phases and lattice distortions than conventional X-ray powder diffraction techniques, a series of Raman measurements were also conducted on pristine and annealed CoMn2O4 samples. Typical unpolarized Raman spectra for the prepared CoMn2O4 at room temperature are shown in Fig. 4.
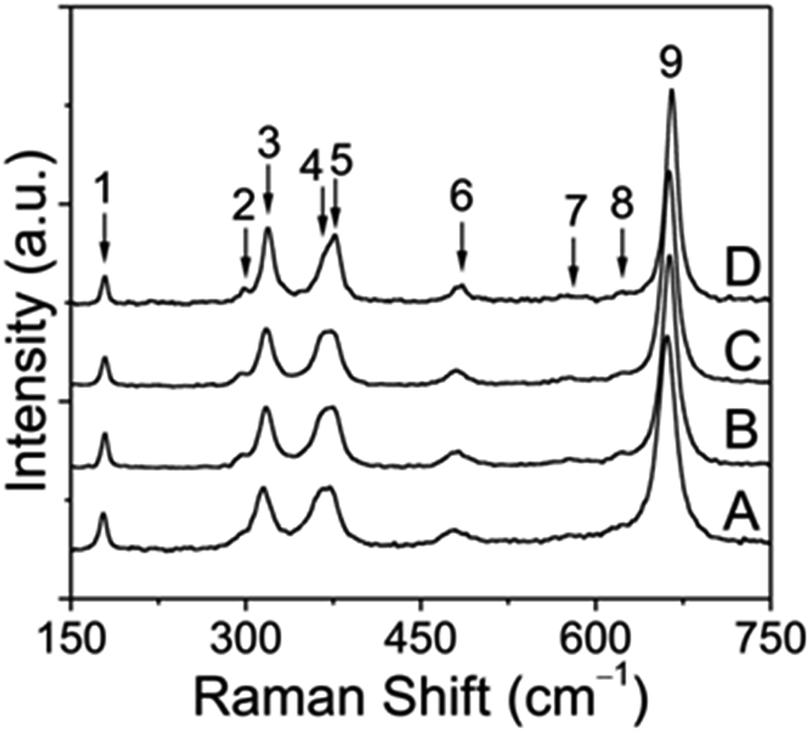 | ||
| Fig. 4 Raman spectra obtained for tetragonal CoMn2O4 nanoparticles: as prepared (A) and annealed to 300 °C (B), 400 °C (C) and 500 °C (D), respectively, followed by aging for 2 h. Spectral intensities have been normalized to the most intense Raman band centered at roughly 660 cm−1 (denoted as peak 9), so that changes in intensity are visible to the naked eye. | ||
According to the group theory, defect-free tetragonal CoMn2O4 with a space group of I41/amd (D4h19) should have ten first-order Raman-modes (namely T(B1g + Eg), L(Eg), υ1(A1g), υ2(A1g + B2g), υ3(B1g + Eg), υ4(B1g + Eg)).40,41 It has to be mentioned that, to the best of our knowledge, no earlier reports on CoMn2O4 have addressed the symmetry assignment of optical phonon modes. This is undoubtedly a challenging task, since optical modes can only be distinguished by polarized Raman spectroscopy on single crystals. Overall, we can see 9 Raman bands in the frequency range of 150–750 cm−1 (see Table S1†). In contrast, non-defective ferrite spinels AFe2O4 (A = divalent metal) with the space group Fd3![[m with combining macron]](https://www.rsc.org/images/entities/i_char_006d_0304.gif) (Oh7) have only 5 Raman-active modes, represented by T(F2g), υ1(A1g), υ2(Eg), υ3(F2g), and υ4(F2g).42 In general, tetragonal CoMn2O4 shows a slight distortion of MnO6 octahedra due to the Jahn−Teller effect, which results in elongation of apical Mn–O bond lengths and contraction of in-plane Mn–O bond lengths as indicated in Fig. 5.
(Oh7) have only 5 Raman-active modes, represented by T(F2g), υ1(A1g), υ2(Eg), υ3(F2g), and υ4(F2g).42 In general, tetragonal CoMn2O4 shows a slight distortion of MnO6 octahedra due to the Jahn−Teller effect, which results in elongation of apical Mn–O bond lengths and contraction of in-plane Mn–O bond lengths as indicated in Fig. 5.
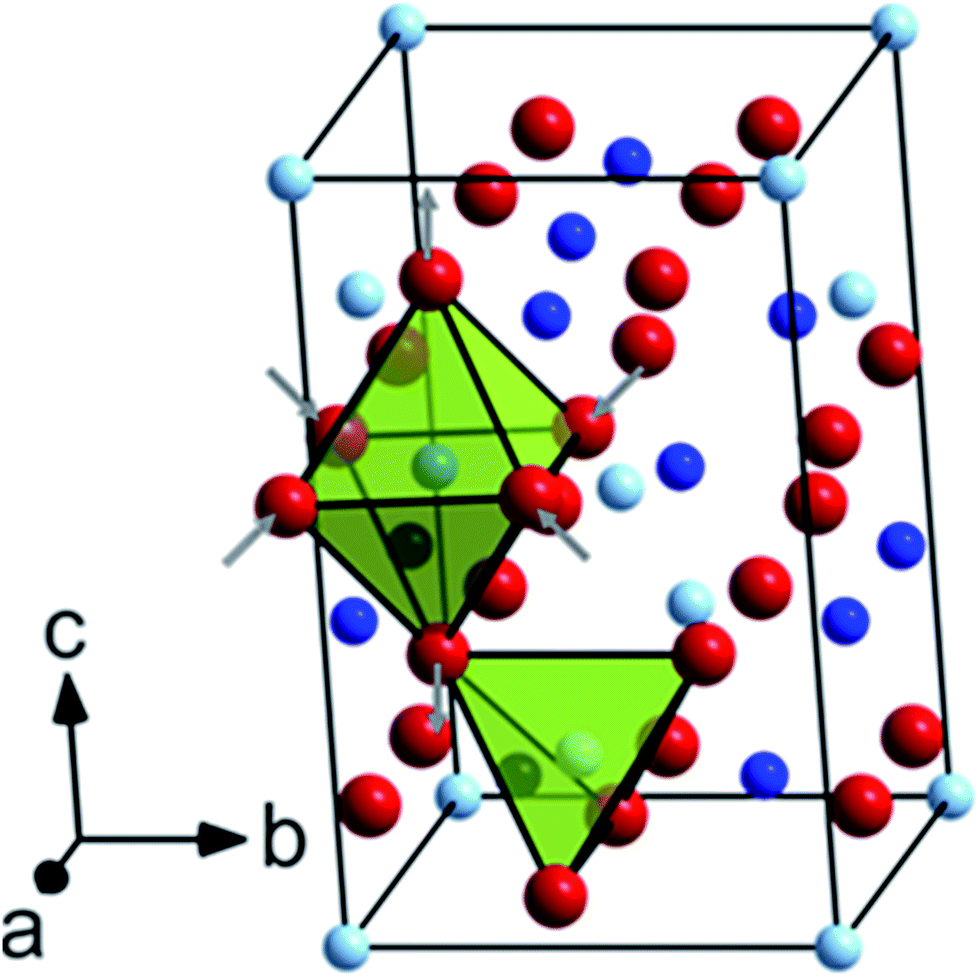 | ||
| Fig. 5 Unit cell of tetragonal CoMn2O4 (I41/amd) viewed along the [412] crystallographic direction. Co atoms occupying the tetrahedral 4(a) Wyckoff sites are shown in pale blue, Mn atoms on octahedral 8(d) Wyckoff sites in blue and O atoms on the 16(h) Wyckoff sites in red, respectively. The different coordination spheres are highlighted by colored polyhedra. | ||
Octahedral deformation, which decreases the symmetry from Fd3 to I41/amd, is by far the most common lattice distortion in spinel structures and can be characterized by the ratio of lattice parameters c/a. From the unpolarized Raman spectra it can be clearly seen that some band positions shift to higher frequencies depending on the annealing temperature, which points to the changes in the tetragonal spinel structure. In the low frequency region, the first Raman band at ∼179 cm−1 can be assigned to the Co–O stretching mode and is characteristic of the CoO4 tetrahedron.43 Furthermore, this band intensity decreased with increasing annealing temperature, which can likely be attributed to the oxidation of a small fraction of Co2+ ions on tetrahedral sites. As a result, it can be inferred from simple redox considerations that both Co3+ and Mn2+ ions must be formed during the thermal treatment. Due to several reasons, including the ionic radius and the crystal field stabilization energy, Mn2+ ions may migrate towards the tetrahedral sites, while thermally induced Co3+ ions preferably occupy the octahedral sites. Indeed, the band between 295 and 298 cm−1, which becomes more intense at higher annealing temperature, is indicative of Mn2+ on tetrahedral sites.44 In the middle frequency region between 300 and 400 cm−1, in total three bands at approximately 315–319 cm−1 (peak 3), 365–369 cm−1 (peak 4) and 375–377 cm−1 (peak 5) can be identified and are probably related to oxygen modes in MnO6 and/or MnO4 sites, because Mn3O4, (Mn2+)IV[Mn23+]VIO4, exhibits three distinctive bands in this region, whereas Co3O4, (Co2+)IV[Co23+]VIO4, exhibits no bands there (Table S2†).
Previous investigations by Malavasi et al.41 have shown that the band between 479 and 484 cm−1 can be associated with the metal–oxygen movement of divalent metal ions in tetrahedral sites. Furthermore, it was evidenced for several AMn2O4 spinels (A = Mg, Mn, Zn) that a significant red shift occurs, if the mass of the divalent A metal ion decreases.41 This fact helps in explaining why a small shift appears with increasing temperature, which we therefore attribute to the partial replacement of Co2+ by the lighter Mn2+ on tetrahedral sites. Lastly, the remaining three bands (peaks 7−9) in the high frequency region between 570 and 670 cm−1 can be considered as vibrational modes involving the motion of oxygen atoms within the MO6 octahedra, as proven for different chromite (ACr2O4), ferrite (AFe2O4) and manganese (AMn2O4) spinel compounds.42,44,45 Interestingly, the two weak bands centered at roughly 578 and 620 cm−1 (peaks 7 and 8) were associated in previous reports with Mn4+ ions on octahedral sites, as shown for LiMn2O4 and LiMn2−xNixO4 spinel compounds.46,47 However, these bands may also arise from the random Mn3+/Co3+ mixing on the octahedral sites because of the metal ion redistribution at elevated temperatures. We will return to this point at a later stage. As can be seen from the XRD analysis, the tetragonal distortion decreases with increasing temperature, for example from c/a = 1.623 (sample S1) to c/a = 1.616 (sample S4), as a result of the incorporation of Co3+ into the octahedral sites. This structural alteration is accompanied by the shortening of metal–oxygen bonds of MO6 polyhedra and causes a progressive red shift of the strongest band (peak 9) from 661 to 665 cm−1. The opposite behavior is predicted for metal ions with larger ionic radii than Mn3+, such as Co2+ and Mn2+. To readdress the possible formation of Mn4+ on octahedral sites, this open question can likely be answered by a closer examination of the full width at half maximum (FWHM) of the aforementioned highest band at different annealing temperatures. In previous work, this band has been interpreted as A1g mode. In addition, it was reported that Mn4+ ions give rise to peak broadening of this mode.47 Our experimental data demonstrate, however, that the FWHM becomes narrower when the annealing temperature increases from 300 to 500 °C. Thus, it can be concluded that (1) most probably no Mn4+ ions are produced by the heat treatment and (2) the structure undergoes a change to a spinel composition of (Co1−x2+ Mnx2+)IV[Mn2−x3+ Cox3+]VIO4, as a consequence of redox-induced cation migration. Similarly, these results also confirm that the pristine CoMn2O4 sample shows the same but less pronounced cation dislocation, and, moreover, that no secondary phases such as Mn2O3 and CoO are created by the employed preparation procedure.
SEM images of CoMn2O4 samples annealed at different temperatures are shown in Fig. 6. It can be observed that the size increases significantly with increased annealing temperature, from 115 ± 3 nm for sample S1 (as prepared) to 5.9 ± 0.2 μm for sample S4 (treated at 500 °C). The obtained cycling performance, Coulombic efficiency and rate performance of the CoMn2O4 based electrodes are shown in Fig. 7 and 8, respectively. In all samples, the initial capacity after the first cycle drops as commonly observed in high capacity metal oxide materials.18,28,30,31,33,34,48–53 It was reported that the theoretical first discharge capacity of CoMn2O4 is 921 mA h g−1,53 while the reversible theoretical capacity is 691 mA h g−1.31,33,53 The reversible theoretical capacity calculation is based on the equation Co + 2Mn + 3Li2O ↔ CoO + 2MnO + 6Li+ + 6e−,31,33,53 while the entire electrochemical process also includes the following: CoMn2O4 + 8Li+ + 8e− → Co + 2Mn + 4Li2O.53 However, after the initially observed decrease in capacity for a number of cycles, the specific capacity increases again. For sample S1, the initial capacity of 829 mA h g−1 drops to 654 mA h g−1 by the 2nd cycle, 377 mA h g−1 by the 10th cycle and 199 mA h g−1 by the 100th cycle. Then, a small increase is observed, and the obtained capacity is 201 mA h g−1 by the 200th cycle, 237 mA h g−1 by the 500th cycle, and 507 mA h g−1 by the 1000th cycle. For sample S2, the initial capacity was higher, 1195 mA h g−1, but it reduced to 547 mA h g−1 in the 2nd cycle. By the 10th cycle, it dropped to 314 mA h g−1, and by the 100th cycle to 183 mA h g−1. In this case as well an increase in capacity was observed with continued cycling, so that the capacity increased to 255 mA h g−1 by the 200th cycle and to 479 mA h g−1 by the 500th cycle. The capacity remains at 433 mA h g−1 by the 800th cycle and 420 mA h g−1 by the 1000th cycle. For the S3 sample, the initial capacity of 1272 mA h g−1 dropped to 553 mA h g−1 by the 2nd cycle, 390 mA h g−1 by the 10th cycle and 324 mA h g−1 and 320 mA h g−1 at the 100th and 200th cycles, respectively. In this sample as well an increase is observed, up to 404 mA h g−1 by the 500th cycle and then there was a decrease again to 291 mA h g−1 by the 800th cycle and 267 mA h g−1 by the 1000th cycle. The best cycling performance is obtained in the case of the S4 sample, with specific capacity decreasing from an initial capacity of 1477 mA h g−1 to 601 mA h g−1 by the 2nd cycle and 325 mA h g−1 by the 100th cycle, and then increasing to 384 mA h g−1 by the 200th cycle and 644 mA h g−1 by the 500th cycle. By the 800th cycle, the capacity remains at 602 mA h g−1, and it is 624 mA h g−1 by the 1000th cycle. Coulombic efficiencies for all samples remain high after several initial cycles, as can be observed from Fig. 7. For sample S1, the initial Coulombic efficiency is 84%, and it increases to over 95% by the 8th cycle, while for samples S2, S3, and S4 initial efficiencies are 48%, 47%, and 43% and they increase to over 95% by the 4th to 5th cycle.
 | ||
| Fig. 6 SEM images of CoMn2O4 particles for different annealing temperatures: (a) as prepared (sample S1), (b) 300 °C (sample S2), (c) 400 °C (sample S3), and (d) 500 °C (sample S4). | ||
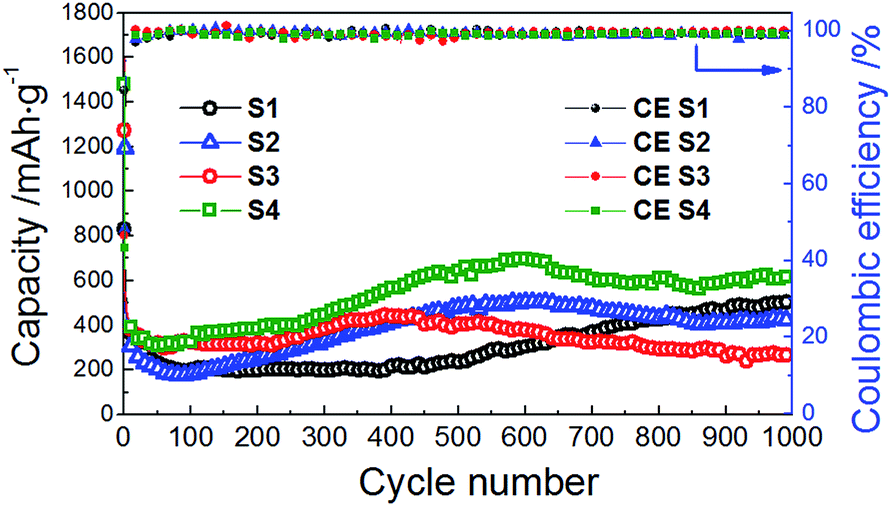 | ||
| Fig. 7 Cycle performance and Coulombic efficiency of the CoMn2O4 anodes for different annealing temperatures at the discharge/charge rate of 200 mA g−1. | ||
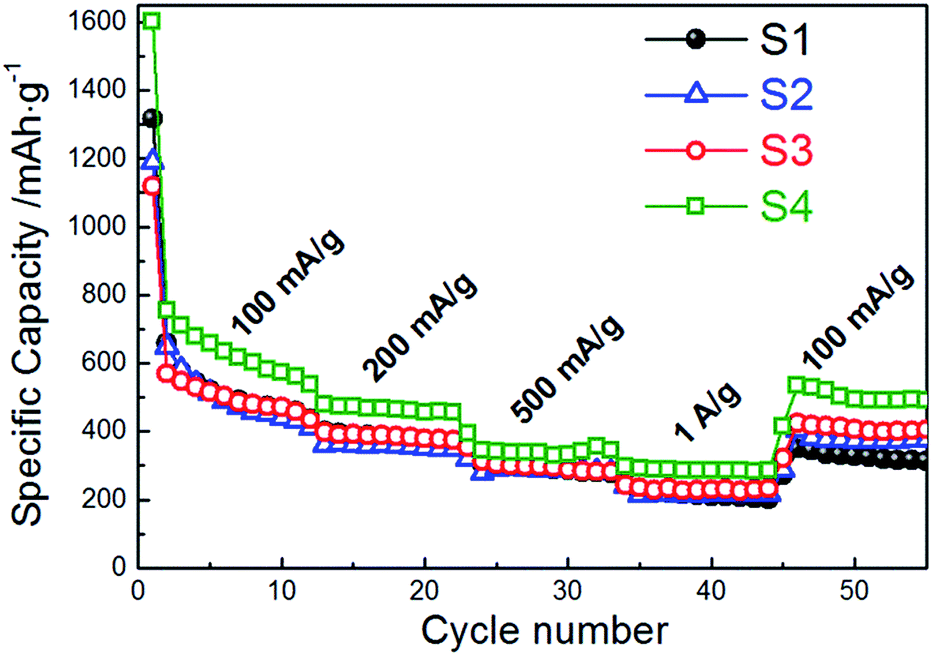 | ||
| Fig. 8 Rate cycle performance of CoMn2O4 anodes for different annealing temperatures at charge/discharge rates from 100 mA g−1 to 1 A g−1. | ||
Thus, the highest specific capacities are observed for sample S4 (thermally treated at the highest temperature of 500 °C) despite its large particle size. In terms of the particle size, it has been shown that in some metal oxides better capacity retention is obtained for larger particle sizes and it was proposed that there is an optimal size for improved capacity retention.51 Existence of an optimal particle size for an optimal battery performance has been demonstrated for ZnMn2O4.30 The optimal particle size is likely material dependent, since for ZnMn2O4 performance can be enhanced with increase in particle sizes in the range of 10–150 nm; however for samples having particle sizes larger than 200 nm the performance deteriorated.1 On the other hand, large 1 μm Cu2O particles performed better than 150 nm ones. No upper limit in regard to the optimal size was observed in our study; results showed continuous improvement of anode performance in terms of capacity and stability with thermally promoted particle growth (Fig. S2†). Our results suggest that larger particles of metal oxides could be responsible for the better state of division of the metal particles upon reduction and hence the improved electrochemical performance of the materials operating under a conversion redox mechanism, similar to a previous report.51
Moreover, it seems that the capacity–particle size correlation exists even after cycling (Fig. S3†); among all samples, sample S3 having the smallest average particle size after 1000 cycles (200 ± 3 nm) exhibited the smallest capacity (267 mA h g−1 in the 1000th cycle) while sample S4 with the largest average particle size after 1000 cycles (847 ± 7 nm) displayed the highest value of capacity (624 mA h g−1 in the 1000th cycle).
Regardless of the initial particle size, an increase in capacity after cycling for a certain period of time is observed in all the samples. Similar increase in capacity was previously observed in ZnMn2O4 hollow microspheres, and it was attributed to a possible activation process in the electrode.28 It was reported to be common in transition metal oxides,50,52 and was attributed to increased interfaces.52 An increase in capacity in a CuxO/SnOx/carbon nanotube composite was attributed to the reduction in particle size with cycling.18 A reduction in the particle size after cycling can be observed in SEM images of anodes after cycling, as shown in Fig. 9 after 1000 cycles and Fig. S4† after 300 cycles. This trend was especially pronounced for samples with the highest crystallinity; the average particle size for sample S4 decreased from 5.9 ± 0.2 μm to 847 ± 7 nm after 1000 cycles and from 1.8 ± 0.1 μm to 145 ± 3 nm for sample S3.
 | ||
| Fig. 9 SEM images of CoMn2O4 electrodes after 1000 cycles: (a) as-prepared S1; (b) 300 °C – S2; (c) 400 °C – S3 and (d) 500 °C – S4. | ||
From the element mapping, shown in Fig. 10 after 1000 cycles, one can observe that in larger particles (sample S4 in particular), there is a less uniform distribution of Co and Mn. This pronounced effect is clearly visible along the boundary of isolated large particles (S4), indicating a defect-rich area with vacant Co/Mn crystallographic sites. This could contribute to the outstanding performance of the sample S4, since the existence of compositional non-uniformity and disorder may result in improved buffering of volume changes.18 Similar observations can be made for the samples after 300 cycles (Fig. S5†).
 | ||
| Fig. 10 TEM images and corresponding element mapping of the CoMn2O4 electrode after 1000 cycles: as-prepared (sample S1), 300 °C (sample S2), 400 °C (sample S3) and 500 °C (sample S4) (from top to bottom). | ||
In addition to good cycling properties, sample S4 also exhibits good rate performance. A specific capacity of 285 mA h g−1 was obtained at the 40th cycle at a charge/discharge rate of 1 A g−1, while at the charge/discharge rate of 100 mA g−1 at the 50th cycle a specific capacity of 494 mA h g−1 is obtained, which is 66% of the specific capacity at the 2nd cycle. For comparison, capacity retention rates at the 50th cycle were 50%, 57%, and 72% for samples S1, S2 and S3, respectively, while the corresponding specific capacities at the 40th cycle and charge/discharge rate of 1 A g−1 were 209 mA h g−1, 218 mA h g−1, and 230 mA h g−1.
To further characterize the samples, cyclic voltammetry measurements were performed, and the obtained results are shown in Fig. 11. It can be observed that in all cases the first cycle exhibits significantly different features compared to later cycles. In the first cycle, several reduction peaks can be observed. The sample S2 exhibits a clear peak at 0.75 V corresponding to SEI formation.31–33,49 The peak in the range of 1.1–1.5 V observable in samples S1 and S2 can be attributed to the reduction of Mn3+ to Mn2+,31–34,49 while the peak at 0.25–0.4 V can be attributed to further reduction of Mn2+ and Co2+ to the corresponding metals.31–34,48,49 In the oxidation cycle, the peak at ∼1.35 V can be attributed to the oxidation of Mn to Mn2+.31,32,34,49 No peaks corresponding to the oxidation of Co to Co2+ at ∼2 V (ref. 32, 34, 48 and 49) can be observed. After the 10th cycle, the C–V curves almost overlap in all cases.
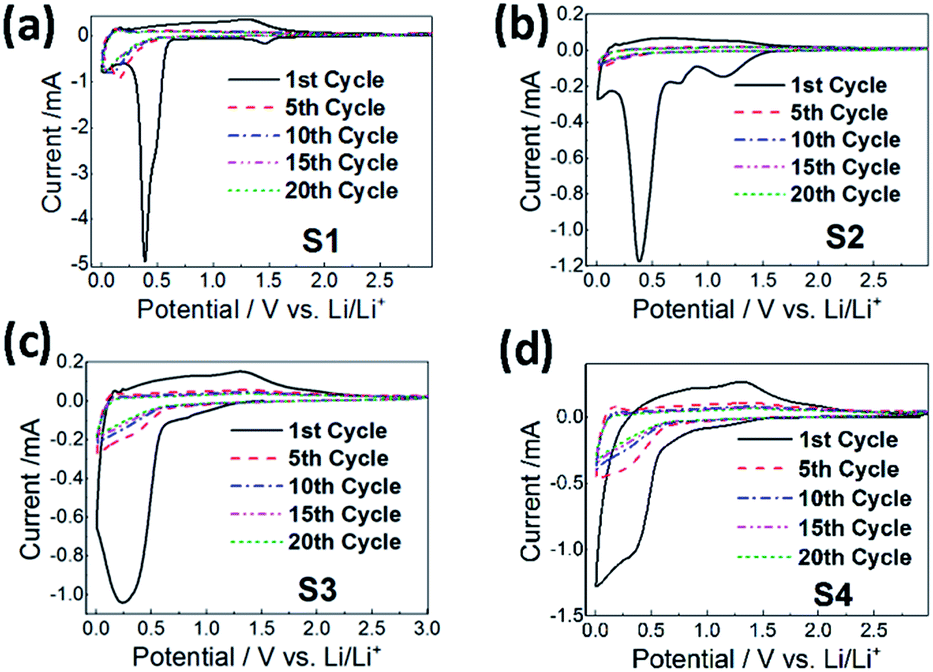 | ||
| Fig. 11 Cyclic voltammetry curves of the CoMn2O4 anodes for different annealing temperatures: (a) as-prepared (sample S1), (b) 300 °C (sample S2), (c) 400 °C (sample S3), and (d) 500 °C (sample S4) at the 1st, 5th, 10th, 15th and 20th cycles. | ||
Electrochemical impedance spectroscopy (EIS) measurements have been performed and the obtained results, together with the equivalent circuit used for fitting, are shown in the ESI (Fig. S6 and Table S3†). The semicircle in the medium frequency region is attributed to the charge transfer resistance Rct, while the high frequency semicircle is attributed to the solid electrolyte interface (SEI) film resistance and/or contact resistance.18,32,34 The diffusion of Li ions into the electrode is related to the inclined line (Warburg impedance).18,32,34 In all samples, Rs increases after the first cycle. An increase in Rs with the number of cycles may indicate the formation of a thicker SEI.32 In all samples, Rct decreases after the first cycle, and this decrease is particularly significant in samples with a large particle size (S3 and S4). A decrease in the cell impedance with cycling can be attributed to the reduction in the particle size, i.e. pulverization process,32 which is in agreement with the observed decrease in particle size after cycling.
Conclusions
CoMn2O4 can be prepared, without any impurity phases, via a fast and facile precipitation route by using the corresponding acetates and NaOH. Particle sizes can be tuned easily by changes in the annealing temperature. Besides microstructural changes, an increase in annealing temperature caused changes within the CoMn2O4 crystal lattice as well. Unit cell parameters increased as a consequence of changes in cation distribution between spinel A- and B sites as found by XRD analysis and confirmed by Raman spectroscopy. CoMn2O4 exhibited a strong dependence of the initial capacity as well as capacity retention on the particle size. The initial particle size and initial capacity increased with increasing annealing temperature. After 1000 cycles, the correlation between particle size and capacity persisted, although the final particle size was not necessarily proportional to the starting one. In the sample exhibiting the best performance and the largest starting and final particle sizes, exceptionally high capacity retention of 104% after 1000 cycles (compared to the 2nd cycle) was obtained.Acknowledgements
M.B. and X.L. have contributed equally to this work. Financial support from the Strategic Research Theme on Clean Energy and Seed Funding for Basic Research Grant (of the University of Hong Kong) is acknowledged. J.P. and A.M.C.N. acknowledge a financial support of a Croatian–Chinese bilateral project entitled: “Oxide materials for next generation Li ion batteries”. The authors would like to thank Prof. K. Y. Chan for the use of the electrochemical workstation.Notes and references
- A. R. Armstrong, C. Lyness, P. M. Panchmatia, M. S. Islam and P. G. Bruce, Nat. Mater., 2011, 10, 223 CrossRef CAS PubMed.
- Y. Yang, C. Xie, R. Ruffo, H. Peng, D. K. Kim and Y. Cui, Nano Lett., 2009, 9, 4109 CrossRef CAS PubMed.
- B. Liu, J. Zhang, X. Wang, G. Chen, D. Chen, C. Zhou and G. Shen, Nano Lett., 2012, 12, 3005 CrossRef CAS PubMed.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002 CrossRef CAS PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS PubMed.
- Y. Ren, A. R. Armstrong, F. Jiao and P. G. Bruce, J. Am. Chem. Soc., 2010, 132, 996 CrossRef CAS PubMed.
- M. Zhang, D. Lei, X. Yin, L. Chen, Q. Li, Y. Wang and T. Wang, J. Mater. Chem., 2010, 20, 5538 RSC.
- W.-M. Zhang, X.-L. Wu, J.-S. Hu, Y.-G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941 CrossRef CAS.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- J. Zhou, H. Song, X. Chen, L. Zhi, S. Yang, J. Huo and W. Yang, Chem. Mater., 2009, 21, 2935 CrossRef CAS.
- Y. Piao, H. S. Kim, Y.-E. Sung and T. Hyeon, Chem. Commun., 2010, 46, 118 RSC.
- J. Zhong, C. Cao, Y. Liu, Y. Li and W. S. Khan, Chem. Commun., 2010, 46, 3869 RSC.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22, E145 CrossRef CAS PubMed.
- Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265 CrossRef CAS PubMed.
- S. Xiong, J. S. Chen, X. W. Lou and H. C. Zeng, Adv. Funct. Mater., 2012, 22, 861 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258 CrossRef CAS.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425 CrossRef CAS PubMed.
- X. Liu, F. Liu, Q. Sun, A. M. C. Ng, A. B. Djurišić, M. Xie, C. Liao, K. Shih and Z. Deng, ACS Appl. Mater. Interfaces, 2014, 6, 13478 CAS.
- M.-S. Park, G.-X. Wang, Y.-M. Kang, D. Wexler, S.-X. Dou and H.-K. Liu, Angew. Chem., Int. Ed., 2007, 46, 750 CrossRef CAS PubMed.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868 CrossRef CAS.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Chem. Mater., 2008, 20, 6562 CrossRef CAS.
- P. Meduri, C. Pendyala, V. Kumar, G. U. Sumanasekera and M. K. Sunkara, Nano Lett., 2009, 9, 612 CrossRef CAS PubMed.
- S. Ding, J. S. Chen, G. Qi, X. Duan, Z. Wang, E. P. Giannelis, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 21 CrossRef CAS PubMed.
- Y.-L. Ding, X.-B. Zhao, J. Xie, G.-S. Cao, T.-J. Zhu, H.-M. Yu and C.-Y. Sun, J. Mater. Chem., 2011, 21, 9475 RSC.
- D. K. Kim, P. Muralidharan, H.-W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948 CrossRef CAS PubMed.
- X. Zhang, F. Cheng, K. Zhang, Y. Liang, S. Yang, J. Liang and J. Chen, RSC Adv., 2012, 2, 5669 RSC.
- L. Xiao, Y. Yang, J. Yin, Q. Li and L. Zhang, J. Power Sources, 2009, 194, 1089 CrossRef CAS.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609 CrossRef CAS PubMed.
- L. Zhou, H. B. Wu, T. Zhu and X. W. Lou, J. Mater. Chem., 2012, 22, 827 RSC.
- F. M. Courtel, H. Duncan, Y. Abu-Lebdeh and I. J. Davidson, J. Mater. Chem., 2011, 21, 10206 RSC.
- L. Zhou, D. Zhao and X. W. Lou, Adv. Mater., 2012, 24, 745 CrossRef CAS PubMed.
- L. Hu, H. Zhong, X. Zheng, Y. Huang, P. Zhang and Q. Chen, Sci. Rep., 2012, 2, 986 Search PubMed.
- L. Wang, B. Liu, S. Ran, L. Wang, L. Gao, F. Qu, D. Chen and G. Shen, J. Mater. Chem. A, 2013, 1, 2139 CAS.
- J. Li, S. Xiong, X. Li and Y. Qian, Nanoscale, 2013, 5, 2045 RSC.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, X. Xu, P. Sun and T. Chen, RSC Adv., 2014, 4, 4727 RSC.
- H. Toraya, J. Appl. Crystallogr., 1986, 19, 440 CrossRef CAS.
- C. Reitz, C. Suchomski, J. Haetge, T. Leichtweiss, Z. Jagličić, I. Djerdj and T. Brezesinski, Chem. Commun., 2012, 48, 4471 RSC.
- C. Reitz, C. Suchomski, V. S. K. Chakravadhanula, I. Djerdj, Z. Jagličić and T. Brezesinski, Inorg. Chem., 2013, 52, 3744 CrossRef CAS PubMed.
- W. G. Fateley, N. T. Mcdevitt and F. F. Bentley, Appl. Spectrosc., 1971, 25, 155 CrossRef.
- L. Malavasi, P. Galinetto, M. C. Mozzati, C. B. Azzoni and G. Flor, Phys. Chem. Chem. Phys., 2002, 4, 3876 RSC.
- M. D. P. Silva, F. C. Silva, F. S. M. Sinfrônio, A. R. Paschoal, E. N. Silva and C. W. A. Paschoal, J. Alloys Compd., 2014, 584, 573 CrossRef CAS.
- V. G. Hadjiev, M. N. Iliev and I. V. Vergilov, J. Phys. C: Solid State Phys., 1988, 21, L199 CrossRef.
- M. Kim, X. M. Chen, X. Wang, C. S. Nelson, R. Budakian, P. Abbamonte and S. L. Cooper, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174424 CrossRef.
- H. D. Lutz, B. Müller and H. J. Steiner, J. Solid State Chem., 1991, 90, 54 CrossRef CAS.
- D. H. Park, S. T. Lim, S.-J. Hwang, J.-H. Choy, J. H. Choi and J. Choo, J. Power Sources, 2006, 159, 1346 CrossRef CAS.
- C. V. Ramana, M. Massot and C. M. Julien, Surf. Interface Anal., 2005, 37, 412 CrossRef CAS.
- G. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 9041 CrossRef CAS PubMed.
- M. H. Kim, Y. J. Hong and Y. C. Kang, RSC Adv., 2013, 3, 13110 RSC.
- Y. Deng, L. Wan, Y. Xie, X. Qin and G. Chen, RSC Adv., 2014, 4, 23914 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- H. Song, L. Shen and C. Wang, J. Mater. Chem. A, 2014, 2, 20597 CAS.
- G. R. Yang, X. Xu, W. Yan, H. H. Yang and S. J. Ding, Electrochim. Acta, 2014, 137, 462 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld refinement results, parameter tables, particle size vs. capacity dependences, SEM and TEM images after 300 cycles, and EIS results. See DOI: 10.1039/c5ta03570h |
| This journal is © The Royal Society of Chemistry 2015 |