
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge storage mechanism of activated manganese oxide composites for pseudocapacitors†
Tzu-Ho
Wu
ac,
David
Hesp
b,
Vin
Dhanak
b,
Christopher
Collins
a,
Filipe
Braga
a,
Laurence J.
Hardwick
*a and
Chi-Chang
Hu
*c
aDepartment of Chemistry, The University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: laurence.hardwick@liv.ac.uk
bDepartment of Physics, The University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
cDepartment of Chemical Engineering, National Tsing Hua University, Hsin-Chu, 30013 Taiwan. E-mail: cchu@che.nthu.edu.tw
First published on 15th May 2015
Abstract
Manganese oxides can undergo an electrochemical activation step that leads to greater capacitances, of which the structural change and mechanism remains poorly understood. Herein we present a wide-ranging study on a manganese oxide synthesised by annealing manganese(II) acetate precursor to 300 °C, which includes in operando monitoring of the structural evolution during the activation process via in situ Raman microscopy. Based on powder X-ray diffraction, X-ray photoelectron spectroscopy, transmission electron and ex situ Raman microscopy, the as prepared manganese oxide was characterised as hausmannite-Mn3O4 with a minor portion of MnO2. The activation process of converting as-prepared hausmannite-Mn3O4 into amorphous MnO2 (with localised birnessite structure) by electrochemical cycling in 0.5 M Na2SO4 was examined. After activation, the activated MnOx exhibited capacitive performance of 174 F g−1 at a mass loading of 0.71 mg cm−2. The charge storage mechanism is proposed as the redox reaction between Mn(III) and Mn(IV) at outer surface active sites, since the disordered birnessite-MnO2 does not provide an ordered layer structure for cations and/or protons to intercalate.
Introduction
Electrochemical supercapacitors have been widely investigated due to their high charge/discharge efficiency, high-rate charge storage and delivery, as well as long cycle life.1 Recently, researchers have investigated approaches to increase the specific energy stored in supercapacitors in order to apply to numerous energy storage applications, such as hybrid electric vehicles, starting assistance to fuel cells, and power sources of electronics and cordless electric tools.1–3 According to the charge storage mechanism, the non-faradaic electrical double-layer capacitors (EDLCs) are based on charge separation (i.e., arrangement of ions/polar molecules) at the electrode/solution interface. The pseudocapacitance originates from the Faradaic redox reactions of electroactive species within electrode materials.The capacitive performance of MnO2 in neutral aqueous electrolytes has received much attention, due to improving cycle life (at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles4–6), acceptable specific capacitance at moderate scan rates for MnO2 thin films (100–400 F g−1),7–10 and low cost.10–13 MnO2 exhibits more than 0.9 V potential window in neutral media, such as alkali metal chloride,10,14 alkali metal sulphate,5,12 and specific lithium salts15 aqueous electrolytes, without considering the significant enlargement of the cell voltage via the asymmetric design.16–18 The charge storage mechanism of MnO2 has been discussed by various analyses, such as X-ray photoelectron spectroscopy (XPS),13 electrochemical quartz crystal microbalance (EQCM),19 and in situ powder X-ray diffraction (PXRD).14 Two mechanisms have been proposed to elucidate the MnO2 charge storage process. The first is based on the surface adsorption of cations on MnO2 active sites,10 while the second involves the intercalation and expulsion of alkali metal cations and/or protons into/from MnO2 structures.20 The synergistic effect of MnO2 and Mn3O4 has been proposed from our previous work,21 which demonstrated that the specific current responses for MnO2/Mn3O4 composites are superior than individual manganese oxides (MnOx), i.e., MnO2 or Mn3O4 measured on single-crystalline MnOOH nanowires. Alternatively, the activation from hausmannite-Mn3O4 to birnessite-MnO2 by electrochemical methods has been proposed by a series of PXRD, X-ray absorption spectroscopic (XAS), and ex situ Raman spectroscopic analyses.22–26 The activation process has been proposed as a complex and irreversible process, and the new formed birnessite-MnO2 is accepted as the material responsible for pseudocapacitive behaviour via the insertion of cations and/or protons.22,26 The structural disorder of electrochemically activated birnessite-MnO2 has been previously remarked upon.23 However, the charge storage mechanism of electrochemically activated birnessite-MnO2 from hausmannite-Mn3O4 in neutral aqueous electrolytes has not yet been verified.
000 cycles4–6), acceptable specific capacitance at moderate scan rates for MnO2 thin films (100–400 F g−1),7–10 and low cost.10–13 MnO2 exhibits more than 0.9 V potential window in neutral media, such as alkali metal chloride,10,14 alkali metal sulphate,5,12 and specific lithium salts15 aqueous electrolytes, without considering the significant enlargement of the cell voltage via the asymmetric design.16–18 The charge storage mechanism of MnO2 has been discussed by various analyses, such as X-ray photoelectron spectroscopy (XPS),13 electrochemical quartz crystal microbalance (EQCM),19 and in situ powder X-ray diffraction (PXRD).14 Two mechanisms have been proposed to elucidate the MnO2 charge storage process. The first is based on the surface adsorption of cations on MnO2 active sites,10 while the second involves the intercalation and expulsion of alkali metal cations and/or protons into/from MnO2 structures.20 The synergistic effect of MnO2 and Mn3O4 has been proposed from our previous work,21 which demonstrated that the specific current responses for MnO2/Mn3O4 composites are superior than individual manganese oxides (MnOx), i.e., MnO2 or Mn3O4 measured on single-crystalline MnOOH nanowires. Alternatively, the activation from hausmannite-Mn3O4 to birnessite-MnO2 by electrochemical methods has been proposed by a series of PXRD, X-ray absorption spectroscopic (XAS), and ex situ Raman spectroscopic analyses.22–26 The activation process has been proposed as a complex and irreversible process, and the new formed birnessite-MnO2 is accepted as the material responsible for pseudocapacitive behaviour via the insertion of cations and/or protons.22,26 The structural disorder of electrochemically activated birnessite-MnO2 has been previously remarked upon.23 However, the charge storage mechanism of electrochemically activated birnessite-MnO2 from hausmannite-Mn3O4 in neutral aqueous electrolytes has not yet been verified.
Raman spectroscopy is a powerful technique for the characterisation of short-range order of surface molecular species (especially for covalent character of the metal–oxygen bond).27,28 Therefore, Raman analyses have been widely used to characterise various manganese oxides, since they possess a variety of oxidation states and phases.28–33 The use of in situ measurement techniques are important in understanding the Mn3O4 activation process and charge storage mechanism in neutral aqueous electrolytes since the dried electrodes used in ex situ techniques may not represent the true state during electrochemical analyses.
A number of previous works have characterised the conversion of various manganese oxides into birnessite-MnO2,22–26,34 with applicable studies relating to electrochemical activation from Mn3O4 to MnO2 that are detailed in Table S1.† Among these reported works, the as-prepared and electrochemically activated MnOx were characterised as pure Mn3O4 and MnO2 according to various ex situ analyses methods, respectively.22–25 In the majority of studies, the MnOx were identified as crystallised hausmannite-Mn3O4 and birnessite-MnO2.22,24,25 Consequently, the crystallinity of activated MnOx is presumed to be dependent on the crystallinity of as-prepared Mn3O4. For example, the applied high temperature thermal reduction of electrolytic manganese dioxide at 1050 °C for 72 h is believed to be a well crystallised Mn3O4, which probably results in the highly crystallised birnessite structure after electrochemical activation.
In this study, the electrochemical activation of hausmannite-Mn3O4 in 0.5 M Na2SO4 was characterised by both in situ and ex situ techniques in order to understand both the activation process and the charge storage mechanism of this promising manganese oxide composite pseudo-capacitive material.
Experimental
The preparation of MnOx material followed our previous work.17 In short, the manganese(II) acetate tetrahydrate (99.99%, from Sigma-Aldrich) precursor solution annealed at a ramping rate of 1 °C min−1 to 300 °C and then left to cool down to ambient temperature. PXRD was measured using either a Cu Kα source (λ = 1.54 Å, Mac Science, MXP 18) for as-prepared MnOx or a Mo Kα source (λ = 0.709 Å, Bruker, D8 advance) for activated MnOx in Debye–Scherrer geometry with a capillary diameter of 0.5 mm. TEM analyses were conducted on a Philips Tecnai F20 G2 electron microscope at 200 kV, and samples for TEM analyses were prepared by drop casting on copper grids followed by solvent evaporation in air at 80 °C. The surface morphologies of MnOx were examined by SEM (JEOL, JSM-6610 at 20 kV). XPS measurements were performed in a standard ultrahigh vacuum surface science chamber (2 × 10−10 mbar) a PSP Vacuum Technology electron energy analyzer (angle integrating ± 10°) and a dual anode Mg Kα (1253.6 eV) X-ray source.For in situ Raman cell configuration, the working electrode was free-standing sample film composed of as-prepared MnOx (30%), ground carbon black (XC-72, Cabot Corporation, USA, 173 m2 g−1; 20%), and poly(vinylidenefluoride-hexafluoropropylene) co-polymeric binder (Kynar-flex, Arkema) powders (50%). The counter electrode was prepared by casting activated carbon (ACS-679 from China Steel Chemical Corporation, Taiwan, 1635 m2 g−1) onto an Al current collector, while activated carbon free-standing film composed of ACS-679 activated carbon (40%) and Kynar-flex powders (60%) was served as the quasi-reference electrode (QRE).35 The three-electrode compartments were sealed in the in situ Raman cell with adequate amount of electrolyte (4–5 drops).36 The mass based on Mn3O4 of working electrode and activated carbon of counter electrode were ca. 1 mg and 15 mg, respectively. Note that the potential of QRE were tested in a beaker cell, the cyclic voltammetric (CV) profile of MnO2 between −0.1 and 1.1 V was extremely similar to the CV measured against an Ag/AgCl (ALS co. Ltd, Japan, 3 M NaCl, 0.209 V versus SHE at 25 °C) in the range between 0 and 1.2 V, indicating that the potential difference of QRE and Ag/AgCl is ca. 0.1 V (Fig. S1†). The electrolyte was 0.5 M Na2SO4 degassed with purified argon gas flow before the measurements. In situ Raman spectra were recorded with Raman microscope (Renishaw inVia), using a He–Ne laser (632.8 nm) focussed through an inverted microscope (Leica), via a 50× objective lens (Leica). In situ Raman spectra were measured at different potentials with an interval of 200 mV, starting from 0.1 V vs. QRE to 1.1 V (positive sweep, PS) and then back from 0.9 V to −0.1 V (negative sweep, NS). Therefore, 12 spectra were obtained from each in situ Raman cycle. For each spectrum, the working electrode was pre-polarised for 3 min and held the potential during the spectral acquisition with the range of 250–750 cm−1. The formation of Mn3O4 triggered by an increase in lattice temperature due to local heating by laser has been widely reported.28,29,33 In order to avoid this factor, the laser exposed to sample surface was controlled via the use of an appropriate filter to keep below a maximum power of 0.37 mW.
For ex situ Raman experiments, the working electrodes were prepared by casting the slurry composed of as-prepared MnOx (70%), XC-72 (20%), and Kynar (10%) onto graphite electrodes. Total mass of each electrode is 0.71 mg, which is used for calculating specific capacitance. An Ag/AgCl and a Pt wire were used as reference and counter electrodes, respectively. The measurements under the three-electrode mode were performed in deaerated 0.5 M Na2SO4 in a sealed beaker cell at 25 °C. The cyclic voltammograms were measured at 25 mV s−1 between 0 and 1.2 V (vs. Ag/AgCl). The MnOx-coated working electrodes were potential-cycled for 5, 10, 50, 100, 150, 200, and 500 cycles to elucidate the activation process. After CV cycling, the MnOx-coated electrodes were gently washed by deionised-water and dried in 85 °C oven overnight.
Results and discussion
Chemical synthesised as-prepared MnOx
The diffraction pattern of as-prepared MnOx (Fig. 1) corresponds to the PXRD pattern of hausmannite-Mn3O4 from the database (JCPDS CARD 24-0734). Reflections at 18.0°, 28.9°, 32.4°, 36.1°, and 59.8° are recorded, which correspond to face (101), (112), (103), (211), and (224) of hausmannite-Mn3O4, respectively. The sub-particles of the MnOx can be observed by TEM images (Fig. 2b and c). From TEM images, the particle size of as-prepared MnOx is ca. 6–8 nm. The d-spacing of 0.499, 0.313, 0.281, 0.253, 0.208, 0.182, 0.157, and 0.146 nm can be found and calculated from the selected area electron diffraction (SAED) pattern of as-prepared MnOx (Fig. 2a), which are consistent with the d-spacing of hausmannite-Mn3O4. Moreover, the lattice spacing of 0.50, 0.31, 0.28, and 0.25 nm can be identified from the TEM image (Fig. 2c), which correspond to face (101), (112), (103), and (211) of hausmannite-Mn3O4, respectively. The diffraction peaks and d-spacing of as-prepared MnOx are compiled and compared to hausmannite-Mn3O4 (JCPDS CARD 24-0734) in Table 1, which verifies that the as-prepared MnOx can be considered as consisting of hausmannite-Mn3O4.13,37–39 The mean manganese oxidation state of MnOx can be determined by the energy separation between the two peaks of Mn 3s core level spectrum.13,37–39 It has been reported that more interaction can occur upon photoelectron ejection since a lower valence state implies more electrons in the 3d orbital. Consequently, the energy separation between the two components of the Mn 3s multiplet will increase.13,38 The energy separation between these two peaks of Mn 3s spectra is 5.79, 5.50, 5.41, and 4.78 eV for reference sample of MnO, Mn3O4, Mn2O3, and MnO2, respectively.13,38,39 The peak separation is 5.23 eV obtained from Mn 3s spectrum of as-prepared MnOx (Fig. 3a). Therefore, the mean manganese oxidation state of as-prepared MnOx is ca. 3.18 according to the relationship of energy separation and mean oxidation state,13,38,39 which implies that as-prepared MnOx possesses a minor portion of Mn(IV) since the mean oxidation state of as-prepared MnOx is slightly higher than Mn3O4 (mean oxidation state = 2.67). XPS is a surface-sensitive measurement, which indicates the mean manganese oxidation state obtained from XPS results is in the proximity to the sample surface. From Mn 2p spectrum of as-prepared MnOx (not shown here), the peaks of Mn 2p3/2 and Mn 2p1/2 are located at 641.91 and 653.31 eV, respectively. From the Lorentz fitting of O 1s spectrum of as-prepared MnOx (Fig. 3b), the peak position at 529.8, 531.3, and 532.5 eV are attributed to Mn–O–Mn (for anhydrous oxide), Mn–OH (hydroxide), and H–O–H bonds (crystalline water), respectively.13,37–39 The Mn–OH bonds contribute 15% (Table 2), which suggests the as-prepared MnOx exhibit better capacitive performance since the hydrous property facilitates ionic exchange.37,39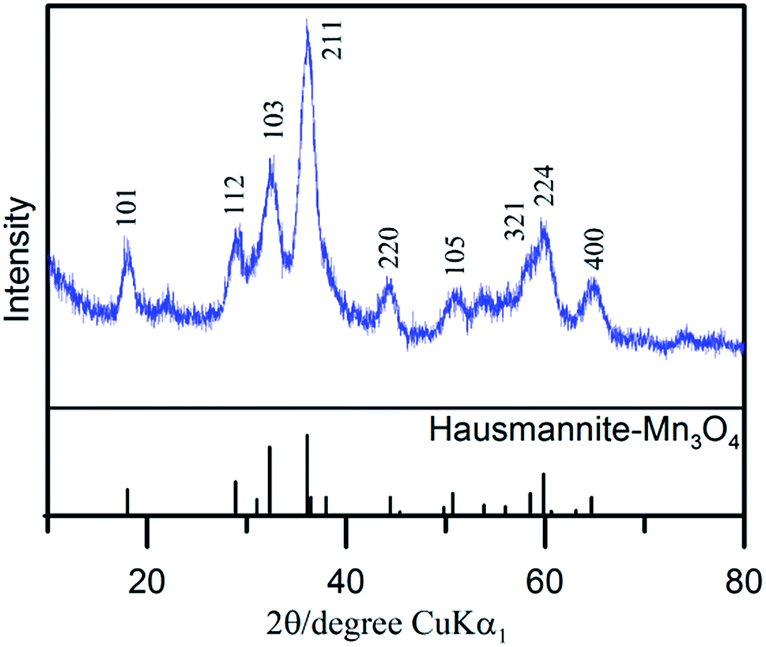 | ||
| Fig. 1 X-ray diffraction pattern of as-prepared MnOx. | ||
 | ||
| Fig. 2 (a and d) SAED pattern and (b, c, e and f) TEM images of (a–c) as-prepared and (d–f) activated MnOx. | ||
| 2θ from XRD pattern (degree) | 2θ from database (degree) | d-spacing calculated from SAED pattern (nm) | d-spacing from Mn3O4 database (nm) | d-spacing calculated from SAED pattern (nm) | d-spacing from MnO2 database (nm) | ||
|---|---|---|---|---|---|---|---|
| As-prepared MnOx | 18.0 | 18.0 | 0.499 | 0.492 | Activated MnOx | 0.497 | |
| 28.9 | 28.9 | 0.313 | 0.309 | 0.308 | |||
| 31.0 | 0.288 | ||||||
| 32.4 | 32.3 | 0.281 | 0.277 | 0.283 | |||
| 36.1 | 36.1 | 0.253 | 0.249 | 0.253 | |||
| 0.242 | 0.244 | ||||||
| 44.3 | 44.4 | 0.208 | 0.204 | ||||
| 0.210 | 0.212 | ||||||
| 49.8 | 0.182 | 0.183 | 0.182 | ||||
| 50.7 | 50.7 | 0.180 | |||||
| 58.4 | 58.5 | 0.157 | 0.158 | 0.158 | |||
| 59.8 | 59.8 | 0.154 | |||||
| 64.7 | 64.7 | 0.146 | 0.144 | 0.146 |
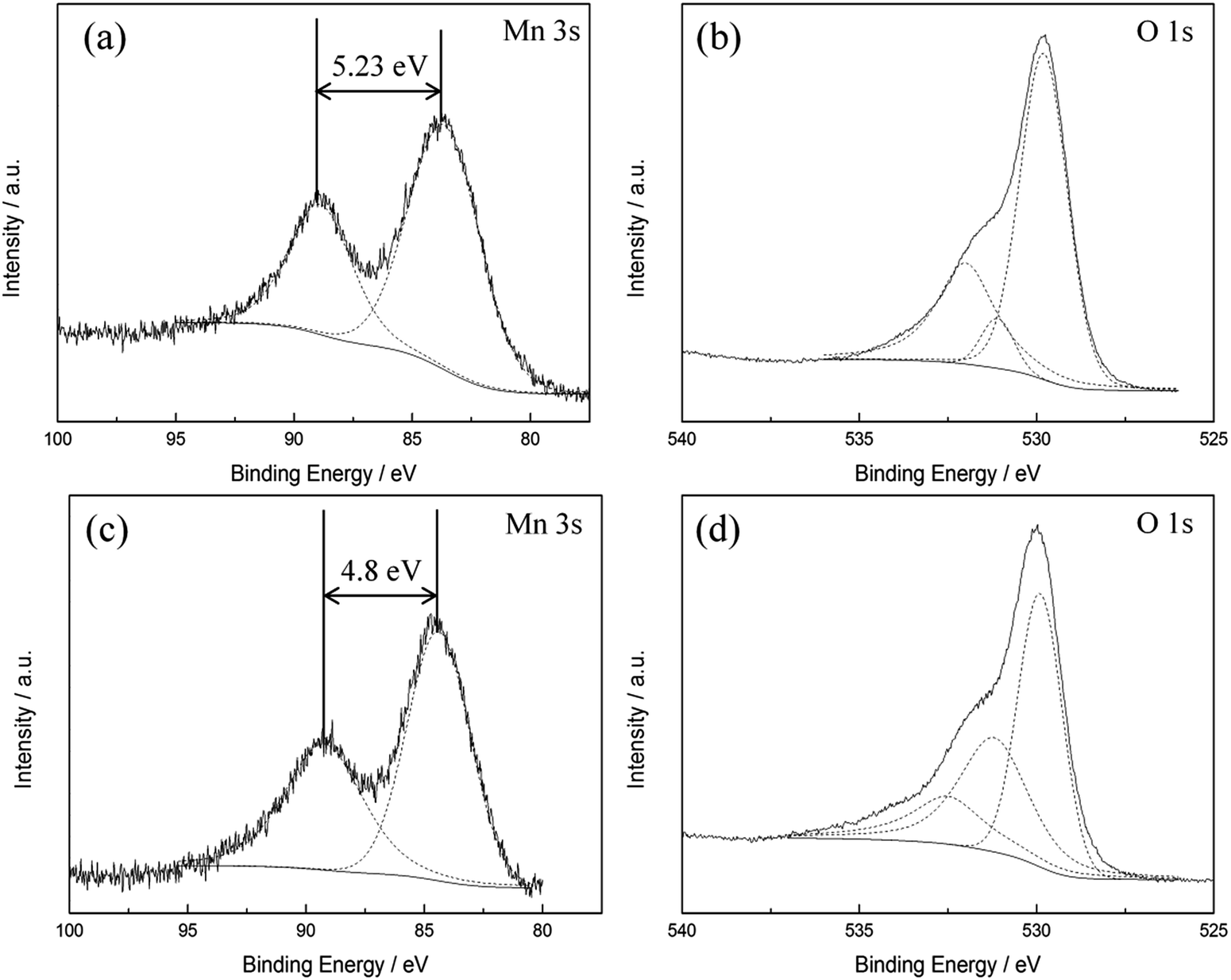 | ||
| Fig. 3 XPS (a and c) Mn 3s, (b and d) O 1s spectra of (a and b) as-prepared and (c and d) activated MnOx. | ||
| O 1 s | Position (eV) | Percentage (%) | Position (eV) | Percentage (%) | ||
|---|---|---|---|---|---|---|
| Mn–O–Mn | As-prepared MnOx | 529.8 | 67 | Activated MnOx | 529.9 | 44 |
| Mn–OH | 531.3 | 15 | 531.2 | 37 | ||
| H–O–H | 532.5 | 18 | 532.5 | 19 |
According to literature,40 the Raman active bands of hausmannite-Mn3O4 are observed at 289, 315, 370, 477, and 660 cm−1. Bands at 310–320 and 360–370 cm−1 are the bending mode of Mn3O4, while 650–660 cm−1 is the Mn–O breathing vibration of Mn2+ in tetrahedral coordination.23,31 In general, the Raman active bands for MnO2 are at 500–510, 575–585, and 625–650 cm−1, which represent Mn–O stretching vibration of MnO6 octahedra, Mn–O stretching vibration of the basal plane of MnO6 sheets, and the symmetric stretching vibration of Mn–O of the MnO6 group, respectively.23,32 The Raman spectrum of as-prepared MnOx is shown in Fig. 4. The intense peak at ca. 658 cm−1 can be observed, which is the most notable band of Mn3O4.23,31 However, a shoulder around 575 cm−1 can also be found, which provides further evidence that as-prepared MnOx might not be pure hausmannite-Mn3O4, since a minor portion of MnO2 can be found in the spectrum. Moreover, the two peaks at 310–320 and 360–370 cm−1 are not able to be resolved from the background noise. The presence of (a minor portion) MnO2 in the as-prepared MnOx might reduce crystallinity of hausmannite-Mn3O4, which might result in the absence of the bending mode of Mn3O4. The results from both Raman and XPS analyses of as-prepared MnOx strongly indicate a minor portion of Mn(IV) is present. According to the above analyses, the as-prepared MnOx can be considered to be hausmannite-Mn3O4 with a minor portion of Mn(IV).
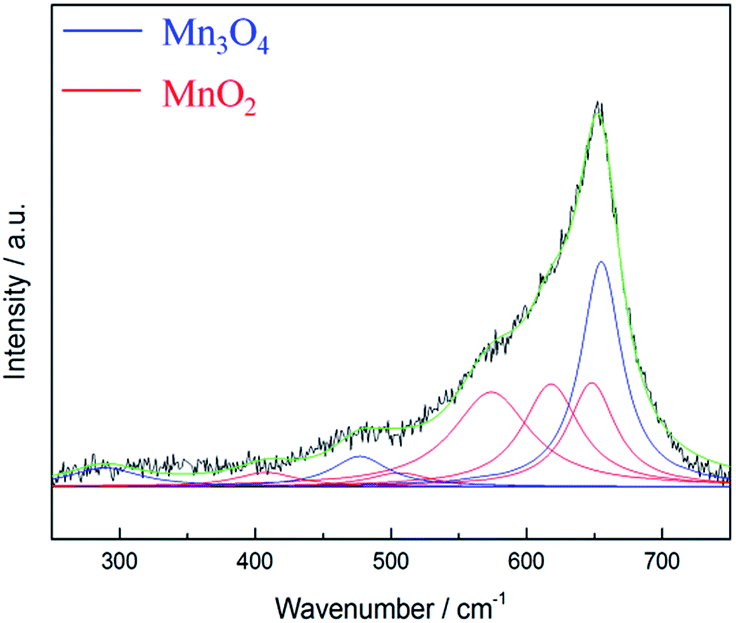 | ||
| Fig. 4 Lorentz fitting of ex situ Raman spectrum of as-prepared MnOx. Green line = envelope of total of the fitted peaks. | ||
Electrochemical activation process of MnOx
CVs measured at a sweep rate of 25 mV s−1 between 0 and 1.2 V (vs. Ag/AgCl) in 0.5 M Na2SO4 for controlled cycles are shown in Fig. 5. For the first few cycles, the anodic peak around 1.05 V on the positive sweep can be considered to be the oxidation of Mn3O4 to higher oxidation state. This anodic peak gradually decreased during cycling, and the cathodic peak around 0.9 V on the negative sweep gradually decreased at the same time. On the other hand, the current responses of the redox peak at ca. 0.4–0.5 V gradually increased during cycling. This peak is related to partial oxidation/reduction between Mn(III) and Mn(IV) according to the Pourbaix diagram.41 The rectangular CV profile at 500th cycle indicates that ideal capacitive performances of MnOx can be obtained by potential cycling which has been proposed as an electrochemical activation method.21 Note that the poor capacitive performance at first few cycles for pure Mn3O4 (i.e., less than 20 F g−1) has been reported.22,24 However, the specific capacitance at 5th cycle reaches ca. 130 F g−1 in this work, which confirms that the as-prepared MnOx is composed of a minor portion of MnO2 and is not pure Mn3O4.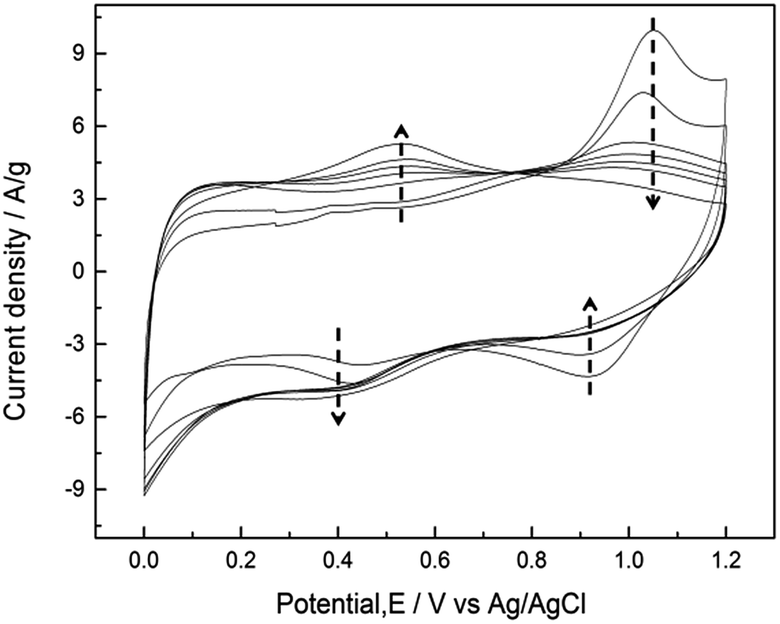 | ||
| Fig. 5 CVs measured at a sweep rate of 25 mV s−1 between 0 and 1.2 V (vs. Ag/AgCl) in 0.5 M Na2SO4 for 5, 10, 50, 100, 150, 200, and 500 cycles. | ||
For the 1st in situ Raman cycle (Fig. 6a), a strong peak located at 658 cm−1 is visible from PS 0.1 V, PS 0.3 V, and PS 0.5 V. This peak is the characteristics of Mn3O4 as described above. From PS 0.9 V to NS −0.1 V, this peak slightly shifts to a lower wavenumber (from 658 to 650 cm−1) with further applying potential. Besides, the intensity of this peak for the 12 spectra remains in the same level. On the other hand, a descending shoulder from ca. 650 to 550 cm−1 is found from PS 0.1 V. The peak at 575 cm−1 was gradually formed during the polarisations, and this peak became significant from PS 0.7 V to NS −0.1 V. Since the peak intensity of 575 cm−1 reached the same level of the peak at ca. 658 cm−1, a new MnOx species (i.e., MnO2) is believed to be formed. The spectra recorded at the 2nd cycle (Fig. 6b) all remain the similar pattern, which indicate that the newly formed MnO2 remains stable in the cycled potential window. Therefore the mean Mn oxidation state during further cycling after activation can only be accessed via other techniques such as X-ray photoelectron spectroscopy or X-ray absorption microscopy.
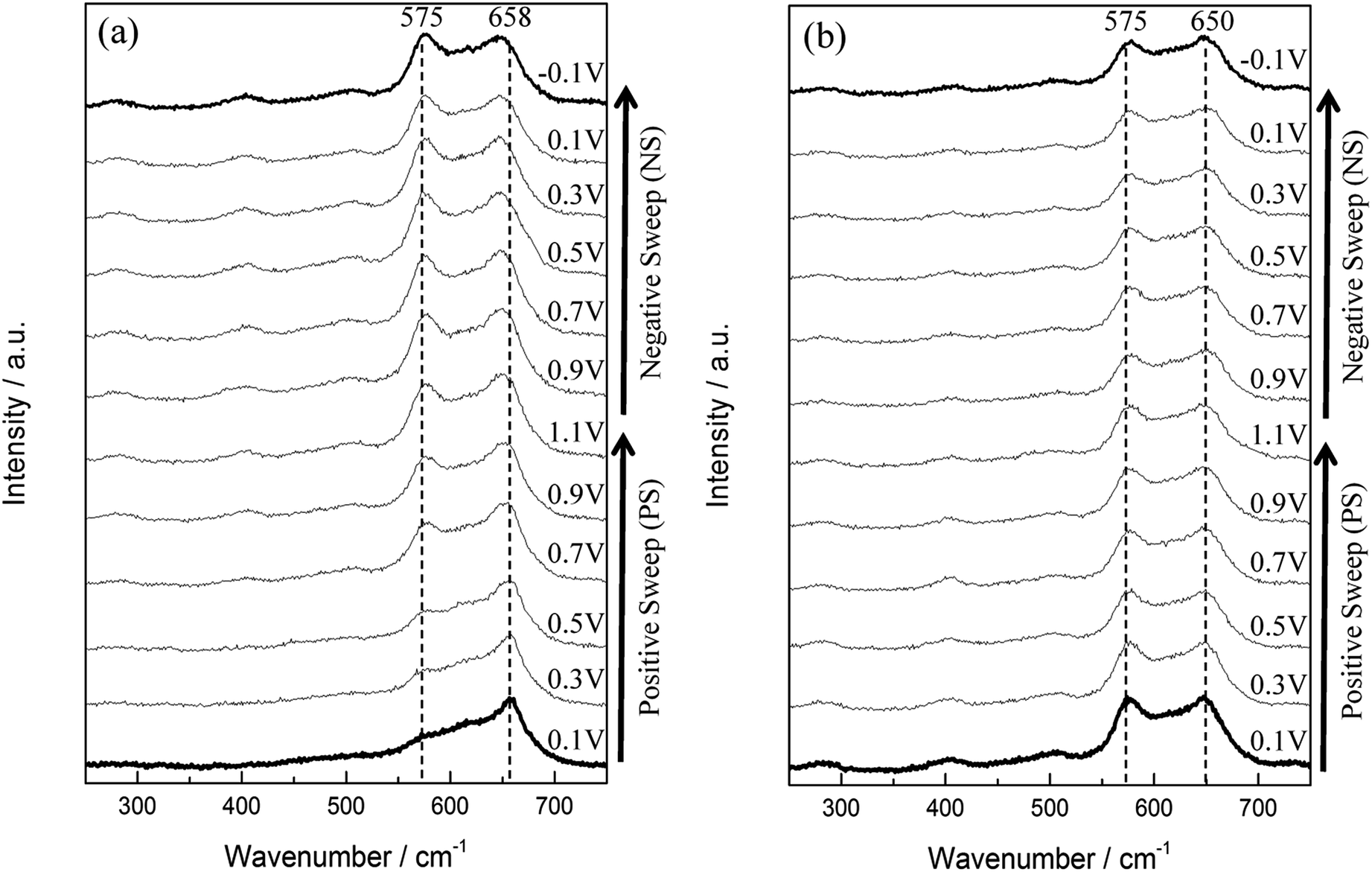 | ||
| Fig. 6 In situ Raman spectra series of (a) the 1st and (b) the 2nd cycle of MnOx. Each cycle started from 0.1 V (PS 0.1 V) and ended at −0.1 V (NS −0.1 V). | ||
Lorentz fitting of Raman spectra is used in order to gain a further understanding of this phenomenon. The peaks at 658, 477, and 289 cm−1 are related to hausmannite-Mn3O4,40 while the peaks at 648, 618, 575, 506, and 408 cm−1 are attributed to birnessite-MnO2.33 Note that all the peak positions are fixed except for the peak at ca. 289 cm−1 (allows from 283 to 289 cm−1). The Mn3O4 peak intensity at 658 cm−1 gradually decreased, while peaks related to birnessite-MnO2 (marked as red line in Fig. 7) gradually enhanced during polarisations. The Mn3O4 peak at 658 cm−1 dominates for the 1st cycle PS 0.1 V and the 1st cycle PS 0.5 V. However, the peak at 648 cm−1 related to birnessite-MnO2 became stronger than the Mn3O4 peak at 658 cm−1 with the positive and negative shifts in electrode potentials. As a result, this peak shifted from 658 (1st cycle PS 0.9 V) to 650 cm−1 (1st cycle NS −0.1 V) and further remained the same position. The peak intensity ratio of MnO2 and Mn3O4 (IMnO2/IMn3O4) is proposed to be an indicator to quantify the degree of activation from Mn3O4 to MnO2 although the different MnOx species show different Raman sensitivities. Here, IMnO2/IMn3O4 is calculated on the basis of the peak intensity of 575 and 658 cm−1 from the fitting results since these two peaks are the best representatives (the most distinguished peaks) for MnO2 and Mn3O4, respectively. IMnO2/IMn3O4 for as-prepared MnOx is 0.42 (Table S2†), and the value increases gradually during the electrochemical polarisations. IMnO2/IMn3O4 is 3.94 for the 2nd cycle PS 0.1 V, which is ca. 8 times higher than as-prepared MnOx (Table 3). The mean manganese oxidation state of activated MnOx is ca. 3.94 based on the energy separation (4.85 eV) of Mn 3s core level spectrum (Fig. 3c). Comparing with the mean manganese oxidation state of as-prepared MnOx (3.18), the results support the activation from Mn3O4 (i.e., Mn(II) and Mn(III)) to MnO2 (Mn(IV)) during the electrochemical activation.
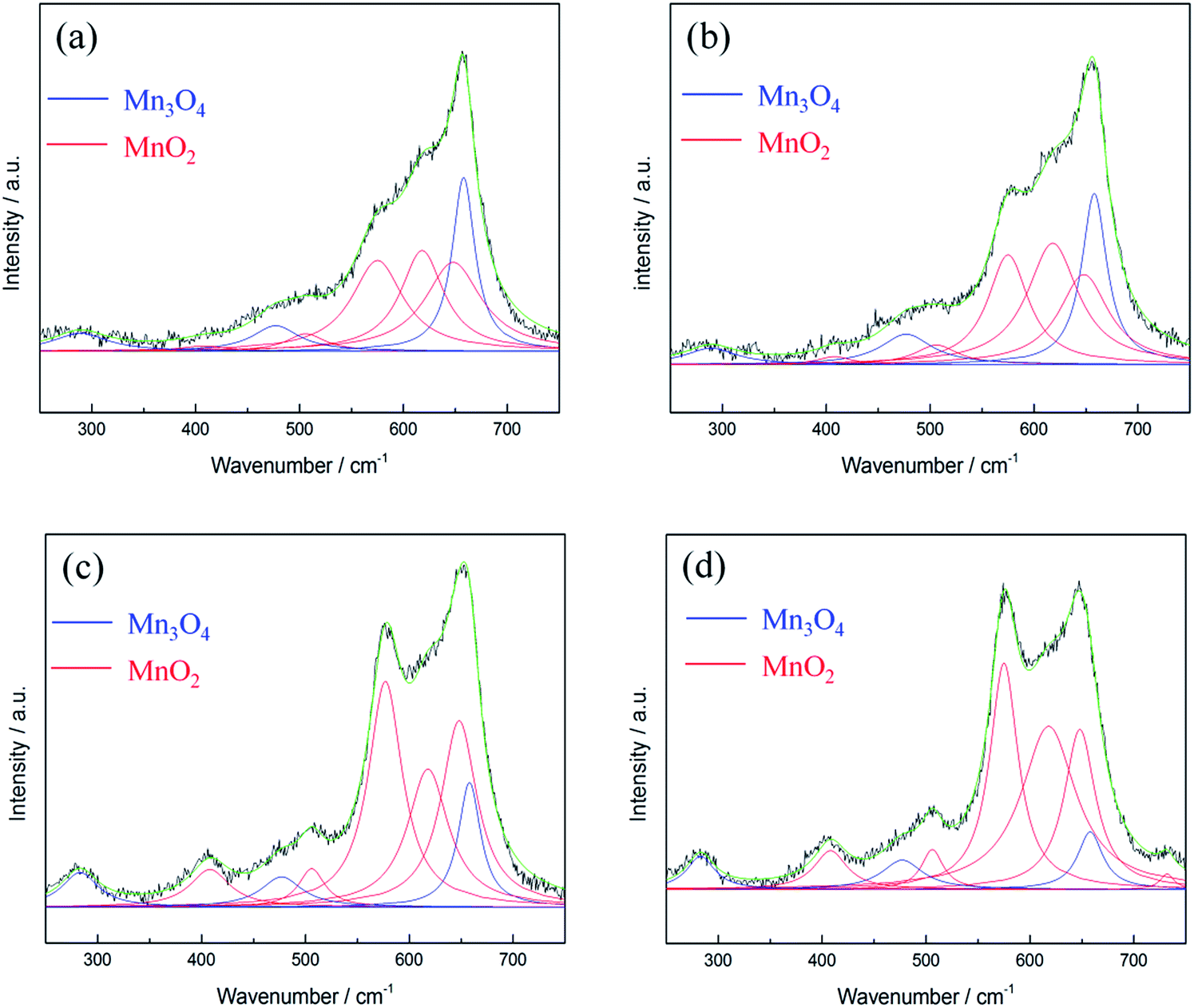 | ||
| Fig. 7 Lorentz fitting of in situ Raman spectra of MnOx (a) 1st cycle PS 0.1 V, (b) 1st cycle PS 0.5 V, (c) 1st cycle PS 0.9 V, and (d) 2nd cycle PS 0.1 V. Green line = envelope of total of the fitted peaks. | ||
| Mn3O4 | MnO2 | MnO2 | MnO2 | MnO2 | Mn3O4 | MnO2 | Mn3O4 | I MnO2/IMn3O4 | |
|---|---|---|---|---|---|---|---|---|---|
| 1st cycle PS 0.1 V | 658 (27) | 648 (66) | 618 (51) | 575 (61) | 506 (45) | 477 (55) | 408 (55) | 289 (65) | 0.52 |
| 1st cycle PS 0.5 V | 658 (29) | 648 (58) | 618 (60) | 575 (47) | 506 (50) | 477 (60) | 408 (40) | 284 (61) | 0.65 |
| 1st cycle PS 0.9 V | 658 (26) | 648 (42) | 618 (48) | 575 (40) | 506 (31) | 477 (48) | 408 (46) | 283 (43) | 1.81 |
| 2nd cycle PS 0.1 V | 658 (29) | 648 (37) | 618 (64) | 575 (33) | 506 (26) | 477 (48) | 408 (38) | 283 (33) | 3.94 |
According to the fitting results, residual Mn3O4 can be still observed even at the 2nd in situ Raman cycle (Fig. 7d). Note that the formed MnO2 is stable in the cycled potential window for the 2nd in situ Raman cycle (Fig. 6b). It therefore be concluded that not all the Mn3O4 is converted to MnO2 in the above electrochemical treatment. Due to the residual Mn3O4, the mean manganese oxidation state for the activated MnOx is not exactly equal to 4.0. Hence, it can be concluded that the major portion of hausmannite-Mn3O4 can be activated to birnessite-MnO2 by the electrochemical treatment. Besides, the residual Mn3O4 in electrochemically activated birnessite-MnO2 results in structure disorder of layered birnessite-MnO2, which reasonably explains why the oxide derived from Mn3O4 showed higher cycling stability because of the coexistence of MnO2 and Mn3O4.21Ex situ Raman spectra were recorded for all CV-cycled MnOx. From Lorentz fitting results, the correlation between specific capacitance of MnOx and CV cycle number is shown in Fig. 8. Note that the current responses related to the activation from Mn3O4 to MnO2 (the anodic peak around 1.05 V on the positive sweep) are evident in the first few cycles, which decreases gradually during the activation process. The ideal pseudocapacitance of MnO2 mainly contributes from the potential window between 0.2 and 0.8 V.19 The current responses related to the oxidation of Mn3O4 cannot be considered pseudocapacitance. Therefore, the calculation in Fig. 8 is based on the baseline of 500th cycle (stable current responses) between 0.8 and 1.2 V. In general, MnO2 is electrochemically active and reversible for supercapacitor applications10 comparing with Mn3O4, which is known as a relatively electrochemical irreversible material among MnOx species. The specific capacitance of MnOx rapidly increased within the initial 50 cycles, which suggests that the activation from Mn3O4 to MnO2 is relatively fast. With further cycling, the specific capacitance of MnOx gradually increased and reached the stable level (ca. 170 F g−1) from the 150th to the 500th cycle. Based on the Lorentz fitting of ex situ Raman spectra (Fig. S2†), the rapid increase of MnO2 content (IMnO2/IMn3O4 = 0.63 and 2.53 for 5th and 100th cycle, respectively) shows the same trend as capacitance increase. With further cycling to 500th cycle, IMnO2/IMn3O4 remained the same level (i.e., 2.45). It should be noted that this ratio is lower than that from in situ measurements (3.94). Accordingly, the activation from hausmannite-Mn3O4 to birnessite-MnO2 can be verified by the in situ and ex situ Raman experiments. The stable electrochemically activated birnessite-MnO2 can be formed and displays promising pseudocapacitance in 0.5 M Na2SO4.
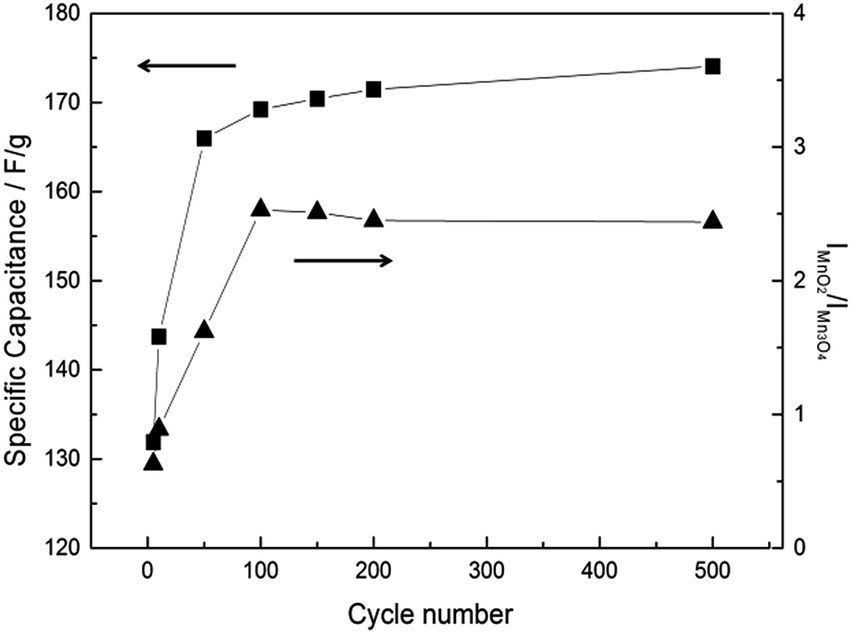 | ||
| Fig. 8 Cycle number dependence of specific capacitance calculated by CV at 25 mV s−1 and intensity ratio (IMnO2 of 575 cm−1/IMn3O4 of 658 cm−1) of MnOx. | ||
Fig. 9 shows the surface morphology change MnOx during electrochemical activation. A compact surface morphology of as-prepared MnOx can be observed in Fig. 9a, while it gradually changes into a porous surface after electrochemical activation (Fig. 9b–d). It has been proposed that the porous surface layer is the re-deposition of dissolved Mn(II) (from hausmannite-Mn3O4 crystalline structure) onto the electrode surface as MnO2 during the activation process via CV cycling.25 The proposed mechanism is reasonable since MnO2 can be successfully formed from Mn(II) precursor by potentiodynamic method.12 Besides, the insignificant electrochemical activation of Mn2O3 comparing with Mn3O4 has been found.25 Therefore, dissolution of Mn(II) from MnOx crystalline is believed to be the key for the electrochemical activation process. In fact, from the investigation of amorphous MnO2 by electrochemical quartz crystal microbalance (EQCM), the porous surface of MnOx (predominate MnO2) is not only from the re-deposition of dissolved Mn(II) during electrochemical activation process but also from the re-deposition (at ca. 0.8–0.2 V) of dissolved Mn(IV) (at ca. 0.8–1.0 V) during CV cycling.19 The normalised area of C 1s and Mn 3s XPS spectra of as-prepared and activated MnOx are listed in Table S3.† In comparison with as-prepared MnOx, the normalised area ratio (C 1s/Mn 3s) of activated MnOx reduced from ca. 2.59 to 1.04, indicating the re-deposition of MnO2 onto the electrode surface suppressing the intensity of carbon black and polymeric binder.25 Note that the intensity (counts/second) of XPS spectra strongly depends on the distance between samples and X-ray beam source. Therefore, normalised area (by atomic sensitivity factor) instead of absolute intensity is used in this work. Moreover, from SEM images (Fig. 9d), the porous nanostructure surface forms at the outer surface, while this structure cannot be found in some deeper regions. This suggests the electrochemical activation starts from outermost layer of Mn3O4 surface and then gradually into the bulk. This finding is consistent with literature, which reported the amorphous and hydrous MnO2 is formed in the outer electrode layers and proceeds to grow at the expense of the remaining bulk crystalline Mn3O4.42
 | ||
| Fig. 9 SEM images of (a) as-prepared MnOx and after (b) 100 (c and d) after 500 CV cycles in 0.5 M Na2SO4. | ||
Electrochemically activated MnOx and its charge–discharge mechanism
The activated MnOx was characterised by PXRD as amorphous MnO2 (Fig. S3†). The SAED pattern of activated MnOx (Fig. 2d) suggests that the electrochemically activated MnOx within the electrode composite possess poor crystallised structure since the diffraction patterns do not have well defined rings.Interestingly, with further analysis, both birnessite-MnO2 and hausmannite-Mn3O4 crystallites can be found in the activated MnOx (Table 1) although no reflections can be observed from diffraction pattern of activated MnOx. The d-spacing of 0.242 and 0.210 nm are corresponding to birnessite-MnO2. Apart from these two d-spacing found in SAED pattern, others are all related to Mn3O4 residual in the activated MnOx. From the TEM images of activated MnOx (Fig. 2e), the particle-like MnOx structure can be still observed. Compare to as-prepared MnOx, the particles and lattices of activated MnOx are not well defined. As a result, the activated MnOx is believed to have disordered layered birnessite structure; however, there is insufficient long-range order to allow for a PXRD pattern to be observed. Therefore, the structure of the electrochemically activated MnOx is determined to be composite material consisting of amorphous MnO2 (with localised birnessite structure) and a minor portion of residual of hausmannite-Mn3O4. Amorphous manganese oxide has been considered to be beneficial for supercapacitors since it provides large interlayer distance and surface area-to-volume ratio. The increase contact between the electrolyte and the electrode material improves the utilisation ratio of the material.43 Besides, the significant increase of Mn–OH (hydroxide) bond in activated MnOx (Fig. 3d and Table 2) indicates that the MnOx became more hydrous after electrochemical activation. As discussed above, hydrous property of MnOx facilitates ionic exchange and exhibits better capacitive performance.37,39
The structure transformation from hexagonal to monoclinic due to cation (Na+) intercalation into birnessite-MnO2 has been reported,44 as well as the subtle change of Raman spectra of birnessite-MnO2 with and without cation (Li+ and Na+) intercalated in MnO2 bilayers.32 The intercalation of alkali-ion into birnessite-MnO2 bilayers causes local lattice distortion, which results in shorter Mn–O chemical bonds (decrease of the interlayer spacing) and partial reduction of Mn4+ ions. As a result, the Mn–O symmetric stretching mode moves toward low wavenumber side upon alkali-ion intercalation.32 However, this peak shift phenomenon due to cation intercalation into layers of birnessite-MnO2 was not observed by in situ Raman during activation process in this work. Moreover, the peak positions of MnO2 correspond to birnessite-MnO2 rather than Na–birnessite-MnO2 from literature.32,33 Moreover, the peak around 732 cm−1, identified as a vibrational mode for non-cation intercalated birnessite-MnO2, can be observed by the Raman spectrum of activated MnOx (Fig. 7d). This indicates that electrochemically activated birnessite structure might not possess a well-defined layer structure (i.e., structure disordering) to allow cation (Na+) intercalation into bilayers.
The XPS Na 1s spectrum of as-prepared MnOx, activated MnOx and a sodium intercalated MnO2 sample (Na0.33MnO2) are shown in Fig. S4.† The Na 1s peak was present at 1072.2 eV (FWHM = 1.85) for activated MnOx and shows ca. 1 eV shift in comparison to Na0.33MnO2 at 1071.2 eV (FWHM = 1.77). The binding energy at 1072−1073 eV is related to the Na–F bonding, which is attributed to the Na+ (from Na2SO4 electrolyte) bonding with co-polymeric binder at F sites.45 Although the Na/Mn ratio of activated MnOx (1.03) is higher than Na0.33MnO2 (Na/Mn = 0.52), the Na element contributes from the Na–F bonding rather than incorporation into the MnOx structure after electrochemical measurements.
Based upon the in situ Raman and Na 1s XPS analyses, cations are believed to reach the outer surface active sites of electrochemically activated birnessite-MnO2 and the capacitive behaviour is attributable to the redox reaction between Mn(III) and Mn(IV). The relatively high specific surface area of as-prepared MnOx (241 m2 g−1) can provide abundant outer surface active sites. As a result, high specific capacitance of birnessite-MnO2 can be obtained (i.e., the specific capacitance measured at 500th is 174 F g−1) by potential cycling, which can be compared with values of similar materials in the literature (Table S4†).22–25 The capacitance normalised to specific surface area (CSA) is ca. 72.3 μF cm−2, which is higher than the double layer capacitance (ca. 10–20 μF cm−2) for high surface area (over 1000 m2 g−1) carbon material.1 The real CSA of electrochemically activated birnessite-MnO2 should be even higher since the calculation is based on the specific surface area of as-prepared MnOx and the material might lose some part of active surface area during electrode preparation. Based on the calculated high CSA, the charge storage of electrochemically activated MnOx is attributed to the redox reaction between Mn(III) and Mn(IV) (pseudocapacitance).
The one-pot synthesised as-prepared MnOx is characterised as hausmannite-Mn3O4 with a minor portion of MnO2. The electrochemically activated MnOx is characterised as a composite material consisting of localised birnessite structure MnO2 and a minor portion of hausmannite-Mn3O4. The improved capacitive performance of activated MnOx is attributed to the synergistic effect of poorly crystallised MnO2 and Mn3O4 in the composite material, and this work describes routes of generating composite materials that possess greater electrochemical performance.
Conclusions
Electrochemically activated MnOx, from one-pot synthesised hausmannite-Mn3O4, exhibits a capacitive performance of 174 F g−1 (at 25 mV s−1) and 1.2 V potential window (0–1.2 V vs. Ag/AgCl), which is superior to reported values of comparable materials. Activation from primarily hausmannite-Mn3O4 to predominantly localised birnessite-MnO2 by potential cycling is verified by both in situ and ex situ Raman microscopy, XPS and TEM analysis. Due to the structural disordering of electrochemically activated birnessite-MnO2 and residual Mn3O4, the charge storage is attributable to the redox reaction between Mn(III) and Mn(IV) at outer surface active sites, rather than cations and/or protons intercalation into layer structures. The improved capacitive performance relates to the synergistic effect of well-dispersed MnO2 and Mn3O4 nanoscale domains present within the activated material. Overall we show that the integrated nanoscale structuring of different phases of manganese oxides offers superior properties compared to bulk parent materials, and as such offers an avenue of future supercapacitor materials development.Acknowledgements
This research is funded by the National Science Council and Ministry of Science Technology of Taiwan under contract no. 101-2221-E-007-112-MY3, 102-2221-E-007-120-MY3, 103-2911-I-007-515, 103-3113-E-006-009, and also supported by the boost program of the Lower Carbon Energy Research centre in NTHU. The financial support of this work, by the Ministry of Science and Technology, Taiwan and The Royal Society, UK, is gratefully acknowledged, as well as the Science without Borders Program, Ministério da Educação, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Brazil; and the Engineering and Physical Sciences Research Council (EPRSC) under grant EP/H000925. We would like to thank Ming-Guan Chen (Department of Chemical Engineering, National Tsing Hua University, Taiwan). From the University of Liverpool UK, we acknowledge T. Whittles & M. Althobaiti (Stephenson Institute for Renewable Energy) for XPS measurements and Ming Liu (Department of Chemistry) for BET and Tobias Heil (Nano Investigation Centre at Liverpool) for TEM analyses. Part of the work was carried out within the cooperative framework set-up between National Tsing Hua University, Taiwan and University of Liverpool, UK.Notes and references
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- P. Simon and Y. Gogotsi, Nature Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. M. Miller and A. F. Burke, Electrochem. Soc. Interface, 2008, 17, 53–57 CAS.
- L. Athouël, F. Moser, R. Dugas, O. Crosnier, D. Bélanger and T. Brousse, J. Phys. Chem. C, 2008, 112, 7270–7277 Search PubMed.
- T. Brousse, P. L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Bélanger and P. Simon, J. Power Sources, 2007, 173, 633–641 CrossRef CAS PubMed.
- T. Brousse, M. Toupin and D. Bélanger, J. Electrochem. Soc., 2004, 151, A614 CrossRef CAS PubMed.
- C. C. Hu and T. W. Tsou, Electrochem. Commun., 2002, 4, 105–109 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef CAS.
- H. Xia, W. Xiao, M. O. Lai and L. Lu, Nanoscale Res. Lett., 2009, 4, 1035–1040 CrossRef CAS PubMed.
- H. Y. Lee and J. B. Goodenough, J. Solid State Chem., 1999, 144, 220–223 CrossRef CAS.
- S. W. Zhang and G. Z. Chen, Energy Mater., 2008, 3, 186–200 CrossRef CAS PubMed.
- C. C. Hu and C. C. Wang, J. Electrochem. Soc., 2003, 150, A1079 CrossRef CAS PubMed.
- M. Toupin, T. Brousse and D. Belanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- N. L. Wu and S. L. Kuo, J. Electrochem. Soc., 2006, 153, A1317–A1324 CrossRef PubMed.
- A. Boisset, L. Athouël, J. Jacquemin, P. Porion, T. Brousse and M. Anouti, J. Phys. Chem. C, 2013, 117, 7408–7422 CAS.
- C. L. Liu, K. H. Chang, C. C. Hu and W. C. Wen, J. Power Sources, 2012, 217, 184–192 CrossRef CAS PubMed.
- T. H. Wu, Y. H. Chu, C. C. Hu and L. J. Hardwick, Electrochem. Commun., 2013, 27, 81–84 CrossRef CAS PubMed.
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2006, 153, 183–190 CrossRef CAS PubMed.
- Y. H. Chu, C. C. Hu and K. H. Chang, Electrochim. Acta, 2012, 61, 124–131 CrossRef CAS PubMed.
- S. C. Pang, M. A. Anderson and T. W. Chapman, J. Electrochem. Soc., 2000, 147, 444 CrossRef CAS PubMed.
- C. C. Hu, C. Y. Hung, K. H. Chang and Y. L. Yang, J. Power Sources, 2011, 196, 847–850 CrossRef CAS PubMed.
- Y. Dai, K. Wang and J. Xie, Appl. Phys. Lett., 2007, 90, 104102 CrossRef PubMed.
- K. W. Nam and K. B. Kim, J. Electrochem. Soc., 2006, 153, A81 CrossRef CAS PubMed.
- D. P. Dubal, D. S. Dhawale, R. R. Salunkhe and C. D. Lokhande, J. Alloys Compd., 2010, 496, 370–375 CrossRef CAS PubMed.
- S. Komaba, T. Tsuchikawa, A. Ogata, N. Yabuuchi, D. Nakagawa and M. Tomita, Electrochim. Acta, 2012, 59, 455–463 CrossRef CAS PubMed.
- C. C. Hu, Y. T. Wu and K. H. Chang, Chem. Mater., 2008, 20, 2890–2894 CrossRef CAS.
- M. Frumar, Z. Polak and Z. Cernosek, J. Non-Cryst. Solids, 1999, 256 & 257, 105–110 Search PubMed.
- F. Buciuman, F. Patcas, R. Craciun and D. R. T. Zahn, Phys. Chem. Chem. Phys., 1999, 1, 185–190 RSC.
- M. C. Bernard, A. H. L. Goff and B. V. Thi, J. Electrochem. Soc., 1993, 140, 3065–3070 CrossRef CAS PubMed.
- M. Sun, B. Lan, T. Lin, G. Cheng, F. Ye, L. Yu, X. Cheng and X. Zheng, CrystEngComm, 2013, 15, 7010 RSC.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- C. Julien, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- C. Julien and M. Massot, Phys. Chem. Chem. Phys., 2002, 4, 4226–4235 RSC.
- R. Ma, Y. Bando, L. Zhang and T. Sasaki, Adv. Mater., 2004, 16, 918–922 CrossRef CAS PubMed.
- P. W. Ruch, D. Cericola, M. Hahn, R. Kötz and A. Wokaun, J. Electroanal. Chem., 2009, 636, 128–131 CrossRef CAS PubMed.
- L. J. Hardwick, M. Hahn, P. Ruch, M. Holzapfel, W. Scheifele, H. Buqa, F. Krumeich, P. Novák and R. Kötz, Electrochim. Acta, 2006, 52, 675–680 CrossRef CAS PubMed.
- C. H. Liang and C. S. Hwang, Jpn. J. Appl. Phys., 2008, 47, 1662–1666 CrossRef CAS.
- M. Chigane and M. Ishikawa, J. Electroanal. Soc., 2000, 147, 2246–2251 CrossRef CAS PubMed.
- C. C. Hu and T. W. Tsou, Electrochim. Acta, 2002, 47, 3523–3532 CrossRef CAS.
- M. Richter, A. Trunschke, U. Bentrup, K. W. Brzezinka, E. Schreier, M. Schneider, M. M. Pohl and R. Fricke, J. Catal., 2002, 206, 98–113 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, Houston, TX, 1996 Search PubMed.
- B. Djurfors, J. N. Broughton, M. J. Brett and D. G. Ivey, Acta Mater., 2005, 53, 957–965 CrossRef CAS PubMed.
- C. H. Wang, S. C. Hsu and J. H. Hu, J. Power Sources, 2014, 249, 1–8 CrossRef CAS PubMed.
- Y. K. Hsu, Y. C. Chen, Y. G. Lin, L. C. Chen and K. H. Chen, Chem. Commun., 2011, 47, 1252–1254 RSC.
- M. Kalapsazova, R. Stoyanova, E. Zhecheva, G. Tyuliev and D. Nihtianova, J. Mater. Chem. A, 2014, 2, 19383–19395 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Literature comparison, potential of quasi reference electrode measurement, Lorentz fitting of ex situ Raman spectra, XPS data and PXRD pattern of MnOx after activation. See DOI: 10.1039/c5ta03334a |
| This journal is © The Royal Society of Chemistry 2015 |