
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dot reduced bimetallic nanoparticles: size and surface plasmon resonance tunability for enhanced catalytic applications†
Jeremy B.
Essner
,
Charles H.
Laber
and
Gary A.
Baker
*
Department of Chemistry, University of Missouri-Columbia, Columbia, MO 65211, USA. E-mail: bakergar@missouri.edu
First published on 8th July 2015
Abstract
We report on a simple and green route toward monometallic (Au or Ag) and alloyed bimetallic AuAg nanoparticles using citric acid-derived carbon nanodots (C-dots) as the reducing and stabilizing agent. Simple variation in the initial C-dot![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal ratio yields a smoothly tunable surface plasmon resonance and the resulting nanomaterials show excellent catalytic activity for 4-nitrophenol reduction which is fully preserved following 5 months of storage. The Au@C-dot catalyst further demonstrated intrinsic peroxidase activity.
:metal ratio yields a smoothly tunable surface plasmon resonance and the resulting nanomaterials show excellent catalytic activity for 4-nitrophenol reduction which is fully preserved following 5 months of storage. The Au@C-dot catalyst further demonstrated intrinsic peroxidase activity.
Metal nanoparticles, especially Au and Ag, have received increasing attention in recent years owing to their attractive electronic, thermal, optical, and chemical properties, making them applicable in a wide range of fields such as catalysis, biomedicine, photovoltaics, antimicrobials, and surface-enhanced Raman spectroscopy (SERS).1–6 In particular, their properties are greatly influenced by the size, shape, crystal structure, and composition of the monometallic nanoparticles (hereafter, referred to as MNPs), which also dictate how the particles interact with incident light. When the size of the nanoparticle is substantially smaller than the wavelength of incident light, it experiences a relatively uniform electric field, resulting in a collective oscillation of conduction electrons. This gives rise to an energy-dependant resonance known as the surface plasmon resonance (SPR). The optoelectronic properties of the MNPs can be altered or enhanced by incorporating other metals into their structure or by the formation of composites or heterostructures. Indeed, bimetallic NPs (BMNPs) have shown enhanced properties over their monometallic counterparts because of a synergetic coupling between the individual metals' SPRs.7–9 The addition of this second metal can improve the electronic and absorption properties, giving rise to SPR bands lying intermediate between the individual MNP SPRs, thereby enhancing photovoltaic, catalytic, and antimicrobial prospects for the material. Recently, Haldar et al. reported on the synthesis of Au@Ag bimetallic core–shell NPs with varying Au core diameters.8 These researchers showed that the catalytic efficiency toward the reduction of 4-nitrophenol (4-NP) varied with the Au core size, however, the bimetallic core–shell NPs were still up to 12 times more active than their monometallic Au counterparts. In similar vein, our group recently demonstrated a green, sunlight-assisted approach toward the generation of bimetallic AgAu alloys decorated onto 3-aminopropyl-functionalized magnesium phyllosilicate nanoclay sheets.9 In this work, the bimetallic-decorated aminoclays possessed superior catalytic and antibacterial activity compared to their pure Ag or Au NP decorated analogues.
Historically, approaches such as laser ablation10 and chemical reduction (e.g., NaBH4,11,12 hydrazine13) were commonly employed to generate MNPs but these methods required either timely, complex, resource-limited equipment, or hazardous chemicals to carry out the reduction. This scenario impelled researchers to shift toward simple, environmentally-friendly, and cost-effective approaches for synthesizing various MNPs, including consideration of phytochemicals found in plant and fruit extracts as the reducing and stabilizing agents.14–23 For example, Nadagouda et al. presented a greener synthetic approach for generating both Au and Ag NPs using blackberry, blueberry, pomegranate, and turmeric extracts, taking advantage of the electron donating capabilities of the antioxidants present in the extracts.18 Use of these greener reducing agents has also recently been extended to the formation of bimetallic AuAg NPs. For example, Kumari et al. used pomegranate juice to generate both alloyed and core–shell AuAg NPs.24 Recently, carbon nanodots (C-dots) prepared by various methods have shown promise as reducing agents towards the formation of Au and Ag NPs,25–35 owing to their intrinsic abilities to act as electron donors and/or acceptors.36 For instance, Luo and co-workers demonstrated that electrochemically-synthesized C-dots functioned as reducing and stabilizing agents in the preparation of Au@C-dot nanocomposites that displayed tuneable particle diameters by varying the chloroauric acid:C-dot ratio.35 Similarly, Shen et al. reported the reductive capabilities of hydrothermally synthesized C-dots for AgNP growth with the resultant AgNP size varying when the silver nitrate:C-dot ratio was altered.34
Since the serendipitous discovery of C-dots over a decade ago, research into their formation and application has grown exponentially due to their many attractive optical and physical properties, including large red-edge effects (excitation wavelength-dependant emission), electron donor/acceptor capabilities, high aqueous solubility, large photostability, and low cytotoxity.37–48 Despite the “green” nature of C-dots, in general, many synthetic approaches often involve unsustainable precursors, harsh acidic/alkaline conditions, high synthetic temperatures, or extensive pre-/post-treatments which are inconsistent with the principles of green chemistry, for example routes involving arc-discharge,38 laser ablation,39 and acid oxidations.40,41 Therefore, greener synthetic strategies such as microwave, thermal, or hydrothermal treatment of various sustainable precursors (citric acid,42,43 urea43) or even low-value waste materials (e.g., grass,44 fruit peels, such as pomelo,45 watermelon,46 banana,47 orange48) are being aggressively pursued. Recently, Dong et al. showed that, via a simple thermal degradation of solid citric acid, one could selectively generate either C-dots or graphene oxide, depending upon the degree of carbonization.42
In this communication, we report on the thermal degradation of solid citric acid to produce C-dots following methods slightly modified from that reported by Dong and co-workers. The resultant C-dots were competent reducing agents for the thermally-assisted formation of AgNPs as well as the ambient-temperature synthesis of AuNPs. Similar to previous reports,34,35 varying the C-dot:metal salt ratio resulted in different MNP sizes and morphologies, an influence reflected in the observed SPRs. Interestingly, the C-dots were also effective for the co-reduction of both Au and Ag salts to synthesize AuxAgy@C-dot BMNPs. To the best of our knowledge, this is the first example of BMNP formation using C-dots as the reducing and stabilizing agent. Due to the synergistic effect of the two metals, the AuxAgy@C-dot BMNPs showed marked improvements in the catalytic reduction of 4-nitrophenol (4-NP) over its monometallic Au@C-dot NPs counterpart. Perhaps more surprisingly, the Ag@C-dot NPs possessed catalytic activities similar to that of the Au0.5Ag0.5@C-dot NPs.
The detailed experimental procedures and characterizations of the C-dots are provided in the (ESI†). Briefly, 50 g of solid citric acid (CA) was thermally decomposed at 250 °C for 2 h. The resultant dark red product was re-dispersed in deionized water, centrifuged, dialyzed, and then lyophilized. The CA-derived C-dots displayed properties akin to those of previously reported C-dots.37,46,48 The citrate-derived C-dots showed an increasing absorbance with decreasing wavelength (Fig. S1A,† dashed black line) which arises from the π–π* transition of aromatic sp2 domains and is likely due to the broad size distribution of C-dots produced.46,48 Similar to other reported C-dots,37,46 the CA-derived C-dots displayed excitation wavelength-dependent emission with a maximum emission occurring near 470 nm under 350 nm excitation (Fig. S1A†). The emission intensity increased as the excitation wavelength was increased from 300 to 350 nm after which it decreased and redshifted. The decrease in emission is further highlighted and quantified by the excitation wavelength-dependent quantum yields (Fig. S1A,† inset). The resultant CA-derived C-dots were slightly larger than those typically reported in the literature (1–10 nm),37 ranging in size from 30–60 nm with some dots reaching 100 nm (Fig. S1B†).
Fourier-transform infrared (FTIR) spectroscopic analysis indicated that the C-dots possessed hydroxyl, carboxyl, and epoxide moieties, accounting for their hydrophilicity and excellent dispersibility in water (Fig. S1C†).42 The large, broad absorption bands between 3600–3000 cm−1 were assigned to the stretching vibrations of O–H (νO–H). Well-defined absorbance bands appeared between 1800 cm−1 and 1600 cm−1 which were ascribed to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (νCO) and the skeletal vibrations of aromatic groups (νCC). The various peaks between 1300–1000 cm−1 arose from the different stretching modes of carboxylic, ester, ether, and alcohol moieties (νC–O–C and νC–OH), while the peaks near 2900, 1400, and 915 cm−1 were attributed to aliphatic carbons (νC–H and νC–H). The presence of these functionalities account for the long-term dispersibility of C-dots in water. Due to these surface functionalities and based on previous literature reports,25–35 we posited that the CA-derived C-dots would likely function as reducing as well as capping agents in the formation of MNPs. After optimizing the reaction conditions (refer to the ESI† for specific details), we found that C-dots were capable of directly reducing HAuCl4 to AuNPs at room temperature whereas Ag reduction required alkaline conditions and elevated temperatures with the most promising results achieved using 20 mM NaOH (pH > 10) and a reaction temperature of 100 °C.
O (νCO) and the skeletal vibrations of aromatic groups (νCC). The various peaks between 1300–1000 cm−1 arose from the different stretching modes of carboxylic, ester, ether, and alcohol moieties (νC–O–C and νC–OH), while the peaks near 2900, 1400, and 915 cm−1 were attributed to aliphatic carbons (νC–H and νC–H). The presence of these functionalities account for the long-term dispersibility of C-dots in water. Due to these surface functionalities and based on previous literature reports,25–35 we posited that the CA-derived C-dots would likely function as reducing as well as capping agents in the formation of MNPs. After optimizing the reaction conditions (refer to the ESI† for specific details), we found that C-dots were capable of directly reducing HAuCl4 to AuNPs at room temperature whereas Ag reduction required alkaline conditions and elevated temperatures with the most promising results achieved using 20 mM NaOH (pH > 10) and a reaction temperature of 100 °C.
For the room temperature reduction of HAuCl4, various ratios of C-dots:HAuCl4 were tested in two fashions: (1) using a constant C-dot concentration for varying Au precursor additions and (2) maintaining a constant HAuCl4 concentration for varying C-dot additions. A given concentration of HAuCl4 (0.3 mM) with increasing amounts of C-dots (0.05–0.60 mg mL−1) resulted in increased SPR intensities accompanied by a blue-shift (indicative of smaller AuNPs) until the C-dot concentration reached 0.30 mg mL−1, after which the SPR intensities remained relatively constant and presented a negligible shift in wavelength (Fig. S2†). On the other hand, for a given concentration of C-dots (0.30 mg mL−1) plus increasing amounts of HAuCl4 (0.10–0.60 mM; 0.05 mM increments), the SPR increased and drastically red-shifted (indicative of larger AuNPs) pointing towards the potential tunability of the resultant AuNPs (Fig. 1B and C). Specifically, between Au concentrations of 0.10 to 0.35 mM, a slight bathochromic shift arose accompanied by an increase in SPR intensities while for Au concentrations greater than 0.35 mM, a large bathochromic shift in the SPR was apparent. At these higher concentrations of Au, the SPR intensities increased up to 0.45 mM, after which the SPRs decreased and substantial peak broadening occurred (indicative of larger, more polydisperse AuNPs). Based on these results, the C-dots appeared to function effectively as both reducing and stabilizing agents. The results also indicate the clear potential for fine-tuning the Au SPR (Fig. 1C), and concurrently the AuNP size, simply by varying the C-dot:Au ratio.
 | ||
| Fig. 1 Representative TEM images of (A) Au@C-dot and (D) Ag@C-dot NPs. Shown are results for 0.3 mg mL−1:0.35 mM and 0.3 mg mL−1:0.30 mM C-dots:metal ratios for Au and Ag reductions, respectively. The SPR shifts of the resultant particles (B–F) show the smooth tunability afforded simply by changing the C-dot:metal ratio. In the case of AuNPs (B and C), the SPR intensity initially increased, reaching a maximum at a 0.3 mg mL−1 C-dot:0.45 mM Au ratio. The increase was also accompanied by a slight bathochromic shift. Higher Au:C-dot ratios resulted in a continuous red-shift with decreasing SPR intensity and substantial peak broadening, indicative of larger, more polydisperse NPs. On the other hand, the AgNPs displayed only a slight bathochromic shift (E and F) in SPR, with increasing intensities as the Ag concentration was varied from 0.15 to 3 mM. The lower images correspond to the Au and Ag samples in (B and C) and (E and F), respectively. | ||
The SPR bathochromic shift and peak broadening were attributed to an increase in particle size and a greater polydispersity, respectively, as evidenced in Fig. S3.† For example, the 0.20 mM, 0.35 mM, 0.45 mM, and 0.60 mM Au concentrations resulted in 17.8 ± 6.4, 26.7 ± 10.0, 47.8 ± 20.7, and 96.7 ± 29.9 nm quasi-spherical particles, respectively. The 0.3 mg mL−1:0.35 mM C-dot:Au ratio is also shown in Fig. 1A. Moreover, for higher concentrations of Au, large hexagonal and trigonal plates formed, with a higher amount of plate formation occurring with increasing Au:C-dot ratios. Although sample stability initially appeared to decrease for higher Au concentrations, the samples could be redispersed easily and the sedimentation was actually linked to the presence of large Au nanoplates which settled from solution within a couple days. Indeed, the 0.45 mM and 0.60 mM Au concentrations afforded 266.7 ± 99.3 and 528.8 ± 214.3 nm plates with approximate quasi-spherical:plate ratios of 1:5 and 1:2, respectively. The increasing formation of large hexagonal and trigonal plates translated to substantial peak broadening within the UV-Vis spectra and a bathochromic shift of the SPR, resulting in a color transition of the solutions from red to blue/purple. Interestingly, the higher Au concentrations still produced a population of small AuNPs in the 5–20 nm size range (Fig. S3C and D† insets), however, these particles were relatively rare in these samples.
To gain additional insight into the reduction process and the photophysical characteristics of the Au@C-dots, the room temperature reduction of 0.3 mg mL−1 C-dots:0.35 mM HAuCl4 samples was monitored via UV-Vis and fluorescence spectroscopy (Fig. S4†). After 180 min, the intensity of the SPR band (absorbance at 544 nm) was still increasing, although the SPR growth rate had begun to slow, indicating that the Au reduction was nearing completion (Fig. S4A† inset; left vertical axis). Throughout the reduction, the SPR remained consistently near 544 nm (Fig. S4A† inset; right vertical axis), elucidating that the increase in absorbance was due to the growth of additional NPs of a similar size rather than new species possessing different optical characteristics. Intriguingly, despite the intrinsic quenching nature of Au, the Au@C-dots retained ∼25% of the original fluorescence from the C-dot parent (Fig. S4B†). The inset plot of Fig. S4B† shows that, initially, the fluorescence rapidly decreased but eventually stabilized. Over the monitoring period, the wavelength of maximum emission from the C-dots remained relatively constant near 460 nm.
Similar to the case for Au reduction, various C-dot:Ag ratios were tested at room temperature for potential reduction to AgNPs. In this case, no AgNP formation was evident even after a week's time. Based on its past use in promoting noble metal reduction,25,34 various concentrations of NaOH (1, 10, and 20 mM) were added and elevated temperatures were employed (30, 50, 70, 90, and 100 °C) in an attempt to drive Ag+ reduction. Although some AgNP formation was evident at lower temperatures and modest NaOH concentrations, the best results (on the basis of narrow and intense SPR bands) were achieved at 100 °C using 20 mM NaOH. Under these conditions, various C-dot:Ag ratios were assayed. The Ag reductions displayed a decreased SPR dependency on C-dot:metal ratios compared to their Au reduction counterparts. When the C-dot concentration was held constant (0.3 mg mL−1) and increasing amounts of Ag+ were added (0.15–3.0 mM), the SPR intensity drastically increased (indicative of increased AgNP formation) whilst showing negligible shift in wavelength (Fig. 1E and F). On the other hand, when the AgNO3 concentration was held constant at 1.0 mM, for increasing amounts of C-dots from 0.05–0.60 mg mL−1, the SPR intensity again increased but displayed a small hypsochromic shift followed by a slight bathochromic shift (Fig. S5†). In spite of this slight SPR shift, an increasing Ag:C-dot ratio apparently generates slightly smaller AgNPs (Fig. S6†). A size analysis, conducted by counting over 200 particles per sample, resulted in mean particle sizes of 11.1 ± 9.9, 6.9 ± 3.4, and 6.0 ± 4.0 nm for the 0.30 mM, 1.0 mM, and 3.0 mM Ag concentrations, respectively. A representative TEM image for the Ag@C-dot sample derived from a 0.3 mg mL−1:0.30 mM C-dot:Ag sample is also shown in Fig. 1D.
Both the Ag@C-dot and Au@C-dot NPs, due to their plasmonic nature, hold prospects for SERS and photovoltaic applications, but to further enhance the applicability of these MNPs@C-dots, we attempted to simultaneously reduce both Ag and Au salts to generate AuxAgy@C-dot BMNPs. The fact that we were only able to achieve Ag reduction to generate Ag@C-dots under basic conditions required we tailor our synthetic approach in order to arrive at BMNPs. Firstly, NaOH was added to HAuCl4 to produce gold hydroxide, Au(OH)4−. The C-dot solution was then heated at 100 °C for 15 min to allow for thermal equilibration. Lastly, Au(OH)4− and AgNO3 solutions were simultaneously injected into the hot C-dot solution and allowed to react for 15 min. In all instances, the C-dot, NaOH, and total metal salt concentrations were held constant at 0.3 mg mL−1, 20 mM, and 1 mM, respectively. Aliquots of 10 mM HAuCl4 and 10 mM AgNO3 were mixed to generate Au:Ag ratios of 90:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80, and 10:90 (Fig. 2A). For example, to generate the 50:50 Au:Ag BMNP sample, 0.5 mL of 10 mM HAuCl4 was added to 2 mL of 100 mM NaOH which was simultaneously injected, alongside 0.5 mL of 10 mM AgNO3, into 7 mL of hot C-dot solution (0.43 mg mL−1). Samples containing 100% Ag were made in analogous fashion, whereas the preparation of 100% Au samples involved no addition of NaOH, in accordance with the Au@C-dot preparation method described earlier. It is noteworthy that the use of Au(OH)4− in the absence of Ag+ at 100 °C led solely to the formation of large gold plates. As the Au:Ag ratio decreased, the SPR uniformly blue-shifted, consistent with the presence of BMNPs (Fig. 2A and B).9,13,24
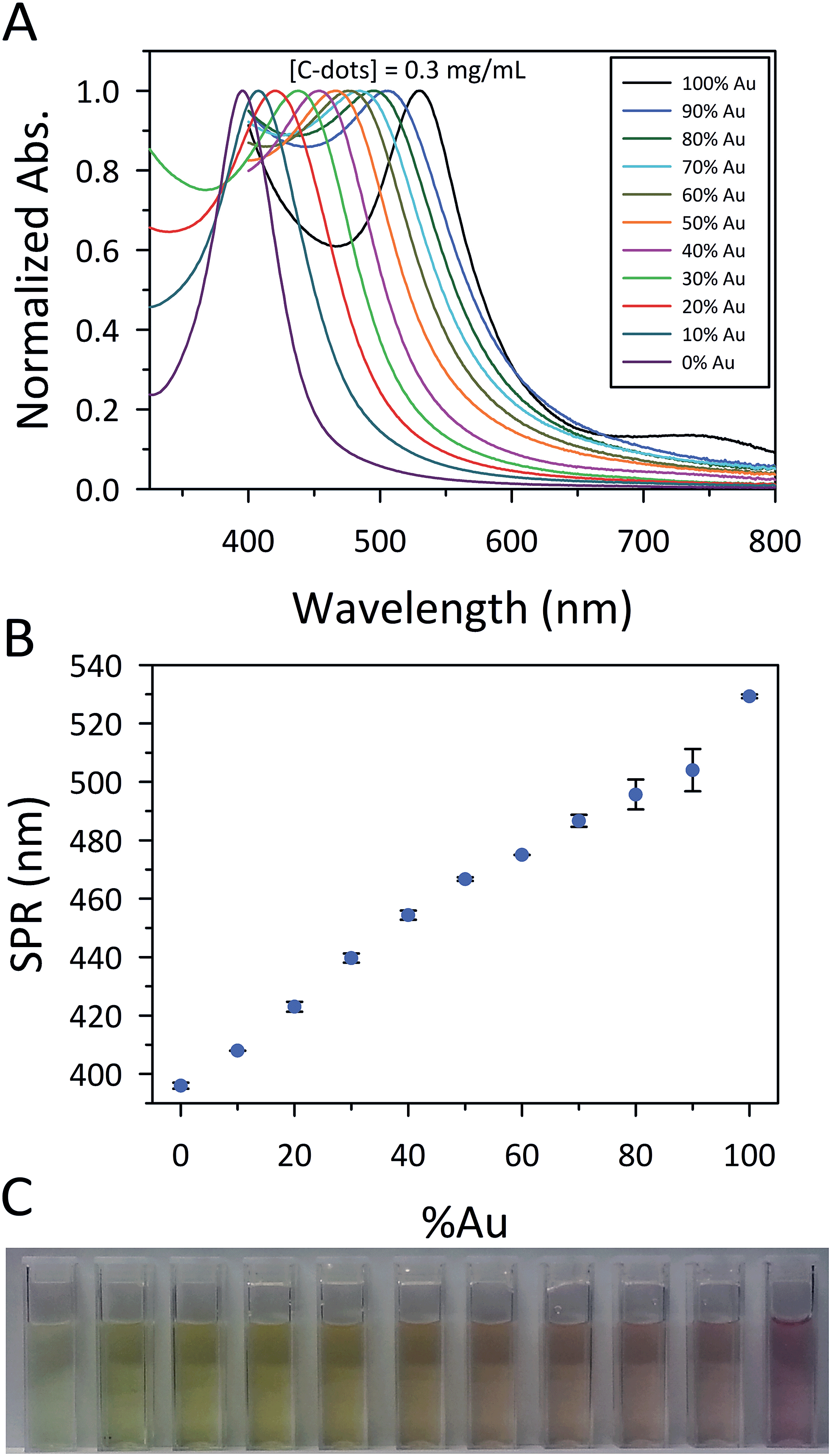 | ||
| Fig. 2 (A) Normalized UV-Vis absorbance of the bimetallic AuxAgy@C-dot NPs showing that as the experimental Au:Ag ratio decreased the resultant SPR displayed a systematic hypsochromic shift. (B) Extracted SPR frequencies plotted versus the %Au, further demonstrating the smooth evolution in SPR wavelength as the Au:Ag ratio was modulated. The sample sets were replicated three times highlighting the reproducibility of the BMNPs (error bars denote one standard deviation from the mean), especially at the lower Au concentrations. (C) Photograph of a series of AuxAgy@C-dot BMNP samples, illustrating a palette of SPR transitions. The cuvettes correspond to the samples in A and B from (left) 100% Ag to (right) 100% Au. | ||
Reminiscent of the room temperature reduction of 0.6 mM Au, use of 1 mM HAuCl4 at 100 °C yielded highly unstable particles which displayed a broad, red-shifted SPR. For this reason, the 100% Au results reported in Fig. 2 were actually generated from 0.25 mM HAuCl4. The effect of Au concentration on the SPR was subsequently investigated for reduction at 100 °C. These results again reiterate the strong control over the SPR made possible simply by varying the ratio of Au:C-dots (Fig. S7†). To verify that the C-dots were indeed responsible for the reduction of the BMNPs, a control reaction was performed for a 50:50 Au:Ag sample in the absence of C-dots (Fig. S8†). The lack of both a SPR band and a visible change in solution color indicated that the C-dots were solely responsible for accomplishing metal ion reduction.
Both the Au@C-dot and Ag@C-dot NPs displayed larger particle sizes than all of their bimetallic counterparts (Fig. 3A–C and S9†). Interestingly, upon the addition of 10% Ag, the average particle size dramatically decreased from 30.0 ± 6.0 nm for the Au@C-dot NPs to 4.1 ± 1.4 nm for the Au0.9Ag0.1@C-dot NPs. Increasing the Ag:Au ratio within the BMNPs led to further decreased particle size. For the Au0.7Ag0.3@C-dot, Au0.5Ag0.5@C-dot, Au0.3Ag0.7@C-dot, and Au0.1Ag0.9@C-dot BMNPs, the average particle sizes were 3.4 ± 0.9, 3.3 ± 1.1, 2.4 ± 0.8, and 1.9 ± 0.7 nm, respectively. Although a small decrease in particle size was present, the dependence of the SPR maximum on the Au:Ag ratio was primarily related to different levels of alloying and therefore synergistic plasmonic cooperation between the two metals.
 | ||
| Fig. 3 Representative TEM images and their corresponding EDX spectra for (A and D) Au@C-dots, (B and E) bimetallic Au0.5Ag0.5@C-dots, and (C and F) Ag@C-dots. Interestingly, upon the slightest addition of silver, the NP's average size decreased by a factor of ∼7 and remained relatively constant across the Au:Ag composition tested. (G) Plot showing the extent of alloying between Au and Ag (%Au measured) within the BMNPs versus the content predicted from the initial Au:Ag stoichiometry (i.e., %Au theoretical). The dashed line represents the ideal case where the measured values directly correspond to reagent metallic ratios. In general, the measured content follows the prediction line. The slight enrichment with Au at higher initial Au% could be tentatively tied to a small amount of galvanic replacement in which Ag0 is etched by Au3+. | ||
Energy-dispersive X-ray (EDX) microanalysis of AuxAgy@C-dot NPs was performed to reveal the average composition of the BMNPs (Fig. 3D–F). As expected, as the Au:Ag ratio decreased, the Au peaks became less prominent whereas the Ag peaks became dominant. Fig. 3G shows that the actual composition of the AuxAgy@C-dot NPs was nominally the same as the starting Au:Ag ratios, although for Au:Ag fractions greater than 20:80, a slight enrichment in Au arose, possibly due to some galvanic replacement of Ag with Au. The Cu peaks present in the EDX spectra simply arose from the grids employed for imaging.
The catalytic activities of the MNPs@C-dots were studied through the model reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) using NaBH4 as the reducing agent. The reaction kinetics of the catalytic conversion were monitored by UV-Vis spectroscopy. The aqueous solution of 4-NP displays little (pale yellow) to no color with an absorbance band at 320 nm. Upon the addition of NaBH4, the solution immediately turns dark yellow due to the formation of the 4-nitrophenolate ion (4-NPO). The formation of 4-NPO results in an increased absorbance accompanied by a bathochromic shift to 400 nm. To test for catalytic activities, UV-Vis spectra were collected every 5 s following injection of NaBH4, monitoring the decreased absorption at 400 nm (i.e., loss of 4-NP). The 4-NP to 4-AP conversion was unaltered in the presence of the CA-derived C-dots (5.8 μg mL−1) even after 30 min, demonstrating that unmodified C-dots are not catalysts for this reaction (Fig. 4B; only shown out to 450 s). Upon addition of MNPs@C-dots to the 4-NPO solution, however, a prompt drop in absorbance at 400 nm was observed within seconds, indicating the rapid conversion of 4-NP to 4-AP (Fig. 4A; S10,† panels A and B). The complete reduction of 4-NP to 4-AP was observed within 120 s, 150 s, and >1000 s for Ag@C-dots, Au0.5Ag0.5@C-dots, and Au@C-dots, respectively. The reduction mechanism can be explained through the Langmuir–Hinshelwood mechanism, in which the analyte of interest (4-NPO) absorbs on the surface of the MNPs, while the MNPs also react with the BH4− to form a metal hydride. The metal hydride then reacts with the sorbed analyte (4-NPO), facilitating its reduction. Due to the slow transfer of electrons from the metal hydride to 4-NPO, it is considered the rate determining step of the reaction.49 This lag phase or induction time (to) in the reaction rate was minimized by degassing the 4-NP solution for 20 min to remove any dissolved oxygen. Despite this preventative measure, lag phases were still observed in the Au@C-dot NP samples. In this study, an excess amount (1000-fold) of NaBH4 (0.2 M, 0.4 mL) compared to 4-NP was used (0.2 mM, 0.4 mL) so that the conversion would follow a pseudo-first-order rate law. Under these conditions, the apparent rate constants (kapp) for each catalyst can be estimated from the slopes of the linear correlation of ln(At/Ao) versus time, where Ao is the initial absorbance and At is the absorbance at time (t). The kapp values for Ag@C-dot, Au0.5Ag0.5@C-dot, and Au@C-dot catalysts were 2.30 ± 0.25 × 10−2 s−1, 1.92 ± 0.43 × 10−2 s−1, and 2.54 ± 0.30 × 10−3 s−1, respectively (Fig. 4B). Thus, the Au0.5Ag0.5@C-dot and Ag@C-dot catalysts showed improved activities (faster 4-NP reduction) over the Au@C-dot NPs, an outcome attributed to their smaller sizes and therefore higher surface areas.
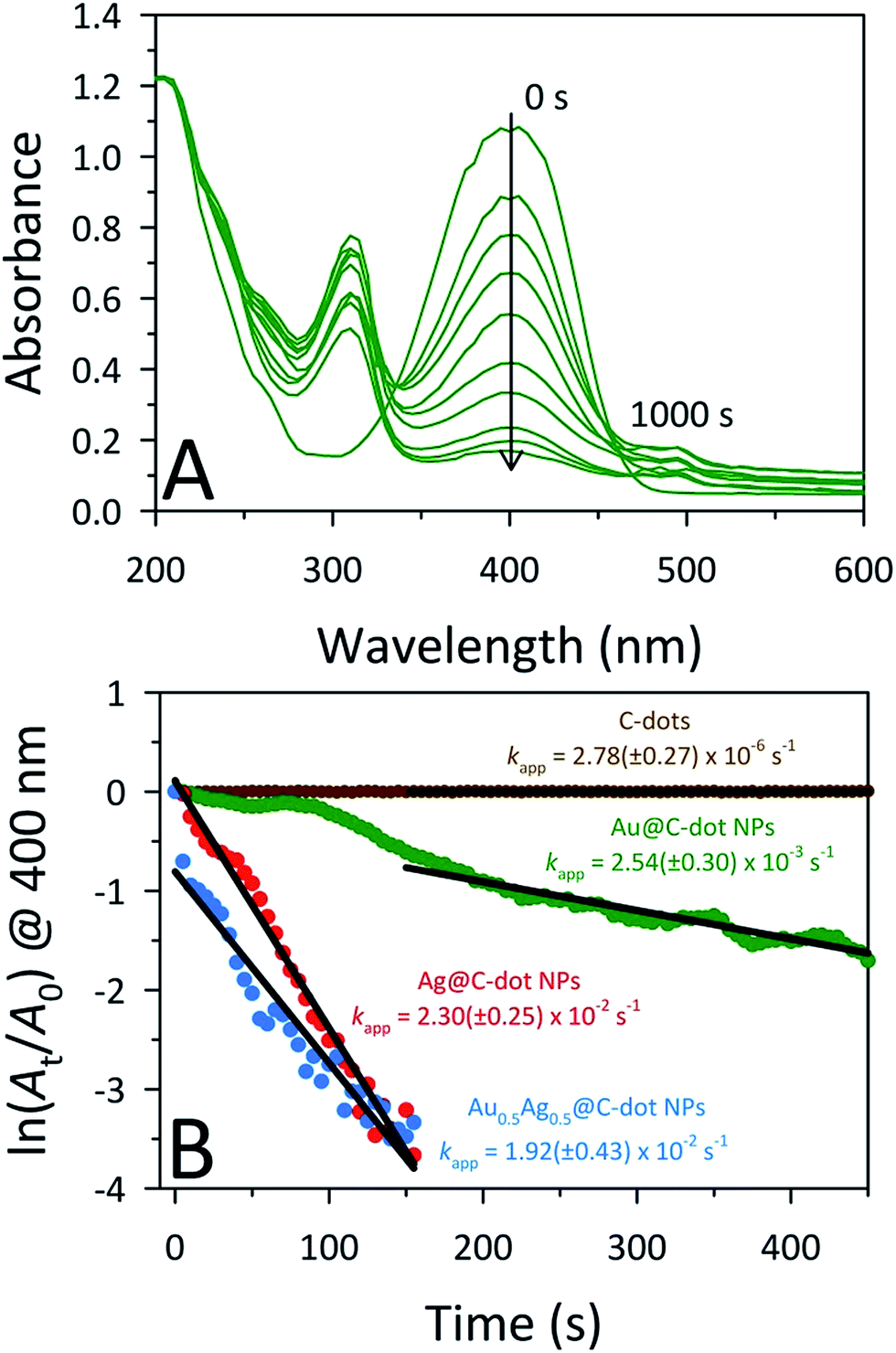 | ||
| Fig. 4 (A) Time-dependent UV-Vis absorption spectra of the NaBH4-assisted reduction of 4-NP catalyzed by the Au@C-dot NPs. Spectra were acquired every 5 s but, for clarity, spectra are shown only for illustrative times. (B) Plots of ln(At/Ao) for 4-NP absorbance at 400 nm against time for various MNP@C-dot catalysts. Each catalyst was tested within days of preparation. | ||
All three types of MNPs@C-dots retained their catalytic activities after months of storage in a lab drawer. Even after 5 months of casual, ambient storage, the apparent rate constants for 4-NP reduction were completely preserved. In fact, surprisingly, the Ag@C-dot and Au0.5Ag0.5@C-dot NPs displayed statistically meaningful increases in their catalytic rates. The kapp for the 5 month aged Ag@C-dot, Au0.5Ag0.5@C-dot, and Au@C-dot catalysts were 3.83 ± 0.30 × 10−2 s−1, 2.69 ± 0.39 × 10−2 s−1, and 2.98 ± 0.22 × 10−3 s−1, respectively (Fig. S10C†). Since the aged Ag@C-dot and Au0.5Ag0.5@C-dot NPs showed improved catalytic rates over the corresponding freshly prepared NPs, UV-Vis studies were conducted on both the aged and freshly prepared NPs to monitor any optical changes (Fig. S11†). The Au@C-dot NPs essentially remained unchanged after 5 months while the aged Au0.5Ag0.5@C-dot and Ag@C-dot NPs displayed increased absorbance at longer wavelengths, possibly due to minor nanoparticle aggregation. The slight particle aggregation upon prolonged storage may have resulted in catalytic hotspots, accounting for the marginal increase in catalytic activity of the aged Ag@C-dot and Au0.5Ag0.5@C-dot NPs. Due to the surprisingly high catalytic rate of the Ag@C-dot NPs and gold's established reputation as a good catalyst for this reaction, the improved catalytic rates of the BMNPs could be attributed to a synergistic effect of Au and Ag nano-domains within the individual particles, in line with results reported recently by Ravula et al.9
Lastly, we examined the potential of the MNP@C-dot samples as peroxidase mimetics toward oxidation of the peroxidase substrate 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) diammonium salt (ABTS) using H2O2. While neither Au0.5Ag0.5@C-dots nor Ag@C-dots displayed any measureable peroxidase activity within 24 h of prospective catalyst addition, the Au@C-dots demonstrated clear activity. As summarized in Fig. S12,† addition of Au@C-dots triggered the formation of green-colored ABTS oxidation product which was monitored spectrophotometrically at 418 nm. A control experiment carried out using citric acid-derived C-dots alone did not yield noticeable peroxidase activity. A dramatic increase in the catalytic velocity was observed in the time-dependent absorption profiles as the Au@C-dot content was increased, validating the intrinsic peroxidase activity of the Au@C-dots in contrast with the naked C-dots. Among other things, these data open the possibility of using hybrid C-dots for selective glucose detection by coupling with glucose oxidase, for example.
In summary, we have demonstrated a simple and green approach towards the synthesis of MNPs/BMNPs using C-dots as the reducing and stabilizing agent. The resultant SPR of the various MNPs and BMNPs showed a systematic tunability indicating that the size of NPs can easily be controlled simply by varying the C-dot:metal ratio. The AuxAgy@C-dot metal compositions were nominally the same as the starting stoichiometric ratios, pointing to the efficiency of this nanoscale preparation. Notably, the Ag@C-dot NPs and AuxAgy@C-dot BMNPs displayed remarkably enhanced catalytic activities over Au@C-dots for 4-nitrophenol reduction, highlighting the potential of these materials in various catalytic applications. Finally, the Au@C-dots showed peroxidase activity not seen in the unmodified C-dots nor in their Ag@C-dot NPs and AuxAgy@C-dot analogs. In the future, these materials might find use in additional areas such as surface-enhanced spectroscopies and plasmon-assisted photovoltaics.
Acknowledgements
We thank Drs Tommi White and Thomas Lam of the MU Electron Microscopy Core Facility for assistance with our nanoparticle imaging.References
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2009, 26, 2885–2893 CrossRef PubMed.
- J. Qi, X. Dang, P. T. Hammond and A. M. Belcher, ACS Nano, 2011, 5, 7108–7116 CrossRef CAS PubMed.
- S. D. Standridge, G. C. Schatz and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 8407–8409 CrossRef CAS PubMed.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- Z. Mu, X.-W. Zhao, Z. Xie, Y. Zhao, Q.-F. Zhong, L. Bo and Z.-Z. Gu, J. Mater. Chem. B, 2013, 1, 1607–1613 RSC.
- H. Chang, H. Kang, J.-K. Yang, A. Jo, H.-Y. Lee, Y.-S. Lee and D. H. Jeong, ACS Appl. Mater. Interfaces, 2014, 6, 11859–11863 CAS.
- K. K. Haldar, S. Kundu and A. Patra, ACS Appl. Mater. Interfaces, 2014, 6, 21946–21953 CAS.
- S. Ravula, J. B. Essner, W. A. La, L. Polo-Parada, R. Kargupta, G. J. Hull, S. Sengupta and G. A. Baker, Nanoscale, 2015, 7, 86–91 RSC.
- I. Lee, S. W. Han and K. Kim, Chem. Commun., 2001, 1782–1783 RSC.
- L. M. Liz-Marzan and A. P. Philipse, J. Phys. Chem., 1995, 99, 15120–15128 CrossRef CAS.
- N. N. Kariuki, J. Luo, M. M. Maye, S. A. Hassan, T. Menard, H. R. Naslund, Y. Lin, C. Wang, M. H. Engelhard and C.-J. Zhong, Langmuir, 2004, 20, 11240–11246 CrossRef CAS PubMed.
- D.-H. Chen and C.-J. Chen, J. Mater. Chem., 2002, 12, 1557–1562 RSC.
- O. V. Kharissova, H. V. R. Dias, B. I. Kharisov, B. O. Pérez and V. M. J. Pérez, Trends Biotechnol., 2013, 31, 240–248 CrossRef CAS PubMed.
- A. K. Mittal, Y. Chisti and U. C. Banerjee, Biotechnol. Adv., 2013, 31, 346–356 CrossRef CAS PubMed.
- A. Bankar, B. Joshi, A. R. Kumar and S. Zinjarde, Colloids Surf., A, 2010, 368, 58–63 CrossRef CAS PubMed.
- A. Bankar, B. Joshi, A. Ravi Kumar and S. Zinjarde, Colloids Surf., B, 2010, 80, 45–50 CrossRef CAS PubMed.
- M. N. Nadagouda, N. Iyanna, J. Lalley, C. Han, D. D. Dionysiou and R. S. Varma, ACS Sustainable Chem. Eng., 2014, 2, 1717–1723 CrossRef CAS.
- H. Xu, L. Wang, H. Su, L. Gu, T. Han, F. Meng and C. Liu, Food Biophys., 2014, 1–7 Search PubMed.
- A. Rao, K. Mahajan, A. Bankar, R. Srikanth, A. R. Kumar, S. Gosavi and S. Zinjarde, Mater. Res. Bull., 2013, 48, 1166–1173 CrossRef CAS PubMed.
- V. Ganesh Kumar, S. Dinesh Gokavarapu, A. Rajeswari, T. Stalin Dhas, V. Karthick, Z. Kapadia, T. Shrestha, I. A. Barathy, A. Roy and S. Sinha, Colloids Surf., B, 2011, 87, 159–163 CrossRef PubMed.
- G. A. Kahrilas, L. M. Wally, S. J. Fredrick, M. Hiskey, A. L. Prieto and J. E. Owens, ACS Sustainable Chem. Eng., 2013, 2, 367–376 CrossRef.
- M. N. Alam, S. Das, S. Batuta, N. Roy, A. Chatterjee, D. Mandal and N. A. Begum, ACS Sustainable Chem. Eng., 2014, 2, 652–664 CrossRef CAS.
- M. Meena Kumari, J. Jacob and D. Philip, Spectrochim. Acta, Part A, 2015, 137, 185–192 CrossRef CAS PubMed.
- L.-M. Shen, Q. Chen, Z.-Y. Sun, X.-W. Chen and J.-H. Wang, Anal. Chem., 2014, 86, 5002–5008 CrossRef CAS PubMed.
- X. Wang, Y. Long, Q. Wang, H. Zhang, X. Huang, R. Zhu, P. Teng, L. Liang and H. Zheng, Carbon, 2013, 64, 499–506 CrossRef CAS PubMed.
- S. Mandani, B. Sharma, D. Dey and T. K. Sarma, Nanoscale, 2015, 7, 1802–1808 RSC.
- S. Liu, B. Yu and T. Zhang, RSC Adv., 2014, 4, 544–548 RSC.
- H. Choi, S.-J. Ko, Y. Choi, P. Joo, T. Kim, B. R. Lee, J.-W. Jung, H. J. Choi, M. Cha, J.-R. Jeong, I.-W. Hwang, M. H. Song, B.-S. Kim and J. Y. Kim, Nat. Photonics, 2013, 7, 732–738 CrossRef CAS PubMed.
- Y. Choi, G. H. Ryu, S. H. Min, B. R. Lee, M. H. Song, Z. Lee and B.-S. Kim, ACS Nano, 2014, 8, 11377–11385 CrossRef CAS PubMed.
- R. Liu, J. Liu, W. Kong, H. Huang, X. Han, X. Zhang, Y. Liu and Z. Kang, Dalton Trans., 2014, 10920–10929 RSC.
- X. Ran, H. Sun, F. Pu, J. Ren and X. Qu, Chem. Commun., 2013, 49, 1079–1081 RSC.
- A. Jaiswal, P. Gautam, S. Ghosh and A. Chattopadhyay, J. Nanopart. Res., 2013, 16, 1–14 Search PubMed.
- L.-M. Shen, M.-L. Chen, L.-L. Hu, X.-W. Chen and J.-H. Wang, Langmuir, 2013, 29, 16135–16140 CrossRef CAS PubMed.
- P. Luo, C. Li and G. Shi, Phys. Chem. Chem. Phys., 2012, 14, 7360–7366 RSC.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774–3776 RSC.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- S. Hu, J. Liu, J. Yang, Y. Wang and S. Cao, J. Nanopart. Res., 2011, 13, 7247–7252 CrossRef CAS.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS PubMed.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS PubMed.
- R. Vikneswaran, S. Ramesh and R. Yahya, Mater. Lett., 2014, 136, 179–182 CrossRef CAS PubMed.
- A. Prasannan and T. Imae, Ind. Eng. Chem. Res., 2013, 52, 15673–15678 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and full experimental details. See DOI: 10.1039/c5ta02949j |
| This journal is © The Royal Society of Chemistry 2015 |