
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability of NdBaCo2−xMnxO5+δ (x = 0, 0.5) layered perovskites under humid conditions investigated by high-temperature in situ neutron powder diffraction†
Mona
Bahout
*a,
Stevin S.
Pramana
b,
James M.
Hanlon
a,
Vincent
Dorcet
a,
Ronald I.
Smith
c,
Serge
Paofai
a and
Stephen J.
Skinner
*b
aInstitut des Sciences Chimiques de Rennes, Equipe « Chimie du Solide et Matériaux », UMR CNRS 6226, Université de Rennes 1, 263 Avenue du Général Leclerc, 35042 Rennes, France. E-mail: mona.bahout@univ-rennes1.fr
bDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK
cThe ISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Didcot, OX11 0QX, UK
First published on 19th May 2015
Abstract
The double perovskites NdBaCo2−xMnxO5+δ (x = 0 and 0.5) were investigated using in situ high temperature neutron powder diffraction in dry argon and wet atmospheres (40% D2O/argon and 40% D2O/air) in order to assess their stability as cathodes in proton conducting fuel cells. The x = 0 oxide loses oxygen on heating in dry argon at T > 400 °C and exhibits an oxygen vacancy order–disorder transition as evidenced by the orthorhombic Pmmm to tetragonal P4/mmm transition. Refinement of site occupancy factors suggests that the oxygen vacancies mainly form in the Nd layers and to a lesser extent at the equatorial positions of the transition metal polyhedra; at 800 °C, δ ∼ 0. When the gas was changed to wet argon at 800 °C and the sample cooled to 260 °C, no structural modification or change in the oxygen content was detected and no impurity phases formed, highlighting the excellent stability of the sample in wet atmospheres. On switching the gas to wet air at 260 °C, thermal analysis and neutron powder diffraction data together reveal that the sample intercalates mainly oxygen rather than proton defects within a two-phase process involving two orthorhombic phases, reflecting the symmetry of the reduced and oxidised materials. On heating, the sample transforms at T ≥ 600 °C to a single tetragonal phase whose symmetry is retained up to 800 °C and on subsequent cooling. The x = 0.5 material prepared in argon adopted a tetragonal P4/mmm structure at RT with δ ∼ 0. Its symmetry remains tetragonal on heating/cooling in wet argon. On changing the gas to wet air at 260 °C, it takes up oxygen via a two-phase process involving two tetragonal phases. Since fast oxidation is the main process that fills the oxygen vacancies of these double perovskites in wet air, a large oxygen deficiency seems to be not the only requirement for effective proton incorporation in this family of materials with basic characteristics.
1. Introduction
Conventional solid oxide fuel cells (SOFCs) based on the oxide ion conducting electrolyte, 8 mol% yttria-stabilized zirconia (8YSZ), need high operating temperatures, between 800 and 1000 °C, to achieve sufficient ionic conductivity in the electrolyte.1 Such high temperatures impose great challenges on the long-term stability and durability of SOFC materials2 and require the use of expensive ceramic interconnects and heat exchangers. For this reason, significant research efforts are currently devoted to reducing the operation of SOFCs to the intermediate temperature (IT) range of ∼400–700 °C, while retaining the benefits of elevated temperatures.2,3 In this view, a proton conducting SOFC (H+-SOFC) based on a proton conducting ceramic electrolyte that can operate between 300 and 600 °C has emerged as a promising candidate.4–8 Proton conduction requires the existence of proton defects in the oxide whose incorporation into the bulk requires the presence of oxygen vacancies.8,9 Indeed, the main reaction leading to the formation of proton defects is the dissociative adsorption of water onto a hydroxide ion and a proton; the hydroxide ion fills an oxide ion vacancy, and the proton forms a covalent bond with lattice oxygen (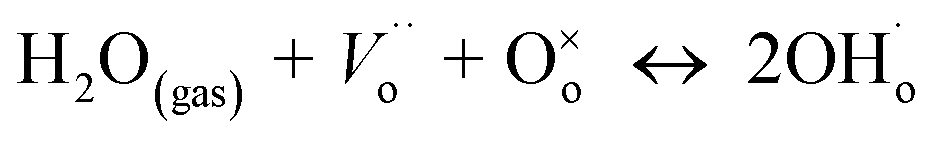 in the Kröger–Vink notation). Proton conduction consists of the hopping of protons between neighboring oxygen positions, termed as the Grotthuss (or free-proton) mechanism.
in the Kröger–Vink notation). Proton conduction consists of the hopping of protons between neighboring oxygen positions, termed as the Grotthuss (or free-proton) mechanism.
Besides the search for suitable electrolytes combining high proton conductivity at moderate temperature with thermodynamic stability, the development of cathode materials for H+-SOFC with fast transfer kinetics in the IT range is another challenging issue.10 The cathode plays a critical role in proton conducting fuel cells because water is produced at the cathode. Most work on H+-SOFC has been carried out using Pt electrodes surrounded by doped cerate (ACeO3) or zirconate (AZrO3) proton conducting electrolytes. Despite their good catalytic activity, Pt electrodes exhibit a large overpotential and are too expensive for large-scale applications.11 Recently, double-layered perovskite oxides, LnBaCo2O5+δ (Ln = lanthanide), have been investigated as cathodes in H+-SOFCs.10,12,13 These oxides were extensively studied as cathode materials for O2−-SOFCs due to their excellent electrochemical properties correlated with their mixed conductivity (O2−/e−).15,16 Their structure consists of A-site ordering into alternate layers of Ln and Ba ions with oxygen vacancies primarily located in the LnOδ layers. A variety of unit cells were reported for LnBaCo2O5+δ compounds at various temperatures and δ ranges; the most common are P4/mmm (ap × ap × 2ap) and Pmmm (ap × 2ap × 2ap), where ap is the lattice parameter of a simple cubic perovskite. The behaviour of LnBaCo2O5+δ compounds in wet air was previously investigated by thermogravimetric analysis (TGA). Chemical compatibility tests with the BaZr0.9Y0.1O3−δ proton conducting electrolyte revealed no impurity phases.12 The oxygen reduction reaction was investigated by electrochemical impedance spectroscopy as a function of water vapor pressure, pH2O, and demonstrated improved performance with increased pH2O which was attributed to the insertion of protonic defects (OHo˙) into the structure.13 Although such behaviour may indicate protonic conductivity in the LnBaCo2O5+δ materials, this needs to be established unambiguously, since there is no direct evidence of the presence of structural protons in this family of materials. Due to the sensitivity of neutron diffraction to both light atom positions and site occupancies, we undertook an in situ high temperature study in dry and wet (D2O) argon and wet (D2O) air on two double perovskite compounds, NdBaCo2−xMnxO5−δ (x = 0 and 0.5), using time-of-flight (TOF) neutron powder diffraction (NPD). The aim was to verify their stability and monitor their structural behaviour under operating conditions relevant to H+-SOFC cathodes. Since the insertion of proton defects into typical proton conducting electrolytes occurs between 300 and 600 °C and dehydration/deprotonation prevails at higher temperatures,7 isothermal neutron diffraction data were collected in the temperature range of 260–800 °C. The x = 0.5 composition has been investigated in order to shed light on the impact of the electronegativity of the B cation as well as that of crystal symmetry and oxygen content on the hydration capacity and stability under wet conditions. It is well known that in perovskite type proton conducting electrolytes, the electronegativity of the B cation impacts the stabilization of proton defects and any symmetry reduction reduces water solubility.7 Whereas the symmetry of the x = 0 material changes between orthorhombic and tetragonal as a function of temperature and gas atmosphere, the symmetry of the x = 0.5 material is tetragonal and remains unchanged on heating/cooling in air or in an inert atmosphere.17
2. Experimental
The preparation of the polycrystalline samples (x = 0, 0.5) has been described elsewhere.18 Phase purity was assessed by powder X-ray diffraction (XRD) collected at room temperature over the range 10 ≤ 2θ ≤ 120°, Δ2θ = 0.02° using a Bruker AXS D8 Advance diffractometer in Bragg–Brentano geometry equipped with a Ge primary monochromator (Cu-Kα1 radiation) and a Lynxeye detector. The initial oxygen content was determined via iodometric titration against a standardized sodium thiosulfate solution (∼0.05 M). A powder sample (∼300 mg) was introduced into a closed argon-flushed glass container and covered by a large excess of KI (∼2 g). Approximately 20 mL of hydrochloric acid twice diluted was added to the warm mixture (∼40 °C) that was stirred under an argon flow. When dissolution was complete, the iodine formed was titrated under an argon flow with Na2S2O3 solution and the calculated oxygen stoichiometry was based on the amount of I2 formed. Thermogravimetric analysis (TGA) was carried out using a Netzsch STA 449 F3 instrument. Samples weighing ∼100 mg, loaded in platinum crucibles, were heated to 800 °C at a rate of 10 °C min−1 in 1 atm of dry air (40 mL min−1) before cooling in air at 10 °C min−1. This cycle was performed to clean the sample and remove absorbed free water, hydrates, hydroxides and carbonates. Subsequent cycles were carried out in dry or wet gas, as detailed in Results and discussion.In situ neutron diffraction data were collected on the high-flux medium resolution POLARIS diffractometer at ISIS, the UK spallation source at the Rutherford Appleton Laboratory. The powder samples were loaded to a height of 4 cm in a double-walled quartz cell which enabled gas to flow through the powder in the neutron beam, and was connected to a panel allowing the switching of gas streams (e.g. dry and wet). Two thermocouples were attached on opposite sides of the inner quartz cell wall just above the sample to control and monitor the furnace temperature. The temperature ramp was always 10 °C min−1. The samples were first heated from 20 to 800 °C in dry argon to clean the surface and remove protonic species (Ba(OH)2, Ba(OH)2·nH2O, and Ba(OH)x(CO3)y·nH2O) and to increase the amount of oxygen vacancies which may favour the insertion of proton defects. Subsequently, the atmosphere was changed to wet argon then to wet air. A humid atmosphere was generated with D2O in preference to H2O (the coherent scattering lengths of H and D are −3.7406 and 6.671 fm, respectively). In addition, the incoherent scattering cross section of D (2.05 barn) is much lower than that of H (80.27 barn).19 The D2O injector was connected to the cell and the humidifier was set at 60 °C to provide a saturated vapor pressure, roughly 0.2 bar at atmospheric pressure. Neutron diffraction data were collected on the x = 0 and 0.5 samples according to the profiles illustrated in Fig. 1, which were chosen to complement the TGA data.
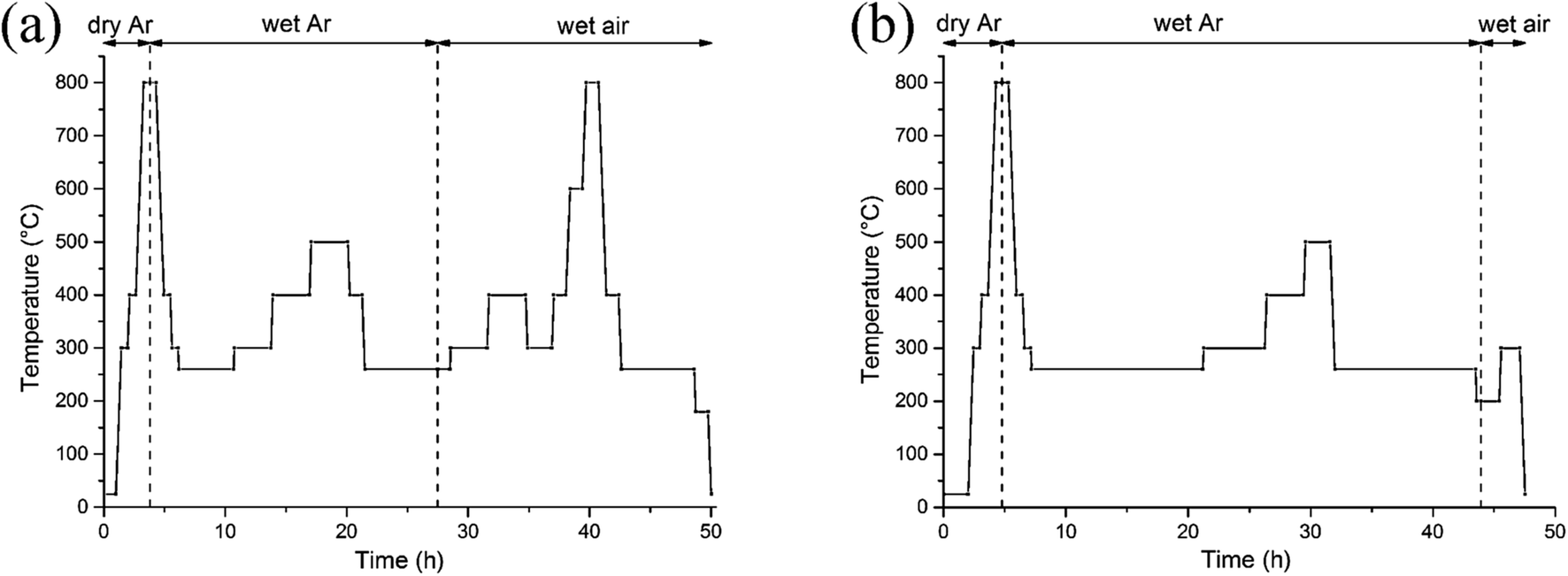 | ||
| Fig. 1 Temperature profiles for (a) NdBaCo2O5+δ and (b) NdBaCo1.5Mn0.5O5+δ. | ||
The main difference in the temperature profiles used for the two compositions lies in more numerous data collected in wet air on x = 0 than on x = 0.5. Since the former exhibits various phase transitions, we investigated any changes in symmetry/crystal structure, oxygen content or hydroxyl intercalation/deintercalation, stability in wet air, kinetics of oxygen exchange and thermodynamic equilibrium as a function of temperature. Data normalisation and file output was performed using the Mantid software package.20 The patterns from two banks of detectors, the backscattering bank (average 2θ = 146.7°, d-spacing range 0.2–2.65 Å) and the 90° bank (average 2θ = 92.6°, maximum d-spacing 4.1 Å) were simultaneously fitted by the Rietveld method using the FullProf refinement program.21 Peak shapes were modeled using the convolution of two back-to-back exponentials with a pseudo-Voigt function. The background due to the amorphous quartz cell was estimated from linear interpolation between manually selected N points (40 ≤ N ≤ 70) chosen to ensure they did not interact with the Bragg reflections of the phase of interest. In addition to the profile parameter (σ1) describing the Gaussian contribution to the Bragg peak profiles, anisotropic strain broadening parameters (i.e. hkl dependent) have been refined through the Shkl microstrain parameters in Stephens’ model.22 The lattice parameters, fractional occupancy of all oxygen sites, atomic positions and atomic displacement parameters (either isotropic or anisotropic) were refined for each pattern. Any additional constraints are noted when describing specific models. Electron diffraction (ED) experiments at room temperature were performed using a Transmission Electron Microscope (TEM) JEOL2100 LaB6 operating at 200 kV. Powders of x = 0 and 0.5 were crushed in dry ethanol and a drop of the suspension was deposited on a copper grid covered by a carbon film. Diffraction patterns were collected with a GATAN Orius 200D Charge Coupled Device (CCD) camera.
3. Results and discussion
3.1. Thermogravimetric analysis
The behaviour of the x = 0 and 0.5 compositions was studied by thermogravimetric analysis in wet air (pH2O = 0.18–0.60 atm) to check the influence of the presence of oxygen vacancies on water uptake in these electron conducting phases. Two series of measurements were performed according to the following procedure: the powders were heated in dry N2 from RT up to 800 °C at a rate of 10 °C min−1 followed by isothermal heating at 800 °C for 1 h, in order to achieve thermodynamic equilibrium and maximize the oxygen vacancy content. The sample was then cooled to 260 °C at a rate of 10 °C min−1 in dry N2 and maintained at 260 °C for 1 h. Afterwards, the gas was changed to dry air and after equilibration the sample was stepwise heated to 800 °C before cooling. To check for possible water insertion, a second treatment that consisted of heating/cooling in wet air was performed after the cycle in dry N2. Thermogravimetric data measured for the x = 0 sample are displayed in Fig. 2. | ||
| Fig. 2 Thermogravimetric analysis (TGA) of NdBaCo2O5+δ in dry N2 and subsequent heating/cooling cycles in dry (black) or wet (blue) air; the heating/cooling rate is 10 °C min−1 and the flow rate is 50 mL min−1. Gas change from N2 to air at 260 °C is indicated by a vertical dotted line. | ||
The large mass decrease subsequent to the heating/cooling cycle in dry nitrogen (Δm/m ∼ 1.75%) corresponding to a loss of ∼0.5 oxygen/f.u. suggests the formation of a large number of oxygen vacancies. As soon as the gas was changed to dry air at 260 °C, a sharp mass increase immediately occurred reflecting rapid oxygen exchange, suggesting that the sample recovered its initial oxygen composition (NdBaCo2O5.5). On heating at T > 260 °C, the mass decreased, reflecting rapid reversible oxygen exchange with the atmosphere.
Switching to wet air after the heating/cooling cycle performed in dry N2 induces an instantaneous mass increase similar to that observed in dry air. After the stepwise heating to 800 °C and subsequent cooling, a small difference in weight with respect to cycling in dry air was observed. The mass of the x = 0.5 sample exhibits little change in dry N2 reflecting the stability of this composition towards oxygen and humidity when stored under ambient conditions. When heated in air at 260 °C, the behaviour is similar to that of the x = 0 sample reflecting immediate chemical oxidation and possible reversible insertion of proton defects (Fig. S.I. 1†).
It is important to stress that the difference in mass between the ‘wet’ and ‘dry’ treatments observed for the x = 0 and 0.5 samples may indicate the intercalation of proton defects and also reveal the adsorption of free molecules of water and protonic entities such as hydroxide or carbonate. For example, in the XRD pattern of the PrBaCo2O5+δ sample hydrated at 500 °C for 12 h in wet air (pH2O ∼ 0.1 bar), Grimaud et al. showed evidence of the formation of barium carbonate.13 Since oxidation is the main process that fills the oxygen vacancies of highly deficient double perovskites in wet and dry air, a large oxygen deficiency seems to be not the only requirement for effective proton incorporation in the LnBaCo2O5+δ family of materials, likely due to a limited number of lowest energy sites for the protons in this A-site ordered double perovskite layered structure. Indeed, water solubility approaches the saturation limit in cubic perovskite-type proton conducting electrolytes such as SrTi0.95Sc0.05O3−δ,23 whereas water solubility is reduced in distorted perovskite-type electrolytes and complex perovskites which may exhibit B-site ordering.23,24 To check the stability of the layered perovskites, NdBaCo2−xMnxO5+δ, x = 0 and 0.5 under humid conditions, diffraction experiments have been conducted.
3.2. Structural analysis at room temperature
 | ||
| Fig. 3 [100] zone axis electron diffraction patterns of (a) NdBaCo2O5+δ (x = 0) and (b) NdBaCo1.5Mn0.5O5+δ (x = 1.5). The patterns are indexed considering the cubic parent perovskite unit cell. The white arrows in x = 0 and x = 0.5 indicate superstructure reflections related to A-site cation ordering along the c-axis, whereas the black arrow in x = 0 indicates a superstructure reflection resulting in cell doubling in b due to oxygen vacancy ordering. | ||
SAED revealed the existence of two main structural features for the x = 0 composition: doubling of two lattice parameters corresponding to an ap × 2ap × 2ap supercell, where ap is the lattice parameter for the cubic perovskite, was observed. The superstructure reflections along c* have stronger intensities than those along b*, in agreement with the ordering of the Nd and Ba cations in alternate layers along the c-axis and ordering between oxygen and vacancies along the b-axis in the lanthanide layer, as reported in the literature.14 For the x = 0.5 composition (Fig. 3b), the cation ordering between Nd and Ba cations is retained, whereas oxygen and vacancy ordering along b* has disappeared likely due to the lower oxygen content due to synthesis under reduced oxygen pressure (i.e. argon atmosphere).18
 | ||
| Fig. 4 (a) Rietveld refinement at room temperature from the backscattering bank of NdBaCo2O5+δ with (b) an emphasis on 1.82 < d < 2.02 Å showing the superstructure peak of 014 associated with the ordering of the oxygen vacancies. Indexing refers to the (ap × 2ap × 2ap) cell. | ||
The orthorhombic crystal structure ap × 2ap × 2ap comprises two crystallographically distinct cobalt sites Co1 and Co2 in octahedral and pyramidal coordination, respectively, and seven crystallographically independent oxygen atoms: O1 and O2 in the Ba layer, O3, O4 and O5 at the equatorial positions of the Co polyhedra, and O6 and O7 in the Nd layer (Fig. 5).
 | ||
| Fig. 5 Crystal structure of NdBaCo2O5.5 highlighting the seven crystallographically distinct oxygen positions. Ordering of oxygen vacancies was observed in the Nd layer, with O6 preferentially occupied and O7 partially occupied. | ||
The atomic displacement parameters (ADPs) of all the atoms were refined isotropically. Due to high correlations with site occupancy, the ADPs of the oxygen atoms in the same layer were initially constrained to be equal. However, this gave very small values for Biso of the oxygen atoms in the Nd layer (O6 and O7) in comparison to the Biso of the other oxygen atoms. Consequently, the Biso of all the oxygen atoms were constrained to be equal. The refinement of site occupancies showed that the oxygen vacancies were primarily on one of the two crystallographically independent oxygen sites located in the Nd layers and may exist at the nearest-neighboring O3, O4 and O5 sites in the Co-layer. The difference in site occupancy between O6 [1.00(5)] and O7 [0.20(4)] is consistent with vacancy ordering.25,26 The occupancy factors of the oxygen sites in the Ba layer (O1 and O2) are within a standard deviation of 1.0 and were therefore fixed to 1.0. Moreover, this value did not vary when the sample was heated/cooled in Ar or air and was therefore maintained fixed in all of the refinements. The total oxygen content corresponded to 5.5(2) atoms/formula unit (f.u.) and agreed with iodometric titration results.18 Therefore, mainly Co3+ ions are present in octahedral CoO6 and pyramidal CoO5 environments. The structural parameters of NdBaCo2O5+δ at RT are listed in Table 1.
| Atom | Position (z), occupation factor (Occ.) | B iso (Å2) |
|---|---|---|
| Space group Pmmm, a = 3.8914(2) Å, b = 7.84422(44) Å, c = 7.59312(42) Å with sites Nd 2p (0.5 y 0.5), Ba 2o (0.5 y 0), Co1 2r (0 0.5 z), Co2 2q (0 0 z), O1 1a (0 0 0), O2 1e (0 0.5 0), O3 4u (0 y z), O4 2s (0.5 0 z), O5 2t (0.5 0.5 z), O6 1g (0 0.5 0.5), O7 1c (0 0 0.5); # constrained, * fixed, χ2 ∼ 0.9, RB ∼ 2.8%, RF ∼ 2.8%. | ||
| Nd | y: 0.271(1) | 0.74(8) |
| Ba | y: 0.250(1) | 0.7(1) |
| Co1 | z: 0.247(2) | 0.7(2) |
| Co2 | z: 0.255(2) | 0.3(2) |
| O1, O2 | Occ: *1.00 | #1.14(5) |
| O3 | y: 0.2393(1), z: 0.2868(1), Occ: 0.98(2) | #1.14(5) |
| O4 | z: 0.306(2), Occ: 0.94(4) | #1.14(5) |
| O5 | z: 0.268(2), Occ: 0.98(4) | #1.14(5) |
| O6 | Occ: 1.00(5) | #1.14(5) |
| O7 | Occ: 0.20(4) | #1.14(5) |
| O-content | 5.5(2) | |
In contrast to NdBaCo2O5+δ, the symmetry of NdBaCo1.5Mn0.5O5+δ (x = 0.5) at room temperature was found to be tetragonal P4/mmm with the unit cell ap × ap × 2ap. This model has a single transition metal site for the (Co/Mn) ions and three O sites (O1, O2 and O3). The crystal structure and the Rietveld plot at room temperature are shown in Fig. 6. The excluded region at 1.49 < d < 1.51 Å was introduced for a small bump likely due to an experimental artifact. The mixed B-site possesses weak overall scattering due to the competing Mn and Co scattering lengths, −3.73 and 2.49 fm, respectively. When the ADPs of the mixed site were refined anisotropically, this appeared to cause slightly negative β33 for (Co,Mn) and large standard deviations for O1 whose occupancy was very low. Therefore the ADP of (Co/Mn) was refined isotropically and the Biso of O1 was fixed at 0. Refinement results indicated that the majority of vacancies were contained in the Nd layer at the O1 site whose occupancy refined to ∼0.088(8), none at the O2 site whose occupancy factor was therefore fixed at 1.0 and very little at the O3 site. The overall oxygen content of ∼5.00(8)/f.u. corresponds to an equal amount of (Co,Mn)3+ and (Co,Mn)2+ cations in a square pyramidal coordination. The change from orthorhombic to tetragonal symmetry between the x = 0 and x = 0.5 compositions and the absence of the superstructure reflections related to vacancy ordering in the latter are consistent with the SAED pattern. The higher symmetry of the x = 0.5 compound is due to the presence of a very small amount of oxygen atoms in the Nd-layer due to the annealing conditions in the inert atmosphere required to prevent the stabilization of the hexagonal perovskite BaMnO3−δ impurity.18 The R factors and refined structural parameters for x = 0.5 at RT are given in Table 2.
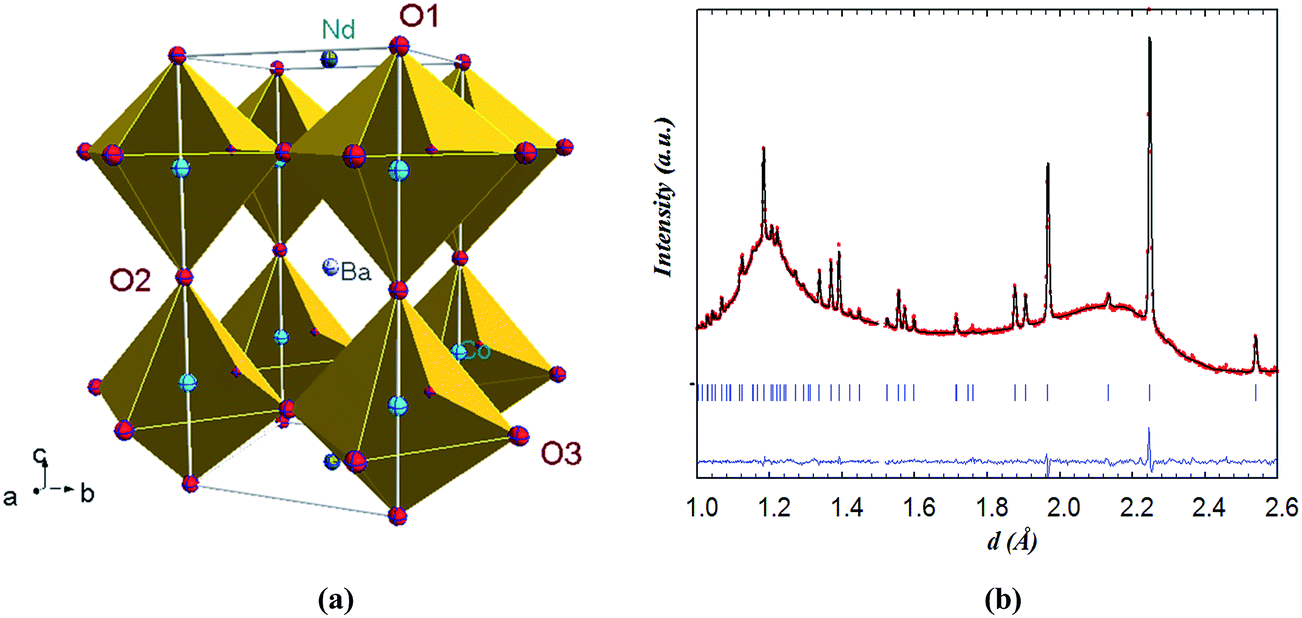 | ||
| Fig. 6 (a) Crystal structure of NdBaCo1.5Mn0.5O5+δ (P4/mmm) with the transition metals highlighted in blue (b) Rietveld refinement at room temperature from the backscattering data bank. The undulating background is due to the quartz sample environment. The excluded region at 1.49 < d < 1.51 Å was introduced for a small bump likely due to an experimental artifact. | ||
| Atom | Position (z), occupation factor (Occ.) | β 11, β22, β33 (× 100) |
|---|---|---|
| a Space group P4/mmm, a = 3.9334(1) Å, c = 7.6197(4) Å with sites Nd 1a (0 0 0), Ba 1b (0 0 0.5), Co 2h (0.5 0.5 z), O1 1c (0.5 0.5 0), O2 1d (0.5 0.5 0.5), and O3 4i (0.5 0 z), # fixed, * refined isotropically, χ2 ∼ 2.1, RB ∼2.2%, RF ∼ 3.0%. | ||
| Nd | 1.4(1), 1.4(1), 0.20(6) | |
| Ba | 1.3(2), 1.3(2), 0.6(1) | |
| Co,Mn | z: 0.251(2) | *0.7(2) |
| O1 | Occ: 0.088(8) | #0.0 (isotropic) |
| O2 | #Occ: 1.0 | 1.5(2), 1.5(2), 0.25(8) |
| O3 | z: 0.1981(2), Occ: 0.98(2) | 1.6(2), 2.4(2), 0.44(5) |
| O content | 5.00(8) | |
3.3. Structural analysis as a function of temperature in an argon atmosphere
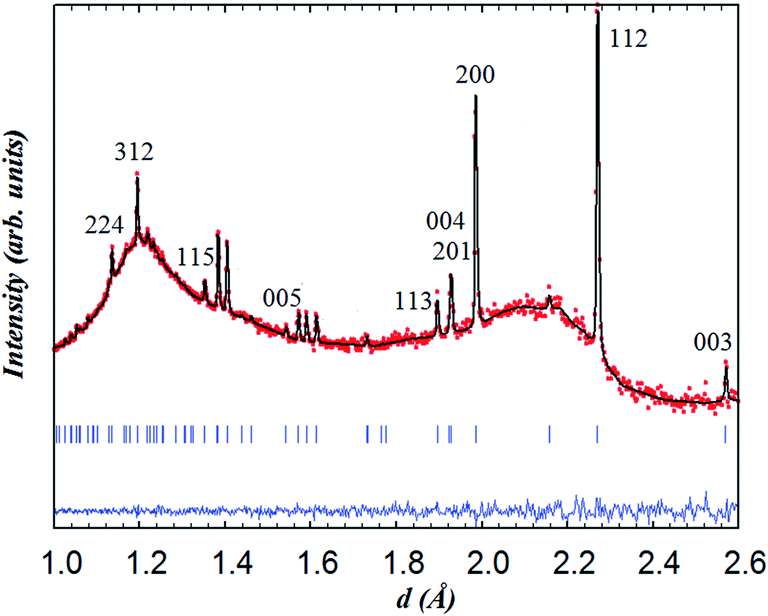 | ||
| Fig. 7 Rietveld refinement for NdBaCo2O5+δ from the data collected from the backscattering bank at 800 °C for 30 min under wet argon. The indexing refers to the (ap × ap × 2ap) cell. The undulating background is due to the quartz sample environment. | ||
| NdBaCo2O5+δ | Dry Ar, 800 °C | Wet Ar, 800 °C |
|---|---|---|
| a (Å) | 3.9657(3) | 3.9712(4) |
| c (Å) | 7.7179(8) | 7.7117(9) |
| Co (z) | 0.246(3) | 0.249(2) |
| O3 (z) | 0.2091(7) | 0.2063(6) |
| Occ. O1 | 0.42(5) | 0.32(2) |
| Occ. O3 | 0.88(3) | 0.90(3) |
| O-content | 4.9(2) | 4.9(1) |
| χ 2 | 0.65 | 0.7 |
| R B% | 4.2 | 3.7 |
| R F% | 10.8 | 11.3 |
The sample was therefore stepwise cooled in wet argon, meanwhile, datasets were collected at different temperatures to allow possible hydration/dehydration to be monitored through the variation of the oxygen content. Rietveld analysis of the datasets collected at relevant temperatures and performed using anisotropic ADPs for the metal positions and isotropic ADPs for the oxygen sites (since anisotropic ADPs gave a negative value of β33 for (O1)) did not show significant variation in the oxygen occupancy. Fig. 8 shows the Rietveld fit of the pattern collected at 260 °C, the lowest temperature reached on cooling in wet argon. Relevant structural parameters are given in Table 4.
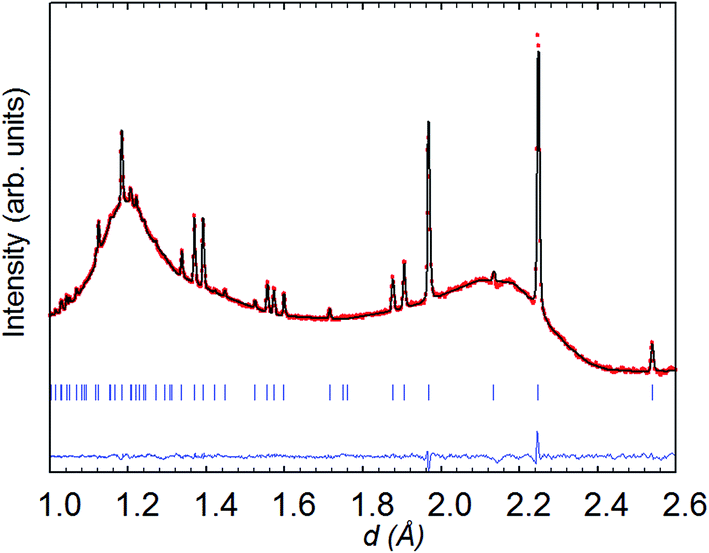 | ||
| Fig. 8 Rietveld refinement (space group P4/mmm) from the backscattering detector bank for NdBaCo2O5+δ at 260 °C in wet (40% D2O) argon; data collected for 6 h. | ||
| Atom | Position (z), occupation (Occ.) | β 11, β22, β33 (× 100) |
|---|---|---|
| a Space group P4/mmm, a = 3.9349(1) Å, c = 7.6197(3) Å with sites Nd 1a (0 0 0), Ba 1b (0 0 0.5), Co 2h (0.5 0.5 z), O1 1c (0.5 0.5 0), O2 1d (0.5 0.5 0.5), O3 4i (0.5 0 z), #Biso constrained to be equal, χ2 ∼ 2.2, RB ∼ 3%, RF ∼ 3.7%. | ||
| Nd | 2.3(1), 2.3(1), 0.54(5) | |
| Ba | 2.0(2), 2.0(2), 0.45(7) | |
| Co | z: 0.2468(7) | 1.9(2), 1.9(2), 0.51(7) |
| O1 | Occ: 0.264(8) | #1.72(4) |
| O2 | #1.72(4) | |
| O3 | z: 0.2035(2), Occ: 0.944(8) | #1.72(4) |
| O-content | 5.04(4) | |
| T (°C) atom | 800 (30 min) | 260 (14 h) | 300 (5 h) | 400 (3 h) | 500 (2 h) | 260 (11 h) | 200 (20 min) |
|---|---|---|---|---|---|---|---|
| a Refinement with anisotropic ADPs for all the atoms except Biso for (Co,Mn) and Biso = 0 for O1. | |||||||
| O1 | 0.072(8) | 0.072(0) | 0.080(8) | 0.072(8) | 0.064(8) | 0.080(8) | 0.064(8) |
| O3 | 0.95(4) | 0.98(1) | 0.99(2) | 0.97(2) | 0.96(2) | 0.99(2) | 0.97(3) |
| O-content/f.u. | 4.9(2) | 4.98(5) | 5.03(7) | 4.94(7) | 4.90(8) | 5.03(7) | 5.0(1) |
Fig. 9 displays the Rietveld refinement of the data collected for 11 h at 260 °C in wet argon with Table 6 listing the corresponding structural parameters.
 | ||
| Fig. 9 Rietveld profile (backscattering detector bank) for NdBaCo1.5Mn0.5O5+δ in wet argon at 260 °C; data collected for 11 h; refinement with anisotropic ADPs for all the atoms except for (Co,Mn) for which Biso was refined and Biso for O1 fixed at 0. | ||
| Atom | Position (z), occupancy (Occ.) | β 11, β22, β33 (× 100) |
|---|---|---|
| # Refined isotropically. * Fixed. | ||
| Nd | 1.9(1), 1.9(2), 0.28(5) | |
| Ba | 1.7(2), 1.7(2), 0.7(1) | |
| Co,Mn | z: 0.252(2) | #0.7(2) |
| O1 | Occ: 0.080(8) | *Biso = 0 |
| O2 | 2.3(2), 2.3(2), 0.32(7) | |
| O3 | z: 0.1986(2), Occ: 0.99(2) | 2.0(2), 2.6(2), 0.81(5) |
| O-content | 5.03(7) | |
The behaviour of the x = 0.5 sample in dry and wet argon is governed by thermal expansion and exhibits some hysteresis at low temperature (e.g. at 260 °C) likely due to low equilibration. The evolution of the structural parameters is displayed in Fig. S.I. 3.†
3.4. Structural evolution in wet air
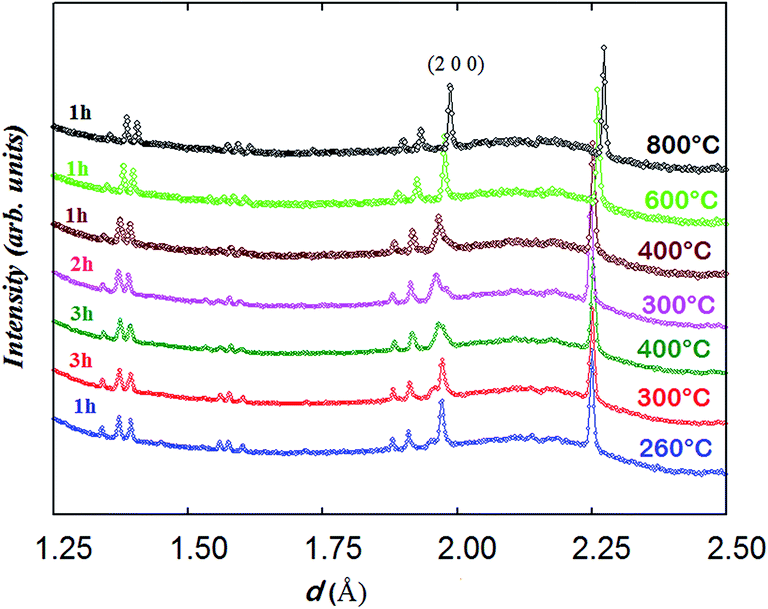 | ||
| Fig. 10 Evolution of the NPD patterns (backscattering detector bank) of NdBaCo2O5+δ obtained in wet air from 260 to 800 °C, index corresponds to the P4/mmm symmetry. The two orthorhombic phases co-exist in the temperature range of 260–400 °C; the collection duration of each dataset is indicated; time increases upwards. | ||
The data collected at 260 °C for 1 h were initially fitted with two tetragonal phases with different lattice parameters and oxygen contents; however, the model did not resemble the observed pattern well with the reliability factors: global χ2 ∼ 1.45, RB ∼ 8.0%, RF ∼ 5.6% for phase 1 and RB ∼ 3.0%, RF ∼ 4.6% for phase 2. The use of two orthorhombic phases yielded lower reliability factors: global χ2 ∼ 1.0, RB ∼ 5.7%, RF ∼ 4.8% for phase 1 and RB ∼ 3.0%, RF ∼ 4.4% for phase 2. It is interesting to mention that using a single orthorhombic phase resulted in a higher reliability factor, χ2 = 1.8.
In contrast to the Pmmm structural model (ap × 2ap × 2ap) which fits the data of the as-prepared material at RT and on heating in dry Ar up to 400 °C, the Pmmm model (ap × ap × 2ap) that assumes disordered vacancies and consists of a single Co site and four different oxygen sites O1, O2, O3 and O4 was used in the two-phase refinements due to the large number of parameters and similarity of the two phases. All the sites were refined with isotropic ADPs. Due to the low occupancy at the O1 site in the Nd layer and similar symmetry to the O2 site in the Ba layer, Biso for O1 and O2 was constrained to be identical. Due to strong correlations between Biso and site occupancy for O3 and O4 (which are localized at the equatorial positions of the Co polyhedra and accommodate very little oxygen vacancies), Biso, occupancy and z positions of O3 and O4 were constrained to be the same in each phase. Relaxing these constraints did not change the quality of the refinement. The refinement of two strain broadening parameters, S400 and S040, was needed to fit accurately the data collected during the intercalation process and must be constrained to be equal for the two banks and the two phases to allow sensible evolution of the weight ratios as a function of time. The O3/O4 sites in phase 2 (oxidised phase) were found to be fully occupied and their occupancy was fixed at 1.0 in all of the two-phase refinements. Oxygen vacancies were mainly localised at the O1 site in the two phases although less in phase 2 than in phase 1. The distortion of the basal plane of the intercalated phase (phase 2) at 260 °C (a = 3.94483(7), b = 3.8960(8) Å) whose oxygen content refined to 5.5(2) atoms/f.u. is more pronounced than in the initial phase 1 (a = 3.9345(8), b = 3.9315(9) Å) whose oxygen-content refined to 4.9(1) atoms/f.u. In addition, the c parameter of phase 2 is larger than that in phase 1, 7.633(2) vs. 7.6181(6) Å, in agreement with the larger oxygen content (or proton defects) in the former. The fit of the pattern collected for 1 h at 260 °C in wet air is displayed in Fig. 11.
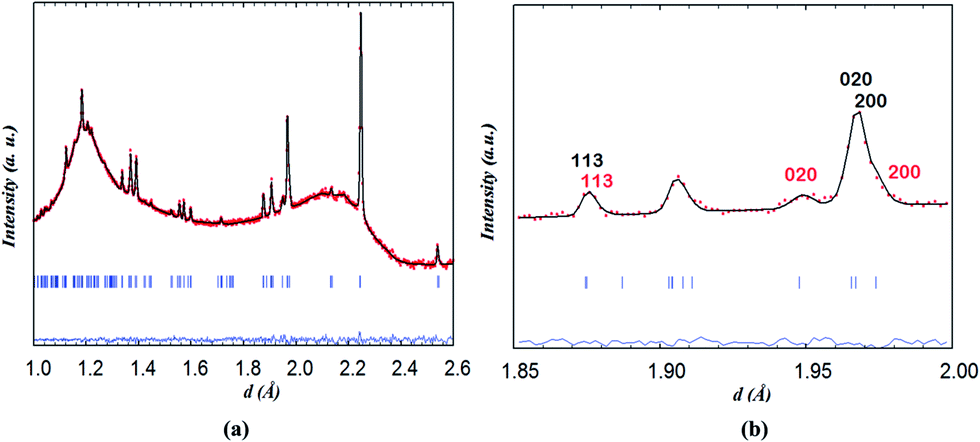 | ||
| Fig. 11 (a) Two-phase fit of neutron diffraction data collected on NdBaCo2O5+δ for 1 h at 260 °C in wet air and (b) emphasis on the contributions of (black) phase 1 and (red) phase 2. | ||
Heating above 260 °C enhances the nucleation and growth of the phase 2 whose δ value remains almost constant, at ∼0.5(2) in the temperature range of 260–400 °C. The oxygen content of phase 1 increases with time although it remains lower than that of phase 2 as illustrated in Fig. 12c. After 7 hours of heating in wet air, the two phases have a similar weight ratio and an “inversion” of the orthorhombicity is produced; the a and b lattice parameters of phase 2 which were significantly different at 260 °C become similar at 400 °C (a = 3.929(4) and b = 3.921(4) Å), whereas phase 1 which was pseudo tetragonal at 260 °C (a = 3.9345(8) and b = 3.9315(9) Å) becomes significantly orthorhombic at 400 °C (a = 3.949(1) and b = 3.9134(9) Å) as shown in Fig. 12a. This behavior may be due to the increased oxygen content in both phases (Fig. 12c) and possible ordering of the oxygen vacancies in phase 1 whose δ value approaches 0.5. The evolution of the c lattice parameters is mainly governed by thermal expansion (Fig. 12b) rather than chemical expansion consecutive to variations in the oxygen content and shows the presence of some hysteresis (e.g. at 400 °C). The c lattice parameter of phase 2 remains larger than that of phase 1, in agreement with the larger oxygen content in the former. Heating at 600 °C promotes the intercalation reaction kinetically and the two phases merge into a single tetragonal phase with δ ∼ 0.4(2), suggesting that the oxygen transport at 600 °C is sufficiently rapid to overcome the kinetic limitations experienced at lower temperatures. The sample remains in a single tetragonal phase at 800 °C and on subsequent cooling. No secondary phases were detected at any temperature. In the temperature range of the coexistence of the two phases, the sample undergoes a fast intercalation reaction between an oxygen deficient (δ ∼ 0) and an oxygen rich phase (δ ∼ 0.5(2)). Variation of the cell parameters, oxygen content and weight fractions is displayed in Fig. 12.
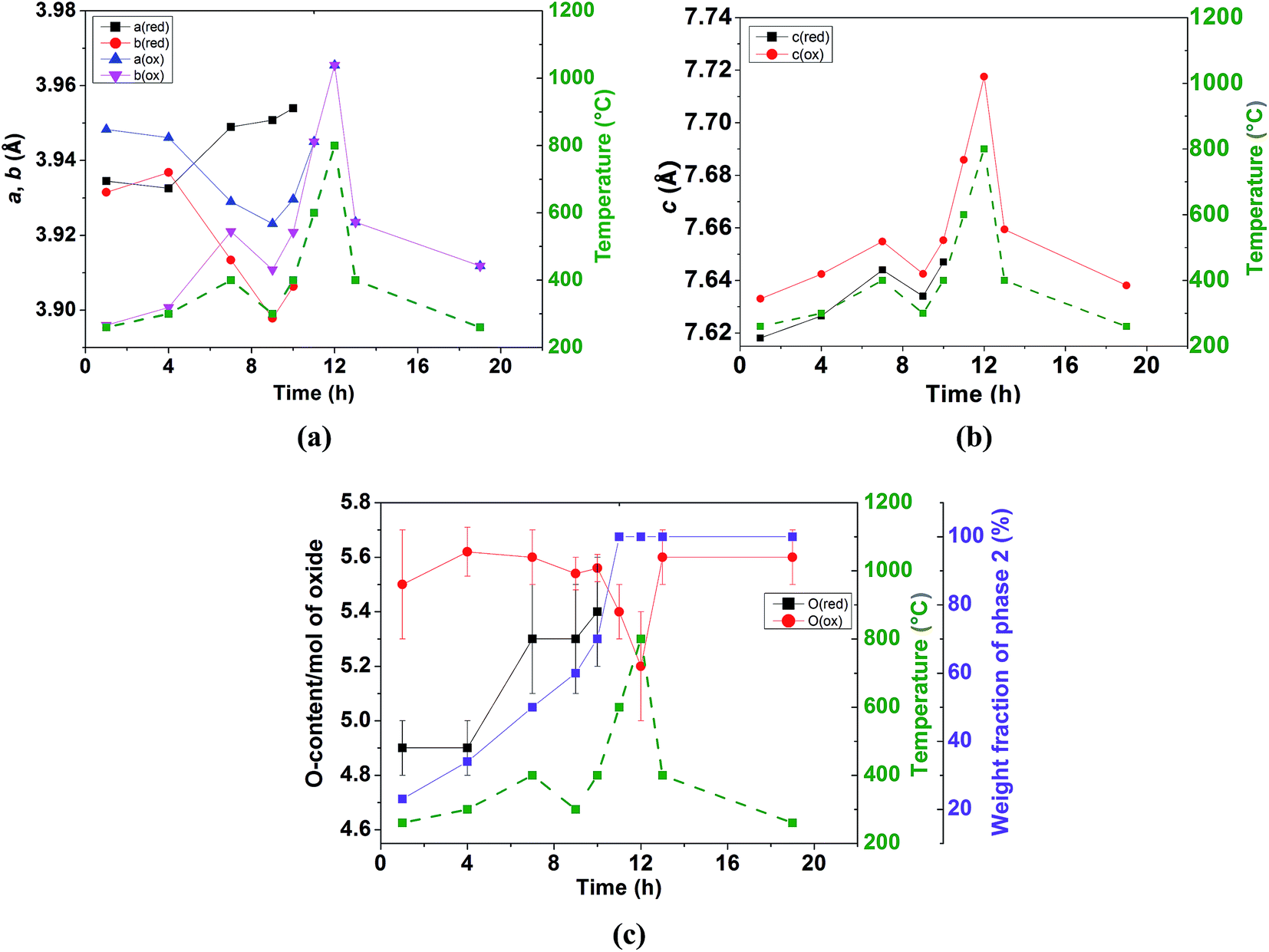 | ||
| Fig. 12 Structural variation of NdBaCo2O5+δ (x = 0) in wet air; (a) a and b lattice parameters, (b) c lattice parameter (c) oxygen content and weight fraction of phase 2; (red) and (ox) relate to phases 1 and 2, respectively. | ||
The two-phase tetragonal model consistent with the diffraction pattern recorded at 200 and 300 °C for 90 minutes is shown in Fig. 13. The main results of the Rietveld refinements are displayed in Table 7. At 200 °C, the smaller a- and larger c-lattice parameters of phase 2 (oxidised phase) with respect to phase 1 (reduced phase) are consistent with the larger oxygen content in the former. This behaviour is exhausted at 300 °C where the proportion of phase 2 has reached ∼80%.
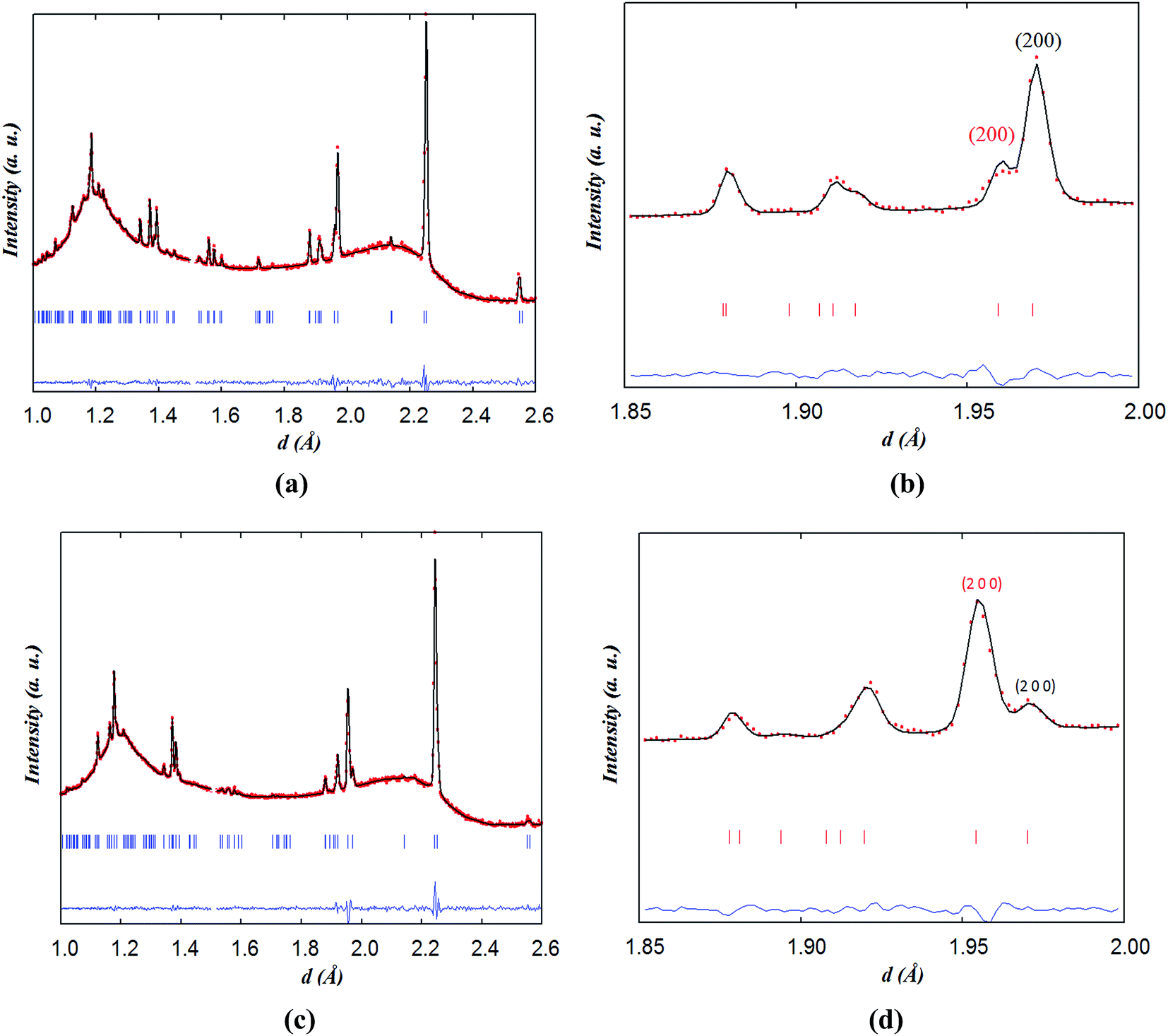 | ||
| Fig. 13 Two-phase fits for neutron data collected on NdBaCo1.5Mn0.5O5+δ in wet air for 90 minutes at (a and b) 200 °C and (c and d) 300 °C using a tetragonal model; (red) intercalated and (black) reduced phases. | ||
| Phase 1 | Phase 2 | |
|---|---|---|
| a The occupation of O3 and O4 have been fixed at 1.0. | ||
| 200 °C (1 h 30 min) | ||
| a (Å) | 3.9380(2) | 3.9179(4) |
| c (Å) | 7.6421(5) | 7.6685(2) |
| Occ. O1 | 0.056(8) | 0.40(6) |
| Oxygen content | 5.12(2) | 5.8(1) |
| Weight fraction (%) | 80(5) | 20(5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| 300 °C (1 h 30 min) | ||
| a (Å) | 3.9405(5) | 3.9090(2) |
| c (Å) | 7.653(2) | 7.6809(5) |
| Occ. O1 | 0.17(4) | 0.42(2) |
| Oxygen content | 5.33(8) | 5.82(3) |
| Weight fraction (%) | 20(5) | 80(5) |
The reason for the existence of the two phases during the exposure of the x = 0 and 0.5 samples to wet air may stem from the two accessible values of δ for the NdBaCo2O5+δ and NdBaCo1.5Mn0.5O5+δ compositions, close to 0.0 and 0.5 with a portion of the sample oxidizing preferentially. The need for two phases to fit the data collected in wet air for 10 h up to 400 °C on the x = 0 sample may indicate that the kinetics of oxidation are relatively slow and that the oxidation process is limited for the time that the samples were held at each temperature between 260 and 400 °C (1–3 hours) because of the low flow rate of the gas (20 mL min−1) and the large quantity of the powder sample used in the neutron experiment (∼4 g). Conversely, the TGA experiment, using a smaller quantity of sample (100 mg) and higher flow rate (50 mL min−1), would suggest that the surface exchange and diffusion of oxygen are fast in the reduced phase under these conditions.
Although our diffraction experiment highlighted the stability of the layered phases of x = 0 and 0.5 under humid conditions, it was difficult to establish the incorporation of proton defects into this oxide family. Water dissociation strongly depends on the basicity of the ceramics and the presence of the Ba cation at the A site. This basicity is however lowered by the substitutions with less basic elements such as Nd which prevent the reaction with acidic gases such as H2O and therefore hydrolysis. It is worthwhile stressing that despite intensive studies on systems that insert a large amount of proton defects, such as the brownmillerite Ba2In2O5 and derived compositions34,35 and orthorhombic perovskite BaCe0.9Y0.1O2.9536 or cubic La0.73Ba0.27ScO2.86537 or SrZr0.95Sc0.05O3−δ38 perovskites, the determination of the proton site is only suggested at very low temperatures, far from the conducting conditions. It should be mentioned that the improvement of the electrochemical properties of the layered cobaltite PrBaCo2O5+δ with increased pH2O reported by Grimaud et al.13 can be related to the formation of secondary phases such as hydroxides which are very good proton conductors and consequently did not reflect the properties of their bulk materials. Indeed, Jalarvo et al.39 showed by in situ synchrotron X-ray diffraction that in the case of the proton conducting perovskites, Sr4(Sr2Nb2)O11·nH2O and Sr4(Sr2Ta2)O11·nH2O, secondary phases such as Sr(OH)2 can dominate proton conduction.
4. Conclusion
The vacancy ordered NdBaCo2−xMnxO5+δ (x = 0, 0.5) layered perovskites have been investigated in dry and wet argon and wet air by in situ high temperature neutron powder diffraction. The materials were found to be stable under strongly humid conditions up to 800 °C. However, the incorporation of proton defects could not be clearly established from our in situ data collected under wet conditions as a function of temperature. This suggests that the basic characteristics of the NdBaCo2−xMnxO5+δ phases owing to the Ba cation along with the presence of large amount of oxygen deficiencies and high oxygen mobility which are the key factors involved in the mechanism of water dissociation/insertion in cubic perovskite proton conducting electrolytes (e.g. acceptor-doped barium zirconates and cerates) appear not to be the most relevant factors for water dissociation/incorporation in the ordered Co-based double perovskites studied.Acknowledgements
One of the authors (M. B.) thanks P. D. Battle (Oxford University) and T. Roisnel (University of Rennes) for much helpful advice during the course of the refinements as well as G. Gauthier (Industrial University of Santander) and F. Tonus (Imperial College London) who provided with very valuable comments. The authors also thank R. Haynes and the rest of the sample environment team at ISIS for their assistance. This research was partially supported by the ANR IDEA-MAT grant (ANR-A2-BS08-0012-01) which provided a Post-doctoral Fellowship for J. Hanlon.References
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- S. C. Singhal and K. Kendall, High Temperature Solid Oxide Fuel Cells: Fundamentals, Design and Applications, Oxford, 2003 Search PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- C. Zuo, S. Zha, M. Liu, M. Hatano and M. Uchiyama, Adv. Mater., 2006, 18, 3318–3320 CrossRef CAS PubMed.
- H. Iwahara, Solid State Ionics, 1996, vol. 86–88, Part 1, pp. 9–15 Search PubMed.
- H. Iwahara, T. Yajima, T. Hibino and H. Ushida, J. Electrochem. Soc., 1993, 140, 1687–1691 CrossRef CAS PubMed.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1997, 97, 1–15 CrossRef CAS.
- T. Norby, Solid State Ionics, 1999, 125, 1–11 CrossRef CAS.
- B. Lin, S. Zhang, L. Zhang, L. Bi, H. Ding, X. Liu, J. Gao and G. Meng, J. Power Sources, 2008, 177, 330–333 CrossRef CAS PubMed.
- H. Uchida, S. Tanaka and H. Iwahara, J. Appl. Electrochem., 1985, 15, 93–97 CrossRef CAS.
- G. Goupil, T. Delahaye, B. Sala, F. Lefebvre Joud and G. Gauthier, Solid State Ionics, 2014, 263, 15–22 CrossRef CAS PubMed.
- A. Grimaud, F. Mauvy, J. M. Bassat, S. Fourcade, L. Rocheron, M. Marrony and J. C. Grenier, J. Electrochem. Soc., 2012, 159, B683–B694 CrossRef CAS PubMed.
- A. Maignan, C. Martin, D. Pelloquin, N. Nguyen and B. Raveau, J. Solid State Chem., 1999, 142, 247–260 CrossRef CAS.
- A. Tarancon, S. J. Skinner, R. J. Chater, F. Hernandez-Ramirez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175–3181 RSC.
- A. Tarancón, A. Morata, G. Dezanneau, S. J. Skinner, J. A. Kilner, S. Estradé, F. Hernández-Ramírez, F. Peiró and J. R. Morante, J. Power Sources, 2007, 174, 255–263 CrossRef PubMed.
- K. V. Kordesch and G. R. Simader, Chem. Rev., 1995, 95, 191–207 CrossRef CAS.
- T. Broux, M. Bahout, J. M. Hanlon, O. Hernandez, S. Paofai, A. Berenov and S. J. Skinner, J. Mater. Chem. A, 2014, 2, 17015–17023 CAS.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef PubMed.
- Mantid, Manipulation and Analysis Toolkit for Instrument Data. Mantid Project, 2013.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- P. W. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1999, 125, 285–302 CrossRef CAS.
- A. S. Nowick and Y. Du, Solid State Ionics, 1995, 77, 137–146 CrossRef CAS.
- S. Streule, A. Podlesnyak, E. Pomjakushina, K. Conder, D. Sheptyakov, M. Medarde and J. Mesot, Phys. B, 2006, 378–380, 539–540 CrossRef CAS PubMed.
- J. C. Burley, J. F. Mitchell, S. Short, D. Miller and Y. Tang, J. Solid State Chem., 2003, 170, 339–350 CrossRef CAS.
- M. Lehtimaki, H. Yamauchi and M. Karppinen, J. Solid State Chem., 2013, 204, 95–101 CrossRef CAS PubMed.
- J. H. Kim, L. Mogni, F. Prado, A. Caneiro, J. A. Alonso and A. Manthiram, J. Electrochem. Soc., 2009, 156, B1376–B1382 CrossRef CAS PubMed.
- R. A. Cox-Galhotra, A. Huq, J. P. Hodges, J.-H. Kim, C. Yu, X. Wang, A. J. Jacobson and S. McIntosh, J. Mater. Chem. A, 2013, 1, 3091–3100 CAS.
- I. Sosnowska, R. Przeniosło, W. Schäfer, W. Kockelmann, R. Hempelmann and K. Wysocki, J. Alloys Compd., 2001, 328, 226–230 CrossRef CAS.
- T. Shimoyama, T. Tojo, H. Kawaji, T. Atake, N. Igawa and Y. Ishii, Solid State Ionics, 2008, 179, 231–235 CrossRef CAS PubMed.
- L. Jantsky, H. Okamoto, A. Demont and H. Fjellvåg, Inorg. Chem., 2012, 51, 9181–9191 CrossRef CAS PubMed.
- M. Matvejeff, M. Lehtimäki, A. Hirasa, Y. H. Huang, H. Yamauchi and M. Karppinen, Chem. Mater., 2005, 17, 2775–2779 CrossRef CAS.
- V. Jayaraman, A. Magrez, M. Caldes, O. Joubert, F. Taulelle, J. Rodriguez-Carvajal, Y. Piffard and L. Brohan, Solid State Ionics, 2004, 170, 25–32 CrossRef CAS.
- I. Ahmed, C. S. Knee, M. Karlsson, S. G. Eriksson, P. F. Henry, A. Matic, D. Engberg and L. Börjesson, J. Alloys Compd., 2008, 450, 103–110 CrossRef CAS PubMed.
- K. S. Knight, Solid State Ionics, 2001, 145, 275–294 CrossRef CAS.
- E. Kendrick, K. S. Knight, M. S. Islam and P. R. Slater, Solid State Ionics, 2007, 178, 943–949 CrossRef CAS PubMed.
- T. Ito, T. Nagasaki, K. Iwasaki, M. Yoshino, T. Matsui, N. Igawa and Y. Ishii, Solid State Ionics, 2006, 177, 2353–2356 CrossRef CAS PubMed.
- N. Jalarvo, C. Haavik, C. Kongshaug, P. Norby and T. Norby, Solid State Ionics, 2009, 180, 1151–1156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta02776d |
| This journal is © The Royal Society of Chemistry 2015 |