
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect self-assembly of Fe2O3/reduced graphene oxide nanocomposite for high-performance lithium-ion batteries†
Lisong
Xiao
*a,
Matthias
Schroeder
b,
Sebastian
Kluge
a,
Andrea
Balducci
b,
Ulrich
Hagemann
c,
Christof
Schulz
ad and
Hartmut
Wiggers
*ad
aInstitute for Combustion and Gas Dynamics – Reactive Fluids (IVG), University of Duisburg-Essen, Duisburg 47057, Germany. E-mail: lisong.xiao@uni-due.de; hartmut.wiggers@uni-due.de; Fax: +49 2033798159
bMEET Battery Research Centre – Institute of Physical Chemistry, University of Münster, Münster 48149, Germany
cInterdisciplinary Center for Analytics on the Nanoscale (ICAN), University of Duisburg-Essen, Duisburg 47057, Germany
dCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Duisburg 47057, Germany
First published on 28th April 2015
Abstract
In this study, Fe2O3/reduced graphene oxide (rGO) nanocomposites were prepared using a direct self-assembly of oppositely charged Fe2O3 nanoparticles (NPs) and graphene oxide (GO) sheets, followed with a low-temperature hydrothermal reduction process. The characterization of the nanocomposite shows that Fe2O3 NPs with an average diameter of about 9 nm are uniformly distributed on well-exfoliated rGO layers. The nanocomposites show a high iron oxide mass loading of 63%. The electrical conductivity of the composite was significantly enhanced by about 6 orders of magnitude in comparison to pure Fe2O3 NPs. The characterization of the composite as an anode material for lithium-ion batteries (LIBs) demonstrated a strong positive synergistic effect with respect to its electrochemical performance. Fe2O3/rGO exhibited a capacity of 600 mA h g−1 at a current density of 0.1 A g−1, and even more than 180 mA h g−1 at 10 A g−1 (approx. 17 C), indicating its superior high-rate performance. In addition, it features high efficiency at high rates and very good cyclic stability over a long cycle life of more than 550 cycles.
Introduction
With increasing demand for next generation portable electronic devices, electric vehicles and renewable energy storage systems, the development of rechargeable batteries such as LIBs with high capacity, excellent rate capability and superior cycling performance is increasingly becoming a crucial prerequisite. To meet the future challenges of lithium ion batteries, tremendous efforts are currently being devoted to develop innovative electrode materials.1–7Electrochemically active transition metal oxides have emerged as promising anode materials for rechargeable LIBs due to their high theoretical capacities compared with commercially used graphite (372 mA h g−1).3,8–11 Among of them, Fe2O3 has been attracting more and more research attention owning to its low processing cost and environmental friendliness.12,13 However, Fe2O3 used as electrode material suffers from poor cycling performance caused by its low electrical conductivity and large volume changes (>200%) during the lithiation/delithiation processes and subsequent pulverization of particles.14–17 To address these issues which are mostly related to electrical conductivity and mechanical stability, fabrication of composite materials consisting of Fe2O3 NPs and rGO has been proposed resulting in significant positive effects on the battery performance.5,13,18–22 The promising properties are based on several nano-specific features: firstly, the small diameter of nanosized Fe2O3 shortens the Li-ion insertion/extraction pathways. Therefore, the transport of Li-ions into the interior of the Fe2O3 NPs is fast, allowing for high charging and discharging rates.23–25 Secondly, the drastically increased surface area of the nanoparticulate active material enhances the storage capacity beyond that of conventional bulk materials.20,24,26 Thirdly, rGO acts as a highly conductive, continuous and flexible matrix in the nanocomposite to accommodate the large volume changes of Fe2O3 NPs and even further keeps fractured Fe2O3 pieces trapped within the conductive matrix, leading to enhanced cycle stability.4,5,27 The new material, therefore, counteracts all three conventional problems: limited storage capacity, limited rate capability and limited durability.
To fabricate such nanocomposites, “in situ synthesis”,13,16,20,21,28–30 “self-assembling”31–35 and “spray-drying”15,36,37 are commonly used methods. For the in situ synthesis of Fe2O3/rGO nanocomposites, GO prepared via Hummer's method38 is generally exfoliated and dispersed in aqueous solutions in the presence of iron salts. Iron ions adsorb at the GO layers and form iron hydroxide particles by hydrolysis/precipitation. In a subsequent hydrothermal process, iron hydroxide is converted to iron oxide and GO is reduced to rGO at comparably high temperature. The self-assembling method allows coupling of previously prepared iron oxide NPs with GO in a solvent. It is based on electrostatic forces between negatively charged GO and positively charged iron oxide NPs. Unfortunately, this method requires additional modification of the usually negatively charged iron oxide nanoparticle surface to render iron oxide a positively charged surface, e.g., by using aminopropyltrimethoxysilane33,35 or by ionic charge exchange on the surface of the NPs by adding additional acid.32,34 After assembling, the iron oxide/GO composite undergoes a hydrothermal process to reduce GO. The spray-drying method deals with wrapping of iron oxide NPs in exfoliated GO layers via spraying a dispersion of iron oxide NPs and GO in a specifically equipped spray dryer. Moreover, additional devices for post-annealing are required for this method.15,36 Thus, it still remains a challenge to develop a simpler method to prepare Fe2O3/rGO nanocomposites for high-performance LIBs that can be scaled up to economically viable production.
We report a new and facile route to directly assemble Fe2O3 NPs with GO layers in an aqueous solution. This self-assembling process is driven by electrostatic interactions induced by the oppositely charged surfaces of Fe2O3 and GO. The Fe2O3 NPs used in this method were synthesized in a flame reactor, which possess positively charged surfaces as-synthesized. Therefore, our method works without using any surfactants or chemical linkers which would otherwise be required for the pre-modification of the Fe2O3 NPs. A low-temperature hydrothermal process in the presence of a reducing agent is then used to assemble a three-dimensionally interconnected structure of Fe2O3/rGO. This process can be easily scaled up to production scale required for battery material generation.
Experiment
Synthesis of Fe2O3 nanoparticles
Fe2O3 NPs with small particle size distribution were synthesized using a low-pressure flat flame reactor in which iron pentacarbonyl (Fe(CO)5, 1000 ppm) was added to a premixed H2/O2/Ar flame. The reactor has an almost one-dimensional and flat flame burning from bottom to top. Low pressure (30 mbar) was used to spatially extend the reaction zone leading to the formation of small iron oxide NPs with a narrow particle size distribution. Details of the synthesis including the underlying reaction mechanism have been described in previous publications.39 The as-synthesized Fe2O3 NPs were collected on a PTFE coated filter and used without any further purification.Synthesis of graphene oxide
GO was synthesized from natural graphite by a modified Hummer's method.38 In brief, graphite (2.0 g) and NaNO3 (1.0 g) were put into a 1 l flask, which was seated in an ice bath. Concentrated H2SO4 (120 ml) and KMnO4 (6.0 g) were added slowly under stirring. Under continuous stirring, the obtained mixture was kept in the ice bath for 2 hours, subsequently heated up to 35 °C, and stirred for another 2 hours. After that, 250 ml of warm water (70 °C) was dropped into the mixture, resulting in a brownish dispersion. At the end, 40 ml of H2O2 aqueous solution (30%) was added and the color of the dispersion turned to brilliant yellow. The mixture was centrifuged and the obtained precipitate was washed three times with 5 wt% HCl aqueous solution and another three times with warm water. Finally, the product was dried overnight under vacuum at 60 °C.Preparation of Fe2O3/rGO nanocomposites
Aqueous dispersions of Fe2O3 nanoparticles and GO (both 4 mg ml−1) were prepared by mixing pure water with the Fe2O3 NPs and GO, respectively. To exfoliate the GO layers, strong ultrasonication was applied for 15 minutes. Subsequently, the Fe2O3 dispersion was added into the GO dispersion under stirring and the obtained mixture was further ultrasonicated for 15 minutes. Afterwards, ethylenediamine (EDA) as the reducing agent with a mass ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to GO was added under stirring and the dispersion was transferred into glassware, which was kept at 80 °C for 12 hours. After the 12 hour hydrothermal process, a hydrogel was obtained, which was collected and washed three times with water. The hydrogel was then dried in a freeze dryer (BETA 2-4 LD plus LT, Martin Christ Gefriertrocknungsanlagen GmbH, Germany) for further characterization and processing.
:1 to GO was added under stirring and the dispersion was transferred into glassware, which was kept at 80 °C for 12 hours. After the 12 hour hydrothermal process, a hydrogel was obtained, which was collected and washed three times with water. The hydrogel was then dried in a freeze dryer (BETA 2-4 LD plus LT, Martin Christ Gefriertrocknungsanlagen GmbH, Germany) for further characterization and processing.
Preparation of the electrodes
For electrochemical characterization, three different types of electrodes were prepared containing either Fe2O3/rGO, pure rGO or pure Fe2O3 NPs. The final electrodes contained 80 wt% active material, 10 wt% conductive agent (Super C 65, Timcal, Switzerland), and 10 wt% binder (PVDF, Kynar® 761, Arkema, France), and had a mass loading of around 0.9 mg cm−2. The binder was dissolved in NMP before the premixed active material and conductive agent were added. Dendritic copper foil (Schlenck, Germany) was used as current collector. The counter and the reference electrodes were made of metallic lithium (Rockwood Lithium, Germany). The electrolyte was EC:DMC 1:1 (w:w) 1 M LiPF6 (LP30, Selectilyte™, BASF, Germany) and a glass fiber separator was used (Whatman GF/D, GE Healthcare, United Kingdom). The cells were built in an argon filled glove box (MBraun, Germany, H2O and O2 content <1 ppm).
Characterization
The phase composition of all samples was determined by X-ray diffraction (XRD, PANalytical X-ray diffractometer X'Pert) with a Cu Kα radiation (λ = 1.5406 Å). Raman spectra were recorded at room temperature on a Renishaw inVia Raman microscope with the laser wavelength of 532 nm. X-ray photoelectron spectroscopy (XPS) was performed on a VersaProbe II by Ulvac-Phi, using monochromatic Al Kα light at 1486.6 eV and an emission angle of 45°. FT-IR spectra were recorded on a Bruker Spectrometer in the wavenumber range of 4000 to 400 cm−1. The morphology and microstructure of the Fe2O3 NPs and the Fe2O3/rGO nanocomposite were studied by scanning electron microscopy (SEM, Philips XL20) and transmission electron microscopy (TEM, Philips CM12). High-resolution transmission electron microscopy (HR-TEM) images were measured on a JEOL JEM-2200FS microscope. Nitrogen adsorption/desorption isotherm measurements were carried out at 77 K on a Quantachrome NOVA2200 analyzer. The mass loading of Fe2O3 NPs in the Fe2O3/rGO nanocomposite was determined by thermogravimetric (TG) analysis (Netzsch 449 F1 Jupiter) with a heating rate of 10 K min−1 under a synthetic air flow (25 ml min−1). To measure the electrical conductivities of the Fe2O3, GO and Fe2O3/rGO, the dry powders were pressed into pellets with a diameter of 5 mm and a thickness of around 0.3 mm, respectively. After sputtering with gold from both sides, the pellets were put into a tempered measurement cell and their AC impedance was recorded using a Solartron 1255 equipped with a Solartron 12961 Dielectric Interface. Impedance spectra were measured between 1 Hz and 1 MHz with an amplitude of 100 mV in the temperature range of 30–100 °C. The surface charge of Fe2O3 and GO in aqueous solutions was investigated by Zeta potential (ζ) measurements on a Zetasizer (Malvern Instruments). The final ζ value of each sample was calculated as an average of three measurements.The electrochemical tests were performed in three-electrode Swagelock® cells (electrode area 1.13 cm2) on a battery tester (Series 4000, Maccor Inc. USA, galvanostatic cycling) and on a multichannel potentiostatic–galvanostatic system (VMP, Biologic Science Instrument, France). All electrochemical tests were performed in a potential window between 5 mV and 3.0 V vs. Li/Li+. Cyclic voltammetry was done with a scan rate of 50 μV s−1. The galvanostatic tests started with three formation cycles at 0.05 A g−1. In the cycling tests the number of cycles increased as the increase of current density (0.1 A g−1: 3 cycles, 0.2 A g−1: 6 cycles, 0.5 A g−1: 15 cycles, 1 A g−1: 30 cycles, 2 A g−1: 60 cycles, 5 A g−1: 150 cycles, 10 A g−1: 300 cycles and 0.1 A g−1: 3 cycles). The capacities are calculated based on the active mass of the electrode.
Results and discussion
The synthesis route to Fe2O3/rGO is schematically illustrated in Fig. 1. The proposed formation mechanism of the Fe2O3/rGO nanocomposite can be seen as an “electrostatic-interaction-induced self-assembling”. After the preparation of the GO and the Fe2O3 suspensions in pure water, their ζ potentials were measured “as is” without changing pH by adding any acid or base. The measurements (Fig. S1†) showed that the GO layers and the Fe2O3 NPs have a ζ value of −37.6 mV and 31.3 mV, respectively, which indicated that GO layers and Fe2O3 NPs were oppositely charged. The electrostatic attraction between these two oppositely charged dispersed compounds provided a driving force for the self-assembling process. A colloidal suspension was observed directly after mixing the GO and Fe2O3 dispersions which implied that the self-assembling of these two components occurred immediately. Ethylenediamine (EDA) was selected as the reducing agent. According to previous reports, EDA can reduce GO at low temperature and meanwhile provide nitrogen doping to the rGO layers.40,41 During the hydrothermal process, the GO sheets covered with Fe2O3 NPs self-assemble into a 3D hydrogel (Fig. 1). The interconnecting networks are caused by hydrophobic and π–π stacking interactions of the graphene layers.32,42,43 | ||
| Fig. 1 Schematic illustration of the synthesis strategy of the Fe2O3/rGO nanocomposite: electrostatic interaction induces self-assembly that is coupled with a low-temperature hydrothermal reduction process. | ||
The XRD patterns of the prepared Fe2O3 and Fe2O3/rGO are shown in Fig. 2. All diffraction peaks from Fe2O3 can be assigned to maghemite (PDF 25-1402, the existence of the maghemite phase has also been proven by our previous Mössbauer spectroscopy study on the NPs39). The broadened diffraction reflexes indicate that the crystallite size of the prepared iron oxide is very small. The diffraction pattern of the Fe2O3/rGO nanocomposite agrees also well with the maghemite phase and do not show any characteristic reflexes neither of graphite (002) at about 26.5° nor of GO (002) at about 10.6° (Fig. S2†), indicating the GO is successfully reduced by EDA under the low-temperature hydrothermal condition. These results also demonstrate that the Fe2O3/rGO nanocomposites possess disordered stacking layers.
 | ||
| Fig. 2 XRD patterns of Fe2O3 NPs and the Fe2O3/rGO nanocomposite. | ||
The reduction of GO is further proven by the Raman spectroscopy (Fig. 3 and S3†). The intensity ratio of the well-documented D and G band in rGO (ID:IG = 1.11, Fig. 3) is enhanced after the hydrothermal process compared with that of GO (ID:IG = 0.85, Fig. S3†), indicating a high-degree reduction of the graphene sheets22,41,44 The ID:IG ratio (1.29) of Fe2O3/rGO (Fig. 3) is even elevated compared to that of pure rGO, which implies more disordered carbon structures in the nanocomposite that could be attributed to the anchoring of NPs on the rGO layers.15,21,45 Furthermore, all the characteristic bands of pristine Fe2O3 in the lower wavenumber range are also present in the Raman spectrum of Fe2O3/rGO, suggesting again the formation of the nanocomposite. The band at 1306 cm−1 in the spectrum of the pristine Fe2O3 does not appear in the Fe2O3/rGO spectrum due to the overlapping with the broad D band (1342 cm−1).
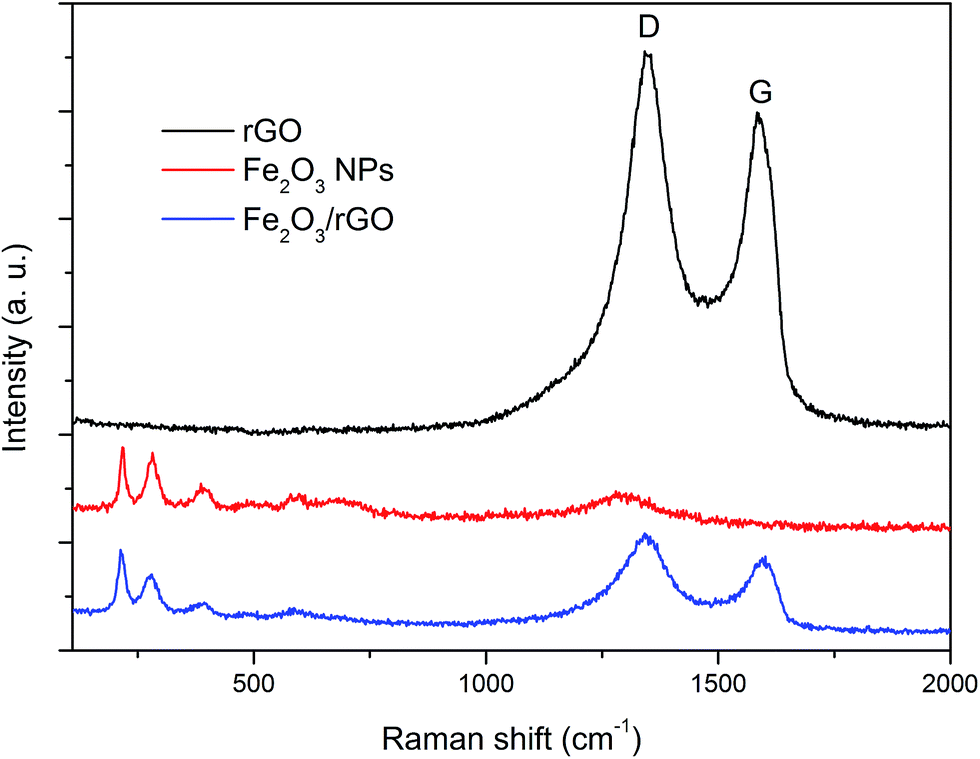 | ||
| Fig. 3 Raman spectra of rGO, Fe2O3 NPs and the Fe2O3/rGO nanocomposite. | ||
XPS measurements were employed to further characterize the elemental components of the Fe2O3/rGO nanocomposite. The high-resolution XPS spectra of Fe 2p, C 1s and N 1s are shown in Fig. 4. Two peaks located at 724.6 and 711.1 eV are exhibited in the Fe 2p spectrum (Fig. 4a), corresponding to Fe 2p1/2 and Fe 2p3/2 of the Fe2O3 nanoparticles, respectively.32 The satellite peak centered at 718.9 eV can be solely attributed to the presence of Fe3+ ions in Fe2O3.32,46 In the C 1s spectrum (Fig. 4b), the peaks at 284.8 and 285.5 eV belong to the sp2-C and sp3-C bonds of rGO, respectively; the peaks at 286.5, 287.6, and 288.7 eV are attributed to C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO configurations, respectively.30,32,43 The low intensity of these three latter peaks (compared to that of GO in Fig. S4†) also indicates the removal of most of the oxygen-containing functional groups after the low-temperature hydrothermal process. In addition, the peak at 285.9 eV is correlated with C in the C–N bonds,40,41 which indicates the nitrogen doping of the rGO layers. The presence of nitrogen in the nanocomposite is also shown in the N 1s spectrum (Fig. 4c) where the N 1s peak can be resolved into two components centered at 400.1 eV (pyrrolic structure) and 401.6 eV (graphitic N).40 For surface characterization of GO, rGO, Fe2O3 and Fe2O3/rGO with FTIR, refer to the ESI (Fig. S5†).
O and O–CO configurations, respectively.30,32,43 The low intensity of these three latter peaks (compared to that of GO in Fig. S4†) also indicates the removal of most of the oxygen-containing functional groups after the low-temperature hydrothermal process. In addition, the peak at 285.9 eV is correlated with C in the C–N bonds,40,41 which indicates the nitrogen doping of the rGO layers. The presence of nitrogen in the nanocomposite is also shown in the N 1s spectrum (Fig. 4c) where the N 1s peak can be resolved into two components centered at 400.1 eV (pyrrolic structure) and 401.6 eV (graphitic N).40 For surface characterization of GO, rGO, Fe2O3 and Fe2O3/rGO with FTIR, refer to the ESI (Fig. S5†).
 | ||
| Fig. 4 High-resolution XPS spectra of: (a) Fe 2p, (b) C 1s and (c) N 1s of the prepared Fe2O3/rGO nanocomposite. | ||
SEM images of the prepared Fe2O3/rGO nanocomposite (Fig. 5a) show that the composite rGO layers are well exfoliated. The Brunauer–Emmet–Teller (BET) analysis of the nitrogen adsorption/desorption isotherm (Fig. S6†) suggests that the nanocomposite has a relatively large specific surface area of 123 m2 g−1. A higher magnification SEM image (Fig. 5b) reveals a continuously rough surface of the composite rGO layers which implies that Fe2O3 NPs are homogeneously distributed on the rGO sheets. The dense loading of Fe2O3 NPs on the rGO sheets is further shown in the TEM images in Fig. 6. The low-magnification image (Fig. 6a) shows the typical morphology of the Fe2O3/rGO nanocomposite, which displays exfoliated and flat rGO sheets highly covered with NPs. These NPs are in contact to each other, and in some cases they overlap. Nevertheless, the most important thing is that these nanoparticles are well hosted by the rGO layer. The accommodation of the rGO layers guarantees that the iron oxide nanoparticles function in a continuous electrical conductive network even during their deformation. At higher magnification, the TEM image (Fig. 6b) shows Fe2O3 NPs with an average particle size of 8.8 nm (this result is consistent with the nitrogen adsorption/desorption measurement of pure Fe2O3 NPs which yielded an average particle size of 9 nm) uniformly distributed on the rGO sheets. Further HR-TEM images of the Fe2O3/rGO nanocomposite see ESI (Fig. S7†). All the measurements of SEM, TEM and HR-TEM indicate that an efficient electrostatic-interaction-induced self-assembling between the Fe2O3 NPs and the rGO sheets has successfully achieved.
 | ||
| Fig. 5 SEM images of the Fe2O3/rGO nanocomposite: (a) low magnification and (b) high magnification. | ||
 | ||
| Fig. 6 TEM images of the Fe2O3/rGO nanocomposite: (a) low magnification and (b) high magnification. | ||
The mass loading of the Fe2O3 NPs in the nanocomposite was quantified by thermogravimetry (TG) (Fig. S8†). The weight loss (<2 wt%) at the temperature below 200 °C can be attributed to the evaporation of adsorbed moisture or gas molecules, while the strong mass loss up to 600 °C can be attributed to the decomposition of the rGO layers. Furthermore, the color of the product changed from black to brownish-red after the TG measurement, which implies that all carbon was burnt away. The XRD analysis of the TG residue (Fig. S9†) shows that the phase of the iron oxide changed to hematite (PDF 33-0664) after the TG measurement. Based on the gravimetric findings the amount of Fe2O3 NPs in the nanocomposite was determined to be approx. 63 wt%.
rGO significantly increases the conductivity of the low electrically conductive NP materials. The electrical measurements of pure Fe2O3 NPs, GO and Fe2O3/rGO nanocomposite indicate a significant decrease of the resistance for the nanocomposite compared to the two raw materials. The corresponding electrical conductivities of Fe2O3 NPs, GO and Fe2O3/rGO nanocomposite are 3.1 × 10−6 S cm−1, 4.9 × 10−8 S cm−1 and 1.1 S cm−1, respectively. The substantial improvement of the electrical conductivity for the nanocomposite is caused by the presence of highly conductive rGO. The observation proves that GO was efficiently reduced in the low-temperature hydrothermal process. A significantly improved electrical conductivity of the active material is mandatory for high rate performance. Details and the analysis of the impedance measurements of the Fe2O3 NPs, GO and Fe2O3/rGO nanocomposite are given in the ESI (Fig. S10†).
To evaluate the mechanism of the electrochemical reactions, pure Fe2O3 NPs, pure rGO and Fe2O3/rGO nanocomposite were investigated with cyclic voltammetry (CV) measurements. Fig. 7 shows the CV profiles of the pure materials (Fig. 7a Fe2O3, Fig. 7b rGO) and of the Fe2O3/rGO nanocomposite (Fig. 7c). All three diagrams show delithiation in a potential region between 1.0 and 2.0 V vs. Li/Li+. However, the lithiation behavior of these three materials is quite different. The pure iron oxide shows the typical two distinct regions:47 one very sharp signal around 0.75 V vs. Li/Li+ and a broad, more diffuse region between 0.5 and 0.005 V vs. Li/Li+. The pure rGO is lithiated in a very broad potential region already beginning at around 1.0 V vs. Li/Li+.48,49 As expected, the profile of the nanocomposite is a combination of the first two profiles, clearly indicating the electrochemical activity of both components within the composite. There are 3 peaks observed in the lithiation process (Fig. 7c): the inconspicuous peak at 1.6 V vs. Li/Li+ and the broad peak at around 1.0 V vs. Li/Li+ correspond to the irreversible reaction with the electrolyte and the stepwise Li insertion to the Fe2O3 structure.15,19,22,50 The sharp cathodic peak at 0.75 V vs. Li/Li+ is attributed to the reduction of Fe3+ to Fe0 by Li.13,17,22,51 The influence of the rGO can be seen in the increased peak current density at the region below 0.5 V vs. Li/Li+. For the delithiation process of the nanocomposite, two peaks are recorded at about 1.58 V and 1.85 V vs. Li/Li+, which correspond to the oxidation of Fe0 to Fe3+.19,20 It is noteworthy that the anodic peak intensity shows only slight decrease during the cycling, suggesting the good reversible oxidation reaction mentioned above.
 | ||
| Fig. 7 Cyclic voltammetries measured between 3.0 and 0.005 V vs. Li/Li+ at 50 μV s−1: (a) pure Fe2O3, (b) pure rGO and (c) Fe2O3/rGO nanocomposite. | ||
The intensity of the signals of the first lithiation half cycle is very high for all three cells and is not maintained in the following cycles (Fig. 7). However, in contrast to the pure iron oxide, the pure rGO and the Fe2O3/rGO nanocomposite reach stable conditions gradually, while the current density for the pure Fe2O3 decreases from cycle to cycle. There is a little decay of the current density for the nanocomposite (Fig. 7c) at the beginning cycles; however, the degree of this decay is far less than it for pure Fe2O3 (Fig. 7a). This improvement is mainly attributed to the stabilizing effect of the rGO to the Fe2O3 nanoparticles. The minor decay of the current density over the beginning cycles is understandable since these cycles still belong to the “initialization period”, when the structure of the electrode and the performance of the battery stabilize gradually.
The strengthening effect of rGO in terms of electrochemical performance is also underlined in the galvanostatic cycling tests shown in Fig. 8. The pure iron oxide displays a capacity of around 150 mA h g−1 only at the current density of 0.1 A g−1 (Fig. 8a). The rGO has a specific capacity in the range of ordinary graphite; however, it shows much better capacity retention at high current densities (10 A g−1 corresponding to a rate of above 25 C for graphite). A capacity of 600 mA h g−1 for this Fe2O3/rGO nanocomposite is reached at a current density of 0.1 A g−1, which is much higher than that of the two raw materials (150 mA h g−1 for pure Fe2O3 and 340 mA h g−1 for rGO). It can still deliver a capacity of over 180 mA h g−1 when the current density rises to 10 A g−1 (approx. 17 C for the nanocomposite, the C-rate value was calculated based on the practical capacity (600 mA h g−1) of the Fe2O3/rGO nanocomposite, which was measured at a low current density of 0.1 A g−1).
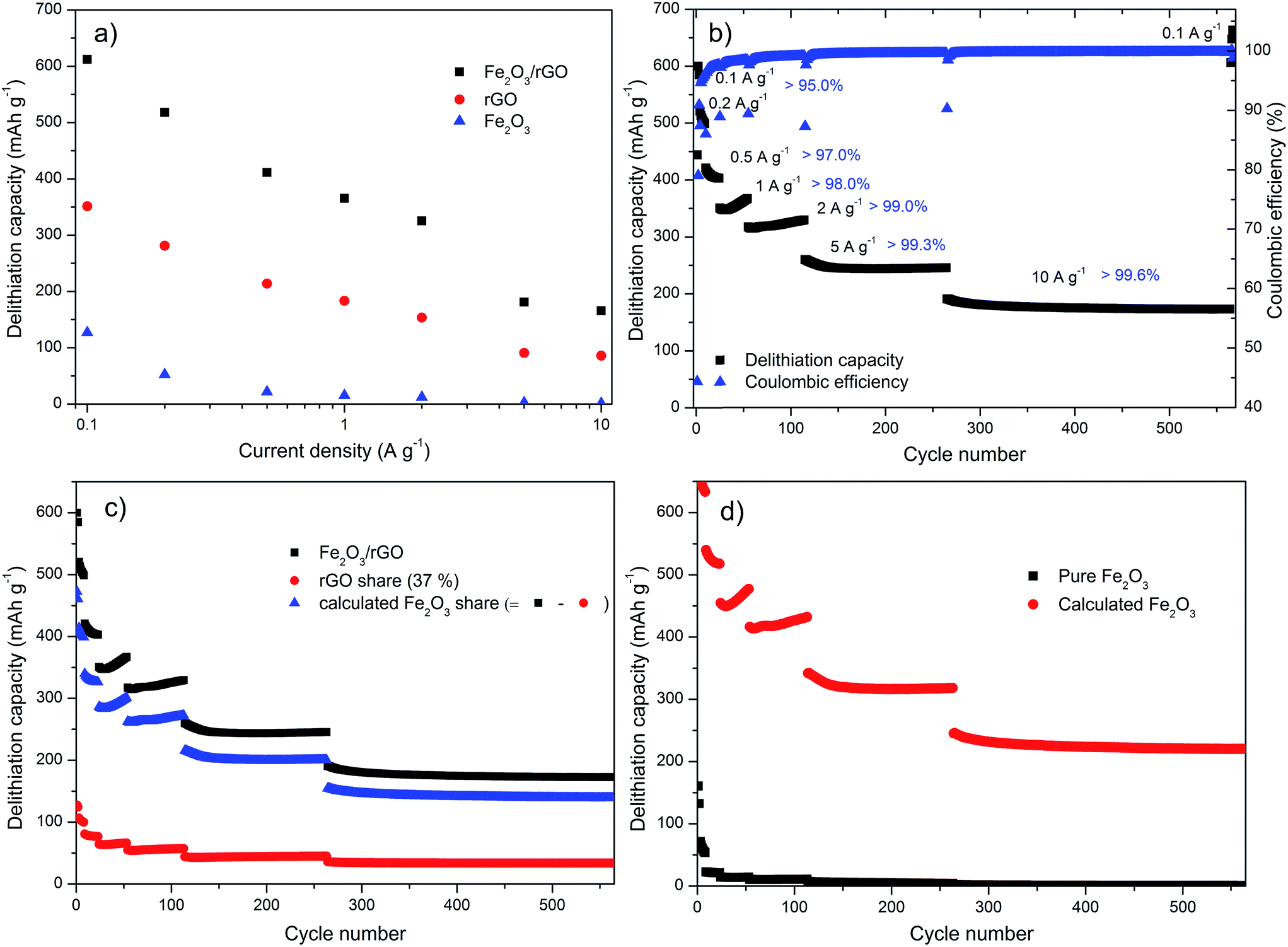 | ||
| Fig. 8 Galvanostatic cycling at selected current densities: (a) delithiation capacity vs. current plot for pure Fe2O3, pure rGO and the Fe2O3/rGO nanocomposite, (b) delithiation capacity and Coulombic efficiency vs. cycle number for the Fe2O3/rGO nanocomposite, (c) delithiation capacity vs. cycle number for the Fe2O3/rGO nanocomposite, the share of the rGO (rGO capacity multiplied by 37%) and the calculated Fe2O3 share (difference between the other two), (d) capacity vs. cycle number for the pure Fe2O3 and the calculated Fe2O3 capacity of (c) (divided by 63%). | ||
In order to further evaluate the rate capability and the long-cycle stability of the Fe2O3/rGO nanocomposite, the lithiation/delithiation tests were performed at various current densities with different cycles (Fig. 8b). Reversible cycling capacities of 600, 510, 410, 360, 320, 250 and 180 mA h g−1 of the nanocomposite electrode are retained at the current densities of 0.1, 0.2, 0.5, 1.0, 2.0, 5.0 and 10 A g−1, respectively. In agreement with the CV measurements (Fig. 7c), the galvanostatic cycling tests (Fig. 8b) showed also little capacity decay of the nanocomposite at the beginning. However, with increasing cycle numbers, the decay gets smaller and smaller, which indicates the electrode becomes more and more stable. Remarkably, the capacities are stable at very high current densities over long cycling tests (5 A g−1 for 150 cycles, 10 A g−1 for 300 cycles), indicating the nanocomposite possesses superior rate performance and very high cyclic stability. Furthermore, a capacity of 640 mA h g−1 can be reached when the current density returns to 0.1 A g−1 after more than 550 cycles, demonstrating an excellent reversibility of the nanocomposite. As also shown in Fig. 8b, the Coulombic efficiency increases in this test, both within a fixed current density and with current density increase. Using a current density of 2 A g−1 it reaches values above 99%. The low initial Coulombic efficiency is most likely caused by the irreversible lithium loss due to the formation of the solid electrolyte interface (SEI) film, the decomposition of electrolyte and irreversible structural changes of the material.15,20,29,52 To address such problems, Wang et al. proposed a lithium-compensation method, which successfully enhanced the initial Coulombic efficiency of a nanostructured GeOx anode from 70% to 99.5%.53 This method would probably also help in our case for the future study.
The last two panels of Fig. 8 are dedicated to the synergetic effect of the nanocomposite compared to the pure materials. Fig. 8c displays the capacity of the Fe2O3/rGO nanocomposite, the capacity share of the rGO (37% (according to the TG result) of the value obtained for the pure rGO in Fig. S11†) and the difference between these two. The difference is attributed to the capacity share of the decorated Fe2O3 NPs, which benefits from the synergetic effects of the nanocomposite. It is obvious that the rGO only contributes a fraction (125 mA h g−1 at 0.1 A g−1) of the total capacity. The benefit of the hybridization can be seen in Fig. 8d. It shows the measured capacity of the pure Fe2O3 and the calculated capacity of the decorated iron oxide (the value of the calculated Fe2O3 share in Fig. 8c divided by 63%). It can be clearly seen that the pure Fe2O3 lacks from a very poor cycling stability. The capacity fades very fast with increased current density and the retention is almost 0 when the cycling test is performed at higher current densities of more than 5 A g−1. In contrast, the decorated iron oxide exhibits very good cycling performance at each current density. It can even provide a capacity of approx. 225 mA h g−1 at the current density of 10 A g−1, which is reversible within the tested 300 cycles. These results strongly demonstrate the synergetic effect provided by the well-distributed Fe2O3 NPs and the high electrically conductive and flexible rGO layers: Fe2O3 nanoparticles ensure the high global capacity of the nanocomposite; rGO acts as a continuous electrical conductive matrix, which can accommodate the high volume change of the NPs and also allow using high currents during the lithiation/delithiation processes, and thus assure the stable contribution of Fe2O3 NPs to the overall capacity; at the same time, rGO contributes a small part to the total capacity.
Further insight into the Fe2O3/rGO nanocomposite can be achieved with the differential capacity (Fig. 9a) and the potential profiles (Fig. 9b). The differential capacity plot shows that the regions identified by the cyclic voltammetry also exist in the galvanostatic cycling. Both the peaks between 0.75 and 1.0 V vs. Li/Li+ and the broader amorphous region below 0.5 V vs. Li/Li+ can be seen here during lithiation, while delithiation takes place in a broad double peak between 1.25 and 2.25 V vs. Li/Li+. The peak at ∼0.75 V vs. Li/Li+, however, has a lower intensity compared to that in the lower potential region. This effect is even stronger at increased current densities until the peak almost disappears above a current density of 2 A g−1. This may be related to a reduced resolution due to the high current density or to a kinetic hindrance of the underlying process. This is also visible in the potential profile, where this process is represented with a plateau that is fading at higher current densities.
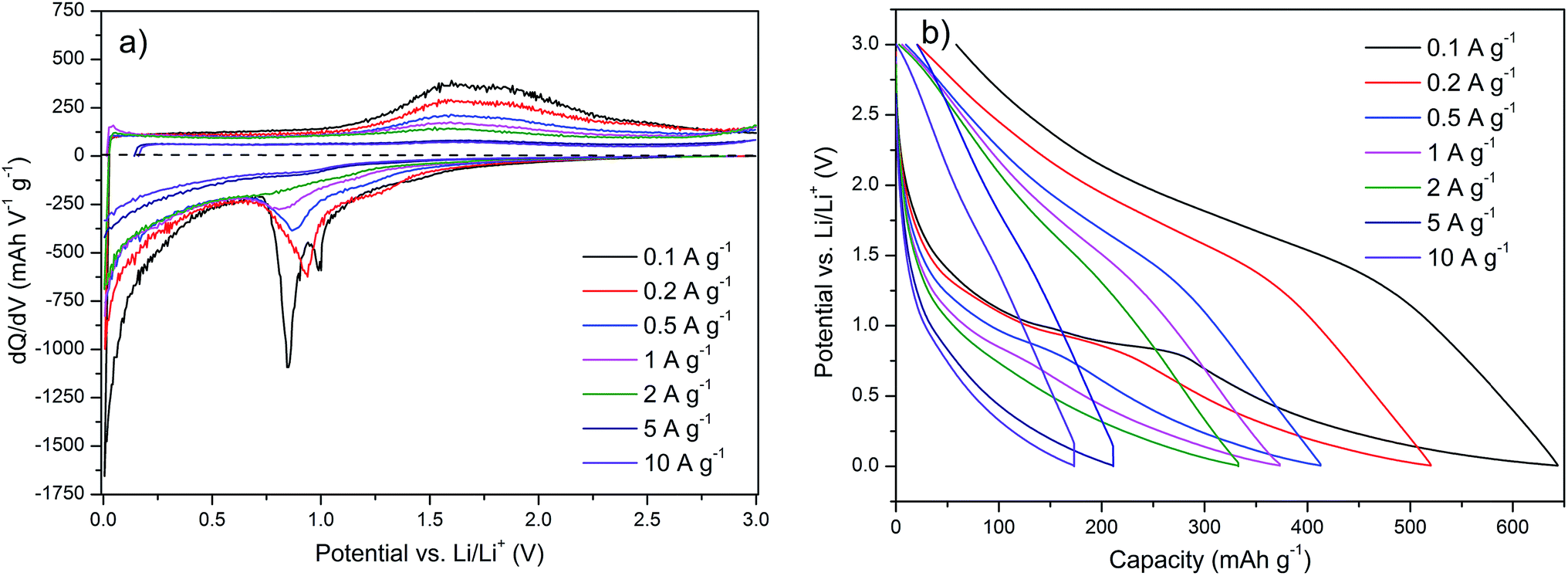 | ||
| Fig. 9 (a) Differential capacity and (b) potential profile of the Fe2O3/rGO nanocomposite (shown in Fig. 8b). | ||
Conclusions
A direct self-assembly of gas-phase produced Fe2O3 NPs and GO layers in aqueous solution driven by electrostatic interactions has been successfully developed in this study. This provides a simple synthesis route without using surfactants or chemical linkers that can be carried out at low hydrothermal temperature. The prepared Fe2O3/rGO nanocomposite demonstrates an outstanding electrochemical performance as an anode material for lithium-ion batteries. It shows a high reversible specific capacity of 600 mA h g−1 (at 0.1 A g−1), superior rate performance (180 mA h g−1 at 10 A g−1 for 300 cycles without noticeable capacity decay) and the long stable cycling life (more than 550 cycles), which is attributed to the synergetic effect between the two components of the nanocomposite: Fe2O3 NPs and rGO. This straightforward synthesis strategy enables an effective pathway to produce Fe2O3/rGO nanocomposite with superior LIBs performance that has the potential to be scaled up. Moreover, according to our experience, metal oxide (MO) NPs produced by this method are positively charged and a similar approach proved successfully with TiO2/rGO and V2O5/rGO nanocomposites. Therefore, it is anticipated that this method can be readily adapted to other MO NPs synthesized from the gas phase to prepare innovative MO/rGO nanocomposites.Acknowledgements
The authors thank M. Timpner, A. Sandmann and C. Liebscher (UDE) for Raman, Zeta potential and HR-TEM measurements, respectively. L. Xiao acknowledges financial support (69995000) of the UDE and fruitful discussions with Dr Pengfei Gao (Uni. Münster). A. Balducci and M. Schroeder wish to thank the University of Münster and the Ministry of Innovation, Science and Research of North Rhine-Westphalia (MIWF) within the project “Superkondensatoren und Lithium-Ionen-Hybrid-Superkondensatoren auf der Basis ionischer Flüssigkeiten” and the BMBF (contract number 03EK3010) for the financial support.References
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- W. W. Sun and Y. Wang, Nanoscale, 2014, 6, 11528–11552 RSC.
- Z. S. Wu, G. M. Zhou, L. C. Yin, W. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS PubMed.
- N. Mahmood, C. Z. Zhang and Y. L. Hou, Small, 2013, 9, 1321–1328 CrossRef CAS PubMed.
- Q. Li, N. Mahmood, J. H. Zhu, Y. L. Hou and S. H. Sun, Nano Today, 2014, 9, 668–683 CrossRef CAS PubMed.
- N. Spinner, L. C. Zhang and W. E. Mustain, J. Mater. Chem. A, 2014, 2, 1627–1630 CAS.
- X. F. Song, J. Pan, L. Xiao, L. Gao and S. Mathur, Nanotechnology, 2013, 24, 205401 CrossRef PubMed.
- B. Wang, H. B. Wu, L. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 4165–4168 CrossRef CAS PubMed.
- N. Mahmood and Y. L. Hou, Adv. Sci., 2014, 1, 1400012 Search PubMed.
- H. C. Chen, C. C. Wang and S. Y. Lu, J. Mater. Chem. A, 2014, 2, 16955–16962 CAS.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS PubMed.
- J. Kan and Y. Wang, Sci. Rep., 2013, 3, 3502 Search PubMed.
- G. W. Zhou, J. L. Wang, P. F. Gao, X. W. Yang, Y. S. He, X. Z. Liao, J. Yang and Z. F. Ma, Ind. Eng. Chem. Res., 2013, 52, 1197–1204 CrossRef CAS.
- J. Qu, Y. X. Yin, Y. Q. Wang, Y. Yan, Y. G. Guo and W. G. Song, ACS Appl. Mater. Interfaces, 2013, 5, 3932–3936 CAS.
- G. Wang, H. Wang, S. B. Cai, J. T. Bai, Z. Y. Ren and J. B. Bai, J. Power Sources, 2013, 239, 37–44 CrossRef CAS PubMed.
- B. Jang, O. B. Chae, S. K. Park, J. Ha, S. M. Oh, H. B. Na and Y. Piao, J. Mater. Chem. A, 2013, 1, 15442–15446 CAS.
- T. Yang, Y. G. Liu, Z. H. Huang, Q. Yang, Y. B. Chen, M. L. Hu, M. Guan and M. H. Fang, RSC Adv., 2014, 4, 41578–41583 RSC.
- S. K. Liu, Z. X. Chen, K. Xie, Y. J. Li, J. Xu and C. M. Zheng, J. Mater. Chem. A, 2014, 2, 13942–13948 CAS.
- Y. Q. Zou, J. Kan and Y. Wang, J. Phys. Chem. C, 2011, 115, 20747–20753 CAS.
- J. X. Zhu, T. Zhu, X. Z. Zhou, Y. Y. Zhang, X. W. Lou, X. D. Chen, H. Zhang, H. H. Hng and Q. Y. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- J. S. Luo, J. L. Liu, Z. Y. Zeng, C. F. Ng, L. J. Ma, H. Zhang, J. Y. Lin, Z. X. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- B. Wang, H. L. Xin, X. D. Li, J. L. Cheng, G. C. Yang and F. D. Nie, Sci. Rep., 2014, 4, 3729 Search PubMed.
- J. J. Zhang, T. Huang, Z. L. Liu and A. S. Yu, Electrochem. Commun., 2013, 29, 17–20 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- Y. Z. Su, S. Li, D. Q. Wu, F. Zhang, H. W. Liang, P. F. Gao, C. Cheng and X. L. Feng, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed.
- Z. L. Ma, X. B. Huang, S. Dou, J. H. Wu and S. Y. Wang, J. Phys. Chem. C, 2014, 118, 17231–17239 CAS.
- L. Xiao, D. Q. Wu, S. Han, Y. S. Huang, S. Li, M. Z. He, F. Zhang and X. L. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 3764–3769 CAS.
- I. T. Kim, A. Magasinski, K. Jacob, G. Yushin and R. Tannenbaum, Carbon, 2013, 52, 56–64 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, S. Ivanovici and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS PubMed.
- H. Zhang, A. J. Xie, C. P. Wang, H. S. Wang, Y. H. Shen and X. Y. Tian, J. Mater. Chem. A, 2013, 1, 8547–8552 CAS.
- W. Wei, S. B. Yang, H. X. Zhou, I. Lieberwirth, X. L. Feng and K. Mullen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- W. F. Chen, S. R. Li, C. H. Chen and L. F. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- T. Yoon, J. Kim, J. Kim and J. K. Lee, Energies, 2013, 6, 4830–4840 CrossRef CAS PubMed.
- Y. S. He, P. F. Gao, J. Chen, X. W. Yang, X. Z. Liao, J. Yang and Z. F. Ma, RSC Adv., 2011, 1, 958–960 RSC.
- T. Yuan, W. T. Li, W. M. Zhang, Y. S. He, C. M. Zhang, X. Z. Liao and Z. F. Ma, Ind. Eng. Chem. Res., 2014, 53, 10849–10857 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- C. Janzen and P. Roth, Combust. Flame, 2001, 125, 1150–1161 CrossRef CAS.
- P. Chen, J. J. Yang, S. S. Li, Z. Wang, T. Y. Xiao, Y. H. Qian and S. H. Yu, Nano Energy, 2013, 2, 249–256 CrossRef CAS PubMed.
- H. Hu, Z. B. Zhao, W. B. Wan, Y. Gogotsi and J. S. Qiu, Adv. Mater., 2013, 25, 2219–2223 CrossRef CAS PubMed.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- N. Mahmood, C. Z. Zhang, F. Liu, J. H. Zhu and Y. L. Hou, ACS Nano, 2013, 7, 10307–10318 CrossRef CAS PubMed.
- Y. M. Zhao, Y. H. Li, R. Z. Ma, M. J. Roe, D. G. McCartney and Y. Q. Zhu, Small, 2006, 2, 422–427 CrossRef CAS PubMed.
- A. Brandt and A. Balducci, J. Power Sources, 2013, 230, 44–49 CrossRef CAS PubMed.
- H. F. Xiang, Z. D. Li, K. Xie, J. Z. Jiang, J. J. Chen, P. C. Lian, J. S. Wu, Y. Yu and H. H. Wang, RSC Adv., 2012, 2, 6792–6799 RSC.
- A. M. Chockla, M. G. Panthani, V. C. Holmberg, C. M. Hessel, D. K. Reid, T. D. Bogart, J. T. Harris, C. B. Mullins and B. A. Korgel, J. Phys. Chem. C, 2012, 116, 11917–11923 CAS.
- A. Banerjee, V. Aravindan, S. Bhatnagar, D. Mhamane, S. Madhavi and S. Ogale, Nano Energy, 2013, 2, 890–896 CrossRef CAS PubMed.
- M. V. Reddy, T. Yu, C. H. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS PubMed.
- A. Brandt, F. Winter, S. Klamor, F. Berkemeier, J. Rana, R. Pottgen and A. Balducci, J. Mater. Chem. A, 2013, 1, 11229–11236 CAS.
- X. L. Wang, W. Q. Han, H. Y. Chen, J. M. Bai, T. A. Tyson, X. Q. Yu, X. J. Wang and X. Q. Yang, J. Am. Chem. Soc., 2011, 133, 20692–20695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta02549d |
| This journal is © The Royal Society of Chemistry 2015 |