DOI:
10.1039/C5TA02446C
(Paper)
J. Mater. Chem. A, 2015,
3, 12404-12412
Bottom-up and top-down methods to improve catalytic reactivity for photocatalytic production of hydrogen peroxide using a Ru-complex and water oxidation catalysts†
Received
4th April 2015
, Accepted 23rd April 2015
First published on 23rd April 2015
Abstract
Hydrogen peroxide (H2O2) was produced from water and dioxygen using [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst and [Ir(Cp*)(H2O)3]2+ (Cp* = η5-pentamethylcyclopentadienyl) as a precursor of a water oxidation catalyst in the presence of Sc3+ in water under visible light irradiation. TEM and XPS measurements of residues in the resulting solution after the photocatalytic production of H2O2 indicated that [Ir(Cp*)(H2O)3]2+ was converted to Ir(OH)3 nanoparticles, which are actual catalytic species. The Ir(OH)3 nanoparticles produced in situ during the photocatalytic production of H2O2 were smaller in size than those prepared independently from hydrogen hexachloroiridiate (H2IrCl6), and exhibited higher catalytic reactivity for the photocatalytic production of H2O2. The photocatalytic production of H2O2 from water and dioxygen was also made possible when Ir(OH)3 nanoparticles were replaced by nickel ferrite (NiFe2O4) nanoparticles, which are composed of more earth abundant metals than iridium. The size of NiFe2O4 nanoparticles became smaller during the photocatalytic production of H2O2 to exhibit higher catalytic reactivity in the second run as compared with that in the first run. NiFe2O4 nanoparticles obtained by the treatment of NiFe2O4 in an aqueous solution of Sc3+ exhibited 33-times higher catalytic reactivity in H2O2-production rates than the as-prepared NiFe2O4. Thus, both the bottom-up method starting from a molecular complex [Ir(Cp*)(H2O)3]2+ and the top-down method starting from as-prepared NiFe2O4 to obtain nanoparticles with smaller size resulted in the improvement of the catalytic reactivity for the photocatalytic production of H2O2 from water and dioxygen.
Introduction
The rapid and unsustainable use of fossil fuels has led to increased attention paid to the development of zero-carbon emission fuels, particularly hydrogen, utilizing renewable energy sources.1–7 Solar energy is obviously the most abundant among the renewable energy sources under consideration. Thus, extensive efforts have been devoted to producing hydrogen by water splitting (eqn (1)), which is highly endergonic with a free energy change of ΔG° = 474 kJ mol−1, which is provided by solar energy.8–12 In this case, however, a method for separating the simultaneously produced H2 and O2 remains to be developed to avoid possible explosion.13 In addition, the storage of hydrogen at reasonable energy density poses a technical and economical challenge due to its low volumetric energy.14,15| | | 2H2O → 2H2 + O2 ΔG° = 474 kJ mol−1 | (1) |
In contrast to hydrogen, hydrogen peroxide (H2O2) is miscible in water, and therefore it can be an ideal energy carrier alternative to hydrogen, because H2O2 can be used as a fuel for one-compartment fuel cells.16–23 The output potential of a H2O2 fuel cell theoretically achievable is 1.09 V, which is comparable with that of a hydrogen fuel cell (1.23 V).16,17 Thus, a combination of H2O2 production using solar energy and power generation with an H2O2 fuel cell provides an ideally sustainable solar fuel.16,17 It is desired to produce H2O2 from H2O and O2 (eqn (2)), which is highly endergonic with the free energy change of ΔG° = 210 kJ mol−1, by using solar energy.16,17
| | | 2H2O + O2 → 2H2O2 ΔG° = 210 kJ mol−1 | (2) |
We have recently reported the photocatalytic production of H2O2 from H2O and O2 by combining the photoreduction of O2 with a Ru-complex photosensitiser and water oxidation with Ir(OH)3 nanoparticles as water oxidation catalysts (WOCs) in the presence of Sc3+ in water under visible light irradiation.24 In order to improve the photocatalytic reactivity of H2O2 production from H2O and O2, it is required to employ more efficient WOCs. Extensive efforts have so far been devoted to developing efficient WOCs using transition metal complexes.25–45 In particular, a series of mononuclear iridium(III) complexes with the η5-pentamethylcyclopentadienyl ligand (Cp*) have been reported to act as efficient WOCs, which are more active than ruthenium complexes.46–49 The Cp* ligand is expected to provide electron rich circumstances useful to stabilise reaction intermediates possessing a high-valent oxidation state in catalytic water oxidation by cerium ammonium nitrate, (NH4)2[Ce(NO3)6], (CAN).46–49 Under the conditions of catalytic water oxidation by CAN, however, the Cp* ligand of Ir complexes has been reported to be oxidised to produce IrO2 or Ir(OH)3 nanoparticles, which act as the actual reactive catalyst for water oxidation.50–54 IrO2 or Ir(OH)3 nanoparticles have been reported to be more active than conventional IrO2 prepared from H2IrCl6.55–61 Thus, in situ formation of a WOC provides a useful way to improve the catalytic reactivity for water oxidation.
We report herein the photocatalytic production of H2O2 from H2O and O2 using an Ir complex, [Ir(Cp*)(H2O)3]2+, as a precursor of a water oxidation catalyst and [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a homogeneous photocatalyst in the presence of Sc3+ in water. The characterisation of the catalytically active species has revealed that [Ir(Cp*)(H2O)3]2+ is converted to Ir(OH)3 nanoparticles, which exhibit high catalytic reactivity for photocatalytic water oxidation. A synthetic strategy for such Ir(OH)3 nanoparticles from H2IrCl6 has not yet been established. Because Ir is a noble metal with limited natural supplies, it is desired to replace the noble metal in WOCs by more earth-abundant metals such as Fe and Ni. In this context, we have also employed nanoparticles composed of earth abundant nickel and iron (NiFe2O4) instead of the Ir complex as a water oxidation catalyst for the photocatalytic production of H2O2. During the reaction, NiFe2O4 nanoparticles were formed from the corresponding as-prepared NiFe2O4. The effect of reaction conditions on the reactivity of the therein-formed nanoparticles from the Ir complex or NiFe2O4 is discussed in this paper.
Results and discussion
Photocatalytic production of hydrogen peroxide with an iridium complex precatalyst
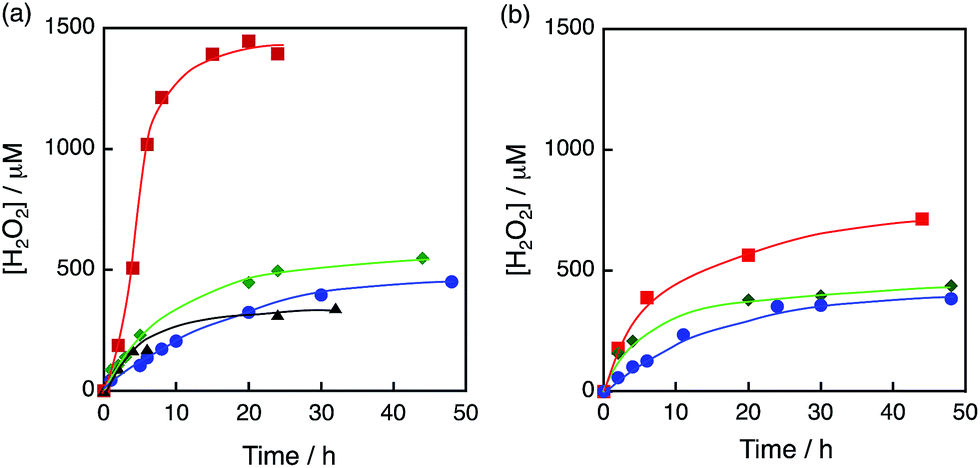
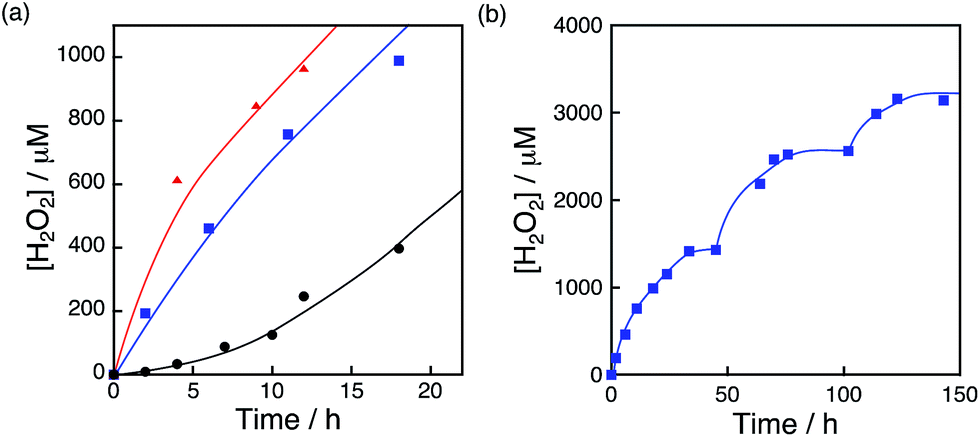
The photocatalytic production of H2O2 was performed using [RuII(Me2phen)3]2+ as a photocatalyst for the two-electron reduction of O2 and various Ir compounds as WOCs in the presence of Sc3+ ions in distilled water as shown in Fig. 1a. Sc3+ ions were reported to prohibit back electron transfer from O2˙− to [RuIII(Me2phen)3]3+, which is formed after photoinduced electron transfer to O2 from an excited state of [RuII(Me2phen)3]2+ (vide infra).24 The initial rate of H2O2 production using [Ir(Cp*)(H2O)3]2+ (red squares in Fig. 1a) was 4.5 times faster than that using Ir(OH)3 nanoparticles (blue circles in Fig. 1a) prepared from H2IrCl6 with the same amount of Ir.54 The rate of H2O2 production using [Ir(Cp*)(H2O)3]2+ was 4.4 times and 2.8 times faster than those using Ir(SO4)2 and [Ir(Cp*)((OH)2bpy)(H2O)]2+ ((OH)2bpy = 4,4′–(OH)2–2,2′-bipyridine), respectively. [Ir(Cp*)(H2O)3]2+ had higher reactivity than Co complexes and Co ions, which have been reported to act as highly active WOCs, as shown in Fig. 1b.62,63 The quantum efficiency determined by using monochromatised light (450 nm) and the solar energy conversion efficiency of the production of H2O2 were determined to be 7.1% and 0.063%, respectively (Fig. S1 and S2†). The sigmoidal behaviour in the initial stage of H2O2 production with [Ir(Cp*)(H2O)3]2+ (Fig. 2, green) indicates that [Ir (Cp*)(H2O)3]2+ acts as a precatalyst to produce catalytically more active species during the photocatalytic production of H2O2.
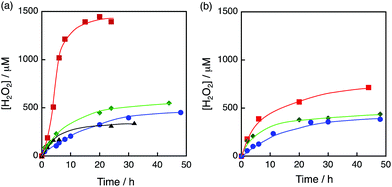 |
| | Fig. 1 Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in the presence of Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) containing (a) various Ir compounds, [Ir(Cp*)(H2O)3]2+ (red squares), [Ir(Cp*)((OH)2bpy)(H2O)]2+ (green diamonds), Ir(OH)3 (blue circles) and Ir(SO4) (black triangles), where Ir content: 100 μM and (b) various Co compounds, [Co(Cp*)(bpy)(H2O)]2+ (red squares), [Co(Cp*)(H2O)3]2+ (green diamonds) and Co(NO3)3 (blue circles), where Co content: 100 μM at 298 K. | |
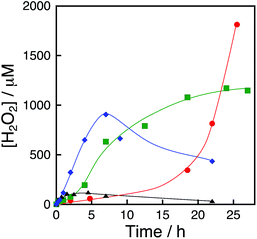 |
| | Fig. 2 Time courses of H2O2 production under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in the presence of [Ir(Cp*)(H2O)3]2+ (100 μM) and Sc3+ (100 mM) in an O2-saturated aqueous solution (3.0 mL) at 333 K (black triangles), 313 K (blue diamonds), 293 K (green squares) and 278 K (red circles). | |
The sigmoidal behaviour was more pronounced when the photocatalytic production of H2O2 was performed at 278 K as shown in Fig. 2 (red circles), where the results at higher temperatures are compared. The initial rate of H2O2 production increases with increasing temperature (Fig. 2), but the maximum H2O2 concentration decreased because of the enhanced decomposition of H2O2.
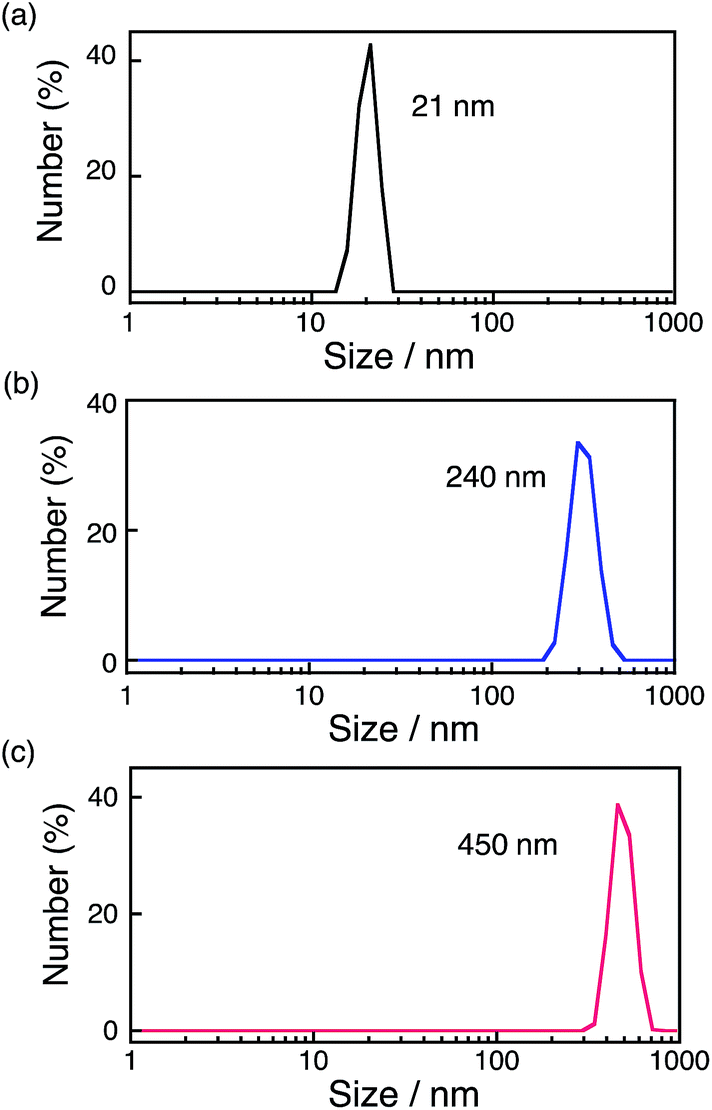
The formation of nanoparticles was observed by transmission electron microscopy (TEM) measurements. TEM images of the particles (Fig. S3†) showed that the diameters of the particles increased by extending the reaction time. The formation of nanoparticles was also confirmed by dynamic light scattering (DLS) measurements as shown in Fig. 3. The size of nanoparticles formed after 12 h photoirradiation at 278 K was 21 nm, whereas the size increased to 240 nm after 36 h photoirradiation. Large-sized particles (450 nm) were obtained after 12 h photoirradiation at 333 K. Thus, the size of the particles depends on the photoirradiation time and temperature. During photocatalytic H2O2 production, the size of the nanoparticles (21 nm) increases to 240 nm after 36 h (Fig. 3a and b) and the rate of the reaction decreases (Fig. 2, green line) under irradiation of visible light at room temperature. The deceleration of the reaction rate may be ascribed to the decrease in the surface area of the nanoparticles with increasing the size of the nanoparticles. [Ir(Cp*)(H2O)3]2+ has been reported to be efficiently oxidised by CAN, and TG/DTA and XPS measurements of nanoparticles produced after the water oxidation suggested that the nanoparticles were composed of Ir(OH)3.61 XPS measurements of the nanoparticles centrifugally recovered from the reaction solution after the H2O2 production reaction were performed for the energy regions of Ir 4f, O 1s and C 1s with reference to commercially available IrO2 (Fig. S4†), which suggested that the formed nanoparticles are also composed of Ir(OH)3. Since the binding energy of Ir 4f5/2 reflects the valence of Ir ions, the value was determined to be 61.9 eV for Ir(OH)3 nanoparticles, which is close to the reported value for IrIII (62.0 eV).62 These values were significantly different from those for Ir0 (61.0 eV) or IrIV (63.7 eV).58,62–67 The binding energy for O 1s of Ir(OH)3 nanoparticles (531.9 eV) shifted from that of IrO2 (530.2 eV) due to the OH moiety as reported previously.54 TEM images of Ir(OH)3 nanoparticles revealed that the size of Ir(OH)3 nanoparticles (10–20 nm) derived from [Ir(Cp*)(H2O)3]2+ was smaller than that of Ir(OH)3 (30–100 nm) derived from H2IrCl6 (Fig. S5†). The higher catalytic reactivity of Ir(OH)3 nanoparticles derived from [Ir(Cp*)(H2O)3]2+ may result from the smaller size of the nanoparticles as compared with those derived from H2IrCl6.
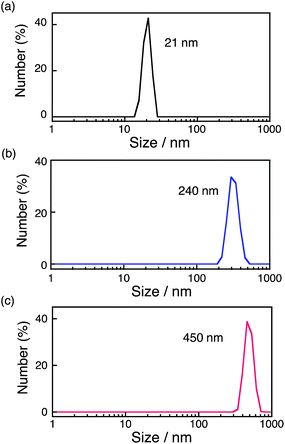 |
| | Fig. 3 Size distributions of the particles obtained by DLS measurements. The particles were formed under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in the presence of [Ir(Cp*)(H2O)3]2+ (100 μM) and Sc3+ (100 mM) in an O2-saturated aqueous solution (3.0 mL) at 298 K for (a) 12 h, (b) 36 h and (c) at 333 K for 12 h. | |
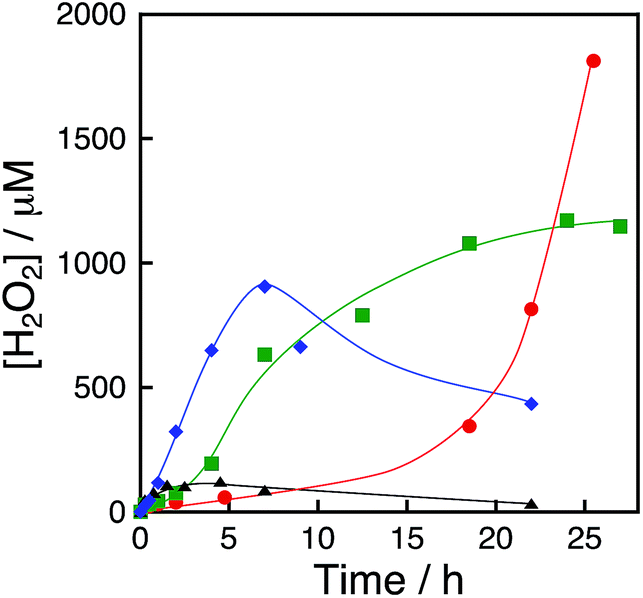
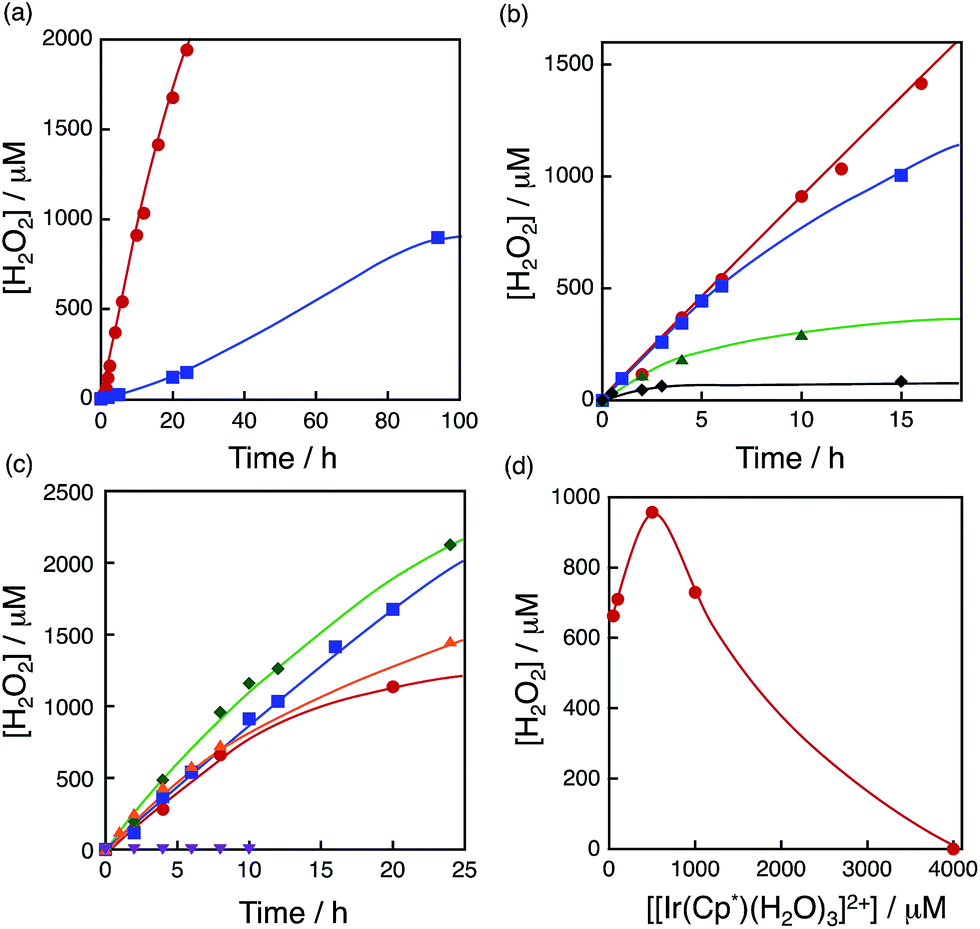
The dependence of photocatalytic reactivity for H2O2 production on the concentration of [RuII(Me2phen)3]2+ was examined as shown in Fig. 4a. The photocatalytic reactivity decreased with decreasing the concentration of [RuII(Me2phen)3]2+, however, the highest TON based on [RuII(Me2phen)3]2+ was determined to be 898 after 94 h photoirradiation when the concentration of [RuII(Me2phen)3]2+ was reduced to 1.0 μM, which is much higher than that reported for the photocatalytic H2O2 production using Ir(OH)3 as a WOC (307).24
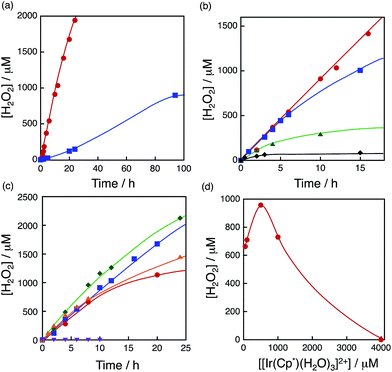 |
| | Fig. 4 (a) Time courses of H2O2 production at different concentrations of [RuII(Me2phen)3]2+ [20 μM (red circles) and 1.0 μM (blue squares)] under irradiation of [RuII(Me2phen)3]2+ with visible light (λ > 420 nm) in the presence of [Ir(Cp*)(H2O)3]2+ (100 μM) and Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) at 298 K. (b) Time courses of H2O2 production at different concentrations of Sc3+ [0 mM (black diamonds), 1.0 mM (green triangles), 10 mM (blue squares) and 100 mM (red circles)] under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) in the presence of [Ir(Cp*)(H2O)3]2+ (100 μM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) at 298 K. (c) Time courses of H2O2 production at different concentrations of [Ir(Cp*)(H2O)3]2+ [50 μM (red circles), 100 μM (blue squares), 500 μM (green diamonds), 1000 μM (orange triangles) and 4000 μM (purple inverse triangles)] at 298 K. (d) Plot of the amount of H2O2 production after 8 h vs. the concentration of [Ir(Cp*)(H2O)3]2+ under irradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) in the presence of Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) at 298 K. | |
The dependence of photocatalytic reactivity for H2O2 production on the concentration of Sc3+ was also examined as shown in Fig. 4b. The photocatalytic reactivity increased with increasing the concentration of Sc3+. This is because Sc3+ inhibits back electron transfer from O2˙− to [RuIII(Me2phen)3]3+, which is generated by photoinduced electron transfer from the excited state of [RuII(Me2phen)3]2+ to O2 as reported previously.24
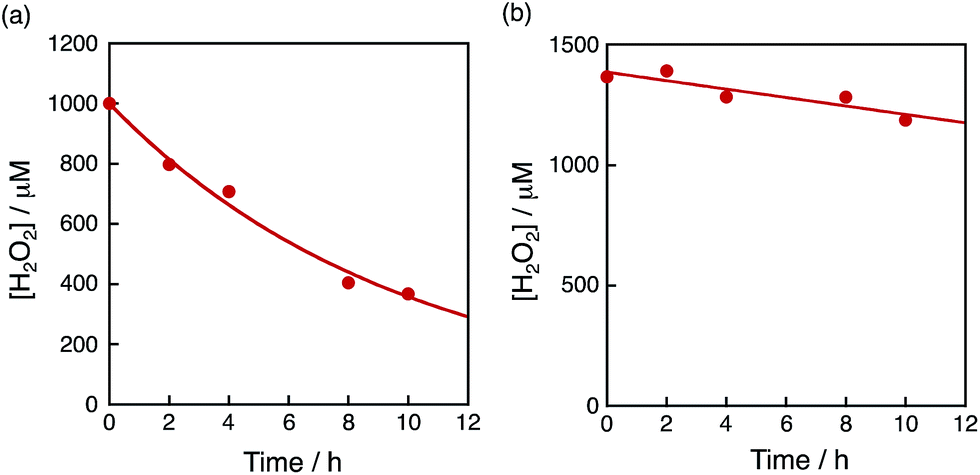
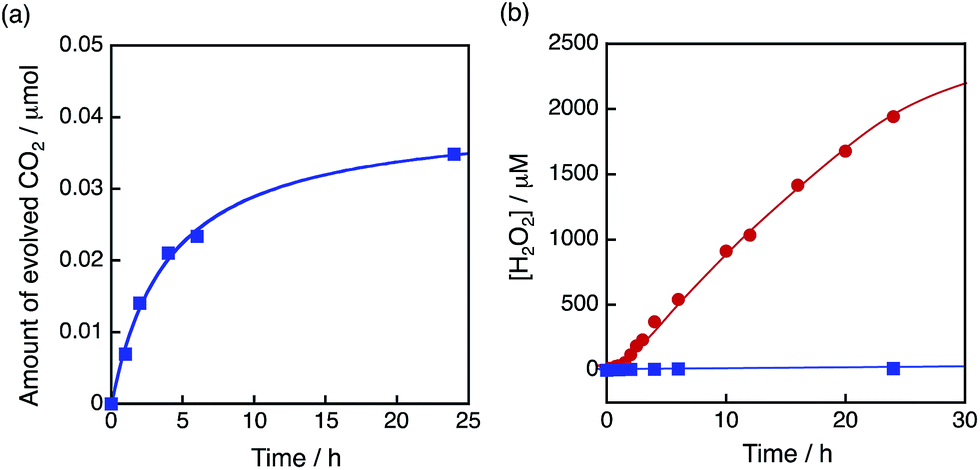
The dependence of photocatalytic reactivity of H2O2 production on the concentration of [Ir(Cp*)(H2O)3]2+ is shown in Fig. 4c. The highest TON based on [Ir(Cp*)(H2O)3]2+ was determined to be 23 after 20 h photoirradiation when 50 μM of [Ir(Cp*)(H2O)3]2+ was employed in the photocatalytic H2O2 production. The photocatalytic reactivity increased with increasing concentration of [Ir(Cp*)(H2O)3]2+, but it decreased through the maximum value with further increase in the concentration of [Ir(Cp*)(H2O)3]2+ as shown in Fig. 4d. The decrease in the rate of H2O2 production may result from the catalytic decomposition of H2O2 with [Ir(Cp*)(H2O)3]2+ as shown in Fig. 5a. When a high concentration of [Ir(Cp*)(H2O)3]2+ (e.g., 1000 μM) was employed in the photocatalytic production of H2O2, a part of [Ir(Cp*)(H2O)3]2+ may remain without the full conversion to Ir(OH)3 nanoparticles. When a low concentration of [Ir(Cp*)(H2O)3]2+ was employed, all of the [Ir(Cp*)(H2O)3]2+ may be oxidised to produce Ir(OH)3 nanoparticles during the photocatalytic reaction. The formed Ir(OH)3 nanoparticles are less reactive toward H2O2 decomposition as compared to [Ir(Cp*)(H2O)3]2+ (Fig. 5). The conversion of [Ir(Cp*)(H2O)3]2+ to Ir(OH)3 during the photocatalytic production of H2O2 may be associated with the oxidation of the Cp* ligand by O2. The full oxidation of Cp* is expected to produce 10 equivalents of CO2 and 8 equivalents of H2O2 (eqn (3)). During the photocatalytic production of H2O2, CO2 evolution was observed as shown in Fig. 6a. However, the yield of CO2 based on eqn (3) is only 1%. Thus, the Cp* ligand is only partially oxidised to CO2. The amount of H2O2 that is expected to be produced from Cp* calculated based on eqn (3) [Fig. 6b (blue line)] was negligible as compared with the observed amount of H2O2 in Fig. 6b (red line). These results indicate that H2O2 was produced by using H2O as an electron source.
| | | C10H15− + 18O2 + H+ → 10CO2 + 8H2O2 | (3) |
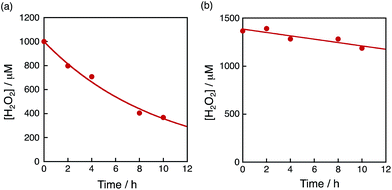 |
| | Fig. 5 Time course of the concentration of H2O2 in the presence of (a) [Ir(Cp*)(H2O)3]2+ (100 μM) and (b) the Ir(OH)3 nanoparticles in H2O (3.0 mL) at 298 K containing H2O2 and Sc3+ (100 mM) under dark conditions. The Ir(OH)3 nanoparticles used were formed after 16 h photoirradiation of [RuII(Me2phen)3]2+ (20 μM) with visible light (λ > 420 nm) in the presence of [Ir(Cp*)(H2O)3]2+ (100 μM) and Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) at 298 K and the resulting aqueous suspension was used as is for the measurements. | |
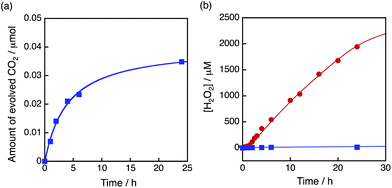 |
| | Fig. 6 (a) Time course of CO2 evolution at 298 K under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in the presence of Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) containing [Ir(Cp*)(H2O)3]2+ (100 μM). (b) Time course of H2O2 production at 298 K under visible light (λ > 420 nm) irradiation of [RuII(Me2phen)3]2+ (20 μM) in the presence of Sc3+ (100 mM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM) containing [Ir(Cp*)(H2O)3]2+ (100 μM) (red circles). Blue squares are the time course of H2O2 production expected from the amount of evolved CO2 based on eqn (3). | |
Photocatalytic production of hydrogen peroxide with NiFe2O4 nanoparticles
The catalytic reactivity of nickel ferrite (NiFe2O4) for water oxidation has been reported to be comparable to that of a catalyst containing Ir, Ru or Co in terms of the oxygen yield and oxygen-evolving rate under ambient reaction conditions.67
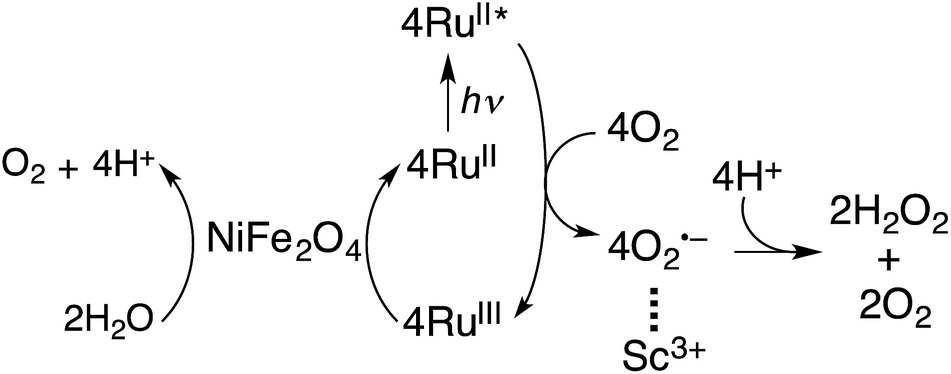
Because NiFe2O4 is composed of much more earth-abundant metals than Ir, NiFe2O4 was employed as a WOC for the photocatalytic production of H2O2 with [RuII(Me2phen)3]2+ in the presence of Sc3+ in water. The overall photocatalytic cycle for H2O2 production is depicted in Scheme 1. Photoinduced electron transfer from the excited state of [RuII(Me2phen)3]2+ to O2 results in the formation of H2O2 and [RuIII(Me2phen)3]3+via the oxidation of NiFe2O4, which oxidises water to form O2 and [RuII(Me2phen)3]2+. Back electron transfer from O2˙− to [RuIII(Me2phen)3]3+ and the decomposition of H2O2 by NiFe2O4 are retarded in the presence of Sc3+ (vide supra, Fig. S6†).24
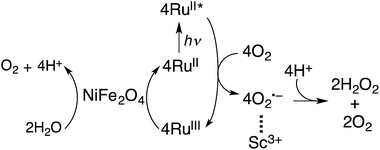 |
| | Scheme 1 Overall photocatalytic cycle for H2O2 production. | |
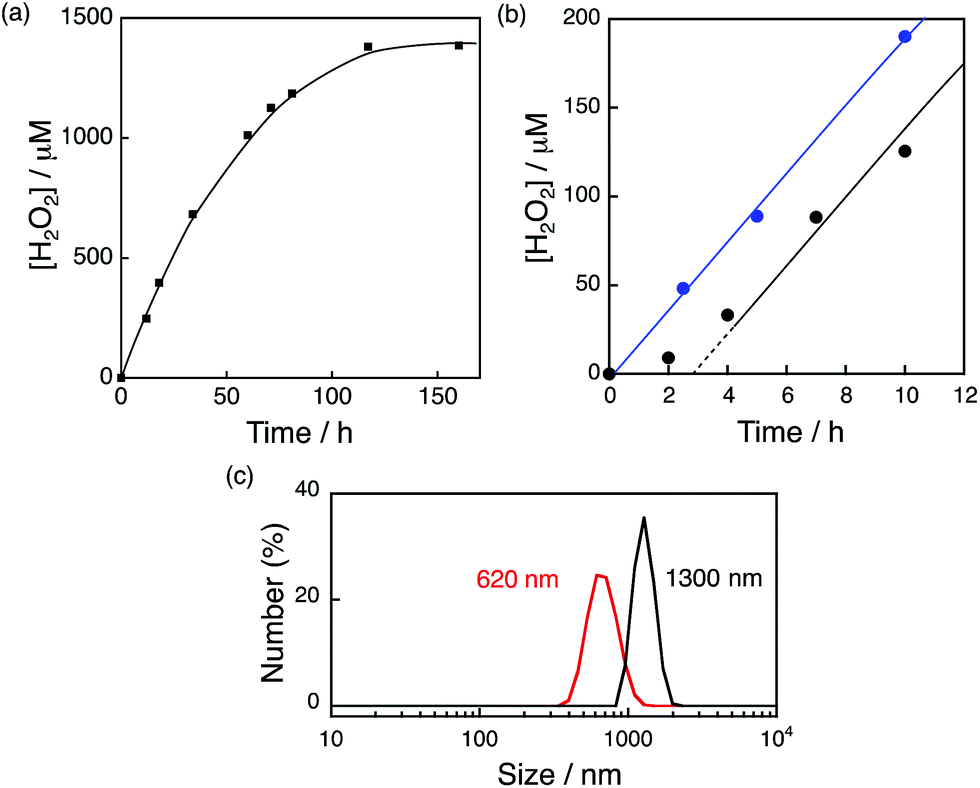
The photocatalytic production of H2O2 was performed using [RuII(Me2phen)3]2+ as a photosensitiser and NiFe2O4 as a WOC (Fig. 7a), which exhibited an induction period at the initial reaction time (black line in Fig. 7b). From the resulting solution, NiFe2O4 nanoparticles were recovered by centrifugation after 12 h photoirradiation and reused as WOCs. With the use of recovered NiFe2O4 as WOCs, the induction period was not observed (blue line in Fig. 7b). The diameter of nanoparticles measured by DLS decreased from 1300 nm to 620 nm after 12 h reaction as shown in Fig. 7c, suggesting that the induction period originates from the decrease in the diameter during the reaction.
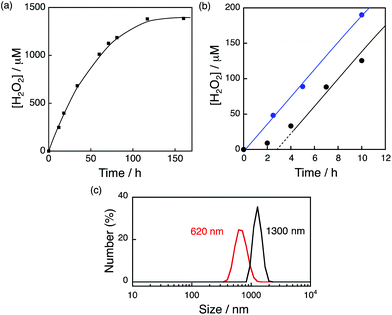 |
| | Fig. 7 (a) Time course of H2O2 production in the presence of NiFe2O4 (0.17 g L−1) and Sc3+ (100 mM) under visible light irradiation (λ > 420 nm) of [RuII(Me2phen)3]2+ (200 μM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM). (b) Initial period of the plot in (a) (black) and the time course of H2O2 production in the presence of NiFe2O4 recovered from the reaction solution after 12 h of visible light (λ > 420 nm) irradiation under the same conditions as in (a) (blue). (c) Size distributions of NiFe2O4 particles determined by DLS in the reaction solution before (black) and after (red) 12 h reaction. | |
In order to determine the conditions necessary for the size change of as-prepared NiFe2O4, DLS measurements of NiFe2O4 in an aqueous solution containing Sc3+ (100 mM) were performed under dark (Fig. S7†). The diameter of NiFe2O4 particles decreased to 710 nm, which is in good agreement with the size observed for the particles in the reaction suspension, although the rate of the size change was significantly reduced to 1/20 of that under photoirradiation. This result indicates that the rate of the size change was accelerated with photoirradiation. Then, the size change of NiFe2O4 was also examined in an aqueous HNO3 (1.0 M) solution (Fig. S8†) because Fe and Ni ions can be soluble in highly acidic solutions. However, the deceleration of the rate of the size change was also observed in the HNO3 solution under dark conditions, as the diameter of NiFe2O4 did not change even after 24 h under dark conditions (Fig. S9†). These results indicate that the presence of Sc3+ is necessary for the size change, thus, the dependence of the rate of the size change on the concentration of Sc3+ ranging from 0.1 to 100 mM was examined under room light at 353 K. The fastest size-decreasing rate was observed for an aqueous solution containing 10 mM of Sc3+ (Fig. S10†). Under these conditions, the size of formed nanoparticles became as small as 91 nm after 12 h (Fig. 8).
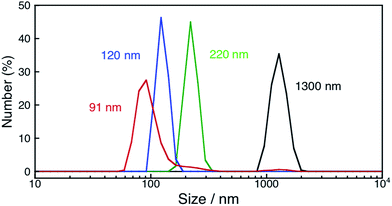 |
| | Fig. 8 Size distributions of NiFe2O4 nanoparticles determined by DLS measurements for an aqueous suspension containing as-prepared NiFe2O4 (0.17 g L−1) (black) and an aqueous suspension containing NiFe2O4 particles (0.17 g L−1) and Sc3+ (10 mM) exposed to room light for 3 h (green), 6 h (blue) and 12 h (red) at 353 K. | |
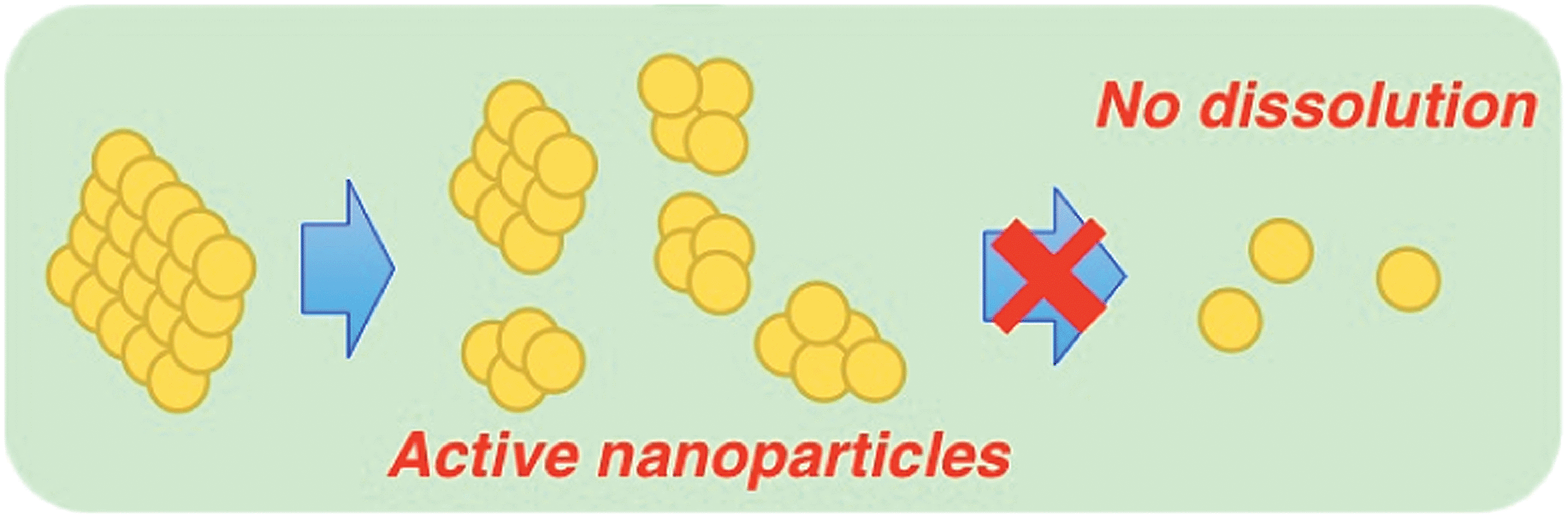
The nanoparticles were characterised by powder XRD to confirm that they kept the ferrite structure (Fig. S11†). It was also confirmed that the as-prepared NiFe2O4 was not dissolved to yield Fe ions. The addition of 1,10-phenanthroline (phen) and the reduced form of β-nicotinamide adenine dinucleotide disodium salt hydrate (NADH) as a reductant to the supernatant containing Fe ions produces [FeII(phen)3]2+, which has strong absorption in the visible region (λmax = 508 nm, ε = 1.1 × 104 M−1 cm−1) and therefore is easily detected (Fig. S12†). It was also supported by the fact that NiFe2O4 nanoparticles used in the reaction solution were recovered by centrifugation in high yield (87%). TEM images of NiFe2O4 particles manifested that the as-prepared NiFe2O4 has the form of aggregated smaller primary particles (Fig. S13†). The nanoparticles were formed by the dissociation of small particles that consist of a few primary particles as depicted in Scheme 2.
 |
| | Scheme 2 Mechanism of NiFe2O4 nanoparticle formation. | |
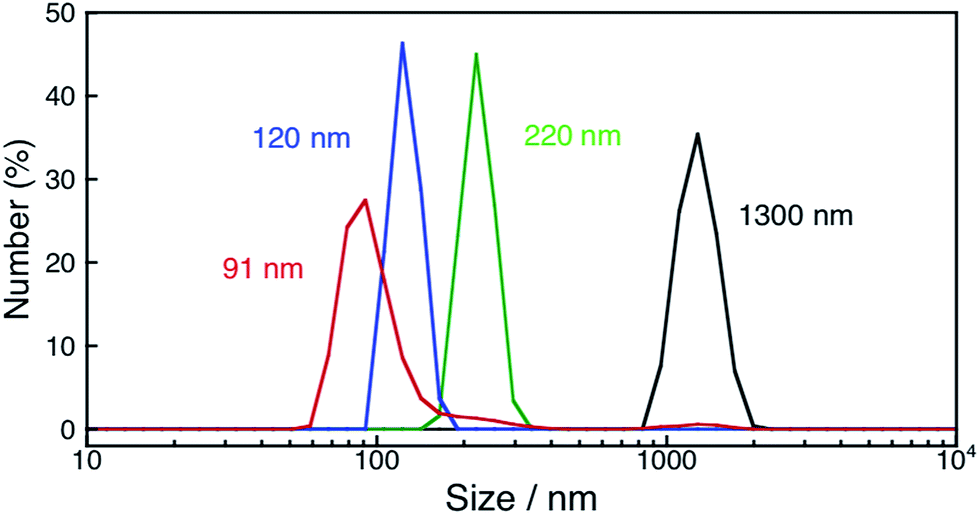
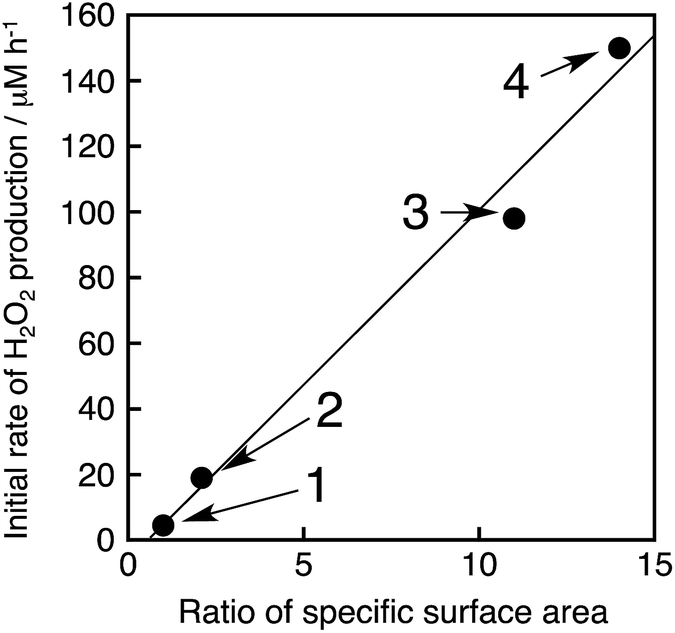
The photocatalytic production of H2O2 was performed using NiFe2O4 nanoparticles as the WOCs in the presence of [RuII(Me2phen)3]2+ and Sc3+ under visible light irradiation (λ > 420 nm) (Fig. 9a and S14†). The quantum efficiency determined by using monochromatised light (450 nm) and solar energy conversion efficiency were determined to be 2.7% and 0.088%, respectively, using NiFe2O4 nanoparticles with a diameter of 90 nm (Fig. S15 and S16†). To reuse the nanoparticles after H2O2 production ceased, an aliquot of an aqueous solution containing a high concentration of [RuII(Me2phen)3]2+ was added to the reaction suspension repeatedly, in which the amount of [RuII(Me2phen)3]2+ added to the starting suspension at each run was calculated in terms of the concentration increase of 200 μM. The concentration of H2O2 in the resulting suspension increased to be as high as 3.3 mM, ensuring the high stability of the nanoparticles as WOCs (Fig. 9b). The initial rate of H2O2 production was accelerated 22 times and 33 times when using NiFe2O4 nanoparticles with diameters of 120 nm and 91 nm, respectively, as compared to the as-prepared NiFe2O4 with a diameter of 1300 nm (Fig. 9a). This increase in reactivity could be due to a simple increase in the surface area, therefore, surface areas for nanoparticles were estimated from the respective diameters by eqn (S5)† and compared with respective initial rates of H2O2 production (Fig. 10). The linear relationship between surface areas and initial rates of H2O2 production observed in Fig. 10 indicates that the reactivity of each active site for water oxidation in the surface of NiFe2O4 remains unchanged irrespective of the particle size.
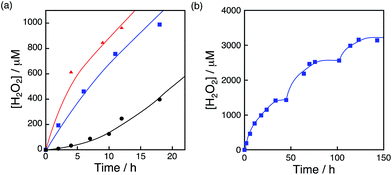 |
| | Fig. 9 (a) Time courses of H2O2 production under visible light irradiation (λ > 420 nm) of [RuII(Me2phen)3]2+ (200 μM) in the presence of Sc3+ (100 mM) and NiFe2O4 (0.17 g L−1) with diameters of 1300 nm (black circles), 120 nm (blue squares) and 91 nm (red triangles) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM). (b) Time course of H2O2 production in the presence of NiFe2O4 (0.17 g L−1) and Sc3+ (100 mM) under visible light irradiation (λ > 420 nm) of [RuII(Me2phen)3]2+ (200 μM) in O2-saturated H2O (3.0 mL, [O2] = 1.2 mM). [RuII(Me2phen)3]2+ was added twice to the reaction suspension at 50 h and 100 h during photoirradiation. The amount of [RuII(Me2phen)3]2+ added each time at 50 h and 100 h to the reaction suspension was calculated in terms of the concentration increase of 200 μM. | |
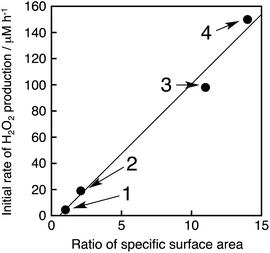 |
| | Fig. 10 Plot of initial rates of the photocatalytic H2O2 production vs. ratios of the specific surface area (R) of NiFe2O4 particles, where R = (specific surface area of the particles)/(specific surface area of non-treated NiFe2O4). The details of the calculation are described in ESI (Page S23).† The circles denoted as 1–4 correspond to NiFe2O4 particles with diameters of 1300 nm, 620 nm, 120 nm and 91 nm, respectively. The methods to produce each size of NiFe2O4 particles and the calculation of initial H2O2 production rates are described in the caption of Fig. S17.† | |
Conclusions
The reactivity of water oxidation catalysts for the photocatalytic production of H2O2 from H2O and O2 with [RuII(Me2phen)3]2+ and Sc3+ was improved by using [Ir(Cp*)(H2O)3]2+ as a precatalyst, which was converted to Ir(OH)3 nanoparticles during the photocatalytic reaction, as compared with that using Ir(OH)3 nanoparticles derived from H2IrCl6. The enhanced catalytic reactivity of Ir(OH)3 nanoparticles results from the smaller size of nanoparticles produced in situ as compared with Ir(OH)3 nanoparticles derived from H2IrCl6. The Cp* ligand of [Ir(Cp*)(H2O)3]2+ was partially oxidised to CO2 during the photocatalytic reaction and remaining organic residues may act as capping reagents to prevent further aggregation of Ir(OH)3 nanoparticles. NiFe2O4 nanoparticles, which are composed of much more earth abundant metals than Ir, also acted as a water oxidation catalyst for the photocatalytic production of H2O2 with [RuII(Me2phen)3]2+ in the presence of Sc3+ in water. In this case, the size of NiFe2O4 nanoparticles decreased during the photocatalytic reaction to increase the catalytic reactivity of water oxidation. Thus, both a bottom-up method starting from a metal complex precatalyst ([Ir(Cp*)(H2O)3]2+) to produce Ir(OH)3 nanoparticles with small size and a top-down method starting from as-prepared NiFe2O4 to obtain smaller NiFe2O4 nanoparticles provide promising strategies to develop more efficient water oxidation catalysts for photocatalytic production of H2O2 from H2O and O2.
Experimental section
Materials
All chemicals commercially available were used without further purification unless otherwise noted. H2IrCl6·nH2O (99.99%) was purchased from Furuya Metal. RuCl3 (38.220 wt% Ru) was purchased from Tanaka Kikinzoku Kogyo K.K. 4,7-Dimethyl-1,10-phenanthroline (Me2phen, 98%), Ag2SO4 (99.9%) and (NH4)2SO4 (99.99%) were supplied by Aldrich Chemicals. Pentamethylcyclopentadiene was obtained from Kanto Chemical Co., Inc. Oxo[5,10,15,20-tetra(4-pyridyl)porphinato]titanium(IV) ([TiO(tpyp)]) and NADH were supplied by Tokyo Chemical Industry Co., Ltd. (TCI). Sc(NO3)3·4H2O (99.9%) was supplied by Mitsuwa Chemicals Co., Ltd. Purification of water (18.2 MΩ cm) was performed with a Milli-Q system (Millipore, Direct-Q 3 UV). [Ir(Cp*)(H2O)3]SO4 was prepared by following the reported method.68
Synthesis of NiFe2O4
NiFe2O4 was synthesised according to the literature.67,69 To an aqueous solution (24 mL) containing NiCl2·6H2O (2.0 mmol, 0.46 g) and Fe(NO3)3·9H2O (4.0 mmol, 1.6 g) was added KOH solution (2.0 M, 24 mL) with magnetic stirring at room temperature (RT). The mixture was then transferred into a Teflon-lined stainless-steel autoclave of 140 mL capacity. The sealed tank was heated to and maintained at 160 °C for 10 h in an oven and cooled to RT. The resulting brown precipitates were collected by filtration and washed with water and ethanol more than 3 times, and finally dried in an oven at 60 °C for 10 h.
Formation of NiFe2O4 nanoparticles
Typically, an aqueous suspension (3.0 mL) containing Sc(NO3)3 and NiFe2O4 (0.50 mg) was stirred continuously for 3 h, 6 h or 12 h at 80 °C under room light to yield 220 nm, 120 nm and 91 nm nanoparticles respectively. The suspension was used for the H2O2 production reaction after the addition of [RuII(Me2phen)3]2+ and Sc(NO3)3. NiFe2O4 nanoparticles used as a sample for powder XRD measurements were prepared by immersing as-prepared NiFe2O4 (5.1 mg) in an aqueous solution (31 mL) of Sc(NO3)3 for 12 h. The resulting powder was collected by centrifugation and washed with water 3 times. The yield of NiFe2O4 nanoparticles was 87%.
Quantitative measurements of FeII and FeIII ions
An aqueous suspension containing as-prepared NiFe2O4 and Sc(NO3)3 was stirred at 80 °C for 12 h for the formation of active NiFe2O4 nanoparticles as discussed in the previous paragraph. After the formation of NiFe2O4 nanoparticles, the supernatant of the suspension was examined for the presence of FeII or FeIII ions. The filtered supernatant was diluted by water so that the solution may contain 300 μM of Fe ions if NiFe2O4 was dissolved completely. UV-Vis spectra were measured using a Hewlett Packard 8453 diode array spectrometer for the diluted supernatant in both the presence of 1,10-phenanthroline (phen) (4.5 mM), and in the presence of phen (4.5 mM) and NADH (1.5 mM) to reduce FeIII that may have formed. Measured UV-Vis spectra were compared with the UV-Vis spectra of [FeII(phen)3]32+ (100 μM).
Photocatalytic reactions
[Ir(Cp*)(H2O)3]SO4 or NiFe2O4 was introduced into distilled water (3.0 mL) containing [RuII(Me2phen)3]SO4 and Sc(NO3)3 in a quartz cuvette with a light path length of 1.0 cm. The solution was bubbled with oxygen gas for ∼30 min. The solution containing photocatalyst was irradiated with a xenon lamp (USHIO Optical Modulex SX-UID 501XAMQ) through a cut-off filter (Asahi Techno Glass L42) transmitting λ > 420 nm at room temperature.
Quantification of produced H2O2
From spectroscopic titration with an acidic solution of the [TiO(tpypH4)]4+ complex (Ti-TPyP reagent), the amount of produced H2O2 was determined.70 The [TiO(tpyp)] complex (34 mg) was dissolved in 1.0 L of 50 mM hydrochloric acid and the solution was used as a Ti-TPyP reagent. An aliquot (e.g., 100 μL) of the reaction solution was diluted with water and 0.25 mL of the sample solution was mixed with 0.25 mL of 4.8 M perchloric acid and 0.25 mL of Ti-TPyP reagent. After 5 min at room temperature, the mixture was diluted to 2.5 mL with water and used for the spectroscopic measurement. The absorbance at λ = 434 nm was measured by using a Hewlett Packard 8453 diode array spectrometer (AS). In a similar manner, a blank solution was prepared by adding distilled water in place of the sample solution in the same volume with its absorbance designated as AB. The difference in absorbance was determined by following the equation: ΔA434 = AB − AS. Based on ΔA434 and the volume of the solution, the amount of hydrogen peroxide was determined according to the literature.70
Determination of the quantum yield
Quantum yields (QYs) of the photocatalytic production of hydrogen peroxide (Φ) were determined under irradiation of monochromatised light using a Shimadzu spectrofluorophotometer (RF-5300 PC) through a monochromator transmitting λ = 450 nm, and estimated as| | | QY (%) = (2 × R/I) × 100 | (4) |
where R (mol s−1) and I (einstein s−1) represent the H2O2 production rate and the light intensity, respectively. Two photons are required for the electronic transition of the [RuII(Me2phen)3]2+ photosensitiser in order to produce hydrogen peroxide through two-electron reduction of one molecule of oxygen. When all of the photons are fully utilized to produce hydrogen peroxide, the QY reaches 100%. Therefore, the coefficient of the right-hand side in eqn (4) is 2 for this photocatalytic system. The total number of incident photons was measured by a standard method using an actinometer and potassium ferrioxalate, K3[FeIII(C2O4)]3, in H2O at room temperature under photoirradiation using a Shimadzu spectrofluorophotometer (RF-5300 PC) through a monochromator transmitting λ = 450 nm (slit width of 5.0 mm) at room temperature. For the same quartz cuvette with a light path length of 1.0 cm with 3.0 mL solution as used in the production of hydrogen peroxide experiments, the rate of photon flux of the incident light (I) was determined to be 7.40 × 10−10 einstein s−1.
Quantification of evolved CO2
[Ir(Cp*)(H2O)3]SO4 (100 μM) was added to distilled water (3.0 mL) containing [RuII(Me2phen)3]SO4 (20 μM) and Sc(NO3)3 (100 mM) in a quartz cuvette (light path length = 1.0 cm). The solution was saturated by bubbling with oxygen gas for ∼30 min. The photocatalyst was irradiated with a xenon lamp (USHIO Optical Modulex SX-UID 501XAMQ) through a cut-off filter (Asahi Techno Glass L42) transmitting λ > 420 nm at room temperature. The amount of evolved CO2 was determined by using a Shimadzu GC-14B gas chromatograph (N2 carrier, active carbon with a particle size of 60–80 mesh at 80 °C) equipped with a TCD detector.
Characterisation of particles
Transmission electron microscopy (TEM) images of iridium hydroxide and nickel ferrite, which were mounted on a copper microgrid coated with elastic carbon, were observed using a JEOL JEM-2100 operating at 200 kV. Dynamic light scattering (DLS) data were obtained using a Zeta Sizer Nano ZS (Malvern Instruments Ltd., USA). Powder X-ray diffraction (XRD) patterns were recorded with a Rigaku MiniFlex 600 X-ray diffractometer. X-ray photoelectron spectra (XPS) were obtained with an ULVAC-PHI ESCA5600 X-ray photoelectron spectrophotometer. The incident radiation was the Mg Kα X-ray (1253.6 eV) at 400 W and the charge neutralizer was turned on for acquisition. The binding energy of each element was corrected by the C 1s peak (284.8 eV) from residual carbon.
Acknowledgements
This work was supported by ALCA (to S.F.) and SENTAN (to T.S. and S.F.) projects from JST, Japan and Grants-in-Aid (nos 24350069 and 25600025 to Y.Y. and 24550077 to T.S.) for Scientific Research from Japan Society for the Promotion of Science (JSPS). D.H. gratefully acknowledges the support from JSPS by Grant-in-Aid for JSPS fellowship for young scientists. We sincerely acknowledge the Research Centre for Ultra-Precision Science & Technology, Osaka University for TEM measurements.
Notes and references
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126 CAS.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407 CrossRef CAS.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695 Search PubMed.
- S. Fukuzumi, D. Hong and Y. Yamada, J. Phys. Chem. Lett., 2013, 4, 3458 CrossRef CAS.
- S. Fukuzumi and Y. Yamada, ChemSusChem, 2013, 6, 1834 CrossRef CAS PubMed.
- J. M. Thomas, Energy Environ. Sci., 2014, 7, 19 CAS.
-
Hydrogen as a Future Energy Carrier, ed. A. Züttel, A. Borgschulte and L. Schlapbach, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403 CrossRef CAS PubMed.
- S. Fukuzumi and T. Suenobu, Dalton Trans., 2013, 18 RSC.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360 CAS.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493 CrossRef CAS PubMed.
- S. Fukuzumi and Y. Yamada, Aust. J. Chem., 2014, 67, 354 CrossRef CAS.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272 CrossRef CAS PubMed.
- S. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20 CrossRef CAS PubMed.
- S. A. M. Shaegh, N.-T. Nguyen, S. M. M. Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225 Search PubMed.
- F. Yang, K. Cheng, X. Xiao, J. Yin, G. Wang and D. Cao, J. Power Sources, 2014, 245, 89 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Chem.–Eur. J., 2013, 19, 11733 CrossRef CAS PubMed.
- Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334 RSC.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756 CAS.
- C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704 CAS.
- D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218 RSC.
- X. Liu and F. Wang, Coord. Chem. Rev., 2012, 256, 1115 CrossRef CAS PubMed.
- R. Cao, W. Lai and P. Du, Energy Environ. Sci., 2012, 5, 8134 CAS.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607 CrossRef CAS PubMed.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed.
- Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398 CrossRef CAS PubMed.
- M. Murakami, D. Hong, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605 CrossRef CAS PubMed.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef CAS PubMed.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522 CrossRef CAS PubMed.
- G. Chen, L. Chen, S.-M. Ng, W.-L. Man and T.-C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789 CrossRef CAS PubMed.
- Y. Liu, S.-M. Ng, S.-M. Yiu, W. W. Y. Lam, X.-G. Wei, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2014, 53, 14468 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC.
- M. Hirahara, A. Shoji and M. Yagi, Eur. J. Inorg. Chem., 2014, 595 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100 CrossRef PubMed.
- A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070 CrossRef CAS PubMed.
- X. Sala, S. Maji, R. Bofill, J. Garcia-Anton, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909 CrossRef CAS.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712 RSC.
- J. D. Blakemore, N. D. Schley, G. W. Olack, C. D. Incarvito, G. W. Brudvig and R. H. Crabtree, Chem. Sci., 2011, 2, 94 RSC.
- N. D. Schley, J. D. Blackmore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473 CrossRef CAS PubMed.
- D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M.-C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024 CrossRef CAS PubMed.
- U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837 CrossRef CAS PubMed.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708 CAS.
- G. S. Nahor, P. Hapiot, P. Neta and A. Harriman, J. Phys. Chem., 1991, 95, 616 CrossRef CAS.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578 CrossRef CAS PubMed.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402 CrossRef CAS.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 CAS.
- N. D. Morris and T. E. Mallouk, J. Am. Chem. Soc., 2002, 124, 11114 CrossRef CAS PubMed.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 4, 645 CrossRef PubMed.
- G. Q. Wei, Y. X. Wang, C. D. Huang, Q. J. Gao, Z. T. Wang and L. Xu, Int. J. Hydrogen Energy, 2010, 35, 3951 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107 RSC.
- M. Hara, K. Asami, K. Hashimoto and T. Masumoto, Electrochim. Acta, 1983, 28, 1073 CrossRef CAS.
- H. Y. Hall and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 135 RSC.
- D. Hong, Y. Yamada, T. Nagatomi, Y. Takai and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 19572 CrossRef CAS PubMed.
- S. Ogo, N. Makihara and Y. Watanabe, Organometallics, 1999, 18, 5470 CrossRef CAS.
- Y. Cheng, Y. Zheng, Y. Wang, F. Bao and Y. J. Qin, Solid State Chem., 2005, 178, 2394 CrossRef CAS PubMed.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, time courses of H2O2 production under different conditions (Fig. S1, S2, S14–S17), TEM images (Fig. S3, S5 and S13), X-ray photoelectron spectra of Ir(OH)3 (Fig. S4), time course of H2O2 decomposition in the presence of NiFe2O4 (Fig. S6), DLS data (Fig. S7–S10), powder XRD patterns (Fig. S11), UV-Vis spectra (Fig. S12) and appendix for the derivation of specific surface area of particles. See DOI: 10.1039/c5ta02446c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence