
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-quality functionalized few-layer graphene: facile fabrication and doping with nitrogen as a metal-free catalyst for the oxygen reduction reaction†
Zhenyu
Sun
*abcd,
Justus
Masa
a,
Philipp
Weide
b,
Simon M.
Fairclough
c,
Alex W.
Robertson
 e,
Petra
Ebbinghaus
f,
Jamie H.
Warner
e,
S. C. Edman
Tsang
c,
Martin
Muhler
b and
Wolfgang
Schuhmann
a
e,
Petra
Ebbinghaus
f,
Jamie H.
Warner
e,
S. C. Edman
Tsang
c,
Martin
Muhler
b and
Wolfgang
Schuhmann
a
aAnalytical Chemistry Center for Electrochemical Sciences (CES), Ruhr-Universität Bochum, 44780 Bochum, Germany. E-mail: zhenyus@iccas.ac.cn; Fax: +49 234 3214115
bLaboratory of Industrial Chemistry, Ruhr-Universität Bochum, 44780 Bochum, Germany
cDepartment of Chemistry, University of Oxford, OX1 3QR, UK
dState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
eDepartment of Materials, University of Oxford, OX1 3PH, UK
fMax-Planck-Institut für Eisenforschung GmbH, Max-Planck-Str. 1, 40237 Düsseldorf, Germany
First published on 5th June 2015
Abstract
Functionalization of graphene is fundamental to facilitating its processing and offers a wide scope for advanced applications. Here we demonstrate a facile, highly efficient and mild covalent functionalization of graphene using HNO3 vapour. This results in functionalized few-layer graphene (FLG) that is high in both quantity and quality. We fully characterized the structure and defect level of functionalized FLG by X-ray photoelectron spectroscopy, high-resolution transmission electron microscopy and Raman spectroscopy. The results from this analysis show the tunability of the surface oxygen functionalities of FLG achieved through controlling the oxidation temperature without affecting the major intrinsic properties of graphene. This allows for further doping for applications, for example with nitrogen as a metal-free catalyst in the oxygen reduction reaction.
1. Introduction
There have been intense research efforts recently focused on the functionalization of graphene to help facilitate processing and enable advanced applications to be realised.1–3 Two strategies are well established and applied to the chemistry of graphene, akin to that of carbon nanotubes: (1) covalent functionalization via oxidation,4 hydrogenation,5 diazonium reaction,6 n-type or p-type chemical doping7,8 and (2) non-covalent functionalization with solvents,9,10 surfactants,11,12 polymers13,14 or biomolecules.15 Covalent modification is of particular interest as it allows fine tuning of the electronic and optical properties of the two-dimensional structure, offering potential for graphene manipulation and further development of novel and multifunctional materials. Currently, the most common approach is the wet chemical oxidation of graphite with an excess of strongly oxidizing reagents (e.g., HNO3, H2SO4, KMnO4), resulting in oxygen functionalities such as epoxide, carboxy, or hydroxy moieties.16 However, in addition to the tedious multi-step processes involved, toxic gases (e.g., NO2, N2O4, and/or ClO2) are potentially generated causing safety concerns and environmental issues. Another major disadvantage is the severe disruption of the π system, leading to extensive degradation of graphene's electronic structure. A large number of irremediable holes and sp3 defects with an average distance of 1–2 nm are formed as a result of the aggressive oxidation, which significantly hinders applications that require high-quality functionalized graphene. Aside from these drawbacks, interfering heteroatomic species such as S, or metal-containing impurities (K and/or Mn) may be covalently bound or strongly physisorbed to graphene oxide (GO), thus complicating further processing.17 Indeed, we recently showed that the presence of trace metal residues can profoundly alter the electrocatalytic properties of the catalyst.18 Therefore, it is important to develop new ways that enable an effective surface modification, while retaining the inherent properties of graphene.Herein, we show that high-quality functionalized few-layer graphene (FLG) can be produced on a large scale through a mild, but effective, gas-phase oxidation protocol using HNO3 vapour. This method has aesthetic appeal due to its simplicity and ease of handling, and also avoids the adventitious introduction of foreign metal atoms. Importantly, controlling the oxidation temperature allows the surface oxygen functionalities to be readily tuned whilst preserving the majority of the intrinsic properties of graphene. Moreover, we show that the functionalized FLG can be further doped with nitrogen resulting in a metal-free catalyst for oxygen reduction.
2. Experimental
2.1. Materials
All chemicals used in this work were of analytical grade and used as supplied. N-Methyl-2-pyrrolidinone (product number 328624) (NMP) and nitric acid (product number 30709) were purchased from Sigma-Aldrich. Graphite powder (product number 332461) was also acquired from Sigma-Aldrich and used without further treatments.2.2. Liquid-phase exfoliation of graphite to make graphene
Pristine layered graphite flakes were used as starting materials for graphene exfoliation. Graphene dispersions were prepared by adding graphite to 400 mL NMP (500 mL round-bottom flask) with the aid of bath ultrasonication (Bandelin Sonorex, 35 kHz), a technique applied to accelerate exfoliation.9 After 48 h of ultrasonication, the dispersions were left to stand overnight to allow any unstable graphite aggregates to form and were subsequently centrifuged (ROTOFIX 32A). After centrifugation at 3000 rpm for 30 min, the top two-thirds of the dispersions were gently extracted by pipetting. Few-layer graphene powder was obtained by vacuum filtration of the centrifuged dispersions onto porous nylon membranes (Whatman, 0.2 μm pore size, 47 mm membrane diameter) followed by drying at 60 °C overnight.2.3. Functionalization of few-layer graphene
A simple gas-phase oxidation strategy was adapted in this procedure. Typically, the filtered few-layer graphene flakes were functionalized using a nitric acid vapor at varying temperatures for 13 h. After cooling, the sample was dried at 60 °C overnight, obtaining oxidized FLG (OFLG). For doping of OFLG with nitrogen, 50 mg of the OFLG was loaded into a tubular quartz reactor, and 10 vol% NH3 in He was introduced at 400 °C for 6 h with a flow rate of 25 sccm.2.4. Characterization
The structure and quality of the functionalized few-layer graphene were evaluated by multiple characterizations, including X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and Raman spectroscopy. XRD was performed with a D/MAX-RC diffractometer operated at 30 kV and 100 mA with Cu Kα radiation. XPS measurements were carried out in an ultra-high vacuum (UHV) setup equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV), operated at 14.5 eV and 30.5 mA. The base pressure in the measurement chamber was maintained at about 5 × 10−10 mbar. The high-resolution spectra were carried out in the fixed transmission mode with pass energy of 200 eV, resulting in an overall energy resolution of 0.6 eV. A flood gun was applied to compensate for the charging effects. The CasaXPS software with a Gaussian–Lorentzian product function and Shirley background subtraction was used for peak deconvolution. SEM observations were carried out using a field emission microscope (FEI Quanta 600 FEG) operated at 20 kV and equipped with an energy-dispersive X-ray spectrometer. Standard TEM was using a JOEL 2100 running at 80 kV. HRTEM images were taken using a double aberration corrected JOEL 2200MCO TEM with CEOS third-order probe and image aberration correctors operated at 80 kV. The images were processed by applying a low pass FFT filter on the images, and regions of amorphous carbon were identified by masking and subtracting the crystalline graphene identified through the FFT from the image.19 TEM samples were prepared by pipetting a few milliliters of the dispersion in ethanol onto holey carbon mesh grids (400 mesh). Raman spectra of graphene films and graphite powder were collected with a Horiba Jobin Yvon LabRam 2 confocal Raman microscope with a HeNe laser excitation at 633 nm (1.96 eV) with a power of 3.7 mW. Deposited thin films were prepared by vacuum filtration of the dispersions in NMP onto porous nylon membranes (Whatman, 0.2 μm pore size, 47 mm membrane diameter) and dried at room temperature. Measurements were taken with 5 s of exposure time using a long working distance objective of 50-fold magnification and aperture 0.5 yielding a beam diameter of ∼600 nm in the focus. The peak maximum intensity ratio ID/IG was obtained by taking the peak intensities following baseline corrections to remove residual fluorescence.3. Results and discussion
To gain insight into the structure and quality of the obtained flakes, a number of characterization methods were applied (vide infra). The X-ray diffraction (XRD) patterns of the resultant samples, in Fig. 1a, indicate good crystallinity of the hexagonal structure regardless of the oxidation up to 275 °C. The diffraction peaks at ∼26.8 and ∼54.8° in all traces are attributed to the (002) and (004) reflections of the graphite structure, respectively.10,13,14 The two peaks in traces B–F in Fig. 1a, appear at the same position as those in trace A, which suggests that the graphite lattice was retained after the chemical treatment. No pronounced peak at 10°, characteristic of GO corresponding to a basal spacing of 0.85 nm, was found. A significant weakening in the intensity of the (004) reflection, relative to graphite, occurred in all cases, which supports the formation of randomly stacked few-layer graphene.10,13,14 Fig. S1† shows the UV-vis spectra of pristine FLG and oxidised FLG. A strong peak at about 269 nm was observed in both cases which is due to the conjugated aromatic domains in the graphitic structure.12 However, we did not see any pronounced absorption peak at 230 nm attributable to conjugated ketones or dienes typical for GO.17 This suggests that the chemical structure of our oxidised FLG is distinct from the conventional GO. X-ray photoelectron spectroscopy (XPS) analysis was further employed to probe the surface oxygen species in the samples (Fig. 1b–f and S2†). A dominant C 1s peak at 284.4 eV, assigned to sp2 graphitic carbon, was observed in all cases (Fig. 1b and S2b–d†). Carbon singly bound to oxygen in phenols and ethers (i.e. C–O) at 285.9 eV, carbon doubly bound to oxygen in ketones and quinones (i.e. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 287.6 eV, carbon bound to two oxygen atoms in carboxyls, carboxylic anhydrides and esters (i.e. –COO) at 288.9 eV, and the characteristic shake-up line of aromatic compounds at 290.9 eV (π → π* transition) may also contribute.20 We further estimated the ratio of C bound to varying O species and graphitic C (sp2), as shown in the inset of Fig. 1b. It shows that the FLG functionalized at 125 °C contains 25% oxidised C and 75% graphitic C. All four ratios tended to decrease with the oxidation temperature below 275 °C suggesting that the removal of metastable O species is predominant, rather than the introduction of new O moieties during the treatment. However, an increase of the ratios was observed at 275 °C indicative of higher amounts of O containing functional groups. Despite this, we note that the oxidation level at 275 °C (28% oxidised C and 72% graphitic C) is still much lower than the one reported for GO in literature (69% oxidised C and 31% graphitic C).21 Deconvolution of the O 1s spectrum manifested three peaks at 530.5, 532.4, 533.4 eV indicating the presence of various oxygen-containing functional groups with doubly and singly bound oxygen (Fig. 1c–f).20 The relative ratio of O singly or doubly bound to C follows a similar trend to the results obtained based on the C 1s XPS spectra. The minimal C/O ratio in the functionalized FLG here is 16.8, which is much higher as compared to GO with a C/O ratio typically in the range of 1.8–2.5.17 This observation confirms the high quality of FLG after functionalization.
O) at 287.6 eV, carbon bound to two oxygen atoms in carboxyls, carboxylic anhydrides and esters (i.e. –COO) at 288.9 eV, and the characteristic shake-up line of aromatic compounds at 290.9 eV (π → π* transition) may also contribute.20 We further estimated the ratio of C bound to varying O species and graphitic C (sp2), as shown in the inset of Fig. 1b. It shows that the FLG functionalized at 125 °C contains 25% oxidised C and 75% graphitic C. All four ratios tended to decrease with the oxidation temperature below 275 °C suggesting that the removal of metastable O species is predominant, rather than the introduction of new O moieties during the treatment. However, an increase of the ratios was observed at 275 °C indicative of higher amounts of O containing functional groups. Despite this, we note that the oxidation level at 275 °C (28% oxidised C and 72% graphitic C) is still much lower than the one reported for GO in literature (69% oxidised C and 31% graphitic C).21 Deconvolution of the O 1s spectrum manifested three peaks at 530.5, 532.4, 533.4 eV indicating the presence of various oxygen-containing functional groups with doubly and singly bound oxygen (Fig. 1c–f).20 The relative ratio of O singly or doubly bound to C follows a similar trend to the results obtained based on the C 1s XPS spectra. The minimal C/O ratio in the functionalized FLG here is 16.8, which is much higher as compared to GO with a C/O ratio typically in the range of 1.8–2.5.17 This observation confirms the high quality of FLG after functionalization.
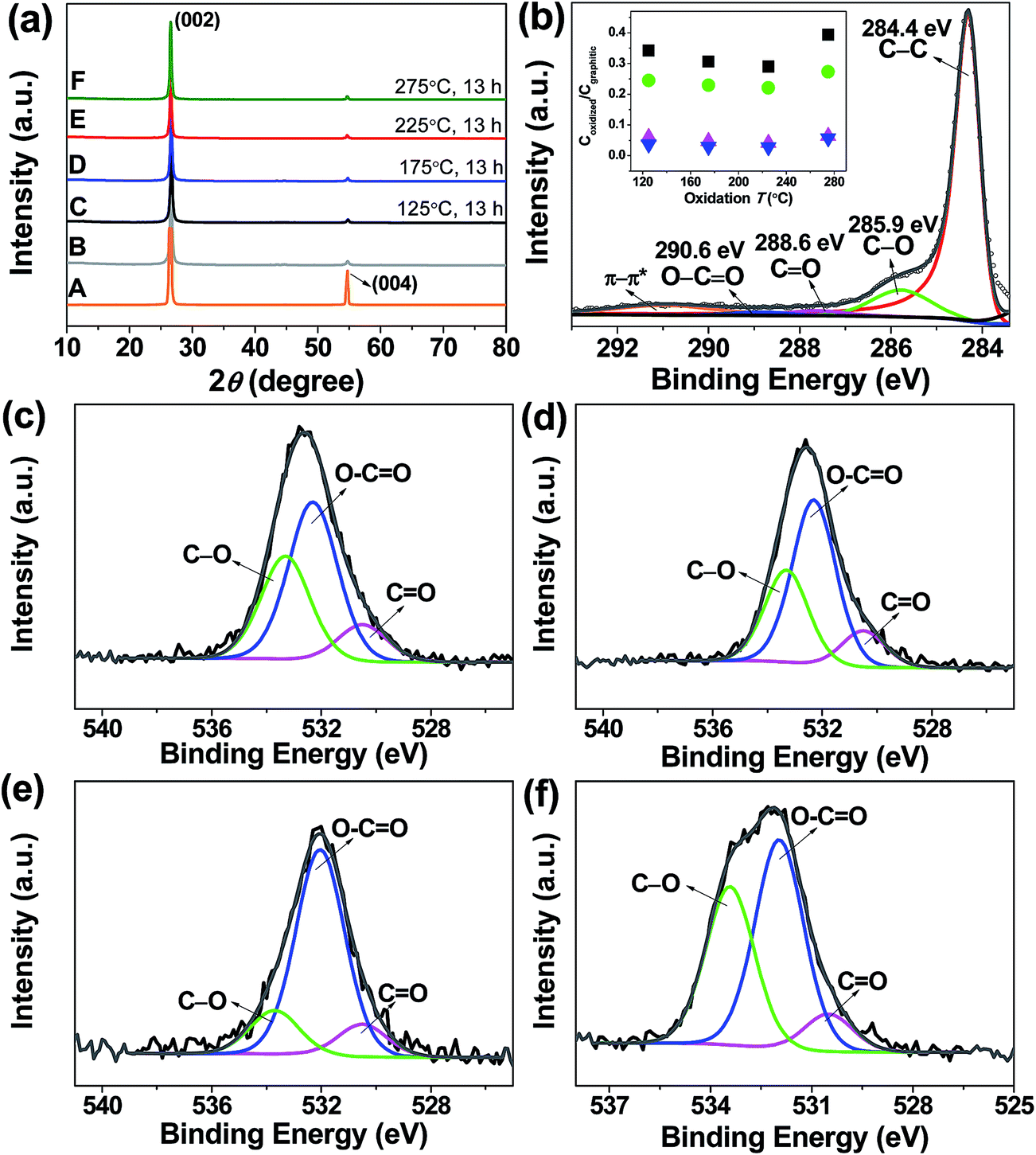 | ||
Fig. 1 (a) XRD patterns of starting graphite (trace A), pristine FLG (trace B) and functionalized FLG obtained at varying temperatures (traces C–F). (b) C 1s XPS spectrum of the functionalized FLG produced at 225 °C. Inset: molar ratio of C singly bound to oxygen ( ), carbon doubly bound to oxygen ( ), carbon doubly bound to oxygen ( ), carbon bound to two oxygen atoms ( ), carbon bound to two oxygen atoms ( ) and the sum of the three C ( ) and the sum of the three C ( ) to graphitic C as a function of oxidation T. O 1s XPS spectra of functionalized FLG at (c) 125 °C, (d) 175 °C, (e) 225 °C and (f) 275 °C. ) to graphitic C as a function of oxidation T. O 1s XPS spectra of functionalized FLG at (c) 125 °C, (d) 175 °C, (e) 225 °C and (f) 275 °C. | ||
Scanning electron microscopy (SEM) (Fig. 2) and transmission electron microscopy (TEM) (Fig. 3a) images show large quantities of graphene-like flakes. The edge-on HRTEM image of functionalized FLG (Fig. 3b) displays a characteristic inter-graphene spacing of 0.34 nm, in contrast to the double layer spacing of GO. An inspection of the HRTEM at high magnification shows the crystalline nature of both FLG (Fig. 3c) and OFLG consisting of an extended, two-dimensional sp2 bound carbon “honeycomb” lattice (Fig. 3d and f and S3a†). Subtracting the crystalline structure from the image, by masking diffraction planes within the FFT, enables the amorphous carbon and defects to be clearly observed (Fig. 3e and S3b†).19 The graphitic structure within the OFLG (Fig. 3d and f and S3a†) comprised up to approximately 75% of the area which suggests a substantial preservation of the original sp2 bonding character. It is noted that, as opposed to most GO observed in literature,22 holes were not seen in these OFLG.
 | ||
| Fig. 2 SEM images of a film formed by vacuum filtration of the OFLG (obtained at 225 °C) dispersion in NMP: (a) the surface and (b) edge of the film. In addition to small flakes of several hundred nanometers, large sheets with dimensions of up to 5 μm in length were detected. The edge of the film consists of interconnected flakes showing a well-defined layered morphology. | ||
 | ||
| Fig. 3 (a) Wide-field TEM image of oxidised FLG. (b) TEM image of a monolayer stacked with a four-layer flake from an oxidised FLG sample. Aberration-corrected HRTEM images. (c) A pristine graphene monolayer. (d)–(f) Oxidised FLG. The arrows shown in image (e) point to the regions of amorphous carbon in image (d) which were highlighted by masking and subtracting the crystalline graphene identified through the FFT from the image. The inset in (f) shows the FFT of the image. The oxidised FLG shown in (a, b and d–f) was obtained at 125 °C. | ||
Raman spectroscopy enabled us to study any generated defects. The Raman spectra of thin films prepared by filtering pristine– (trace B, Fig. 4a) and functionalized– (traces C–F, Fig. 4a) graphene dispersions all show an intense G band, a second-order two phonon mode 2D (also called G′) band and a disorder-related D peak. In stark contrast to the weak and broad 2D peak demonstrated for GO, the doublet 2D shape consisting of two components 2D1 and 2D2 typical of graphite, the 2D line for functionalized FLG here can be described as a single symmetrical Lorentzian peak in agreement with the Raman spectrum of randomly stacked flakes comprising fewer than 5 layers.9–14 The defect level, as a function of oxidation temperature, was investigated in terms of the integrated Raman peak intensity (AD/AG) and the average peak intensity of the D band relative to the G band (ID/IG). It was found that AD/AG and ID/IG increased with oxidation temperature up to 125 °C beyond which both tended to decrease with temperature. Note that the maximum value of ID/IG is around 0.4 which is far less than that of GO (ID/IG ≈ 1.47)23 and high-quality reduced GO (ID/IG ≥ 0.5).24 Interestingly, the values of AD/AG and ID/IG for the FLG oxidised at 275 °C are both lower than those of pristine FLG, which may be due to the removal of some sp3 defects by total oxidation. The in-plane crystallite size of graphene (La) was estimated based on the integrated area ratio of the D band and G band (AD/AG) using the equation 560(AD/AG)−1/E4, where E is the laser energy (1.96 eV).25 We found that La was first reduced upon treatment with HNO3 vapour at 125 °C but started to rise with the increase of T. We further calculated the distance between defects (LD), and the defect density of graphene (nD) using the approximations of LD2 (nm2) = 4300(ID/IG)−1/E4 and nD (cm−2) = 1014/πLD2, respectively.26LD showed a similar trend to La as a function of T. Though a prominent increase in nD was observed for the functionalized FLG at 125 °C (nD = 4.3 × 1010 ± 1.6 × 109 cm−2) compared to pristine FLG (nD = 3.0 × 1010 ± 2.7 × 109 cm−2), the value fell gradually with further increase of T reaching 2.7 × 1010 ± 1.6 × 109 cm−2, which is even lower than that of pristine FLG. Raman mapping of ID/IG over 25 regions (Fig. S4†) displays low defect densities of functionalized FLG with an overall large domain size. Given that the area occupied by a carbon atom in graphite is (33/2/4)d2,24,26 where d is the carbon–carbon bond length (0.1421 nm), the unperturbed domain size of the functionalized FLG at 125 °C is correlated to about 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 carbon atoms. This is three times larger than those (≤9900 carbon atoms) observed for chemically derived graphene.24 These results strongly suggest the high quality of functionalized FLG produced using the approach proposed in this work.
300 carbon atoms. This is three times larger than those (≤9900 carbon atoms) observed for chemically derived graphene.24 These results strongly suggest the high quality of functionalized FLG produced using the approach proposed in this work.
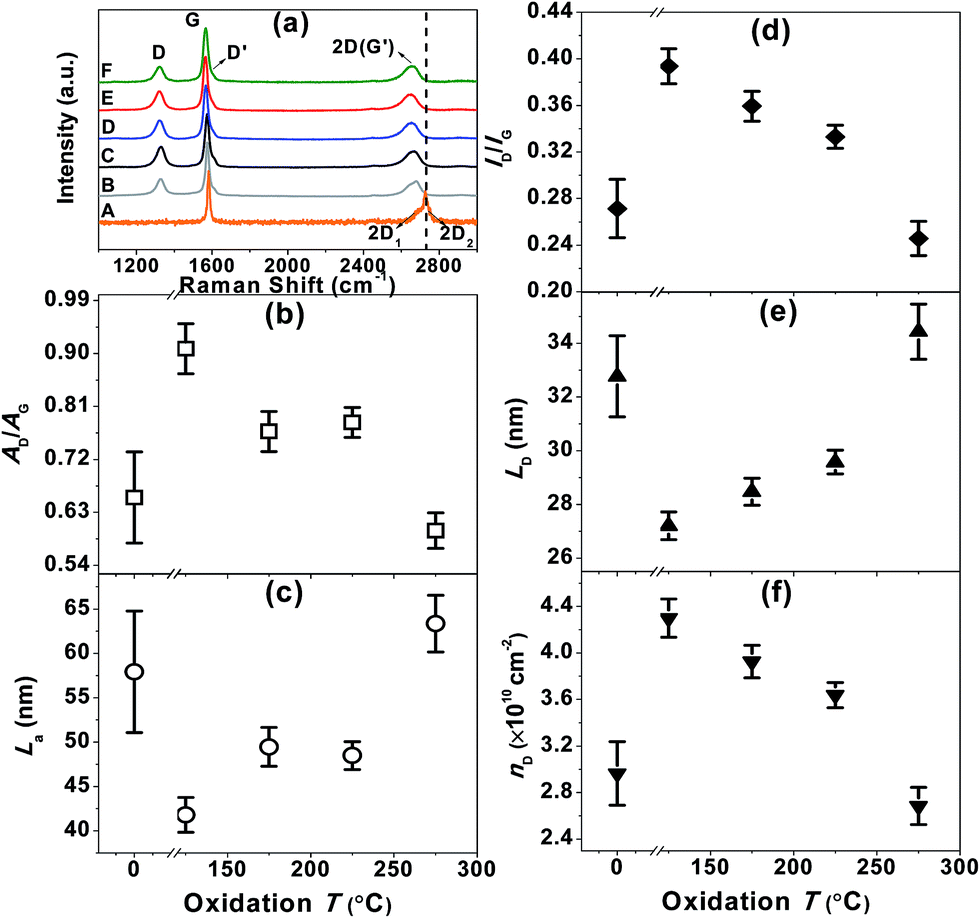 | ||
| Fig. 4 (a) Raman spectra (633 nm excitation) of the starting graphite (trace A), FLG (trace B) and functionalized FLG (traces C–F). The spectra were normalized based on the G mode peak intensity. (b) AD/AG, (c) La, (d) ID/IG, (e) LD and (f) nD as a function of oxidation T. | ||
Three steps are generally involved in the modified Hummers' method that is most commonly used for the preparation of GO.27 Bulk graphite is first converted to a sulfuric acid–graphite intercalation compound, which is then transformed to pristine graphite oxide (PGO). In the end, GO is formed by the reaction of PGO with a solvent, involving the hydrolysis of covalent sulfates and loss of interlayer registry. In contrast to this conventional reaction pathway, here, the nitronium ion (NO2+) in nitric acid may initially attack the defect sites of the graphene surface, yielding a high concentration of hydroxyl or ether-type oxygen groups. This conjecture can be confirmed by the dominant C–O functional species derived from the C 1s spectra, being consistent with what has been observed for highly ordered pyrolytic graphite after treatment in 0.5 M HNO3.28 XPS results also suggested the presence of a small fraction of carbonyls (most probably in ketones).28 These carbonyls are formed where C–C bonds have been cleaved and can further convert into carboxyls and carboxylic anhydrides.17 Based on the average OFLG flake size (Fig. 2a and 3a), the ratio of the edge carbon atoms to the basal plane atoms in the samples is ∼1/200. Therefore, we conclude that some C–O and CO atoms are located on the edges of vacancy defects in the basal planes.
We further demonstrated that the oxygen-functionalized FLG can also be modified with nitrogen upon exposure to NH3, yielding a metal-free catalyst for the oxygen reduction reaction. XPS and EDX mapping during SEM observations confirmed the formation of nitrogen-containing groups in the sample (Fig. S5 and S6†). The N 1s region showed three nitrogen-functionalized carbon groups, namely pyridinic (398.6 eV, N1), pyrrolic (399.8 eV, N2), and quaternary (401.3 eV, N3), respectively (Fig. S5 and S6†).18,29 Evaluation of oxygen reduction by rotating disc electrode (RDE) voltammetry revealed that the N-doped FLG prepared by pretreatment with HNO3 vapour at 225 °C exhibits significantly better performance, with a higher limiting current density but a lower onset potential than pristine FLG (Fig. 5a). This enhancement can be attributed to the presence of pyridinic N which reduces the adsorption energy of O2, with graphitic N being able to promote the electro-reduction of O2 to H2O2via a two-electron pathway.29,30 The onset potential is ≈0.755 V (vs. RHE), comparable to the value of 0.743 V (vs. RHE) reported for chemically synthesized N-graphene,30 whilst the current densities at 0.7, 0.6, 0.5 and 0.2 V being approximately 0.4, 1.2, 1.5 and 2 mA cm−2, respectively, are higher than those (≈0.2, 0.5, 0.9 and 1.3 mA cm−2 at 0.7, 0.6, 0.5 and 0.2 V, respectively) demonstrated for N-graphene.30 We should point out that the N content in the sample is only 0.52 at%, much less than the N doping level (in the range of 3–5 at%) of graphene shown in literature.30 The Koutecky–Levich (K–L) plots showed a linear relationship between 1/i, 1/ik, and 1/w1/2 as follows:
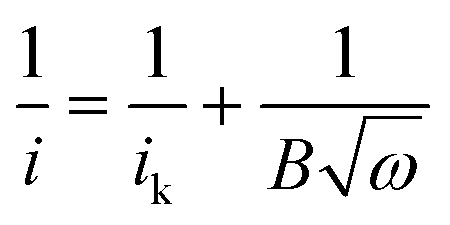 485 C mol−1); A, the geometric area of the electrode (0.1256 cm2); ν, the kinematic viscosity of the electrolyte (0.01 cm2 s−1), and C* the solubility of oxygen in the electrolyte, which for the case of KOH (0.1 M) was taken to be 1.2 × 10−6 mole cm−3 (Fig. 5b).29–32 The average number of electrons transferred during oxygen reduction on the basis of K–L analysis was found to slightly increase with overpotential, as depicted by the noticeable increase in the slope of the K–L lines from 0.2 V to 0.7 V (Fig. 5b), corresponding to 3.2 electrons at 0.75 V to 3.9 electrons at 0.2 V (Fig. 5c). The results indicate that oxygen reduction proceeds concurrently through both two sequential two-electron reactions with formation of OOH− intermediate, and through a direct four-electron reaction. Indeed, the features of the voltammograms are consistent with a series mechanism involving the reduction of oxygen to a peroxide intermediate at low overpotentials and its further reduction to OH− at higher potentials, as previously reported for N-doped graphene29,30 and other metal-free catalysts.18,31 Chemical disproportionation of the peroxide intermediate is also expected to proceed in parallel with its further electrochemical reduction, ultimately converting all the oxygen to OH−.30,31 Chronoamperometric measurements at 0.6 V showed no evident loss in activity after 8 h (Fig. 5d), indicating that the catalyst is able to sustain a stable performance over a long period of time.
485 C mol−1); A, the geometric area of the electrode (0.1256 cm2); ν, the kinematic viscosity of the electrolyte (0.01 cm2 s−1), and C* the solubility of oxygen in the electrolyte, which for the case of KOH (0.1 M) was taken to be 1.2 × 10−6 mole cm−3 (Fig. 5b).29–32 The average number of electrons transferred during oxygen reduction on the basis of K–L analysis was found to slightly increase with overpotential, as depicted by the noticeable increase in the slope of the K–L lines from 0.2 V to 0.7 V (Fig. 5b), corresponding to 3.2 electrons at 0.75 V to 3.9 electrons at 0.2 V (Fig. 5c). The results indicate that oxygen reduction proceeds concurrently through both two sequential two-electron reactions with formation of OOH− intermediate, and through a direct four-electron reaction. Indeed, the features of the voltammograms are consistent with a series mechanism involving the reduction of oxygen to a peroxide intermediate at low overpotentials and its further reduction to OH− at higher potentials, as previously reported for N-doped graphene29,30 and other metal-free catalysts.18,31 Chemical disproportionation of the peroxide intermediate is also expected to proceed in parallel with its further electrochemical reduction, ultimately converting all the oxygen to OH−.30,31 Chronoamperometric measurements at 0.6 V showed no evident loss in activity after 8 h (Fig. 5d), indicating that the catalyst is able to sustain a stable performance over a long period of time.
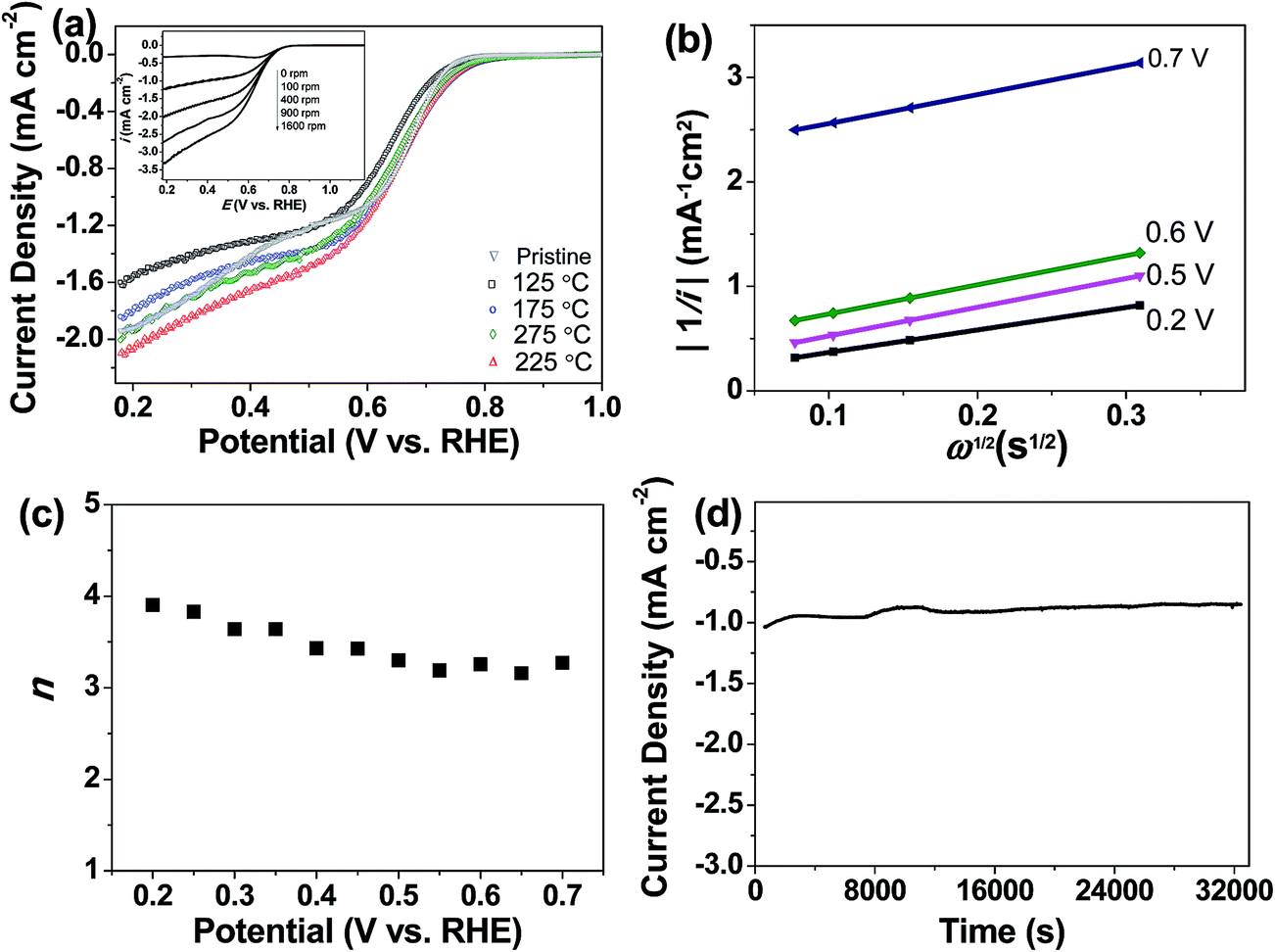 | ||
| Fig. 5 (a) Background corrected voltammograms recorded at 400 rpm and 10 mV s−1 in oxygen-saturated KOH (0.1 M) showing oxygen reduction by N-doped FLG pretreated with HNO3 vapour at different temperatures. Inset: voltammograms recorded at different rotation speeds at a scan rate of 10 mV s−1 in oxygen-saturated KOH (0.1 M) for the N-doped FLG pretreated with HNO3 vapour at 225 °C. (b) K–L plot extracted from the inset in (a). (c) Number of electrons transferred at different potentials. (d) Long-term chronoamperometric stability test at 0.6 V vs. RHE obtained at a rotation speed of 400 rpm in oxygen-saturated KOH (0.1 M). | ||
4. Conclusions
In conclusion, we have developed a very simple and mild but effective gas-phase oxidation strategy using HNO3 vapour for functionalization of graphene. XPS, HRTEM and Raman spectroscopy suggest the substantial preservation of the intrinsic properties of graphene after oxidation. The C/O ratio in the functionalized FLG (C/O ratio ∼16.8) is much higher than GO with a C/O ratio typically in the range of 1.8–2.5. Controlling the oxidation temperature allows us to readily tune the surface oxygen functionalities. This strategy opens up a new route to produce high-quality oxidised few-layer graphene which can thus be used as a functional platform for advanced applications.References
- J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53–64 CrossRef CAS PubMed.
- A. Hirsch, J. M. Englert and F. Hauke, Acc. Chem. Res., 2013, 46, 87–96 CrossRef CAS PubMed.
- M. Quintana, E. Vazquez and M. Prato, Acc. Chem. Res., 2013, 46, 138–148 CrossRef CAS PubMed.
- M. B. C. Brodie, Ann. Chim. Phys., 1855, 45, 351–353 Search PubMed.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- I. Gierz, C. Riedl, U. Starke, C. R. Ast and K. Kern, Nano Lett., 2008, 8, 4603–4607 CrossRef CAS PubMed.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- Z. Y. Sun, X. Huang, F. Liu, X. N. Yang, C. Rösler, R. A. Fischer, M. Muhler and W. Schuhmann, Chem. Commun., 2014, 50, 10382–10385 RSC.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. M. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- Z. Y. Sun, J. Masa, Z. M. Liu, W. Schuhmann and M. Muhler, Chem.–Eur. J., 2012, 18, 6972–6978 CrossRef CAS PubMed.
- Z. Y. Sun, S. Pöller, X. Huang, D. Guschin, C. Taetz, P. Ebbinghaus, J. Masa, A. Erbe, A. Kilzer, W. Schuhmann and M. Muhler, Carbon, 2013, 64, 288–294 CrossRef CAS PubMed.
- Z. Y. Sun, J. Vivekananthan, D. A. Guschin, X. Huang, V. Kuznetsov, P. Ebbinghaus, A. Sarfraz, M. Muhler and W. Schuhmann, Chem.–Eur. J., 2014, 50, 5752–5761 CrossRef PubMed.
- F. Liu, J. Y. Choi and T. S. Seo, Chem. Commun., 2010, 46, 2844–2846 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- J. Masa, A. Q. Zhao, W. Xia, Z. Y. Sun, B. Mei, M. Muhler and W. Schuhmann, Electrochem. Commun., 2013, 34, 113–116 CrossRef CAS PubMed.
- A. W. Robertson, A. Bachmatiuk, Y. A. Wu, F. Schäffel, B. Rellinghaus, B. Büchner, M. H. Rümmeli and J. H. Warner, ACS Nano, 2011, 5, 6610–6618 CrossRef CAS PubMed.
- W. Xia, C. Jin, S. Kundu and M. Muhler, Carbon, 2009, 47, 919–922 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS PubMed.
- J. Q. Liu, C. F. Chen, C. C. He, J. Zhao, X. J. Yang and H. L. Wang, ACS Nano, 2012, 6, 8194–8202 CrossRef CAS PubMed.
- S. Eigler, M. Enzelberger-Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Röckert, J. Xiao, C. Papp, O. Lytken, H. P. Steinrück, P. Müller and A. Hirsch, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS PubMed.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 CrossRef PubMed.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS PubMed.
- R. Burgess, C. Buono, P. R. Davies, R. J. Davies, T. Legge, A. Lai, R. Lewis, D. J. Morgan, N. Robinson and D. J. Willock, J. Catal., 2015, 323, 10–18 CrossRef CAS PubMed.
- L. F. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. H. Tang, H. Gong, Z. X. Shen, J. Y. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- J. Masa, A. Q. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS PubMed.
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015 DOI:10.1021/cr5003563.
Footnote |
| † Electronic supplementary information (ESI) available: UV spectra, XPS spectra, Raman maps, HRTEM images of oxidised FLG, N 1s XPS spectrum, SEM image of N-doped FLG. See DOI: 10.1039/c5ta02248g |
| This journal is © The Royal Society of Chemistry 2015 |