
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ditopic calix[4]pyrrole amide derivative: highlighting the importance of fundamental studies and the use of NaPh4B as additive in the design and applications of mercury(II) ion selective electrodes†
Angela F. Danil
de Namor
*,
Abdelaziz
El Gamouz
,
Salman
Alharthi
,
Nawal
Al Hakawati
and
John R.
Varcoe
Department of Chemistry, Faculty of Physics and Engineering Sciences, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: A.Danil-De-Namor@surrey.ac.uk
First published on 15th May 2015
Abstract
The use of calixpyrroles as cation monitoring systems is very limited. The importance of fundamental studies for providing the basis for the selection of an appropriate ionophore for use in the design of ion selective electrodes cannot be overemphasised. Thus a new ditopic receptor, namely meso-tetramethyl-tetrakis-(4N,N-diethylacetamide) phenoxymethyl]calix[4]pyrrole (CPA), bearing cation and anion recognition sites has been synthesized and characterized using NMR measurements in acetonitrile. This new receptor interacts with Ca(II), Sr(II), Ba(II), Pb(II) and Cd(II) perchlorate salts and Hg(II) (nitrate as counter-ion). Among the cations tested, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1: complexes were found between CPA and the above mentioned cations except Hg(II) for which a 1:2 (ligand:metal cation) complex is found. These fundamental studies provided the basis for the design of a Hg(II) ion selective electrode (ISE). A detailed investigation on possible interactions between the analyte and the individual components of the membrane in solution as well as in membranes prepared in the absence of the receptor, revealed that NaPh4B, an additive frequently used in the preparation of mercury ion selective membranes, interacts with Hg(II). The implications of these findings on previously reported (Hg(II) ISE) are discussed. The optimum working conditions of the new electrode on its potentiometric response were established. Selectivity coefficients of Hg(II) relative to other cations were calculated. The advantages of this electrode relative to others reported in the literature are discussed. Analytical applications include its use as an indicator electrode as well as for the quantitative determination of this cation in an amalgam.
:1: complexes were found between CPA and the above mentioned cations except Hg(II) for which a 1:2 (ligand:metal cation) complex is found. These fundamental studies provided the basis for the design of a Hg(II) ion selective electrode (ISE). A detailed investigation on possible interactions between the analyte and the individual components of the membrane in solution as well as in membranes prepared in the absence of the receptor, revealed that NaPh4B, an additive frequently used in the preparation of mercury ion selective membranes, interacts with Hg(II). The implications of these findings on previously reported (Hg(II) ISE) are discussed. The optimum working conditions of the new electrode on its potentiometric response were established. Selectivity coefficients of Hg(II) relative to other cations were calculated. The advantages of this electrode relative to others reported in the literature are discussed. Analytical applications include its use as an indicator electrode as well as for the quantitative determination of this cation in an amalgam.
Introduction
Monitoring water quality is a process that requires sensors activated with materials capable of selectively recognising pollutants. The drive for a mercury(II) monitoring system is based on the following facts: (i) both inorganic as well as organic mercury species are strongly toxic and their presence in the food chain is undesirable. They are likely to be transferred to the global food chain where their concentration increases over time, a process known as bio-magnification, which has been observed for mercury species that result from common practices in industrial processes. (ii) Mercury can be released into the environment from different sources such as chloro-alkali, incineration of coal, dental, medical, gold mining, button cell battery manufacturing, other industrial waste and even natural sources (e.g. volcanoes) This means that mercury can be present in different forms as an environmental pollutant, dominantly in the form of inorganic Hg(II), with atmospheric deposition being the dominant source of mercury over most of the landscape. Once it is in the environment it goes through a complex conversion cycle between atmosphere, ocean and land, resulting in the formation of different toxic mercury compounds.1–3 Therefore, even though methyl-mercury is recognised as the most toxic form of mercury, it is mainly the product of the transformation of Hg(II). So in order to control or reduce the amount of methyl-mercury entering the food chain, it is critical to monitor the amount of Hg(II) in water.Calixpyrroles or meso-substituted octamethyl porphyrinogens are generally crystalline stable materials which were first synthesized by Baeyer4,5 in the 19th century by the condensation reaction involving pyrrole and acetone in the presence of HCl. This work was followed by Brown et al.6 who proceeded with the production of modified calix[4]pyrroles, while Floriani et al.7,8 investigated the metallation of such macrocycles. However, their anion binding properties were discovered by Sessler and co-workers.9–11 Detailed thermodynamic studies on these systems and their anion complexes have been conducted by Danil de Namor and co-workers.12–19 With the exception of the work by Floriani7,8 on the complexation of iron with deprotonated meso octamethyl calix[4]pyrrole, which dates back to the early nineties, no other work can be tracked in the literature concerning the interaction of calix[4]pyrrole and its derivatives with cations. We have previously reported the thermodynamics of complexation of sulphur containing hetero-calixpyrroles with the Hg(II) cation.13,16 More recently an ion selective electrode based on one of these ligands has been reported based on research carried out at Surrey.20
Within this context we have now synthesised and characterised meso-tetramethyl-tetrakis-[(4N,N diethyl acetamide)phenoxymethyl]calix[4]pyrrole, CPA and its complexation properties with a variety of cations, mainly alkali, alkaline-earth and heavy metal cations. Fundamental studies were carried out to gain information on the sites of ligand–ion interaction through 1H NMR measurements, while the composition of the complexes as well as qualitative information on the stability and therefore the selectivity or hosting ability of the receptor for the cation were obtained from conductance data. The outcome of this investigation was used to explore the capability of the receptor for the design of a Hg(II) ion selective electrode. Optimal working conditions for the potentiometric response of the electrode in terms of its membrane composition and the pH of the aqueous solution were investigated together with the characteristics of this electrode regarding linear working range, detection limit, lifetime and interferences. The advantages of this new electrode relative to those based on ionophores (such as crown ethers derivatives,21–26 calixarene derivatives,27–29 cryptand 22,30 tetra-aza macrocycles31–33 as well as other compounds34–53) are discussed.
Experimental section
Chemicals
Pyrrole (99.0%), 4-hydroxyacetophenone (99.0%), methane sulfonic acid (99.0%) potassium carbonate (≥99%), anhydrous MgSO4 (≥99.5%), 18-crown-6 (≥99.0%), N,N-diethyl chloroacetamide (97.0%), dichloromethane (DCM, ≥99.5%), methanol (≥99.8%) and acetonitrile (99.9%), were purchased from Sigma-Aldrich. and tetrahydrofurane (99.9%), (Fisher Scientific) were first refluxed in a nitrogen atmosphere with calcium hydride (≥95%) (Sigma-Aldrich) for several hours and then distilled. N,N-Dimethylformamide (99.8%); HPLC grade, (Sigma Aldrich) was dried over 3 Å molecular sieves and the middle distilled fraction was collected. Cations (lithium (≥95.0%), sodium (≥98.0%), rubidium (≥99.0%), magnesium (≥99.99%), calcium (≥99.0%), strontium (≥99.0%), barium (≥97.0%), lead (≥99.99%), cadmium (99.0%), zinc (≥98.0%), nickel (≥98.0%)) perchlorates and mercury(II) nitrate (≥99.99%) (Aldrich) were dried over P4O10 (≥99.99%) under vacuum for several days before use. The absence of a signal from residual water in the 1H NMR spectra of CPA with the cations in CD3CN provided an indication that the cation salts used were anhydrous. TMS (≥97.0%), CD3CN (≥99.8%), CD3OD (≥99.8), d6-acetone (≥99.9%), d6-DMSO (99.9%), CDCl3 (≥99.9%) and d7-DMF (99.9%) were purchased from Cambridge Isotope Laboratories. For the preparation of the ion selective membrane, polyvinyl chloride (PVC, 99.9%), diethyl sebacate (DOS) (≥97.0%), 2-nitrophenyl octyl ether (NPOE) (99.0%), sodium tetraphenylboron (NaPh4B) (≥99.5%) and oleic acid (OA) (≥99.0%), KCl (99.0%), NaCl (≥99.0%), silver wire (≥99.9%), HNO3 (70.0%), NaOH (98.0%), oleic acid (≥99.0%), ethylenediaminetetraacetic acid, EDTA (≥99.0%), were purchased from Aldrich. Amalgam alloy (21.55% Ag, 13.05% Cu 15.4% Sn and 50% Hg) was purchased from Nordiska ental.Synthesis of meso-tetramethyl-tetrakis-[(4N,N diethylacetamide)phenoxymethylcalix[4]pyrrole, (CPA)
Meso-tetramethyl-tetrakis-(4-hydroxyphenyl)calix[4]pyrrole (1.7 g, 2.26 mmol), potassium carbonate (2.5 g, 18.1 mmol) and 18-crown-6 (0.125 g) were vigorously stirred and refluxed for 1 h in freshly refluxed MeCN (150 ml) under a nitrogen atmosphere. Then N,N-diethyl chloroacetamide (2.46 ml, 17.9 mmol) was added drop-wise and the reaction was refluxed overnight. The reaction was monitored by TLC using DCM/MeOH (9:1 mixture) as the developing solvent system. After cooling down the reaction, the solvent was removed under vacuum. The solid obtained was re-dissolved in DCM and extracted several times with water to remove the excess of potassium carbonate. The organic phase was separated and dried over anhydrous MgSO4, then filtered through a gravimetric funnel using a filter paper. The DCM was evaporated in a rota-evaporator and the oily product was recrystallized in MeCN to give white needle crystals (70% yield). These were dried under vacuum at 80 °C. Microanalysis was carried out (C72H88N8O8) with calculated %: wt C, 72.46; H, 7.43; N, 9.39; O, 10.72. Found %: C, 71.76; H, 7.47; N, 10.11. 1H NMR (500 MHz, CDCl3) (298 K), δ (ppm): 7.65 (s, broad, 4H, NH), 7.04 (d, 8H, ArH), 6.82 (d, 8H, ArH), 5.73 (d, 8H, pyrrole –H), 4.65 (s, 8H, O–CH2–CO), 3.41 (q, 16H, N(CH2–CH3)2), 1.94 (s, 12H, CH3-bridge), 1.24 (q, 12H, N(CH2–CH3(1)2)), 1.15 (q, 12H, N(CH2–CH3(2)2)). 13C NMR (125.76 MHz), (298 K), δ (ppm): 167.43 (C1), 157.21 (C8), 137.04 (C5), 129.15 (C7), 114.25 (C6), 106.65 (C2), 68.04 (C4), 44.41 (C9), 42.08 (C11), 40.61 (C11′), 28.63 (C3), 14.90 (C12), 13.15 (C12′) (see ESI† for C labelling).
NMR measurements of the CPA–ion complexes
For these measurements, the macrocycle was dissolved in the deuterated solvent. This technique was used to characterise the ligand, provide information about the ion–ligand interaction and establish the sites of coordination of the ligand upon complexation with the ion. 1H NMR, 13C NMR, 13C DEPT NMR, HSQCDEPT NMR and COSY NMR were recorded. To assess whether the ion complexes with the ligand or not, a known concentration of the ion salt (∼8 × 10−3 mol dm−3) was added to the NMR tube containing a 10 times lower concentration of the ligand, to ensure that the whole ligand has complexed with the appropriate anion or cation salt.All NMR measurements were recorded at 298 K using a Bruker AC-500 E pulsed Fourier transform NMR spectrometer. Typical operating conditions for 1H measurements involved a “pulse” or flip angle of 30°, spectral frequency (SF) of 500.25 MHz, delay time of 1.60 s, acquisition time (AQ) of 1.819 s, and line broadening of 0.55 Hz. Solutions of the samples of interest were prepared in the appropriate deuterated solvent, and then placed in 5 mm NMR tubes using TMS as the internal reference. For the 13C NMR measurements, the operating conditions were as follows: spectral frequency of 75.469 MHz, spectral width of 301 ppm, delay time of 0.279 s, acquisition time of 0.721 s and line-broadening of 1.4 Hz with TMS as internal reference. The same NMR equipment was used to establish possible analyte-additive and analyte–plasticizer interactions.
Conductance measurements
For these measurements, a Wayne-Kerr Autobalance Universal Bridge (type B642) was used.54 The conductivity cell constant was determined by the method of Jones and Bradshaw.55 Conductometric titrations of the metal-cation salt (as perchlorates salts, except mercury where nitrate was the counter-ion used), with the ligand (CPA) were carried out in MeCN at 298.15 K. To perform these measurements, the conductometric cell was filled with accurately weighed amounts of the cation salt solution prepared in the appropriate solvent (25 ml) and left under stirring to reach thermal equilibrium. Then, a solution of the ligand prepared in the same solvent was stepwise added into the vessel. After each addition, stable conductivity readings were taken and molar conductance values were calculated. Molar conductances, Λm (Scm2 mol−1) against the ligand–cation concentration ratio [L]/[Mn+], were used to determine the stoichiometry of the complex.Preparation of the CPA based mercury(II) selective electrode
The Hg(II) selective membrane electrode based on the CPA ionophore was made using a procedure similar to that previously described in the literature.34,56 The membrane electrode with different compositions of CPA (1 and 2%) were prepared by dissolving different amounts of PVC, using DOS or o-NPOE as plasticizers. Anion additives used were NaPh4B, or OA, in freshly distilled THF (3 cm3). Membranes without CPA were also prepared to assess possible interactions between the individual components of the membrane and the analyte. The composition ratios were changed within the range most commonly encountered in the literature (1–7% ionophore, 28–33% PVC, 60–69% plasticizer and 0.03–2% ionic additive).57 The resulting mixture was poured to a glass dish (d = 30 mm). The solvent was evaporated off at room temperature. After 2 d, a resulting homogeneous PVC membrane (about 0.2 mm thickness) was cut in circles with a diameter of ∼4 mm and mounted into a PVC tube with a PVC paste (PVC dissolved in THF solvent as adhesive). An internal Ag/AgCl reference electrode was placed in the PVC tube which was filled with a solution of Hg(NO3)2 (1.0 × 10−2 mol dm−3) and KCl, (0.1 mol dm−3). The membrane electrochemical cell was set up as follows:Scanning electron microscopic experiments. Micrographs of membrane surfaces were taken by S-3200 N microscope (Oxford instrument) equipped with an X-ray detector for EDAX analysis, a Back Scatter Robinson Detector for imaging, and a Secondary electron detector.
Construction of the Hg(II)-ISE electrochemical cell. A Ag/AgCl with a flexible connector (MF-2052, RE-5B) filled with a NaCl aqueous solution (3 mol dm−3) was used as an external reference electrode while the silver wire coated with a thin layer of silver chloride was used as the internal electrode. The reference electrode was used in conjunction with the CPA based electrode.
Preparation of standard solutions of the mercury(II) salt. A stock solution of the mercury ion salt (1 × 10−2 mol dm−3) was freshly prepared by dissolving an accurate amount (0.171 g) of the salt (as nitrate) in deionized water (50 cm3) while working solutions in the range 5.0 × 10−8 to 5 × 10−2 mol dm−3 were prepared by appropriate dilution of the stock solution with deionized water.
Potential measurements of Hg(II)-ISEs based on CPA. The performance of the proposed CPA based Hg(II)-ISE was investigated by carrying out calibration curves. This procedure was followed in order to determine the composition which gives the best potential response. Different volumes of standard solution of the Hg(II) ion salt were successively added to a vessel containing deionized water (75 cm3) to cover the concentration range from 6.67 × 10−8 to 4.32 × 10−2 mol dm−3. The additions of standard solutions were carried out from the low to the high concentration. The potential measurements were made in magnetically stirred solutions and stable potential readings were taken after a period of time after each addition. The ion activities of the metal salt solutions were calculated from the ion concentrations according to Debye–Huckel procedure.24,58 Cell potential values E(mV) were plotted as a function of the log10 of the mercury ionic activity. The response characteristics of the ISE were evaluated from the calibration graph.
Effect of soaking time on the response of Hg(II)-ISE. The effect of soaking time on the performance of the CPA based Hg(II)-ISE was examined. The electrode was soaked in aqueous solution containing Hg(NO3)2 (1 × 10−2 mol dm−3) the same as the test solution for different periods of time ranging from 6 to 48 h. The slope values obtained by plotting E(mV) as a function of log10[Hg(II)] were also plotted as a function of soaking time. Thereafter, the optimum soaking time for the Hg(II)-ISE was determined.
Effect of pH of the aqueous solution of the Hg(II) salt on the electrode response. The effect of pH of the test solution on the response of the Hg(II)-ISE was examined. The potential was measured in the 2.0–12.0 pH range at two concentrations of Hg(II) solutions (1.0 × 10−2 and 1.0 × 10−3 mol dm−3). These experiments were achieved through following the variation of potential of the proposed electrode with the change in the solution pH for both concentrations used; the pH value was adjusted by the addition of a very small volume of HNO3 or NaOH (each 0.1–1.0 mol dm−3). The potential readings were plotted against the corresponding pH values. The optimum pH values obtained at both concentrations of Hg(II) solutions (1.0 × 10−2 and 1.0 × 10−3 mol dm−3) were used for further studies.
Determination of selectivity coefficients of ions relative to Hg(II). In this work, potentiometric selectivity coefficients (KpotHg,M) of Hg(II) relative to other cations were determined by using two IUPAC recommended methods, namely, the separate solution method (SSM)59 and the matched potential method (MPM).60 These two methods were carried out under the following experimental conditions: for the SSM method, the concentration of the Hg(NO3)2 (primary ion) was 2.23 × 10−4 mol dm−3; the same concentration was used for the interfering ions. For the MPM method, the concentration of Hg(NO3)2 was 1.0 × 10−2 mol dm−3 while 1.0 × 10−1 mol dm−3 was the concentration of interfering ions.
Assessment of the dynamic response time of the Hg(II)-ISE. Measurements of the dynamic response time of the electrode were conducted at different Hg(NO3)2 concentrations (1 × 10−7, 1 × 10−6, 1 × 10−5, 1 × 10−4, 1 × 10−3 and 1 × 10−2 mol dm−3). Potentials resulting from the addition of each concentration of the primary ion (Hg(II)) solution (50 cm3) were plotted against dynamic response times of the electrode. The reversibility of the electrode was investigated by successive immersions of the Hg(II) ISE at two different concentrations of the Hg(II) salt solution from the lower to the higher concentration and vice versa. Finally, the potential values against response times were plotted.
Determination of the lifetime of the Hg(II)-ISE. The lifetime of the Hg(II)-ISE was monitored over a period of time by measuring the potential values of the calibration solutions of Hg(NO3)2. Calibration curves were obtained after each examined time in the 6.67 × 10−8 to 4.32 × 10−2 mol dm−3 concentration range. The slope values obtained by plotting E(mV) as a function of log[Hg(II)] were also plotted against the period of time tested.
Determination of Hg(II) ion concentration with EDTA in aqueous solution. The Hg(II)-ISEs (1%) with o-NPOE (68%) as plasticizer and OA as ionic additive (1%) were used as an indicator electrode in the potentiometric titration of the mercury(II) ion salt with EDTA at the standard temperature. The aqueous Hg(II) nitrate solution (60 cm3, 1.0 × 10−3 mol dm−3) was titrated with aliquots of EDTA (0.5 cm3, 5.0 × 10−3 mol dm−3). The amount of mercury(II) ion was accurately determined from the end point of the resulting titration by plotting E(mV) as a function of log[Hg(II)].
Analysis of mercury(II) in a real sample. The proposed Hg(II)-ISE as indicator electrode was tested for the potentiometric determination of mercury(II) in a dental amalgam filling. Thus an accurate weight of the dental amalgam alloy (43.1% Ag, 26.1% Cu and 30.8% Sn) (0.6 g) was dissolved in a minimum volume (20 ml) of HNO3 (60%). The solution was intensely heated to near dryness. The acid treatment and the evaporation process of the solution were repeated at least three times. The residue obtained was dissolved in a small amount of water and filtered. The clear solution was quantitatively transferred into a volumetric flask (50 cm3) and filled to the mark with distilled water after adjusting the pH value of the solution to 5.6.
Thereafter, the standard addition method was applied where small increments (0.1 cm3) of the standard solution of Hg(NO3)2 (3.0 × 10−2 mol dm−3) were added to the dental amalgam solution (50 cm3) at the standard temperature. The changes in potential readings were recorded after each addition of the standard solution. The Hg(II) concentration in the dental amalgam solution was calculated. The results obtained with the CPA based electrode were compared with data obtained by atomic absorption spectrometry (AAS).
Results & discussion
For the structural characterization of CPA, NMR experiments were carried out. In a first stage 1D 1H NMR proton and 13C NMR were run in CD3CN (Chemical shifts shown in the Experimental section). However the ID 1H NMR was not sufficient to attribute the protons of the different units, which become non-equivalent because of the loss of symmetry after substitution, therefore we proceeded with 13C NMR-HBBD, 13C NMR-DEPT and HMQC 2D NMR experiments to establish a correlation of 1H and 13C signals. Comparative analysis of 13C NMR-HBBD and 13C NMR-DEPT spectra given in ESI† help to identify the number of signals attributed to five quaternary carbons (one sp3 and four sp2, including a carbonyl function at 166.01 ppm and another one integrating two carbons [C1–C4 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]), three methine [(CH)3 all sp2 all integrating two carbons each], three methylene one oxygenated and two link to a nitrogen atom [(CH2)2N(CH2–O)] and three methyl [(CH3)3]. The assignment of the N–H functionality was made directly from the 1H NMR. Consequently the formula [C1–(C)4–(CO)–(CH)6–(CH2)2–(CH2–O)–(CH3)3–NH] = C18H22N2O2 × 4 = C72H88N8O8 was deduced, which was found to be in accord with the elemental analysis. However, the complete assignments, especially in the overlapped regions (1H at 3.35 ppm) had to be made from the 2D data. The 1H–13C correlation (ESI†) was used for the assignment of hydrogens to carbons in a straight forward manner. The set of protons at 3.35 ppm showed correlation with two carbons C-11 (40.56 ppm) and C-11′ (39.27 ppm), this indicates clearly that this set of protons integrates the two methylene of the acetamide arm group (2CH2–N). In addition the correlation of C-6 and C-7 to H-4 and H-5 benzene ring protons made the assignment of the pyrrole protons easy.
O]), three methine [(CH)3 all sp2 all integrating two carbons each], three methylene one oxygenated and two link to a nitrogen atom [(CH2)2N(CH2–O)] and three methyl [(CH3)3]. The assignment of the N–H functionality was made directly from the 1H NMR. Consequently the formula [C1–(C)4–(CO)–(CH)6–(CH2)2–(CH2–O)–(CH3)3–NH] = C18H22N2O2 × 4 = C72H88N8O8 was deduced, which was found to be in accord with the elemental analysis. However, the complete assignments, especially in the overlapped regions (1H at 3.35 ppm) had to be made from the 2D data. The 1H–13C correlation (ESI†) was used for the assignment of hydrogens to carbons in a straight forward manner. The set of protons at 3.35 ppm showed correlation with two carbons C-11 (40.56 ppm) and C-11′ (39.27 ppm), this indicates clearly that this set of protons integrates the two methylene of the acetamide arm group (2CH2–N). In addition the correlation of C-6 and C-7 to H-4 and H-5 benzene ring protons made the assignment of the pyrrole protons easy.
Having characterised the receptor, 1H NMR measurements were carried out to assess cation–ligand interactions. These were carried out in CD3CN given that in this dipolar aprotic solvent ion salts are predominantly dissociated.
1H NMR measurements of CPA with metal cations and anions in CD3CN at 298 K
Hardly any chemical shift changes (Δδ ppm) were observed in the 1H NMR spectrum of CPA by the addition of alkali metal cations (Li(I), Na(I), K(I), Rb(I) and Cs(I)) as perchlorates in CD3CN at 298 K. The addition of Ag(I) salt to CPA also did not lead to significant chemical shift changes in any of the protons.As far as the alkaline-earth metal cations are concerned, downfield chemical shift changes relative to the free ligand (ppm) were observed by the addition of Mg(II) (H-5, 014), Ca(II) (H-4, 0.15, H-5, 0.16), Sr(II) (H-4, 0.10, H-5, 0.15) and Ba(II) (H-5, 0.13) salts to CPA in CD3CN. Therefore, it can be concluded that these cations being of the hard Lewis acid type may interact with CPA through the phenolic and the carbonyl oxygens of the amide moiety (hard Lewis base).61 As far as the heavy metal cations are concerned, the most significant downfield chemical shift changes for Pb(II) are observed in H-5 (0.21 ppm) and H-6 (0.20 ppm) while for Cd(II) the signal corresponding to H-6 disappeared. The addition of Hg(II) to the NMR tube containing a solution of CPA in CD3CN led to significant downfield chemical shift changes in the methylene protons (H-6, 0.16 ppm) between the phenolic oxygen and the carbonyl group of the acetamide arm group and the one next to the nitrogen atom of the acetamide nitrogen (H-7, 0.14 ppm, H-7′, 0.16 ppm) as well as in the methyl terminal protons of acetamide arm (H-8′, 0.13 ppm). Upfield chemical shift changes observed in the NH proton of the pyrrole units were found to be less significant and may result from the conformational changes of CPA upon complexation. These results suggest that the interaction of Hg(II) with CPA takes place through the amide functional group. We therefore conclude that Pb(II), Cd(II) and Hg(II) interact with the donor atoms of the acetamide functional groups.
Among the anions tested (halides, nitrate, hydrogen sulphate), dihydrogen phosphate, trifluoromethane sulphonate, thiocyanide strong interaction was found only with the fluoride ion where the most remarkable downfield chemical shift change was found for H-1 (4.52 ppm) as a result of the interaction of this anion with the NH functional groups of the pyrrole units. Preliminary studies in different dipolar aprotic solvents revealed a change in the composition of the anion complex in moving from one solvent to another. Therefore 1H NMR titrations were carried out in three solvents: CD3CN, d6-DMSO and d7-DMF at 298 K.
1H NMR titration of CPA with the fluoride anion in deuterated dipolar aprotic solvents
In an effort to study the behavior of the receptor in solution, 1H NMR studies of CPA were carried out in CD3CN, d6-DMSO and d7-DMF in the absence and presence of the fluoride anion. Volumes of TBAF were added to the solution of CPA. The 1H NMR spectrum (CD3CN, 500 MHz, 298 K) of CPA receptor showed one broad singlet at 7.98 ppm (integrating 4H) which was assigned to the four chemically equivalent NH protons' resonance. Addition of 0.5 equivalent of tetrabutylammonium fluoride (TBAF) to a solution of the receptor leads to an equilibrium favoring the cone-like conformation via the formation of the 1:1 fluoride–CPA complex. The conformational change of the receptor was easily observed in the 1H NMR spectrum, where two different signals were observed for the NH protons, one at 7.98 ppm, corresponding to the free receptor, and the other at 12.00 ppm, corresponding to the fluoride complex. This finding was taken as evidence that the exchange between the complex and the free receptor is slow on the 1H NMR timescale. After the addition of 1.0 equivalent of TBAF, only the signals at 12 and 12.5 ppm, corresponding to the CPAF− complex were observed in the 1H NMR spectrum under these conditions, as expected for a highly stable complex.
Surprisingly, the pyrrole NH resonance was found to split into two peaks in the presence of fluoride. It was considered that a likely cause for this splitting might be due to coupling between the NH protons and the 19F nucleus of the bound fluoride anion. The coupling constant was found to be 49.5 Hz as was previously reported in the literature.62 The stoichiometry of complexation was determined from plots of the integrals of the most significant downfield chemical shift changes in the protons of the anion complex (H-1, H-2, H-3, H-6) as shown in Fig. 1(a).
 | ||
| Fig. 1 (a) 1H NMR titration of CPA with the fluoride anion in CD3CN at 298 K. (b) 1H NMR titration of CPA with the fluoride anion in d6-DMSO at 298 K. (c) 1H NMR titration of CPA with the fluoride anion in d7-DMF at 298 K. | ||
The results obtained from the 1H NMR titration of CPA with the fluoride anion salt in d6-DMSO and d7-DMF (Fig. 1(b) and (c)) clearly indicate that in the former solvent, CPA interacts with the anion with the formation of a 2:1 (CPA:F−) complex in the latter solvent the composition of the complex is 1:1. 1H NMR data for the free ligand in these solvents show that DMSO interacts with calixpyrrole through hydrogen bond formation between the basic oxygen atoms of the solvent and the NH moiety of the pyrrole ring. This was reflected in the significant downfield chemical shift changes observed in H-1 in moving from CD3CN to d6-DMSO (1.47 ppm) while insignificant upfield chemical shift changes were observed for the remaining protons. To a lesser extent d7-DMF also interacts with CPA. A downfield shift in H-1 was observed (1.31 ppm) with additional ones in H-4 (0.10 ppm) and H-6 (0.11 ppm). Sessler and co-workers63 reported the X-ray structure of the parent calix[4]pyrrole in a 1,2 alternate conformation in which two molecules of d7-DMF are hydrogen bonded to two adjacent pyrrole units. However these authors found that in solution (benzene) only 1:1 complexes are formed. The same situation was found in DMSO. Apparent stability constants in benzene for the parent calix[4]pyrrole with d6-DMSO (logKs = 1.21) and with d7DMF (logKs = 1.05) were reported. As far as CPA is concerned, a possible explanation of the different compositions of the fluoride complexes in these solvents must be attributed to their different molecular geometries. Thus DMSO is characterized by a pyramidal geometry (sp3 hybridization with a lone pair of electrons) while DMF has a planar structure (sp2 hybridization). It is also known that DMSO can be associated with the formation of head to tail dimers through dipole–dipole interactions.64,65 As a result the binding sites of the receptor to enter interaction with the anion are reduced. Therefore two calixpyrrole units rather than one are required to enhance the number of binding sites required to enter complexation with the anion in d6-DMSO.
In an attempt to test the ability of this receptor to interact simultaneously with Hg(II) and F−, the binding properties of CPA with Hg(II) were examined by 1H NMR titration experiments in CD3CN in the absence and the presence of F− (Fig. 2).
 | ||
| Fig. 2 Shifts and integrals of NH protons of the CPA/F (1:1) complex titrated with Hg(II) in CD3CN at 298 K. | ||
From the results of this experiment the following conclusions can be drawn. As stated above, the addition of 1 equivalent of TBAF to CPA in CD3CN caused a remarkable downfield chemical shift change of the NH protons and upfield shifts of the remaining protons (H-2; H-3; H-4,5; H-6, H-7,7′ and H-8,8′) of this receptor. Further addition of TBAF does not make any significant change in the chemical shifts of the protons, suggesting the formation of a 1:1 (CPA:F−) complex. In the first three additions of the Hg(II) salt solution to the fluoride complex a substantial upfield chemical shift change of the NH protons of the complex (∼3.6 ppm) was observed. Inspection of Fig. 2(a) shows the formation of a 2:1 (Hg(II):CPA) complex. The integrals of the NH protons of the 1:1 fluoride complex with the Hg(II) salt is shown in Fig. 2(b). The break observed in this figure at the Hg(II):CPA ratio of 0.5 clearly indicates that two units of the fluoride complex interacts with only one metal cation. These may be the result of an exchange between the two TBA+ cations with the bivalent mercury ion. It is reasonable to assume that the TBA+ cations are large hydrophobic ions of low charge density relative to Hg(II) and therefore these are likely to be easily displaced by the latter cation. In addition, ion–ion interactions are stronger than ion dipole interactions. Consequently Hg(II) will be more attracted by the negatively charged fluoride complex than by the donor atoms of the amide functionality of the receptor.
Conductometric titrations of ions with CPA in MeCN
Conductometric titrations of all metal cation salts (perchlorate as the counter-ion) investigated, except Hg(II) (as nitrate), led to a defined change in the curvature at the 1:1 (ligand:metal ion) stoichiometry, suggesting the formation of complexes of moderate stability. As far as Hg(II) is concerned. The conductometric titration curve (Fig. 3), which is a plot of the molar conductance, Λm against the ligand:metal cation ratio breaks at 0.5 indicating that two metal cations interact with one unit of receptor. A decrease in conductance due to the formation of the 1:2 complex (eqn (1)) is observed| 2Hg(II)(MeCN) + CPA(MeCN) ↔ [Hg2CPA]4+(MeCN) | (1) |
 | ||
| Fig. 3 Conductometric titration curve of Hg(II) (as nitrate) with CPA in MeCN at 298.15 K. | ||
In the presence of an excess of the ligand, it seems that one of the mercury cations in the complex is transported to the free ligand with the formation of a 1:1 complex (eqn (2)) as shown by the increase in conductance.
| [Hg2CPA]4+ + CPA(MeCN) ↔ 2[HgCPA]2+(MeCN) | (2) |
As far as the F− anion is concerned, the conductometric titration curve consisted of a combination of two linear segments with a well-defined change in the curvature at the 1:1 (CPA:F−) molar ratio, suggesting the formation of a relatively strong complex.
Based on the evidence obtained from the fundamental studies described above, it is concluded that CPA has a higher hosting ability for Hg(II) relative to other ions investigated, therefore a Hg(II) selective liquid membrane electrode based on this receptor was designed. Before discussing the characteristics of the ISE we assessed whether or not interactions are found between the analyte and the individual components of the PVC based membrane (additive and plasticizer) in the absence of the receptor.
Selection of additive and plasticizer for the preparation of the Hg(II) ion selective membrane
Two additives (sodium tetraphenylboron, NaPh4B and oleic acid, OA) were tested to find out whether or not these interact with the analyte and interfere in the selective behaviour of the ionophore for the analyte. Thus 1H NMR data obtained by the addition of Hg(II) (nitrate as counter-ion) shown in Table 1.| NaPh4B | H-1 | H-2 | H-3 | |
|---|---|---|---|---|

|
NaPh4B free | 6.99 | 7.26 | 6.85 |
| Hg(II) with NaPh4B4 | 7.37 | 7.60 | 7.14 | |
| Δδ | 0.38 | 0.34 | 0.29 |
These results clearly indicate that there is a strong interaction between the cation and this additive. This is reflected in the significant downfield chemical shift changes observed in all the aromatic protons. In an attempt to observe the behaviour of NaPh4B, this salt was introduced in a PVC based membrane in the absence of the receptor. Potentiometric measurements carried out demonstrated that the membrane responds to Hg(II) showing a Nernstian behaviour (slope 29.1 ± 0.6 mV/pHg(II) in the 3.25 × 10−5 to 2.5 × 10−3 mol dm−3 concentration range with a response time of about 10 s). This was further corroborated by Scanning Electron Microscopy (SEM) carried out on the membrane with the aim of identifying the locations of the Hg(II) cation on the PVC membrane in the absence of the receptor and to understand the changes to the membrane morphology. Fig. 4(a1) and (a2) represent the SEM micrographs of the membrane surface before and after it has been used in the Hg(II) ISE.
 | ||
| Fig. 4 (a1 & a2) PVC membrane (a1) prepared before being used in the electrode and (a2) after it has been used for several times in the ion selective electrode; (b1 & b2) EDAX spectra of the membranes showing the elemental analysis. | ||
Fig. 4(a 1) shows the typical clear and fibrous PVC membrane surface without exhibiting any dense areas or defects. The Fig. 4(a2) shows the Hg(II) as white dense patches on the membrane which have been detected in the EDAX spectrum in Fig. 4(b2), where the new peaks characteristic of Hg (compared to Fig. 4(b1) are noted and this indicates the binding of Hg(II) to the Ph4B− anion sites). A change in the morphology of the membrane as a function of the ageing time was also observed. It is therefore concluded that a PVC membrane containing NaPh4B is able to detect Hg(II) in the absence of the CPA receptor. These findings as demonstrated below have implications on the working characteristics of the electrode in the presence of the receptor and open serious questions about whether or not the electrode responses that have been previously reported with this additive were due to the receptor or to the presence of NaPh4B particularly when fundamental studies have not been carried out. No interaction was found between the mercury(II) cation and oleic acid by 1H NMR measurements. In addition, blank experiments in the absence of the receptor were carried out. It was found that when the membrane components were 68% o-NPOE, 30% PVC, and 1% OA, the electrode membrane displayed no response towards Hg(II) and therefore this was the additive selected for the preparation of the membrane containing the CPA receptor in the below experiments. In the same way, we tested two plasticizers, DOS and o-NPOE. Two PVC membranes were produced using OA as additive and either DOS or o-NPOE. No interaction was detected with the DOS plasticizer, in the concentration range 1.3 × 10−3 to 1.5 × 10−2 mol dm−3, and with O-NOPE in the concentration range of 7.1 × 10−4 to 1.34 × 10−3 mol dm−3.
Effect of membrane composition on the potentiometric response characteristics of the CPA based Hg(II)-ISE
The characteristics of the electrode such as its Nernstian behaviour, liner range (L-R), detection limit (D-L) and response time were investigated using five electrode membranes (E1–E5, composition electrode in Table 2) of varying compositions: two different plasticizers (o-NPOE or DOS) (E1 and E2) in the absence of additive (E3), in the presence of an increased amount of the receptor (E4). Also included data obtained in the presence of the receptor when NaPh4B was used an additive (E5). Calibration curves for the five electrodes tested are shown in Fig. 5.| Slope (mV/pHg) | L-R mol dm−3 | D-L mol dm−3 | Response time (s) |
|---|---|---|---|
| E1 | 1% CPA, 68% DOS, 30% PVC, 1% OA | ||
| 25 ± 1 | 9.12 × 10−7 to 1.78 × 10−3 | 4.46 × 10−7 | 14 |
| E2 | 1% CPA, 68% o-NPOE, 30% PVC, 1% OA | ||
| 30 ± 1 | 1.81 × 10−7 to 1.78 × 10−3 | 1.81 × 10−7 | 10 |
| E3 | 1% CPA, 68% o-NPOE, 30% PVC, no additive | ||
| 6 ± 1 | 2.51 × 10−5 to 1.58 × 103 | 1.78 × 10−5 | 19 |
| E4 | 2% CPA, 68% o-NPOE, 30% PVC, 1% OA | ||
| 25 ± 1 | 3.98 × 10−7 to 9.46 × 10−4 | 5.62 × 10−7 | <10 |
| E5 | 1% CPA, 68% o-NPOE, 30% PVC, 1% NaPh4B | ||
| 26 ± 1 | 1.6 × 10−6 to 1.98 × 10−3 | 4.67 × 10−6 | ∼6.5 |
 | ||
| Fig. 5 Effect of membrane composition on the potentiometric response characteristics of the CPA based Hg(II)-ISE. Membrane compositions are in Table 2. | ||
The nature of plasticizers used (o-NPOE and DOS) (E1 and E2) effects the characteristics of the Hg(II)-ISE. The results showed that the latter (E2) had a better response when compared with the former (E1). The linear working response was within a wide range of Hg(II) concentration from 1.81 × 10−7 to 1.78 × 10−3 mol dm−3 with a detection limit of 6.0 × 10−8 mol dm−3 and response time in about 10 s. In addition, the slope obtained for this electrode (E2) was found to be 30 ± 1 mV dec−1, which is close to the Nernstian response (29.5 mV dec−1 at 298 K). The difference in the response characteristics between E1 and E2 may be attributed to the dielectric constant of the plasticizers used which in turn may have an effect on the membrane permittivity and the mobility of the ionophore molecules.24 Accordingly, o-NPOE was chosen to be the optimum plasticizer for further studies with the CPA based electrode. Clearly in the absence of additive (E3) the response of the electrode in the presence of the receptor is indeed very weak.
Comparison of the potentiometric response characteristics of the membrane electrode E4 with respect to E2 revealed that the best performance was observed for the latter. The increase of the amount of ionophore led to a lower electrode response. This may be also ascribed to a decrease in the solubility of the ionophore in the membrane. As previously stated58 the ionophore should be soluble in the membrane matrix and should be sufficiently lipophilic to avoid leaching from the membrane into the sample solution.24 As far as E5 is concerned the data in Table 2 show that the presence of NaPh4B decreases the detection limit of the electrode by a factor higher than 10 which may be due to a possible interaction between this salt and the receptor. It is well established that the parent calix[4]pyrrole does not complex the tetraphenyl borate anion, but the possibility exists that its derivative containing aromatic group interacts with this anion through pi–pi interactions. From the above mentioned results, the membrane electrode E2 was chosen for further investigation under different experimental conditions.
The surface analysis of the PVC membrane casted with THF containing CPA, OA as additive and o-NPOE as plasticizer was characterised by SEM coupled with EDAX. Fig. 6(a) and (b) and 7(a) and (b) show SEM-EDAX of the PVC–CPA matrix before and after it has been in contact with Hg (NO3)2. A detectable transformation in the membrane shows the interaction with the cation. The EDAX analysis displays Hg(II) peaks, and this depicts the selective recognition and binding of CPA to the Hg(II) cation.
 | ||
| Fig. 6 (a) Scanning electron microscopy image of a CPA ionophore–PVC porous membrane; E2 (b) EDAX spectrum showing the membrane elemental composition. | ||
 | ||
| Fig. 7 (a) SEM of CPA ionophore–PVC membrane (E2) after it has been used in the ISE for Hg(II) selectivity; (b) EDAX analysis of the membrane showing the presence of Hg(II). | ||
Effect of pH of the aqueous solution on the response of the CPA based Hg(II)-ISE
As can be seen from Fig. 8, the pH effect on the response of the electrode was investigated under the same conditions stated above over a 1.5–12.0 pH range of aqueous solutions of Hg(NO3)2 (1.01 × 10−3 and 1.01 × 10−2 mol dm−3). The results show that the electrode potential remains constant in the 4.3–8.5 pH range using 1.01 × 10−3 mol dm−3 Hg(II) solution. At pH lower than 4.30, the increase of the acid nature of the solution gives rise to an increase in the electrode potential. At pH higher than 8.5, the potential values were found to decrease sharply due to the formation of hydroxyl-complexes of Hg(II) in solution. By altering the concentration of the mercury salt to 1.01 × 0−2 mol dm−3, a similar profile was observed. Therefore for further studies, an aqueous solution of Hg(NO3)2 at pH 5.6 was used. | ||
| Fig. 8 Effect of the pH on the response of the CPA based electrode (E2). | ||
Effect of the concentration of the internal solution on the response of Hg(II)-ISE based on CPA
The concentration of the internal reference solution has a remarkable influence on the electrode response (as shown in Fig. 9). The optimum concentration of Hg(NO3)2 was found to be 1 × 10−2 mol dm−3. A slope value of 29 ± 1 mV dec−1 close to the theoretical Nernstian slope was found. However when the concentration was decreased to 1 × 10−3 mol dm−3 Hg(NO3)2 the electrolyte was unable to transfer the charge from the detected ion in the ISE membrane to the measuring system and as a result the electrode response decreased. Therefore, the concentration of 1 × 10−2 mol dm−3 Hg(NO3)2 was used for further studies reported in this paper.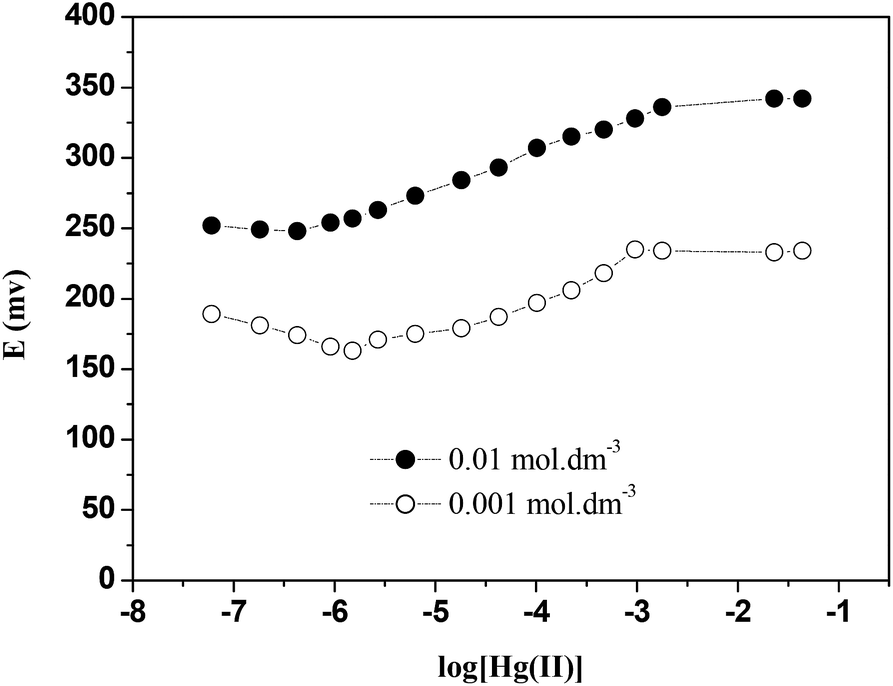 | ||
| Fig. 9 Effect of internal solution concentration on the response of the ISE (E2). | ||
Effect of soaking time on the performance of Hg(II)-ISE based on CPA
Fig. 10 shows the effect of conditioning of the ISE for different periods of time on the slope values obtained by plotting E(mV) as a function of log[Hg(II)]. These results revealed that the soaking time of the ISE has a considerable effect on its response characteristics. It was found that an optimum soaking time was 24 hours, the slope value (27 ± 2 mV dec−1) obtained is close to the theoretical Nernstian slope (29.5 mV dec−1). After longer periods of soaking, the Hg(II)-ISE did not show further improvements in response. Therefore, it can be seen that the conditioning for the proposed (Hg(II)-ISE) is a necessary step to increase the number of active sites of the ionophore used. | ||
| Fig. 10 Effect of soaking time on the Nernstian behaviour of the Hg ISE at 298 K. | ||
Response time and reversibility for Hg(II)-ISE based on CPA
The response time for this ISE (E2) was tested with Hg(NO3)2 solutions by changing from lower to higher concentrations within a range of 1 × 10−7 to 1 × 10−2 mol dm−3. The results (Fig. 11) showed that the response time of the CPA based Hg(II)-ISE was 10 s. The reversibility of the electrode was examined by alternatively dipping it in aqueous solutions of Hg(NO3)2 at two different concentrations (1 × 10−4 and 1 × 10−3 mol dm−3). Results are shown in Fig. 12. It can be observed that the proposed electrode exhibited a satisfactory reversibility although the time required for attaining a stable potential (or equilibrium state) by changing from a low to a high concentration was longer than when changing from a higher to a lower concentration. Therefore, the response time was found to be about of 8 s when the concentration of Hg(NO3)2 solution was changed from 1.0 × 10−3 to 1.0 × 10−4 mol dm−3, but the response time increased to 13 s on the switch from 1.0 × 10−4 to 1.0 × 10−3 mol dm−3. | ||
| Fig. 11 Dynamic response of the CPA based Hg(II)-ISEs for step changes in mercury(II) concentration at 298 K. | ||
 | ||
| Fig. 12 Reversibility of the CPA based Hg(II)-ISE. | ||
Lifetime of the CPA based Hg(II)-ISE
The lifetime of the ISE was monitored with time as shown in Fig. 13. It was found that this electrode exhibited a good stability in the response up to a period of 68 d. Indeed the mean drift of slope observed over this period was found to be less than 0.02 mV dec−1. Therefore, it can be concluded that the lifetime of the CPA based Hg(II)-ISE is 68 d. | ||
| Fig. 13 Lifetime of the CPA based mercury(II) ion selective electrode. | ||
Potentiometric selectivity coefficients of the CPA based Hg(II) ion selective electrode
The potentiometric selectivity coefficients of the CPA based Hg(II)-ISE were determined for a variety of interfering metal ions such as Na(I), K(I), Ag(I), Mg(II), Ca(II), Ba(II), Sr(II), Mn(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), Pb(II), Fe(III) and Al(III) using two methods: the Separate Solution Method (SSM)59 and the Matched Potential Method (MPM).60 The values of selectivity coefficients obtained under optimum experimental conditions are given in Table 3. Although the trend observed is the same for data derived from these two methods, the values differ given that different assumptions are made in these methods.| Interferent (Mn+) | Values of
|
|
|---|---|---|
| Separate solution methoda SSM | Matched potential methodb MPM | |
| a Conditions: primary ion (A): 2.23 × 10−4 mol dm−3 Hg(NO3)2 interfering ions (B): 2.23 × 10−4 mol dm−3. b Conditions: reference solution: 1.0 × 10−4 mol dm−3 Hg(NO3)2 primary ions (A): 1 × 10−2 mol dm−3 Hg(II); interfering ions (B): 1 × 10−1 mol dm−3. | ||
| Li+ | −1.71 | −1.38 |
| Na+ | −2.28 | −2.20 |
| K+ | −2.30 | −2.28 |
| Rb+ | −3.92 | −3.07 |
| Mg2+ | −2.38 | −2.40 |
| Ca2+ | −3.98 | −3.37 |
| Sr2+ | −3.13 | −2.91 |
| Ba2+ | −3.88 | −2.99 |
| Mn2+ | −5.00 | −4.16 |
| Fe3+ | −2.42 | −2.68 |
| Co2+ | −4.26 | −3.77 |
| Ni2+ | −1.89 | −1.55 |
| Cu2+ | −3.17 | −2.94 |
| Ag+ | −0.46 | −0.99 |
| Zn2+ | −4.07 | −3.63 |
| Cd2+ | −2.39 | −2.55 |
| Al3+ | −3.03 | −2.91 |
| Pb2+ | −2.61 | −2.75 |

It can be seen from the  values (by both methods) that the CPA based Hg(II) selective electrode is more selective towards mercury over all of the interfering ions studied. Also
values (by both methods) that the CPA based Hg(II) selective electrode is more selective towards mercury over all of the interfering ions studied. Also  values for most ions examined except for the Ag(I) ion were of the order of −1.38 or smaller.
values for most ions examined except for the Ag(I) ion were of the order of −1.38 or smaller.
However, the  values for the Ag+ interfering ion obtained from the SSM and MPM methods were −0.46 and −0.99 respectively which does not seem to be a strong interference. The reliability of the electrode as an analytical tool was tested and this is now discussed.
values for the Ag+ interfering ion obtained from the SSM and MPM methods were −0.46 and −0.99 respectively which does not seem to be a strong interference. The reliability of the electrode as an analytical tool was tested and this is now discussed.
Analytical applications of the CPA based Hg(II)-ISE
The CPA based Hg(II) ISE was used as (i) an indicator electrode for the potentiometric titration of Hg(II) with EDTA and (ii) the determination of mercury in an amalgam sample. Thus Fig. 14 shows the potentiometric titration curve resulting from the titration of an aqueous solution of Hg (NO3)2 (1 × 10−3 mol dm−3) with EDTA (0.005 mol dm−3) using the optimized electrode. A sharp end point was obtained. | ||
| Fig. 14 Potentiometric titration of Hg(NO3)2 with EDTA using the CPA based electrode as indicator electrode at 298 K. | ||
Table 4 shows the results obtained for the determination of mercury(II) in a filling dental amalgam together with those obtained by atomic absorption spectrometry. An excellent agreement is found between the two methods. Furthermore, the species such as Cu(II), Zn(II) and Sn(II) present in the amalgam samples have no interfering effect. Therefore, it was concluded that the CPA based Hg(II)-selective electrode could be successfully employed for the quantitative determination of Hg in a real dental amalgam sample.
| Sample | Supplier | Ionophore used | Mercury(II) content, mol dm−3 | ||
|---|---|---|---|---|---|
| Practical con. using AAS | Practical con. using Hg-ISE | Compatibility (%) | |||
| Dental amalgam alloy | Nordiska Dental AB Industries, Sweden | CPA | 1 × 10−5 | 9.51 × 10−6 M | 95.1 |
Advantages of the CPA based ISE relative to existing ionophore based Hg(II) electrodes
A great deal of the published material involves the use of the NaPh4B (or related salts) as an additive,20–25,28,29,31,34–39,41,43,44,46–52,66–75 which as shown in this paper interacts with Hg(II) and this has implications on the characteristics reported for these electrodes. In the absence of fundamental studies, the response of the electrode may result from the response of the additive rather than the ionophore (this may be occurring in the prior studies mentioned). Other ISEs for the detection of Hg(II) used plasticisers other than o-NPOE or DOS.40,42,45,53,76 Given that a plasticizer may have an effect on the potential response of the electrode a comparison between the research carried out with different plasticiser is not strictly valid. The only reported Hg(II) ISE in which the same additive and plasticiser have been used is that reported by Somer and co-workers77 using tridodecyl methyl ammonium iodide as the ionophore. However the linear range for this electrode (5 × 10−5 to 1 × 10−1 mol dm−3) with a DL of 1 × 10−5 mol dm−3 is less sensitive than the one reported in this paper.Final remarks
Despite the efforts being made worldwide on the design of Hg(II) ISEs by the use of cyclic and acyclic ligands a comprehensive review at the state of the art in this particular area of research leads to the conclusion that an important drawback is the lack of a link between fundamental studies and their applications. This is clearly demonstrated in this paper where we show that the currently (and almost universally used) tetraphenyl borate additive salts (used in the design of Hg ISEs incorporating PVC membranes) act by themselves as selective detectors for this cation. It may well be that in some cases the additive and receptor can have a cooperative effect (offering an improvement in the characteristics of the electrode) but this need to be investigated. Quite clearly this is not the case in the system investigated in this paper.While calix[4]pyrroles and derivatives have been extensively used for the design of ISEs and chemically modified electrodes, most efforts have been directed to anions rather than cations.78 However the possibility of altering the selectivity through structural modification (as demonstrated in this paper) opens the way for further developments on the use of these receptors for the design of cation selective electrodes. Further studies are being carried out to assess this electrode for monitoring fluoride in water as well as its selectivity in the presence of mercury(II) fluoride.
Acknowledgements
The fundamental studies reported in this paper are part of the PhD thesis of A El Gamouz (University of Surrey, 2009). This work was financially sponsored by the European Commission under Contracts INCO-CT-2004-509153 and INCO-CT-2004-509159 coordinated by AFDdeN, the Government of Saudi Arabia and the Leverhulme Trust (Emeritus Fellowship awarded to AFDdeN). JRV is an EPSRC Leadership Fellow funded from grant EP/I004882/1. The authors thank Prof. John F. Watts, Director of Research in the Department of Mechanical Engineering Sciences, University of Surrey, and his staff for their invaluable assistance in the use of equipments available in the MicroStructural Studies Unit and the Surface Analysis Laboratory.Notes and references
- US EPA, Health Effects, Mercury, http://www.epa.gov/hg/effects.htm.
- J. Gavis and J. F. Ferguson, Water Res., 1972, 6, 989 CrossRef CAS.
- B. M. Miskimmin, J. W. M. Rudd and C. A. Kelly, Can. J. Fish. Aquat. Sci., 1992, 49, 17 CrossRef CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1886, 19, 2184 CrossRef PubMed.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1872, 5, 1094 CrossRef PubMed.
- W. H. Brown and N. W. French, Can. J. Chem., 1958, 36, 371 CrossRef CAS.
- C. M. Kretz, E. Gallo, E. Solari, C. Floriani, C. A. Villa and C. Rizzoli, J. Am. Chem. Soc., 1994, 116, 10775 CrossRef CAS.
- D. Jacoby, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Chem. Soc., Chem. Commun., 1991, 220 RSC.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140 CrossRef CAS.
- J. L. Sessler and P. A. Gale, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 6, p. 257 Search PubMed.
- W. E. Allen, P. A. Gale, C. T. Brown, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 1996, 118, 12 Search PubMed.
- A. F. D. de Namor and M. Shehab, J. Phys. Chem. B, 2003, 107, 6462 CrossRef.
- A. F. D. de Namor, I. Abbas and H. H. Hammud, J. Phys. Chem. B, 2006, 110, 2142 CrossRef PubMed.
- A. F. D. de Namor, M. Shehab, R. Khalife and I. Abbas, J. Phys. Chem. B, 2007, 111, 12177 CrossRef PubMed.
- A. F. D. de Namor, I. Abbas and H. H. Hammud, J. Phys. Chem. B, 2007, 111, 3098 CrossRef CAS PubMed.
- A. F. D. de Namor and I. Abbas, J. Phys. Chem. B, 2007, 111, 5803 CrossRef PubMed.
- A. F. D. de Namor and R. Khalife, J. Phys. Chem. B, 2008, 112, 15766 CrossRef PubMed.
- A. F. D. de Namor and M. Shehab, J. Phys. Chem. A, 2004, 108, 7324 CrossRef.
- A. F. D. de Namor and M. Shehab, J. Phys. Chem. B, 2005, 109, 17440 CrossRef PubMed.
- I. Abbas, Int. J. Chem., 2012, 4, 23 CAS.
- M. T. Lai and J. S. Shi, Analyst, 1986, 111, 891 RSC.
- M. Javanbakht, M. R. Ganjali, H. Eshghi, H. Sharghi and M. Shamsipur, Electroanalysis, 1999, 11(2), 81 CrossRef CAS.
- A. R. Fakhari, M. R. Ganjali and M. Shamsipur, Anal. Chem., 1997, 69, 3693 CrossRef CAS.
- V. K. Gupta, S. Jain and U. Khurana, Electroanalysis, 1997, 9, 478 CrossRef CAS PubMed.
- Z. Brzozka and M. Pietraszkiewicz, Electroanalysis, 1991, 3, 855 CrossRef CAS PubMed.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1991, 91(8), 1721 CrossRef CAS.
- J. Lu, X. Tong and X. He, J. Electroanal. Chem., 2003, 540, 111 CrossRef CAS.
- R. K. Mahajan, R. Kaur, V. Bhalla, M. Kumar, T. Hattori and S. Miyano, Sens. Actuators, B, 2008, 130, 290 CrossRef CAS PubMed.
- R. K. Mahajan, R. Kaur, L. Kaur, V. Sharma and M. Kumar, Anal. Sci., 2004, 20(5), 805 CrossRef.
- W. H. Mahmoud, G. A. W. El-Inany, F. M. E. Omar and R. M. El-Tohamy, Int. J. Acad., Sci. Res., 2013, 1(1), 8 Search PubMed.
- A. K. Singh, R. P. Singh and S. Mehtab, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60(1), 9 CrossRef CAS.
- A. C. Ion, I. Ion, D. N. Stefan and I. Barbu, Mater. Sci. Eng., C, 2009, 29, 1 CrossRef CAS PubMed.
- X. Yang, D. B. Hibbert and P. W. Alexander, Anal. Chim. Acta, 1998, 372, 387 CrossRef CAS.
- M. H. Mashhadizadeh and I. Sheikhshoaie, Talanta, 2003, 60, 73 CrossRef CAS.
- S. S. M. Hassan, M. B. Saleh, A. A. A. Gaber, A. H. Mekheimer and N. A. A. Kream, Talanta, 2000, 53, 285 CrossRef CAS.
- M. Bagheri, M. Mashhadizadeh, S. Razee and A. Momeni, Electroanalysis, 2003, 15, 1824 CrossRef CAS PubMed.
- W. Szczepaniak and J. Oleksy, Anal. Chim. Acta, 1986, 189, 237 CrossRef CAS.
- R. K. Mahajan, R. K. Puri, A. Marwaha, I. Kaur and M. P. Mahajan, J. Hazard. Mater., 2009, 167, 237 CrossRef CAS PubMed.
- V. K. Gupta, S. Chandra and H. Lang, Talanta, 2005, 66, 575 CrossRef CAS PubMed.
- H. A. Arida, J. P. Kloock and M. J. Schoning, Sensors, 2006, 6, 435 CrossRef CAS PubMed.
- M. B. Saleh, E. M. Soliman, A. A. Gaber and S. A. Ahmed, Anal. Lett., 2006, 39, 259 CrossRef PubMed.
- A. K. Hassan, Mod. Chem. Appl., 2013, 1(4), 111 Search PubMed.
- J. Jumal, B. M. Yamin, M. Ahmad and L. Y. Heng, APCBEE Proc., 2012, 3, 116 CrossRef CAS PubMed.
- T. F. Tahir, A. Salhin and S. A. Ghani, Sensors, 2012, 12(11), 14968 CrossRef CAS PubMed.
- R. K. Mahajan, I. Kaur and T. S. Lobana, Talanta, 2003, 59(1), 101 CrossRef CAS.
- V. K. Gupta, A. K. Singh, M. A. Khayat and B. Gupta, Anal. Chim. Acta, 2007, 590, 81 CrossRef CAS PubMed.
- A. A. Ensafi, S. Meghdadi and A. R. Allafchian, IEEE Sens. J., 2008, 8, 248 CrossRef CAS.
- R. K. Mahajan, P. Sood, P. Mahajan and A. Marwaha, Ann. Chim., 2007, 97(9), 959 CrossRef CAS PubMed.
- W. H. Mahmoud and E. A. Assirey, J. Mater. Sci. Eng. A, 2011, 1, 1010 Search PubMed.
- X. Yu, Z. Zhou, Y. Wang, Y. Liu, Q. Xie and D. Xiao, Sens. Actuators, B, 2007, 123, 352 CrossRef CAS PubMed.
- L. P. Marin, E. O. Sanchez, G. M. Miranda, P. A. PerEz, J. A. Chamaro and H. L. Valdivia, Analyst, 2000, 125, 1787 RSC.
- G. Ye, Y. Chai, R. Yuan and J. Dai, Anal. Sci., 2006, 22(4), 579 CrossRef CAS.
- S. S. Hassan, W. H. Mahmoud, A. H. Mohamed and A. E. Kelany, Anal. Sci., 2006, 22(6), 877 CrossRef CAS.
- Operating Instructions from the Wayne Kerr, Co. Ltd., Sussex, England, 1982.
- G. Jones and C. Bradshaw, J. Am. Chem. Soc., 1933, 55, 1780 CrossRef CAS.
- T. Rosatzin, E. Bakker, K. Susuki and W. Simon, Anal. Chim. Acta, 1993, 280, 197 CrossRef CAS.
- E. Bakker, P. Buhlmann and E. Pretsch, Chem. Rev., 1997, 97(8), 3083 CrossRef CAS PubMed.
- L. P. Singh and J. M. Bhatnagar, J. Appl. Electrochem., 2004, 34(4), 391 CrossRef CAS.
- R. P. Buck, E. Lindner, W. Kutner, G. Inzelt and IUPAC, Pure Appl. Chem., 2004, 76(6), 1139 CrossRef CAS.
- V. P. Y. Gadzekpo and G. D. Christian, Anal. Chim. Acta, 1984, 164, 279 CrossRef CAS.
- B. G. Cox and H. Schneider, Coordination and Transport Properties of Macrocyclic Compounds in Solution, New York, Elsevier, 1992 Search PubMed.
- K. A. Nielsen, L. M. Gomis, G. H. Sarova, L. Sanguinet, D. E. Gross, F. Fernandez-Lazaro, P. C. Stein, E. Levillian, J. L. Sessler, D. M. Guidi, A. S. Santos and J. O. Japessen, Tetrahedron, 2008, 64, 8449 CrossRef CAS PubMed.
- W. E. Allen, P. A. Gale, C. T. Brown, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 1996, 118, 12471 CrossRef CAS.
- Y. I. Khurgin and A. N. Isaev, Bull. Russ. Acad. Sci., 1992, 41, 1046 CrossRef.
- W. S. Mac Gregor, Ann. N. Y. Acad. Sci., 2006, 141, 1 Search PubMed.
- J. Lu, X. Tong and X. He, J. Electroanal. Chem., 2003, 540, 111 CrossRef CAS.
- J. Ruzicka and J. C. Tjell, Anal. Chim. Acta, 1970, 51, 1 CrossRef CAS.
- X. Yang, D. B. Hibbert and P. W. Alexander, Anal. Chim. Acta, 1998, 372, 387 CrossRef CAS.
- M. Mazloum, M. K. Amini and I. M. Baltork, Sens. Actuators, B, 2000, 63, 80 CrossRef CAS.
- B. Dalkıran, A. D. Ozel, S. Parlayan, E. Canel, U. Ocak and E. Kılıc, Monatsh. Chem., 2010, 141, 829 CrossRef PubMed.
- R. K. Mahajan, R. K. Puri, A. Marwaha, I. Kaur and M. P. Mahajan, J. Hazard. Mater., 2009, 167, 237 CrossRef CAS PubMed.
- U. Ocak, M. Ocak, A. Basoglu, S. Parlayan, H. Alp and H. Kantekin, J. Inclusion Phenom. Macrocyclic Chem., 2010, 67, 19 CrossRef CAS.
- A. K. Singh, G. Bhattacharjee and R. Singh, Sens. Actuators, B, 2004, 99, 36 CrossRef CAS PubMed.
- A. C. Ion, I. Ion, D. N. Stefan and L. Barbu, Mater. Sci. Eng., C, 2009, 29, 1 CrossRef CAS PubMed.
- M. Ghaedi, A. Shokrollahi, M. Montazerzohori and S. Gharaghani, Acta Chim. Slov., 2006, 53, 428 CAS.
- M. Javanbakht, M. R. Ganjali, H. Eshghi, H. Sharghi and M. Shamsipur, Electroanalysis, 1999, 11(2), 81 CrossRef CAS.
- G. Somer, S. Kalaycı and G. Ekmekci, Sens. Actuators, B, 2001, 81, 122 CrossRef CAS.
- A. F. D. de Namor, O. A. Webb, A. El Gamouz, W. A. Hamdan and M. Al-Nuaim, Calixpyrroles: From Fundamental Studies to the Development of Ion Selective Electrodes, in Advanced Synthetic Materials in Detection Science, ed. S. M. Reddy, Royal Society of Chemistry, 2014, ch. 6, pp. 172–196 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta022215k |
| This journal is © The Royal Society of Chemistry 2015 |