
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel polyvinylimidazolium nanoparticles as high-performance binders for lithium-ion batteries†
Jiayin
Yuan
*,
Simon
Prescher
,
Ken
Sakaushi
* and
Markus
Antonietti
Max Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, D-14424 Potsdam, Germany. E-mail: jiayin.yuan@mpikg.mpg.de; ken.sakaushi@mpikg.mpg.de; Fax: +49-331-5679502; Tel: +49-331-5679552
First published on 4th March 2015
Abstract
A series of polyvinylimidazolium-based nanoparticles were prepared via precipitation polymerization and tested as binders in cathodes of lithium ion batteries. Compared with the commercial standard poly(vinylidene fluoride), the as-synthesized nanoparticles present a superior performance. This “little change” of a polymer in otherwise unaltered devices gives not only a higher specific capacity but also an outstanding long-term electrochemical durability at least for 1000 charge–discharge cycles.
Electrochemical energy storage and conversion devices, such as batteries, fuel cells, supercapacitors, and solar-fuel devices, are the focus of current research due to their key role in the use of renewable energy, one of the main challenges of our society.1–9 These devices are commonly based on electrochemically active powdered materials, which are often poorly electron conductive. Therefore, conductive additives, such as carbon powders, are added to increase the conductivity of the electrodes and to achieve acceptable electrochemical performances. The electrode material and the carbon powders are glued together into a stable electrode by a binder polymer.6,7,9–11 The impact of polymer binders on the overall behavior of the complete electrode system, particularly with regard to the resulting mechanical properties and interactions with electrolytes and other active components, is highly relevant for the performance and the cycling stability of the device.3,12–14 So far, only a few literature reports have addressed this issue, but recent work has already illustrated that the polymer binder itself has the potential to tremendously improve the performance of the electrochemical devices.15–18 Functionality, polarity, viscosity, flexibility, stability, and in some cases conductivity appear to be crucial factors. For example, the viscosity of the polymer binder solutions is known to strongly affect both the processing and drying behavior of electrode slurries and the resulting electrode morphology. In general, many studies suggest that poly(vinylidene fluoride) (PVDF), which is a standard choice and for instance applied to commercially available lithium-ion batteries (LIBs),19,20 is not the most appropriate binder for electrodes, but is taken as a benchmark, nevertheless, which is due to the assumed high chemical stability.21 Some drawbacks have however already been recognized, such as the limited surface activity to bind eventually all electrode components as well as defluorinating side reactions with the electrolyte and the carbon component of the electrode.22,23 The pursuit for more functional and efficient binders is therefore still an open race with high potential benefits. In more simple words: a better binder literally improves the storage capacity and lifetime of the billions of batteries around, without really relying on any technology or processing change.
The present manuscript relates to the investigation of a series of densely crosslinked polymer nanoparticles bearing imidazolium-type ionic liquid (IL) units as an innovative binder for the cathodes in LIBs. The IL-species structured in a nanoparticle form can combine the high surface-to-volume ratio of nanoparticles with the superior physical properties of ILs, such as high electrochemical stability. The synthetic route to these polymer nanoparticles is illustrated in Fig. 1. Polyvinylimidazolium-based polymers have found versatile applications in various fields from heterogeneous catalysis, solid polymer electrolytes, to gas separation.24–34 This broad application spectrum is in fact related to the dense packing of the IL species in the polymer chains. Such ILs play an important role in the fabrication of various functional materials, such as nanocomposites.35–37 Due to a broad electrochemical window of the imidazolium-based IL species, polyvinylimidazolium-based polymers can in principle withstand a broad range of operation voltage in electrochemical energy storage/conversion devices.38–44 Considering the high polarity and the excellent surface activity already known, polyvinylimidazolium-based polymers are a potentially favorable choice of polymer binders. To our knowledge, only in one study, vinylimidazolium-based crosslinked copolymer nanoparticles of 40 nm have been tested as a binder for the silicon anode in lithium ion batteries (LIBs).45 Yet when applied to the cathode, these binders were claimed to decompose under operation. These pioneering observations, though exciting and encouraging, due to a possibly restricted choice of the polymer nanoparticle candidates did not reveal the real potential of the polyvinylimidazolium nanoparticle binders.
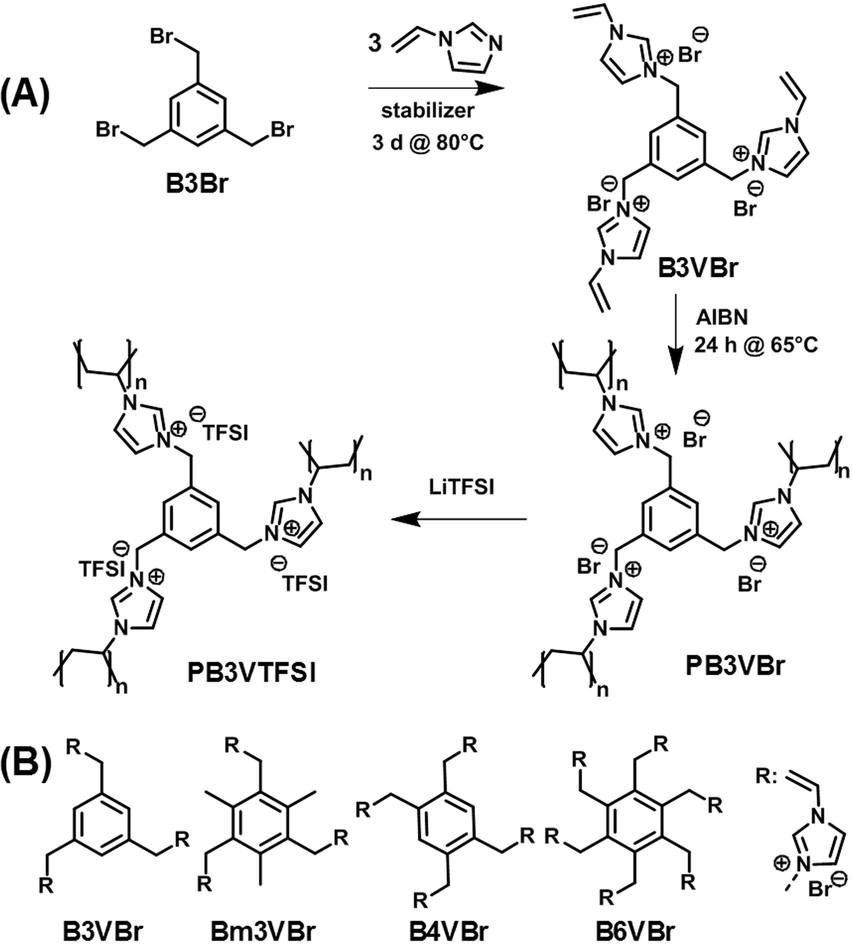 | ||
| Fig. 1 (A) Synthetic route to the polyvinylimidazolium nanoparticle binder PB3VTFSI. (B) Chemical structures of the imidazolium-based monomers applied in this work. | ||
Instead of solid, compact nanoparticles, we built up nanoparticles from a crosslinked, hyperbranched IL network made by precipitation polymerization of the pure cross-linker. Such a polymerization is known to result in non-spherical, broadly distributed, fractal structures, which were called Staudinger-microgels in earlier days,46,47 and have open pores as well as high surface area. It is our opinion that such an open particle can buffer mechanical stress and undergo geometrical deformation to accommodate the size-expansion of cathode materials in battery operation, while providing sufficient access to ions for transport at their surface. In addition, adherence of such hyperbranched particles, for entropy reasons, is known to be quite high.
The synthetic route to a typical polyvinylimidazolium nanonetwork is shown as an example in Fig. 1A. Initially, 1,3,5-tribromomethyl benzene (B3Br) reacted with excess of 1-vinylimidazole to produce monomer B3VBr bearing three vinylimidazolium bromide units. The following polymerization of monomer B3VBr was carried out to form in fact molecular, but three dimensionally interconnected, networks. Anion exchange from Br to bis(trifluoromethane)sulfonimide (TFSI) further improves the electrochemical stability of these polymer nanoparticles, as imidazolium-based ILs when paired with TFSI exhibit exceptional stability in a broad electrochemical voltage window. Following this design principle, 4 different monomers (Fig. 1B) were chosen in this work to investigate the structure–function relationship. They are denoted as B3VBr, Bm3VBr, B4VBr, and B6VBr, respectively, where “B” stands for the benzene core and nVBr for a specific number of the vinylimidazolium bromide IL units attached to the phenyl ring. Bm3VBr carries additionally three methyl groups.
The success in the monomer synthesis was verified by proton and carbon nuclear magnetic resonance spectra (ESI†). The degree of functionalization was found to be quantitative, except B6VBr, which contains 5 mol% of –CH2Br residues. We attribute this incomplete functionalization to the enhanced steric hindrance. The resultant crosslinked nanoparticles were further analyzed by Fourier transform infrared spectroscopy (FT-IR) (Fig. 2A and B and ESI†) to confirm the radical crosslinking reaction as well as the anion exchange process. In Fig. 2A, monomeric B3VBr shows a distinct vinyl band at 1654 cm−1. The intensity of this band drops significantly (indicated by a dotted arrow) after radical polymerization, revealing that the majority of the vinyl-moieties joined the polymer network formation and created PB3VBr. Through anion exchange, the as-synthesized PB3VTFSI presents prominent bands νa(SO2) at 1344, νa(CF3) at 1182, νs(SO2) at 1129, and νa(S–N–S) at 1052 cm−1 (indicated by black arrows), which are characteristic vibration bands for TFSI anions. Since sulfur is present exclusively in the TFSI anion, we applied elemental analysis of sulfur to these nanoparticles to quantify the anion exchange degree. The elemental analysis result indicates the degree of anion exchange in the sequence of PB3VTFSI (75%) = PBm3VTFSI (75%) > PB4VTFSI (72%) > PB6VTFSI (66%) (Table S1†). The lower degree of anion exchange in PB6VTFSI is associated with the hindered accessibility to the Br anion trapped in a more densely crosslinked network introduced by the six vinyl units in the B6VBr monomer. The amount of exchange of Br− at the same time measures the accessibility and mobility of all ions involved in our special network particle structure, and at least around 70% of the ions contribute to binding and ion transport.
 | ||
| Fig. 2 (A and B) FITR spectra of B3VBr, PB3VBr and PB3VTFSI. (C) TEM image of the polyvinylimidazolium nanoparticle binder PB3VTFSI. | ||
The size and shape of the binder nanoparticles were investigated by transmission electron microscopy (TEM) and dynamic light scattering (DLS), respectively. Fig. 2C shows a TEM image of the PB3VTFSI binder, where dark objects of irregular shapes, corresponding to the crosslinked polyvinylimidazolium nanoparticles, are observed. Similar morphologies were also observed in other polyvinylimidazolium nanoparticles used in this work. These nanoparticles feature a large distribution of size and shapes, which is indeed expected for such systems. The branched fractal structure as well as the high adhesion makes the size evaluation via TEM difficult. Thus the nano-network size and size distribution were characterized by the DLS measurements in solution. The number-averaged diameters for these nanoparticles are found in the range of 10–30 nm, however with the expected rather broad size distribution (see Table S1†).
These polyvinylimidazolium nanoparticles were then examined as a binder to a lithium ion battery setup using LiFePO4 as the active cathode material. It is well-known that LiFePO4 can intercalate/de-intercalate Li+ at ∼3.4 V vs. Li/Li+ with a theoretical specific capacity of 168 mA h g−1.48,49 The cathodes were prepared by casting a mixture of LiFePO4, carbon additive, binder, and acetonitrile on an Al foil, followed by a drying process. We did not apply mechanical pressing, although it typically further improves the electrochemical performance, in order to focus exclusively on the intrinsic properties of binders. Fig. 3A shows the charge–discharge curves running at a current density of 1 C (=170 mA g−1) using PVDF as a reference, representing the default value for the specific capacity of 120 mA h g−1 in our preparation. The nanoparticle binders PB4VTFSI, PB3VTFSI, and PBm3VTFSI favorably delivered a higher specific capacity of 140, 145 and 153 mA h g−1, respectively, reaching 91% of the maximum value of 168 mA h g−1 and suggesting that they indeed improve the overall cathode performance. This is supported by the mixing behaviour of all cathode components in the presence of different binders in the primary dispersion state. As shown in Fig. 4, when PVDF is used as a binder, dispersion instability was optically detected after 3 days, while the polyvinylimidazolium-based mixtures remained well-dispersed after 5 days. The dispersion-apparent optical density (ODapp) test, which measured the absorbance at λ = 500 nm of the dispersions after dilution of the original dispersion in acetonitrile by 500 times, shows that the polyvinylimidazolium-based dispersion has a much higher absorbance (56.1%, ODapp ∼ 280) than the PVDF-based system (31.8%, ODapp ∼ 159).50 Taking consideration of the isolating nature of PVDF and the well-known interfacial activity of polyvinylimidazolium-based polymers to adopt and attach to eventually all surfaces, we assume that the polyvinylimidazolium networks indeed form a homogeneous, comparably better conductive matrix with the carbon additives embedding LiFePO4 to enhance the charge flow and transfer. It should be noted that PB6VTFSI provides an unfavorable capacity of only 83 mA h g−1, which we attribute to the defects in synthesis, i.e. the chemically reactive residual –CH2Br units and a low degree of anion exchange, as discussed above.
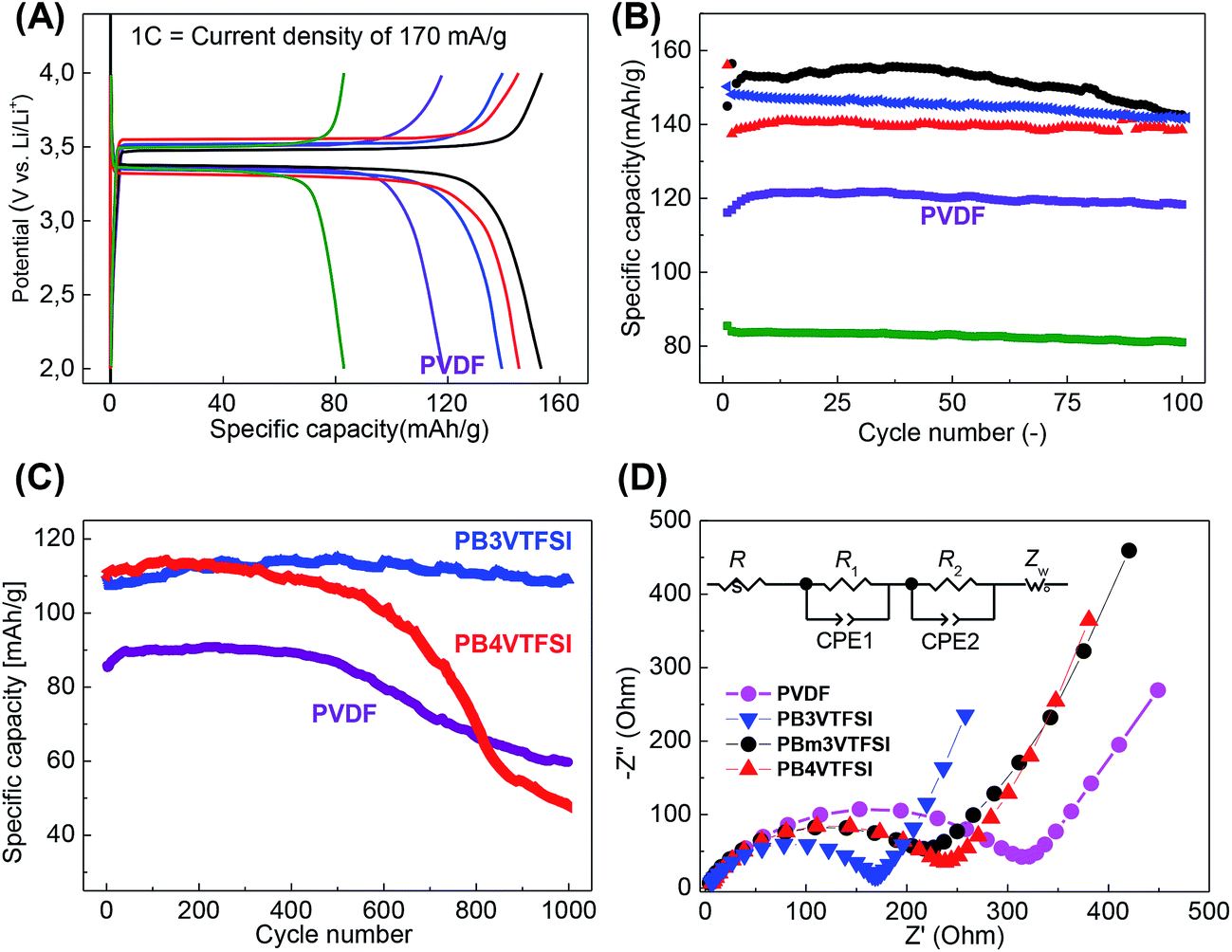 | ||
| Fig. 3 Electrochemical properties of LiFePO4 using different polyvinylimidazolium binders. PB3VTFSI (blue), PBm3VTFSI (black), PB4VTFSI (red), PB6VTFSI (green), and PVDF (purple). (A) Charge–discharge curves at a current density of 1 C (=170 mA g−1). (B) Cycling performance up to 100 cycles at 1 C. (C) Cycling performance up to 1000 cycles at 5 C (=850 mA g−1). (D) Electrochemical impedance spectra with their equivalent circuit description. | ||
 | ||
| Fig. 4 Photographs of 3 vials containing a mixture of LiFePO4 (80 wt%), carbon additive (10 wt%), and binder (10 wt%), in acetonitrile after 5 min, 3 and 5 days, to illustrate the dispersion stability. The binder materials in the vials from left to right are PVDF, PBV3TFSi and PB4TFSI. | ||
The electrodes carrying PVDF, PB3VTFSI, PBm3VTFSI and PB4VTFSI were further tested up to 100 cycles at 1 C to approach their cycling performance. As shown in Fig. 3B, they all present a higher specific capacity than PVDF. A close view reveals more details in their different cycling features. Although PBm3VTFSI initially bears the best specific capacity, after 100 cycles, a clear degradation of its specific capacity by 8% is found, with a more severely rapid fall after 40 cycles. This degradation can be explained by its chemical structure. PBm3VTFSI incorporates three methyl moieties at positions 2, 4, and 6 of each phenyl ring. The methyl moiety is known to undergo electrochemical oxidation at voltages around 1.5 V, depending on the other substituents on the aromatic ring and the chemical environment.51,52 In comparison, PB3VTFSI and PB4VTFSI exhibit more robust stability. PB4VTFSI lost ca. 5% of its capacity, while PB3VTFSI eventually retained its full binding capability.
The battery tests were then extended to 1000 cycles for PB3VTFSI, PB4VTFSI and PVDF at higher charge/discharge currents, 5 C = 850 mA g−1 (Fig. 3C). This is a harsh, more realistic condition in the future daily life usage of LIBs, i.e. full charge–discharge in only 24 min. The PVDF and PB4VTFSI cathodes were found to degrade after 400 cycles under these violent conditions. In contrast, PB3VTFSI showed an extraordinary stable cycling performance all along the measurement. The reason for the degradation of PVDF could be the swelling of the electrolyte and the following loss of electrical contact between the active material and the current collector.53 The rate capability of PB3VTFSI was compared with PVDF, and we found that PB3VTFSI shows a better rate capability than PVDF (Fig. S8†). This test on one side verifies polyvinylimidazolium nanoparticles as a top candidate to replace PVDF as binders for LIBs; on the other side it motivates us to understand the inherent mechanism of the unusual function of the polyvinylimidazolium binder in the electrochemical environment.
We investigated the electrochemical processes by using electrochemical impedance spectroscopy (EIS) (Fig. 3D, Table S2, and Fig. S9†).54 As a two-phase mechanism is expected to dominate the insertion/de-insertion process of Li+ into LiFePO4, we first carried out the EIS measurements for pristine LiFePO4 particles in order to use a solid solution phase to fit the impedance spectra with the Warburg impedance.55–59 The obtained Nyquist plots were fitted by an equivalent circuit with a two-step ion insertion/de-insertion process, which is a typical equivalent circuit for the insertion mechanism (Fig. 3D inset).60–62Rs, R1 and R2 are the electrolyte resistance, interfacial charge transfer resistance in the high frequency region and interfacial charge transfer resistance in the low frequency region, respectively. The constant phase element (CPE) is described by ZCPE = 1/(jω)pTCPE which is constituted of a CPE constant (TCPE) and a CPE index (p). Zw is the Warburg impedance, which corresponds to the diffusion of ions into a bulk.62 The results of EIS measurements showed that the nanoparticle binders reduce both resistances R1 and R2 which correspond to the Li+ transfer from the outer Helmholtz plane (OHP) to the inner Helmholtz plane (IHP) and the ad-ion diffusion from IHP to the electrode surface, respectively (Table S2†). Chemically this can be translated to a promoted transport of the Li+ through all physical layers of the electroactive material, potentially related to the fact that the polyvinylimidazolium binders give a favourable polarizing medium to desolvate the Li+, a process which has the highest activation energy during the Li+ insertion process into a solid material,63–65 plus that the substitution of PVDF avoids the formation of non-conductive polymeric insulation layers which diminish effective ion transport.
Conclusions
In conclusion, new polymer nanonetwork particles were applied as a binder material in the cathode of LIBs. Our experimental data support that the polyvinylimidazolium nanoparticle binders with our specific structure design enhances dramatically the cathode performance. As a new generation binder material, they desirably deliver a higher specific capacity than PVDF, while at the same time the best system demonstrated a robust cycling stability even above 1000 cycles under harsh electrochemical conditions. In ongoing work, the superior performance of the polyvinylimidazolium nanoparticle as a binder is extended to other electrochemical devices, such as metal–O2 batteries, fuel cells, supercapacitors and solar-fuel devices. This will be reported in the future elsewhere.Acknowledgements
The authors would like to thank the Max-Planck Society (MAXNET Energy program) for the financial support, and M&T Olivine Co., LTD for providing high-quality LiFePO4. We thank Dr J. S. Lee for his support.Notes and references
- J.-M. Tarascon, Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 2010, vol. 368, pp. 3227–3241 Search PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- Y. S. Hu, Y. G. Guo, R. Dominko, M. Gaberscek, J. Jamnik and J. Maier, Adv. Mater., 2007, 19, 1963–1966 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337–1344 CrossRef CAS PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Z.-S. Wu, L. Chen, J. Liu, K. Parvez, H. Liang, J. Shu, H. Sachdev, R. Graf, X. Feng and K. Müllen, Adv. Mater., 2014, 26, 1450–1455 CrossRef CAS PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele, S. Pejovnik and J. Jamnik, J. Power Sources, 2003, 119–121, 770–773 CrossRef CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS PubMed.
- S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65–68 CrossRef CAS.
- C. Erk, T. Brezesinski, H. Sommer, R. Schneider and J. Janek, ACS Appl. Mater. Interfaces, 2013, 5, 7299–7307 CAS.
- W. Li, Q. Zhang, G. Zheng, Z. W. Seh, H. Yao and Y. Cui, Nano Lett., 2013, 13, 5534–5540 CrossRef CAS PubMed.
- Z. W. Seh, Q. Zhang, W. Li, G. Zheng, H. Yao and Y. Cui, Chem. Sci., 2013, 4, 3673–3677 RSC.
- S. Xin, L. Gu, N.-H. Zhao, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed.
- Y.-Q. Wang, L. Gu, Y.-G. Guo, H. Li, X.-Q. He, S. Tsukimoto, Y. Ikuhara and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 7874–7879 CrossRef CAS PubMed.
- B. Guo, X. Wang, P. F. Fulvio, M. Chi, S. M. Mahurin, X.-G. Sun and S. Dai, Adv. Mater., 2011, 23, 4661–4666 CrossRef CAS PubMed.
- H. Maleki, G. Deng, A. Anani and J. Howard, J. Electrochem. Soc., 1999, 146, 3224–3229 CrossRef CAS.
- A. D. Pasquier, F. Disma, T. Bowmer, A. S. Gozdz, G. Amatucci and J. M. Tarascon, J. Electrochem. Soc., 1998, 145, 472–477 CrossRef.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2012, 36, 1629–1648 CrossRef.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49, 291–314 CrossRef CAS.
- Y. Men, D. Kuzmicz and J. Yuan, Curr. Opin. Colloid Interface Sci., 2014, 19, 76–83 CrossRef CAS.
- G. B. Appetecchi, G. T. Kim, M. Montanino, M. Carewska, R. Marcilla, D. Mecerreyes and I. De Meatza, J. Power Sources, 2010, 195, 3668–3675 CrossRef CAS.
- J. Zhao, X. Shen, F. Yan, L. Qiu, S. Lee and B. Sun, J. Mater. Chem., 2011, 21, 7326–7330 RSC.
- J. Texter, Macromol. Rapid Commun., 2012, 33, 1996–2014 CrossRef CAS PubMed.
- D. England, N. Tambe and J. Texter, ACS Macro Lett., 2012, 1, 310–314 CrossRef CAS.
- X. Feng, X. Sui, M. A. Hempenius and G. J. Vancso, J. Am. Chem. Soc., 2014, 136, 7865–7868 CrossRef CAS PubMed.
- K. Zhang, X. Feng, X. Sui, M. A. Hempenius and G. J. Vancso, Angew. Chem., Int. Ed., 2014, 53, 13789–13793 CrossRef CAS PubMed.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS PubMed.
- Q. Ji, I. Honma, S.-M. Paek, M. Akada, J. P. Hill, A. Vinu and K. Ariga, Angew. Chem., Int. Ed., 2010, 49, 9737–9739 CrossRef CAS PubMed.
- S. Tang, G. A. Baker and H. Zhao, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC.
- M. Armand, F. Endres, D. R. Macfarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473–13476 RSC.
- H. Matsumoto, H. Kageyama and Y. Miyazaki, Chem. Commun., 2002, 1726–1727 RSC.
- J.-H. Shin, W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 1016–1020 CrossRef CAS.
- J.-H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152, A978–A983 CrossRef CAS.
- J.-K. Kim, L. Niedzicki, J. Scheers, C.-R. Shin, D.-H. Lim, W. Wieczorek, P. Johansson, J.-H. Ahn, A. Matic and P. Jacobsson, J. Power Sources, 2013, 224, 93–98 CrossRef CAS.
- H. Xing, C. Liao, Q. Yang, G. M. Veith, B. Guo, X.-G. Sun, Q. Ren, Y.-S. Hu and S. Dai, Angew. Chem., Int. Ed., 2014, 53, 2099–2103 CrossRef CAS PubMed.
- J. von Zamory, M. Bedu, S. Fantini, S. Passerini and E. Paillard, J. Power Sources, 2013, 240, 745–752 CrossRef CAS.
- H. Staudinger and E. Husemann, Ber. Dtsch. Chem. Ges., 1935, 68, 1618–1634 CrossRef.
- M. Antonietti and C. Rosenauer, Macromolecules, 1991, 24, 3434–3442 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- D. Ager, V. A. Vasantha, R. Crombez and J. Texter, ACS Nano, 2014, 8, 11191–11205 CrossRef CAS PubMed.
- R. Tomat and A. Rigo, J. Appl. Electrochem., 1984, 14, 1–8 CrossRef CAS.
- S. D. Ross, M. Finkelstein and R. C. Petersen, J. Am. Chem. Soc., 1967, 89, 4088–4091 CrossRef CAS.
- S. S. Zhang and T. R. Jow, J. Power Sources, 2002, 109, 422–426 CrossRef CAS.
- C. Ho, I. D. Raistrick and R. A. Huggins, J. Electrochem. Soc., 1980, 127, 343–350 CrossRef CAS.
- A. Yamada, H. Koizumi, N. Sonoyama and R. Kanno, Electrochem. Solid-State Lett., 2005, 8, A409–A413 CrossRef CAS.
- A. S. Andersson, B. Kalska, L. Häggström and J. O. Thomas, Solid State Ionics, 2000, 130, 41–52 CrossRef CAS.
- N. Meethong, H. Y. S. Huang, S. A. Speakman, W. C. Carter and Y. M. Chiang, Adv. Funct. Mater., 2007, 17, 1115–1123 CrossRef CAS.
- C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 2008, 7, 665–671 CrossRef CAS PubMed.
- G. Kobayashi, S.-i. Nishimura, M.-S. Park, R. Kanno, M. Yashima, T. Ida and A. Yamada, Adv. Funct. Mater., 2009, 19, 395–403 CrossRef CAS.
- P. G. Bruce and M. Y. Saidi, J. Electroanal. Chem., 1992, 322, 93–105 CrossRef CAS.
- P. G. Bruce and M. Y. Saidi, Solid State Ionics, 1992, 51, 187–190 CrossRef CAS.
- Y. Mizuno, M. Okubo, D. Asakura, T. Saito, E. Hosono, Y. Saito, K. Oh-ishi, T. Kudo and H. Zhou, Electrochim. Acta, 2012, 63, 139–145 CrossRef CAS.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120–A1123 CrossRef CAS.
- K. Xu, J. Electrochem. Soc., 2007, 154, A162–A167 CrossRef CAS.
- Y. Yamada, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2009, 25, 12766–12770 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data. See DOI: 10.1039/c5ta01374g |
| This journal is © The Royal Society of Chemistry 2015 |