
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen cycling in γ-Mg(BH4)2 with cobalt-based additives†
O.
Zavorotynska
a,
I.
Saldan
ab,
S.
Hino‡
a,
T. D.
Humphries
a,
S.
Deledda
a and
B. C.
Hauback
*a
aPhysics Department, Institute for Energy Technology, P.O. Box 40, NO-2027 Kjeller, Norway. E-mail: Bjorn.Hauback@ife.no
bDepartment of Physical and Colloid Chemistry, Ivan Franko National University of Lviv, 6 Kyryla and Mefodia Str., UA-79005 Lviv, Ukraine
First published on 24th February 2015
Abstract
Magnesium borohydride (Mg(BH4)2) is an attractive candidate as a hydrogen storage material due to its high hydrogen content and predicted favorable thermodynamics. In this work we demonstrate reversible hydrogen desorption in partially decomposed Mg(BH4)2 which was ball milled together with 2 mol% Co-based additives. Powder X-ray diffraction and infrared spectroscopy showed that after partial decomposition at 285 °C, amorphous boron-hydride compounds were formed. Rehydrogenation at equivalent temperatures and hydrogen pressures of 120 bar yielded the formation of crystalline Mg(BH4)2 in the first cycle, and amorphous Mg(BH4)2 with other boron–hydrogen compounds upon the third H2 absorption. Reversibility was observed in the samples with and without Co-based additives, although the additives enhanced hydrogen desorption kinetics in the first cycle. X-ray absorption spectroscopy at Co K-edge revealed that all the additives, apart from Co2B, reacted during the first desorption to form new stable species.
Introduction
Magnesium borohydride Mg(BH4)2 is a particularly interesting complex hydride for hydrogen storage due to its high gravimetric hydrogen capacity (14.9 wt%). However, hydrogen desorption kinetics and reversibility present a challenge, and cycling stability of Mg(BH4)2 remains an open question. DFT calculations have predicted that H2 desorption from the α-polymorph of Mg(BH4)2 is feasible within 20–75 °C.1–3 Nevertheless, the material decomposes in the 200–500 °C temperature range through a complex reaction pathway, which depends on the experimental conditions applied.4–9 These discrepancies are attributed to kinetic barriers and/or intermediate phases that were not taken into account in the calculations.10 Rehydrogenation of completely decomposed Mg(BH4)2 to a mixture of Mg(BH4)2 and MgB12H12 requires 900 bar H2 and more than 70 h at 400 °C.11,12 Alternatively, amorphous Mg(BH4)2 in a 50% yield can be obtained by reactive milling of its decomposition product MgB2 under 100 bar of H2 for 100 h.13 Milder conditions can be employed for rehydrogenation of partially decomposed Mg(BH4)2: 2–6 wt% of H2 were re-absorbed at pressures below 400 bar and 300 °C.7,14–16 To the best of our knowledge, hydrogen cycling in Mg(BH4)2 has not been reported yet.In order to improve the hydrogen desorption and absorption performance of Mg(BH4)2, a wide range of methodologies has been tried, including high-energy ball-milling,17 preparation of composite materials,18,19 dispersion in porous matrices,20,21 and addition of transition metal (TM) compounds.16,17,22–25 Because of a remarkable effect of Ti-based additives on hydrogen desorption kinetics and reversibility in NaAlH4,26 various TM compounds have been extensively explored in order to enhance the hydrogen storage properties of other complex hydrides, including metal borohydrides (see, for example, lit. 12,22,27–29 and references therein).
Small quantities (≥2 mol%) of TM additives, such as NbCl5, NbF5, TiCl3, TiO2, Ti–Nb, TiF3, ScCl3, CoCl2, ZnF2, have been shown to significantly reduce the temperature of hydrogen release of various polymorphs and composites of Mg(BH4)2 during the first decomposition.11,16,17,23,24,30,31 Only few of these studies have explored the effect of the additives on the rehydrogenation. Bardaji et al.23 did not observe rehydrogenation of Mg(BH4)2 decomposed above 450 °C in the presence of Ti- and Nb-based additives. Newhouse et al.11 were able to reform Mg(BH4)2 from its decomposition products in the presence of 5 mol% of TiF3 and ScCl3 under 900 bar H2, although rehydrogenation was also observed in the samples without additives. In our previous study we reported that Ni-based additives improved hydrogen absorption (and desorption) kinetics in partially decomposed Mg(BH4)2 composites at moderate conditions (up to 120 bar H2 at 220–260 °C).16
The positive effect of additives on the decomposition temperature (Tdec) appears to be related to chemical reactions with the hydride. CoCl2 in a LiBH4 + Mg(BH4)2 composite reacted to form H2 and LiMgCl3.30 TiCl3 in Mg(BH4)2 was converted to TixMg1−3x/2(BH4)2 (x = 0–0.67) after ball-milling, and subsequently to TiB2 during the first dehydrogenation stage (100–150 °C).32 In the same work, TiO2 was found to convert slowly to lower oxidation states at the temperature around the main dehydrogenation peak of Mg(BH4)2 (350 °C). In this case, the additive effect17 was related to the reduction of the TiO2. The changes in the Ti-additives in Mg(BH4)2 upon rehydrogenation and cycling were not investigated. Ni-additives in γ-Mg(BH4)2 were found to form new compounds with amorphous Ni3B-like structure during Mg(BH4)2 decomposition.16 Those in situ formed catalysts were suggested to be responsible for the improved hydrogen desorption and absorption kinetics. Whether those compounds can act as catalysts in further cycles remains an open question.
In this work several cobalt additives were investigated towards improving the hydrogen de- and reabsorption in Mg(BH4)2 over three cycles. Particular attention was paid to the changes in the local structure of cobalt upon hydrogen cycling. Hydrogen desorption and re-absorption was performed at mild conditions, i.e. 280–285 °C and 115–125 bar H2 as the absorption pressure. Desorption was performed at an initial back pressure of 2.7 bar H2. These conditions were chosen for several reasons. Firstly, we aimed at finding the reversible hydrogen capacity of Mg(BH4)2 when desorption is carried out using a H2 back pressure close to the working conditions of a fuel cell. Secondly, lower working temperatures were expected to hinder the formation of irreversible boron–hydrogen compounds such as closed-cage boron hydrides. Finally, we wanted to investigate the stability of the TM-additives (or in situ formed catalysts) and to find whether their effect on the hydrogen de-/absorption behavior of Mg(BH4)2 could be observed beyond the first desorption.
Experimental details
All procedures were carried out in a glove box under a continuously purified Ar atmosphere (O2, H2O < 1 ppm) if not mentioned otherwise.Commercial γ-Mg(BH4)2 (Sigma-Aldrich, 95%, Fig. S1 (ESI†)) was used for all composites. Infrared (IR) analysis of the compound showed that it did not contain any molecular vibrations due to water or solvent impurities (Fig. S2 (ESI†)). CoF3,§ CoCl2 (99.999%), and Co3O4 (Co(II), Co(III) oxide nanoparticles, <50 nm particle size, 99.5%) were purchased from Sigma-Aldrich, whereas Co2B was purchased from American Elements. Powder X-ray diffraction (PXD) analysis of the as received CoF3 has shown it to be a mixture of CoF2 and CoF3. The list of the samples studied in this work is shown in Table 1. The starting powder mixtures consisted of Mg(BH4)2 and 2 mol% of an additive (Coadd). The milling was carried out in stainless steel vials and balls with a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ball-to-powder weight ratio. Pure Mg(BH4)2 and the Mg(BH4)2 + Coadd mixtures were milled for 1 hour at 280 rpm under 1 bar Ar pressure.
:1 ball-to-powder weight ratio. Pure Mg(BH4)2 and the Mg(BH4)2 + Coadd mixtures were milled for 1 hour at 280 rpm under 1 bar Ar pressure.
Hydrogen desorption and absorption isotherms were obtained in a calibrated Sieverts-type apparatus built in-house. For the desorption measurements, samples were heated to the required temperature at a back pressure of 50 bar H2 to hinder H2 desorption during heating. At isothermal conditions, the pressure was reduced to 2.7 bar. For the absorption measurements, the required hydrogen absorption pressure was set at room temperature and then the sample was heated to the required temperature. The amount of desorbed/absorbed hydrogen was determined from the pressure changes in the closed system. The temperature variation during the isothermal step was within ±2.5 °C during the first 30 min and not more than ±0.5 °C for the remaining duration of the measurement. Desorption measurements were carried out until the isotherms approached a plateau. In most cases the desorption reaction at the final stages was very slow and was stopped before completion. The total amount of gas released during these desorption steps was taken into account when the reacted fractions y were calculated (y = Δwtfinal/Δwt). In absorption measurements the plateau was reached. The desorption and absorption curves for the samples S0 and S1 were compared with several theoretical models of the solid-state reaction kinetics,33 as described in lit.33,34 For this, the reacted fraction y was plotted vs. time normalized to the time of y = 0.5, (t/t0.5). Theoretical models used are described in the Table S1 (ESI†), and were plotted basing on the y and t/t0.5 data reported in lit.33
Aliquots of the samples were taken after the 1st desorption and 1st absorption for composition analysis. The experimental conditions and the amounts of hydrogen desorbed/absorbed are summarized in the Table 2.
| S0 | S1 | S2 | S3 | S4 | S5 | |
|---|---|---|---|---|---|---|
| a Desorbed or absorbed wt% of H2 measured at isothermal conditions. | ||||||
| 1 st cycle | ||||||
| des | ||||||
| T, °C | 284 | 285 | 284 | 284 | 288 | 288 |
| p range, bar | 2.7–3.3 | 2.8–3.7 | 2.6–3.1 | 2.7–3.5 | 2.6–3.7 | 2.8–4 |
| Δwt%a | 4.1 | 4.2 | 3.2 | 3.1 | 4.4 | 3.9 |
| Isotherm time, h | 19 | 20 | 18 | 10 | 39 | 21 |
|
||||||
| abs | ||||||
| T, °C | 282 | 283 | 281 | 282 | 284 | 285 |
| p range, bar | 117 | 125 | 117 | 120 | 125 | 122 |
| Δwt% | 2.1 | 2.2 | 1.9 | 1.9 | 1.9 | 1.6 |
| Isotherm time, h | 16 | 5 | 4 | 3.5 | 3 | 2 |
|
||||||
| 2 nd cycle | ||||||
| des | ||||||
| T, °C | n/a | 286 | 285 | 286 | 285 | 284 |
| p range, bar | 2.8–3.0 | 2.4–2.5 | 2.7–2.9 | 3.0–3.1 | 2.7–2.9 | |
| Δwt% | 2.2 | 1.9 | 1.9 | 1.4 | 1.7 | |
| Isotherm time, h | 23.5 | 21 | 25.5 | 19 | 22 | |
|
||||||
| abs | ||||||
| T, °C | n/a | 286 | 279 | 283 | 285 | 289 |
| p range, bar | 114 | 118 | 112 | 117 | 117 | |
| Δwt% | 1.9 | 1.9 | 1.7 | 1.6 | 1.4 | |
| Isotherm time, h | 6 | 8 | 9 | 6.5 | 5 | |
|
||||||
| 3 rd cycle | ||||||
| des | ||||||
| T, °C | n/a | 286 | 285 | 286 | 286 | 285 |
| p range, bar | 2.7–2.9 | 2.3–2.4 | 2.7–2.8 | 2.7–2.9 | 2.7–2.9 | |
| Δwt% | 1.8 | 1.7 | 1.5 | 2.0 | 1.4 | |
| Isotherm time, h | 26 | 17 | 23 | 46 | 25 | |
|
||||||
| abs | ||||||
| T, °C | n/a | 282 | 278 | 283 | 285 | 285 |
| p range, bar | 113 | 118 | 122 | 111 | 116 | |
| Δwt% | 1.6 | 2.3 | 1.6 | 1.4 | 1.2 | |
| Isotherm time, h | 5 | 22 | 14 | 4 | 4 | |
X-ray absorption spectroscopy (XAS) was performed at beamline I811 at Max II Laboratory synchrotron facility in Lund, Sweden.35,36 Mg(BH4)2 + Coadd samples were analyzed as-milled, after the 1st desorption, 1st absorption, and the 3rd absorption (cycling). Samples were measured without dilution. The spectra of the reference powders CoF3, Co3O4, CoCl2, Co2B, used in the preparation of the composites, and additionally CoF2 (Aldrich), CoO (>99.99%, Aldrich), Co powder (99.5%, Alfa Aesar), were measured diluted with boron nitride (2 mol% of Coadd). The powders were pressed into pellets and mounted in aluminum sample holders. To avoid contact with air, the sample holders were covered by Kapton tape adhered by air-tight grease. XAS spectra were collected in the energy range 7.7–8.7 keV comprising Co K-edge at 7.7089 keV. The incident X-ray beam cross section on the sample was 0.5 × 0.5 mm2. XAS data were collected in transmission and fluorescence mode. Two to four quick scans and two consecutive step scans were obtained for each sample. The step scans were measured in pre-edge and edge regions (XANES) with counting time 0.5 s and in the Extended X-ray Absorption Fine Structure (EXAFS) region with 20 s per step. The fluorescence detector was placed at 90° to the incident beam. The spectrum of the Co foil was used as a reference for data calibration and alignment. Data analysis was carried out with DEMETER software pack (ATHENA and ARTEMIS).37 The spectra were pre-processed in ATHENA (background subtracted, aligned, the step scans were averaged and used for final plots and fitting). Calculation of the theoretical scattering paths in the EXAFS region and fitting were performed with the FEFF6 code38 using ARTEMIS. The details of the fitting are described in the ESI.†
Synchrotron powder X-ray diffraction (SR-PXD) data were collected at the Swiss-Norwegian Beam Line (station BM01A) at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France with a wavelength of 0.70135 Å. The samples were contained in glass capillaries of 0.5 mm diameter. 2D images were collected with an exposure time of 10 s using a PILATUS image plate detector. The capillary was rotated 10° during the exposure. The 2D datasets were integrated into one-dimensional powder diffraction patterns with the program FIT2D.39 PXD data were also obtained with a laboratory Bruker AXS D8 Advance diffractometer equipped with a Göbbel mirror and a LynxEye 1D strip detector. In this case, patterns were obtained in a Debye–Scherrer geometry using Cu Kα radiation (λ = 1.5418 Å) and rotating glass capillaries, filled and sealed under Ar atmosphere. The PXD data were analyzed using DIFFRAC.SUITE software (BRUKER) and the Rietveld refinement program MAUD.40 All PXD measurements were performed at room temperature.
Combined differential scanning calorimetry-thermogravimetric analysis (DSC-TGA) measurements were performed with a Netzsch STA 449 F3 Jupiter instrument in the 25–450 °C temperature range. Samples were measured in Al2O3 crucibles with pierced lids at a heating rate of 2 °C min−1 in a 50 mL min−1 Ar flow.
Attenuated total reflection Fourier transformed infrared (ATR FT-IR) spectra were collected with a Bruker Alpha-Platinum infrared spectrometer upon a diamond crystal. The spectra were obtained in the range of 4000–400 cm−1 with a resolution of 2 cm−1. The samples were measured without any dilution inside an argon-filled glove box. IR spectra were ATR-corrected and normalized using commercial spectroscopic software OPUS.
Results
Kinetics of hydrogen desorption and absorption
Hydrogen desorption and absorption isotherms are shown in Fig. 1. In the first cycle, a comparison was made between pure as-received (S0) and pure milled (S1) γ-Mg(BH4)2 to examine the effect of ball-milling alone. The effect of ball-milling alone on the Mg(BH4)2 decomposition in the 1st cycle was evident after 1 h (Fig. 1a). In the samples milled with or without additives, 90% of the decomposition reaction at 285 °C was complete within ca. 3 h, in contrast to 8 h in the non-milled S0. The average reaction rates, r, calculated for the intermediate reaction step (0.3 < y < 0.7, Table 3), were four times faster in the milled sample. The effect of the additives was clearly visible especially at the initial stages of desorption. After 1 h, y equaled 0.4 and 0.7 in pure Mg(BH4)2 (S0, S1) and in the S2–S5 with additives, respectively. At the intermediate stage, the desorption r in the samples with additives were also slightly larger than those in the pure milled sample (Table 3). The nature of the additives had minor effects. Desorption reactions were not completed due to the very slow rates at the final stages, but most of decomposition possible at the given conditions was achieved. At these desorption conditions only 3–4 wt% H2 desorption capacity was obtained (for the gravimetric values see Table 1). | ||
| Fig. 1 Hydrogen desorption and absorption isotherms for the S0–S5 samples during the three cycles. | ||
| Cycle | Reaction rates (fraction per h) | |||||
|---|---|---|---|---|---|---|
| S0 | S1 | S2 | S3 | S4 | S5 | |
| des 1 | 0.1 | 0.4 | 0.5 | 0.5 | 0.6 | 0.6 |
| des 2 | n/a | 0.1 | 0.2 | 0.2 | 0.1 | 0.1 |
| des 3 | n/a | 0.4 | 0.6 | 0.5 | 0.3 | 0.3 |
| abs 1 | 0.3 | 1.2 | 1.1 | 0.5 | 0.9 | 1.4 |
| abs 2 | n/a | 0.3 | 0.2 | 0.3 | 0.4 | 0.6 |
| abs 3 | n/a | 0.3 | 0.3 | 0.4 | 0.5 | 0.6 |
The 1st cycle absorption isotherms (Fig. 1b) showed that milling of Mg(BH4)2 alone enhances H2 absorption kinetics by 2–5 times. Absorption curves and the r in the Table 3 show that all additives except Co3O4 (S5) hampered the absorption kinetics in the 1st cycle.
Decomposition of all samples in the 2nd cycle (Fig. 1c) was much slower than that during the 1st cycle. R at 0.3 < y < 0.7 indicate that CoF3 (S2) and Co2B (S3) slightly enhanced the desorption kinetics when compared to pure milled Mg(BH4)2 (S1). 90% of decomposition was reached within 10 h and 99% completion required 20–30 h. The absorption kinetics rates fell at least twice by comparison with the first cycle (Fig. 1d and Table 3). Co3O4 (S5) and CoCl2 (S4) seemingly enhanced the absorption kinetics, whereas Co2B did not appear to have any significant effect, and CoF3 slowed down the reaction kinetics. Complete re-absorption required 2–7 h.
Desorption kinetics in the 3rd cycle were faster than that of the 2nd cycle (Fig. 1e). The average r at the intermediate reaction step increased by several times (Table 3). CoF3 and Co2B had a slightly enhancing effect, whereas CoCl2 and Co3O4 slightly negative. The absorption kinetics was similar to that of the previous cycle. Complete absorption required 2.5–5 h (S1, S3, S4 and S5) and 10 h in the sample with CoF3 (S2).
Overall, the desorption kinetics rates in the 1st cycle were the fastest, in the 2nd they decreased and in the 3rd increased again. Most of the additives slightly enhanced the kinetics in the 1st cycle and had almost no effect in the 2nd. In the last cycle CoF3 and Co2B slightly improved the r, whereas other additives hampered the kinetics. Similarly, the absorption kinetics in the 1st cycle were the fastest, then dramatically decreased, and in the last cycle the rates were similar to those of the 1st cycle. The additives mostly slowed down the absorption kinetics, only Co3O4 enhancing it distinctively in all the 3 cycles. It is worth noting that the shapes of the curves in Fig. 1 seem different for all cycles, especially in desorption, indicating that different mechanisms were governing the reactions. This could explain the inconsistent change in the reaction rates with the cycling.
Fig. 1 illustrates that milling alone had the greatest effect upon the reaction kinetics, especially during the absorption step. To investigate how the absorption and desorption mechanisms were changed by the ball-milling process, the experimental data were plotted alongside several theoretical models for different reaction rate-limiting mechanisms (Fig. 2).33,34 The experimental desorption curve for non-milled Mg(BH4)2 (Fig. 2a, curve S0-des) at the initial stages was similar to the phase-boundary-controlled model, whereas for y > 0.2 it resembled a diffusion-controlled-type reaction. During the early stages of the first desorption of as-milled Mg(BH4)2 (Fig. 2a, curve S1-des 1) the decomposition seemed to be controlled by processes at the interfaces, whereas after y > 0.5 by diffusion. In the subsequent cycles (Fig. 2a, curves S1-des 2, 3) the desorption appeared to be limited both by processes at the interfaces and by diffusion, whereas in the final stages only by diffusion.
 | ||
| Fig. 2 Comparison of desorption (a) and absorption (b) isotherms of non-milled and milled Mg(BH4)2 (S0, S1) with several theoretical models for different bulk rate-limiting mechanisms.33,34 The models considered here are: D1, D2, and D3: 1-, 2-, and 3-dimensional diffusion-controlled reactions, respectively; D4: diffusion-controlled reaction starting on the exterior of a spherical particle; R2 and R3: inwards phase-boundary-controlled reactions for a circular disk or a cylinder (R2), and sphere (R3); A2 and A3: reactions controlled by nucleation and/or growth following Avrami–Erofe'ev equations (see also Table S1 (ESI†)).41,42 | ||
The absorption reactions for S0 and S1 resembled Avrami–Erofe'ev models for nucleation and growth-controlled mechanisms in the first two cycles, and a phase-boundary-controlled reaction in the last cycle (Fig. 2b, curve S1-abs 3). The modeling considers simplified one-step reactions to and from Mg(BH4)2. Since none of the isotherms strictly follow a particular reaction mechanism, it can be suggested that desorption and absorption for these systems have complex mechanisms consisting of several competing reactions at the conditions applied in this work. Therefore the in-depth modeling of the experimental data with the theoretical models and the refinement of the kinetics parameters was not further pursued.
DSC-TG analysis
Whereas milling and additives demonstrated significant effects on reaction kinetics during first desorption and absorption (Fig. 1), DSC analysis illustrated that phase-transition and decomposition temperatures were also altered (see Fig. 3a). The endothermic peaks in the 150–200 °C temperature region correspond to the phase transitions: from γ-Mg(BH4)2 (cubic Id![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) a) to ε-Mg(BH4)2 (not indexed) and from ε-Mg(BH4)2 to β-Mg(BH4)2 (orthorhombic Fddd).5,43–45 In all milled samples (S1–S5), the heat-flow intensity ratio of the two phase transition peaks was reversed when compared to the non-milled sample S0. There was also a shift in the phase transition temperatures in the samples with additives, the most notable being a decrease of 15 °C in the sample containing CoF3 (S2).
a) to ε-Mg(BH4)2 (not indexed) and from ε-Mg(BH4)2 to β-Mg(BH4)2 (orthorhombic Fddd).5,43–45 In all milled samples (S1–S5), the heat-flow intensity ratio of the two phase transition peaks was reversed when compared to the non-milled sample S0. There was also a shift in the phase transition temperatures in the samples with additives, the most notable being a decrease of 15 °C in the sample containing CoF3 (S2).
 | ||
| Fig. 3 DSC (a) and TG (b) curves for samples S0–S5 (ΔT/Δt = 2 °C min−1). | ||
The two broad endothermic events at 250–300 and 300–370 °C, associated with a large weight loss in the TG profile (Fig. 3b) are due to H2 release.4–9 In the samples milled with additives (S2–S5), the intensities of these H2-release events were reversed. The first hydrogen release event is the most relevant to our isothermal desorption studies. It is somewhat broader in the samples milled with additives (S2–S5) than that in as-milled Mg(BH4)2 (S1). This may indicate some structural disorder induced by milling with additives. The temperature of this event was also slightly shifted to lower temperatures by up to 10 °C in S2.
TG profiles (Fig. 3b) showed that the Mg(BH4)2 + Coadd mixtures had decreased mass change of 2–3 wt% compared to pure Mg(BH4)2. The total mass change did not exceed the theoretical hydrogen capacity (Table 1), which suggests a negligible, if any, release of boranes or other heavy gases (BF3, HF, HCl etc.). Interestingly, the onset of weight loss began as low as 100 °C, which is much lower than the decomposition temperature (∼200 °C) reported previously.4–9
Reversible formation of Mg(BH4)2
The solid products of desorption and absorption were analyzed by PXD and IR. After ball-milling with additives, PXD profiles of all samples showed diffraction peaks corresponding to γ-Mg(BH4)2 and the additives (Fig. 4 and S3 (ESI†), bm curves). The PXD of S1 displayed only peaks from γ-Mg(BH4)2 (Fig. S2 (ESI†)). High background at low angles indicates the presence of an amorphous phase(s) in all samples (including in the as received and as-milled γ-Mg(BH4)2). γ-Mg(BH4)2 was previously reported to slowly become amorphous upon storage.46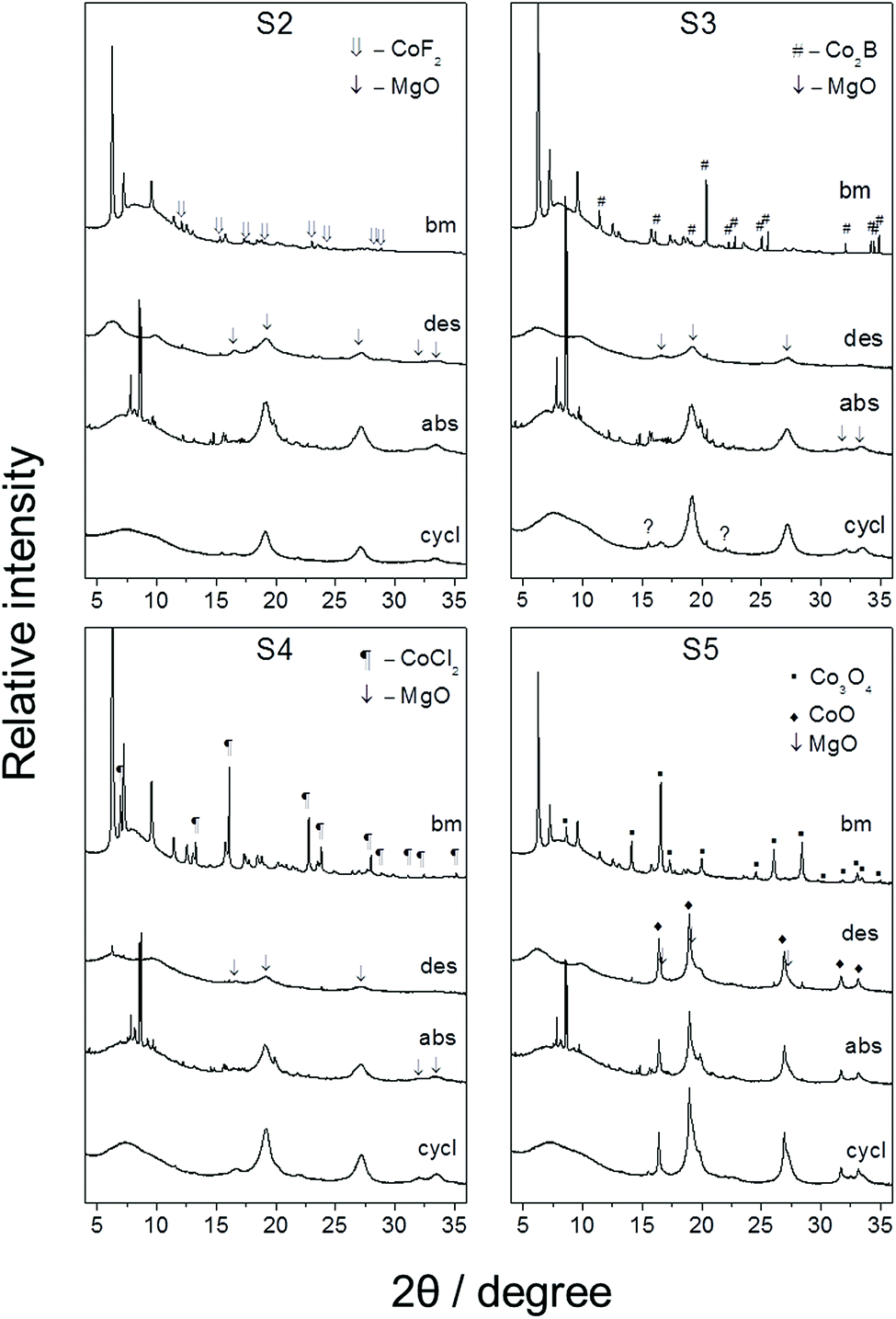 | ||
| Fig. 4 SR-PXD patterns (λ = 0.70135 Å) of the mixtures: bm – after ball-milling, des – after the 1st desorption, abs – after the 1st absorption, cycl – after the 3rd absorption. The unmarked peaks in the bm and in the abs patterns belong to γ-Mg(BH4)2, and β-Mg(BH4)2, respectively. | ||
IR spectroscopy can characterize both amorphous and crystalline phases since the spectra show presence of molecular groups. The spectra of S0–S5 after milling, 1st desorption, 1st absorption and after cycling were very similar therefore Fig. 5 illustrates only the spectra of S1, as an example. In the 2900–600 cm−1 region, Mg(BH4)2 exhibits absorption bands corresponding to the internal vibrations of (BH4)− ions. The B–H fundamental stretching modes are centered at 2270 cm−1, whereas the bending modes are located at ca. 1420, 1260 and 1130 cm−1. Absorption due to the overtones and combinations of (BH4)− bending are observed at 2660 and 2390 cm−1. The peak at 420 cm−1 can be tentatively assigned to the librations of (BH4)−. According to Raman spectroscopy measurements and DFT calculations, Mg(BH4)2 does not exhibit normal modes of vibrations in the 670–1120 cm−1 region. Therefore the peaks at 913 and 706 cm−1, together with part of the broad absorption at 1420 cm−1, may be attributed either to the vibrations of Mg(BH4)2 not observed earlier, or to impurities, for example, borates.47 These impurities must be amorphous since no corresponding crystalline phases were found in the PXD patterns.
 | ||
| Fig. 5 IR-ATR spectra of Mg(BH4)2 after ball-milling (a), 1st desorption (b), 1st H2 absorption (c), and 3rd H2 absorption (d). The peaks are marked as follows: solid line: peaks present in the initial spectrum of Mg(BH4)2, dotted line: attributed to C1, dash-dot-dot line: attributed to C2. C1 and C2 are unidentified compounds, as discussed in text. The spectra are Y-offset for better presentation. | ||
After the 1st desorption, all samples formed an amorphous phase(s), exhibiting only two broad diffraction halos at low angles in the PXD patterns, as well as nanocrystalline MgO (Fig. 4 and S3 (ESI†), des). The peaks corresponding to MgO were very broad suggesting very small particle sizes. The IR spectra of the amorphous phases still showed peaks corresponding to Mg(BH4)2 stretching and bending modes, such as the combination at 2270, 1260 and 1130 cm−1 (Fig. 5b). This indicates that Mg(BH4)2 was still present in the samples. Besides those peaks, the absorptions at ca. 1600, 900 (broad) and peaks at 770, 748, 695, and 490 cm−1 appeared. The peaks at 1420, 913 and 706 cm−1, tentatively attributed earlier to the impurities, disappeared from the IR spectra after the 1st desorption, which implies that the impurities can be the source of MgO.
After the 1st hydrogen absorption, crystalline β-Mg(BH4)2 was formed as identified by PXD (Fig. 4 and S3 (ESI†), abs). By comparing the intensities of the peaks at 8.2, 8.56, 8.66 and 9.2° 2θ in the PXD patterns of S2–S5 (Fig. S4 (ESI†)), it can be inferred that a fraction of ε-Mg(BH4)2 was also present. Since the structure for ε-Mg(BH4)2 has not been reported, its presence was inferred by comparison of the patterns to the literature.5,45 IR spectra of the samples after hydrogen absorption (Fig. 5c) confirmed the PXD results. The peaks for Mg(BH4)2, including the peak at 420 cm−1 (allegedly corresponding to (BH4)− librations) regained intensity, whereas the absorptions at 1600, 770, 748, 695, and 490 cm−1 almost vanished. As such, it appears that the latter peaks belong to the same compound, which is plausibly a reversible phase. We will subsequently refer to this phase as C1. C1 most probably have also the absorption in the B–H stretching region (2550–2000 cm−1), and around 900 cm−1. New peaks at 2540, ca. 2400, and 1023 cm−1 appeared. These peaks likely belong to another compound(s), which we will refer to as C2.
After the 3rd hydrogen absorption, no evidence of crystalline Mg(BH4)2 was identifiable in the PXD patterns (Fig. 4 and S3 (ESI†), cycl). However, the intensity of the broad amorphous halos at low angles increased. IR spectra (Fig. 5d) indicated that the vibrational features of Mg(BH4)2 were partially restored, including the peak at 420 cm−1. As such, it can be concluded that the rehydrogenated Mg(BH4)2 is amorphous. Concomitantly, the absorption peaks for C2 became stronger in intensity and the broad absorption at 1650 cm−1 appeared.
These results of reaction products analysis can be summarized as follows:
(1) After the 1st desorption, IR spectroscopy identified in the samples some amorphous Mg(BH4)2 along with the reversible amorphous product C1;
(2) After the 1st absorption, crystalline β-Mg(BH4)2 with addition of some ε-Mg(BH4)2 was formed, C1 almost completely disappeared, and another compound(s) C2 was formed;
(3) After the 3rd absorption, the peaks for C2 became more pronounced, amorphous Mg(BH4)2 and an additional phase(s) with the broad absorption at 1650 cm−1 were produced.
Many of the literature reports have focused on the decomposition pathway of Mg(BH4)2, and numerous compounds have been suggested as intermediates.4–9,14,48 These compounds include Mg(B3H8)2, MgB5H9, MgB5H8, MgB10H10, MgB12H12. So far, out of these, only Mg(BH4)2, possibly Mg(B3H8)2,49 and MgB12H12 (ref. 50) have been isolated as pure non-solvated solid compounds. This study shows that after the partial decomposition of Mg(BH4)2, at least one reversible phase was formed. This phase (C1) has BH groups with the stretching vibrations coinciding with (BH4)−, i.e. in the 2500–2000 cm−1 region and without identifiable peaks corresponding to the stretching of bridged hydrogen groups (at 1800–2200 cm−1). On the other hand, C1 showed a broad absorption at ca. 1600 cm−1, which is typically assigned to the ring modes in pure boron hydrides51,52 or M–H–B stretching in the transition metal borohydrides containing M–H–B bonds with BH4− in bidentate orientation towards metal atoms.53 After rehydrogenation, the Mg–B–H compound(s), C2, displaying the vibrational features characteristic for those of the higher boranes (B–H stretching at >2400 cm−1) were formed in addition to β-Mg(BH4)2. C2, similarly to C1, did not have clear IR absorptions which could be assigned to the bridged B–H–B groups. MgB12H12 has been reported as one of the decomposition products of Mg(BH4)2.54,55 However, the IR spectra in Fig. 5 do not support the formation of MgB12H12 at the temperatures employed in this work due to the absence of a strong peak at 2470 cm−1 (B–H stretching), and a medium-intensity peak at 720 cm−1 ((B–B skeletal breathing), Fig. S5 (ESI†)). In fact, comparison of the spectra for S1 at different stages of cycling with a range of reference compounds (Fig. S5 (ESI†)) shows clear similarities with the spectra of only pure Mg(BH4)2. Therefore the nature of the C1 and C2 remains unclear.
PXD and IR analyses seem to indicate that the additives did not have much effect on hydrogen desorption and absorption reaction products. However, they may have affected the relative ratios of the reaction products, as can be seen from the relative intensities of the peaks and amorphous haloes in the PXD patterns. S3 after cycling also contained a small amount of unidentified phase(s).
Local structure of additives
PXD analysis has given an average picture of the chemical state of the additives in the composites after ball-milling, desorption, absorption, and cycling. Crystalline additives were identified in all as-milled composites, which means that reaction had not occurred between the additives and Mg(BH4)2 during milling. One exception was the CoF3 additive in S2 (initially a mixture CoF3 and CoF2), which had been completely reduced to CoF2. After hydrogen desorption, the intensities of the PXD peaks of all additives were significantly weaker, including those of Co2B (S3) (which is not expected to react with Mg(BH4)2). After the 1st and 3rd absorption, the PXD pattern of S4 did not show peaks of CoCl2. In the patterns of S5, the peaks of Co(II,III) oxide changed to those of CoO.A more detailed characterization of the additives is provided by XAS. XANES part of the XAS spectrum characterizes oxidation and coordination states of the absorbing atom, and EXAFS part of the spectrum provides information on the distance between absorbing and neighboring atoms in the nearest coordination shells. XANES spectra and Fourier Transform (FT) of the EXAFS are shown in Fig. 6.
 | ||
| Fig. 6 Normalized X-ray absorption spectra (XANES, a–d) and Fourier transform of k3-weighted χ(k) function (EXAFS, e–h) of Co additives in the S2–S5 samples after milling (mil), 1st hydrogen desorption (des), 1st absorption (abs) and 3rd absorption (cyc) together with the commercial Co-based additives as references (ref). | ||
XANES spectra of as-milled S2 were different from those of the CoF2 and CoF3 references (Fig. 6a). Further changes were observed after cycling in H2. Most notable were the differences in the pre-edge and white-line intensities (at 7708.8 and 7724 eV, respectively). Pre-edge peaks indicate 1s → p electronic transitions in the absorber, and are generally weak or absent in octahedrally coordinated cobalt atoms. In CoF3 and CoF2, cobalt has octahedral coordination (slightly distorted in CoF2) and hence the pre-edge peak is weak. An increase in the intensity of the pre-edge peak of as-milled S2 and the further increase in the samples after cycling in hydrogen indicated that the coordination environment around cobalt was altered.
The FT-EXAFS spectra for S2 are shown in Fig. 6e. Peak maxima correspond to the single and multiple scattering from the nearest neighbors located at the distance R plus phase shift Δφ. Phase shifts were obtained by FEFF calculations of the reference compounds and are reported in Fig. S6 (ESI†). The spectra of CoF2 and CoF3 references were rather similar due to the fact that as-received CoF3 was in fact, as mentioned earlier, a mixture of CoF3 and CoF2 compounds. Because of this, we used CoF2 reference for comparison with S2. The spectrum of as-milled S2 was already somewhat different from the references. The first peak at R = 1.6 Å broadened and its maxima shifted to 2.0 Å. This peak was assigned to the scattering from the six nearest fluorine atoms located at two distances reff = 2.01 and 2.06 Å, where reff is the scattering path length obtained in FEFF calculations. Reff of the single-scattering pathways is equal to the interatomic distance. The broadening of the peak and its shift to higher R means that milling induced inhomogeneity in the first coordination shell of cobalt. The peaks in the 2.5–4.2 Å R-range in the spectrum of CoF2 (Fig. S6 (ESI†)) are composed of several contributions from different scatters with predominant contributions from cobalt atoms at reff = 3.2 and 3.7 Å and from F atoms at reff = 3.6–3.8 Å. The intensity of those peaks in the spectrum of as-milled S2 decreased, but the peak positions and relative intensities were still similar to those of the CoF2 reference. This suggests that the Co-additive in S2 preserved its local environment although milling with Mg(BH4)2 introduced some disorder.
The EXAFS spectrum of S2 after decomposition is significantly altered and shows only one distinct peak at R = 2.0 Å with a shoulder at 1.7 Å, and some very weak features at 3.0–4.4 Å. These changes were largely preserved after absorption and cycling. The observed changes mean that after desorption, absorption and cycling, part of the nearest neighbors of cobalt were at reff ≈ 2.1 Å, and some of them moved to reff = 2.4 Å, i.e. a new coordination environment formed around cobalt. The fact that the spectra of S2-des, S2-abs, and S2-cycl did not display significant features at R > 4 Å means that there is a high degree of disorder in the medium to long range. This conclusion is supported by the PXD patterns, where peaks corresponding to the crystalline additive in the samples after desorption, absorption and cycling were not observed.
XANES spectra of Co2B in the S3 after milling, hydrogen desorption and absorption (Fig. 6b) differed slightly in the edge energy E0 and post-edge region. ΔE0 of +1 eV found between the cycled and the rest of the samples, including Co2B reference can be indicative of a slight oxidation of Co. The FT-EXAFS spectrum of as-milled S3 was similar to the Co2B reference (Fig. 6f). In this metal-rich boride, scattering from four B atoms in the first coordination shell at reff = 2.1 Å is weak and contributes only slightly to the shoulder at R = 1.8 Å. The most intense peaks are due to scattering from Co atoms. The strongest peak centered at R = 2.3 Å results mostly from Co atoms at reff = 2.4–2.7 Å, (Δφ = 0.3 Å, see Fig. S6†). The R-region 3.0–5.5 Å also exhibits single and multiple scattering paths from cobalt in distant coordination shells. Cycling in hydrogen produced only slight variations in the peak intensities and peak structure of all S3 samples (Fig. 6f). At the same time, PXD indicated very little crystalline Co2B in the S3 after desorption and no crystalline additive after absorption. It can be concluded that the local environment around cobalt was preserved but the long-range order was destroyed.
XANES spectra of the S4 (Fig. 6c) followed the behavior of the S2 spectra, the only difference being that the changes in Co coordination geometry and oxidation state occurred after desorption, not milling. FT-EXAFS of the as-milled sample (Fig. 6g) was very similar to the spectrum of CoCl2 reference with a small variation in the peaks intensities. The most intense peaks at R = 2.0 and 3.1 Å were assigned to the scattering from Cl and Co at reff = 2.5 and 3.6 Å respectively (Fig. S6 (ESI†)). The spectrum of S4 changed after desorption revealing an alternate chemical structure, the EXAFS pattern having a shoulder at R ∼ 1.7 Å and peaks at R ∼ 2.1 and ∼3.2 Å. The low peak intensities at R > 3 Å conjectures a high degree of disorder around cobalt in this (or these) new species. The spectra of the sample after desorption, rehydrogenation and cycling were very similar to each other, suggesting that CoCl2 reacted into chemically stable species after the first dehydrogenation of S4.
In Co3O4 there are two Co environments. 2/3 of the Co are octahedrally coordinated Co3+, while 1/3 are tetrahedrally coordinated Co2+. The pre-edge peak in the XANES spectra of Co3O4 (Fig. 6d) originates from tetrahedrally coordinated cations. It can be compared with the very weak pre-edge feature in the spectrum of CoO, where all Co cations have octahedral coordination. XANES spectra of as-milled S5 showed some difference with respect to the Co3O4 reference (Fig. 6d). There was a small decrease in the intensity of the pre-edge peak and increase in the intensity of the white line, which means a variation in the coordination geometry. XANES spectra of S5 after desorption, absorption and cycling showed an increase in the intensity of the pre-edge peak and a negative shift in E0, which suggests a change in the coordination state and reduction of Co. Significant changes were also observed in the post-edge region, which implies the formation of a new Co environment. More details about this environment were revealed by EXAFS (Fig. 6h). FT-EXAFS spectra of as-milled S5 were very similar to the Co3O4 reference. The spectrum of Co3O4 is also a combination of the scattering environments from the Co2+ (Co1) and Co3+ (Co2) in two different crystallographic sites. The first peak at R = 1.5 Å in the spectrum of Co3O4 (and as-milled S5) is due to the scattering from oxygen atoms around Co1 and Co2 which are at a similar distance reff = 1.9 Å (Fig. S6 (ESI†)). The next two strong peaks at R = 2.4 and 3.0 Å are due to the scattering from Co atoms in the second coordination shell of the Co1 and Co2. These are the 6 atoms at reff = 2.9 Å around Co2 and 12 atoms around Co1 at reff = 3.4 Å. The peaks at R > 3 Å are composed of several single and multiple scattering pathways. EXAFS spectra of S5 after desorption, absorption, and cycling were significantly modified. In the first coordination shell, a new peak appeared at ca. 2 Å. This was not observed either in the Co3O4 or in the CoO references. The region at R > 2 Å was on the contrary very similar to that of CoO reference.
In summary, the EXAFS spectra of the reference compounds (Fig. S6†) show that in Co halides and oxides the first peak maxima are located at R < 2 Å and correspond to the scattering from the first nearest neighboring anion (O, F, Cl). The Co–anion bond distances in these compounds are in the range of reff ≈ 1.9–2.5 Å. In cobalt boride and cobalt metal, the first peak maxima are located at R > 2 Å and originate from the scattering from the nearest Co atoms at the distances reff ≈ 2.5–2.7 Å. In Co2B there is also a shoulder at lower R corresponding to the weak scattering from boron neighbors at reff ∼ 2.1 Å.
The second coordination shell in halides and oxide is formed by Co atoms with Co–Co distances in the range of 2.9–3.6 Å. This gives rise to the peaks at R ∼ 2.5–3.5 Å. In Co3O4, Co atoms are located at two different crystallographic positions. Co atoms in the second coordination shell are located at reff = 2.9 Å and 3.4 Å for Co2 and Co1, respectively. This produces two peaks in the EXAFS spectrum, unlike a single peak in CoO, where all Co atoms are equivalent.
FT-EXAFS spectra of the additives in the composites after desorption (Fig. 6, e–h blue curves) showed that the first peak maxima of the CoF3/CoF2 additive was now shifted to higher R values, although a shoulder was still present at the R < 2 Å where the signal from nearest F, O and B appears. In the spectrum of S4 (CoCl2), the new shoulder at the R < 2 Å indicated that Co had acquired new neighbors at the distance closer than the chlorine atoms in the parent CoCl2 structure. The maximum at R ∼ 2.1 Å was only slightly closer than that for Co–Co distances in Co2B and Co. In the Co3O4 reference, a completely new peak appeared at R ∼ 2.2 Å, i.e. corresponding to Co–Co distances in metal and boride. This implies that the halide and oxide additives had transformed into new compounds with Co atoms surrounded by B in the first coordination shell (weak shoulders at R < 2 Å) and Co in the second coordination shell (1st peak maxima at R > 2 Å). The EXAFS spectra of the additives after desorption are similar to those obtained upon further cycling, suggesting that the additives formed after desorption were stable. The peaks at R > 3 Å were strongly reduced in intensity in the spectra of all additives, apart from Co3O4, suggesting the formation of a disordered environment in the medium to long range. The spectra of Co3O4-containing samples seem to be composed by the spectra of CoO and another compound with neighbors at R ∼ 2.1 Å as in cobalt metal and boride. This new species or mixture of them were preserved with cycling.
Discussion
We have shown that Mg(BH4)2 can be reversibly cycled at least three times if it is not decomposed past a 4 wt% H2 threshold. The initial γ-polymorph transforms to β and eventually to the amorphous phase. Isothermal decomposition of pure γ-Mg(BH4)2 at the practical hydrogen backpressure ca. 3 bar yielded 4 wt%, however, it was possible to re-absorb only up to 2 wt% of H2. The desorbed and absorbed H2 gravimetric capacity during the subsequent cycles decreased. There are several possible reasons for incomplete rehydrogenation: formation of MgO, low absorption pressure and formation of several decomposition products, of which only some being reversible. In the evidence of the latter, after the first rehydrogenation not only crystalline β-Mg(BH4)2 was detected by IR spectroscopy, but also other amorphous phase(s). The decomposition phase(s) identified during cycling have (i) B–H stretching vibrations typical for the boron-hydrides with the H/B ratio > 1 (∼2300 cm−1); (ii) Mg–H–B bonds or B–H–B rings absorbing at ca. 1600 cm−1; and (iii) B–B bonds, possibly of boron rings, or fragments, with the ring modes at 770–748–695 and 490 and cm−1. The vibrational profiles of these phases differ from those of B12H12, B10H10 and B3H8 salts of Na, K, and Cs. The phases formed after rehydrogenation, in addition to Mg(BH4)2, have B–H stretching at ca. 2400 and 2540 cm−1. The quantity of those phases increased with cycling. It is plausible that they are formed in a competing reaction together with Mg(BH4)2. In this case, alteration of reaction conditions, for example, lower decomposition or rehydrogenation temperature may increase the final yield of Mg(BH4)2. Cobalt-based additives were partially chosen because of the inhibiting properties toward the formation of higher boranes, but they did not have an unambiguous effect in this particular case. In fact, the IR spectra of the samples with and without these additives after milling, desorption, absorption, and cycling were very similar.The drawback of decreasing the reaction temperature is a decrease in the reaction kinetics. The decomposition kinetics of as-received γ-Mg(BH4)2 at the conditions chosen in this work were already impractically slow, while rehydrogenation kinetics, on the contrary, were significantly faster. Kinetic modeling of the desorption and absorption curves of pure Mg(BH4)2 suggested that the decomposition reaction is controlled by several mechanisms, including chemical reactions at the interphases and diffusion processes, whereas re-hydrogenation is also limited by nucleation in addition to the diffusion-controlled growth of the new (rehydrogenated) phase. The major effect on the hydrogen desorption and absorption first cycle kinetics was introduced by mere milling. Mechanical milling seemingly eliminated the diffusion problem during the first cycle. The treatment might have decreased the particles size and the hydrogen diffusion pathways, enhancing the overall reaction kinetics. Those effects diminished with cycling in H2 at high temperature, probably due to sintering of the smaller particles.
Phase-boundary reactions, however, remained the main rate-limiting step for the samples with and without milling, especially for the decomposition reaction, as kinetic modeling suggested. Thus the catalyst which affects breaking and formation of the B–H bonds in Mg(BH4)2 can further improve the reaction kinetics. Co-additives used in this study were shown to slightly enhance the desorption kinetics during the first decomposition of Mg(BH4)2. The nature of the additives seemingly did not make a difference during the first desorption. EXAFS analysis revealed that after the first desorption, cobalt oxide and halides formed new compounds whereas Co2B preserved its local cobalt environment. It is then possible that upon reaction, all additives formed species similar to cobalt boride. Those boride phases affected the dehydrogenation kinetics of Mg(BH4)2 in the same way. Similar results were observed upon cycling of Mg(BH4)2 containing Ni-based additives, which all formed nickel boride species.16 EXAFS spectra have indicated that the Co3O4 additive may have transformed into the mixture of oxide and boride after the first desorption. CoO was in fact the only crystalline additive-related phase observed by PXD in the Mg(BH4)2 + Coadd samples upon cycling. Coexistence of the oxide and boride could explain why Co3O4 had affected both desorption and absorption kinetics.
The kinetics modeling has shown that overall the desorption and absorption reactions in Mg(BH4)2 in the chosen conditions have complex mechanisms possibly with several competing reactions, as also suggested by the analysis of the reaction products.
Conclusions
Desorption and absorption of hydrogen in Mg(BH4)2 for 3 cycles was demonstrated. Isothermal decomposition of pure γ-Mg(BH4)2 and γ-Mg(BH4)2 + Coadd at 285 °C and about 3 bar H2 pressure yielded up to 4 wt% of H2, and the re-absorption at this temperature and 120 bar H2 resulted in the uptake of ca. 2 wt% of H2 in the first cycle. After re-absorption crystalline β-Mg(BH4)2 was formed. With further cycling the reversible hydrogen capacity of the samples decreased with an increase in the amount of nano-crystalline MgO and unidentified boron–hydrogen compounds. Amorphous Mg(BH4)2 was formed during the 3rd H2 absorption.Ball-milling of γ-Mg(BH4)2 significantly increased desorption and absorption reaction rates in the first cycle. Cobalt additives further enhanced decomposition kinetics and hampered (but Co3O4) rehydrogenation kinetics in the first cycle thus stabilizing the decomposition products. The additives however were not found to impact significantly the decomposition temperature of Mg(BH4)2 during the first desorption. EXAFS has proven to be a very helpful technique in the characterization of the TM additives, especially with respect to the formation of the amorphous species, which can not be detected by PXD. XAS data indicated that all cobalt-based additives used in this study were chemically modified into boride-like species, and the Co2B additive became amorphous. Those new species did not show a significant impact on the kinetics of hydrogen desorption and absorption in the second and third cycle.
Acknowledgements
The work was financed by the European Fuel Cells and Hydrogen Joint Undertaking (http://www.fch-ju.eu) under collaborative project “BOR4STORE” (Grant agreement no. 303428). Partial financial support from SYNKNØYT program in the Research Council of Norway is greatly acknowledged. XAS measurements were conducted at the beam line I811, MAX-lab synchrotron radiation source, Lund University, Sweden. Funding for the beamline I811 project was kindly provided by The Swedish Research Council and Knut och Alice Wallenbergs Foundation. Part of this work was carried out the Swiss-Norwegian beam-line of the European Synchrotron Radiation facility (ESRF-SNBL, BM01A). The skillful assistance of the beam line personnel at SNBL and I811 at MAX-lab is gratefully acknowledged.References
- V. Ozolins, E. H. Majzoub and C. Wolverton, Phys. Rev. Lett., 2008, 100, 135501 CrossRef CAS.
- V. Ozolins, E. H. Majzoub and C. Wolverton, J. Am. Chem. Soc., 2009, 131, 230–237 CrossRef CAS PubMed.
- M. J. van Setten, G. A. de Wijs, M. Fichtner and G. Brocks, Chem. Mater., 2008, 20, 4952–4956 CrossRef CAS.
- N. Hanada, K. Chlopek, C. Frommen, W. Lohstroh and M. Fichtner, J. Mater. Chem., 2008, 18, 2611–2614 RSC.
- M. Paskevicius, M. P. Pitt, C. J. Webb, D. A. Sheppard, U. Filso, E. M. Gray and C. E. Buckley, J. Phys. Chem. C, 2012, 116, 15231–15240 CAS.
- M. D. Riktor, M. H. Sorby, K. Chlopek, M. Fichtner, F. Buchter, A. Züttel and B. C. Hauback, J. Mater. Chem., 2007, 17, 4939–4942 RSC.
- G. L. Soloveichik, Y. Gao, J. Rijssenbeek, M. Andrus, S. Kniajanski, R. C. Bowman Jr, S.-J. Hwan and J.-C. Zhao, Int. J. Hydrogen Energy, 2009, 34, 916–928 CrossRef CAS.
- Y. Yan, H.-W. Li, H. Maekawa, M. Aoki, T. Noritake, M. Matsumoto, K. Miwa, S. Towata and S. Orimo, Mater. Trans., 2011, 52, 1443–1446 CrossRef CAS.
- J. Yang, X. Zhang, J. Zheng, P. Song and X. Li, Scr. Mater., 2011, 64, 225–228 CrossRef CAS.
- M. J. van Setten, W. Lohstroh and M. Fichtner, J. Mater. Chem., 2009, 19, 7081–7087 RSC.
- R. J. Newhouse, V. Stavila, S.-J. Hwang, L. E. Klebanoff and J. Z. Zhang, J. Phys. Chem. C, 2010, 114, 5224–5232 CAS.
- E. Rönnebro, Curr. Opin. Solid State Mater. Sci., 2011, 15, 44–51 CrossRef.
- C. Pistidda, S. Garroni, F. Dolci, E. G. Bardaji, A. Khandelwal, P. Nolis, M. Dornheim, R. Gosalawit, T. Jensen, Y. Cerenius, S. Surinach, M. Dolors Baro, W. Lohstroh and M. Fichtner, J. Alloys Compd., 2010, 508, 212–215 CrossRef CAS.
- M. Chong, A. Karkamkar, T. Autrey, S. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330–1332 RSC.
- H. W. Li, K. Kikuchi, Y. Nakamori, N. Ohba, K. Miwa, S. Towata and S. Orimo, Acta Mater., 2008, 56, 1342–1347 CrossRef CAS.
- I. Saldan, S. Hino, T. D. Humphries, O. Zavorotynska, M. Chong, C. M. Jensen, S. Deledda and B. C. Hauback, J. Phys. Chem. C, 2014, 118, 23376–23384 CAS.
- H. W. Li, K. Kikuchi, Y. Nakamori, K. Miwa, S. Towata and S. Orimo, Scr. Mater., 2007, 57, 679–682 CrossRef CAS.
- E. G. Bardaji, Z. Zhao-Karger, N. Boucharat, A. Nale, M. J. van Setten, W. Lohstroh, E. Roehm, M. Catti and M. Fichtner, J. Phys. Chem. C, 2011, 115, 6095–6101 CAS.
- J. Yang, H. Fu, P. Song, J. Zheng and X. Li, Int. J. Hydrogen Energy, 2012, 37, 6776–6783 CrossRef CAS.
- S. Sartori, K. D. Knudsen, F. S. Hage, R. H. Heyn, E. G. Bardaji, Z. Zhao-Karger, M. Fichtner and B. C. Hauback, Nanotechnology, 2012, 23, 1–7 CrossRef PubMed.
- P. E. de Jongh, M. D. Allendorf, J. J. Vajo and C. Zlotea, MRS Bull., 2013, 38, 488–494 CrossRef CAS.
- H.-W. Li, Y. Yan, S. Orimo, A. Züttel and C. M. Jensen, Energy, 2011, 4, 185–214 CAS.
- E. G. Bardaji, N. Hanada, O. Zabara and M. Fichtner, Int. J. Hydrogen Energy, 2011, 36, 12313–12318 CrossRef CAS.
- A. Al-Kukhun, H. T. Hwang and A. Varma, Int. J. Hydrogen Energy, 2012, 37, 17671–17677 CrossRef CAS.
- A. Amieiro-Fonseca, S. R. Ellis, C. J. Nuttall, B. E. Hayden, S. Guerin, G. Purdy, J. P. Soulie, S. K. Callear, S. D. Culligan, W. I. F. David, P. P. Edwards, M. O. Jones, S. R. Johnson and A. H. Pohl, Faraday Discuss., 2011, 151, 369–384 RSC.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253–254, 1–9 CrossRef CAS.
- J. Wang, H. W. Li and P. Chen, MRS Bull., 2013, 38, 480–487 CrossRef CAS.
- T. D. Humphries, G. N. Kalantzopoulos, I. Llamas-Jansa, J. E. Olsen and B. C. Hauback, J. Phys. Chem. C, 2013, 117, 6060–6065 CAS.
- T. D. Humphries, J. W. Makepeace, S. Hino, W. I. F. David and B. C. Hauback, J. Mater. Chem. A, 2014, 2, 16594–16600 CAS.
- J. Chen, Y. Zhang, Z. Xiong, G. Wu, H. Chu, T. He and P. Chen, Int. J. Hydrogen Energy, 2012, 37, 12425–12431 CrossRef CAS.
- Z. G. Zhang, H. Wang, J. W. Liu and M. Zhu, Thermochim. Acta, 2013, 560, 82–88 CrossRef CAS.
- D. Matsumura, T. Ohyama, Y. Okajima, Y. Nishihata, H.-W. Li and S. Orimo, Mater. Trans., 2011, 52, 635–640 CrossRef CAS.
- J. H. Sharp, G. W. Brindely and B. N. Narahari Achar, J. Am. Ceram. Soc., 1966, 49, 379–382 CrossRef CAS.
- K. J. Gross, R. K. Carrington, S. Barcelo, A. Karkamkar, J. Purewal, S. Ma, H.-C. Zhou, P. Dantzer, K. Ott, T. Burrell, T. Semeslberger, Y. Pivak, B. Dam and D. Chandra, http://energy.gov/eere/fuelcells/downloads/recommended-best-practices-characterization-storage-properties-hydrogen-0, US Department of Energy, 2012, p. 579.
- S. Carlson, M. Clausen, L. Gridneva, B. Sommarin and C. Svensson, J. Synchrotron Radiat., 2006, 13, 359–364 CrossRef CAS PubMed.
- T. M. Grehk and P. O. Nilsson, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468(1), 635–638 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- A. P. Hammersley, ESRF Internal Report, 1997, ESRF97HA02T.
- L. Lutterotti, D. Chateigner, S. Ferrari and J. Ricote, Thin Solid Films, 2004, 450, 34–41 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- B. V. Erofe'ev, Cont. Rend. Acad. Sci. URSS, 1946, 52, 511–514 Search PubMed.
- J.-H. Her, P. W. Stephens, Y. Gao, G. L. Soloveichik, J. Rijssenbeek, M. Andrus and J.-C. Zhao, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 561–568 CAS.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed., 2011, 50, 11162–11166 CrossRef CAS PubMed.
- W. I. F. David, S. K. Callear, M. O. Jones, P. C. Aeberhard, S. D. Culligan, A. H. Pohl, S. R. Johnson, K. R. Ryan, J. E. Parker, P. P. Edwards, C. J. Nuttall and A. Amieiro-Fonseca, Phys. Chem. Chem. Phys., 2012, 14, 11800–11807 RSC.
- V. Ban, A. V. Soloninin, A. V. Skripov, J. Hadermann, A. Abakumov and Y. Filinchuk, J. Phys. Chem. C, 2014, 118, 23402–23408 CAS.
- J. Li, S. Xia and S. Gao, Spectrochim. Acta, 1995, 51A, 519–532 Search PubMed.
- Y. Yan, H.-W. Li, Y. Nakamori, N. Ohba, K. Miwa, S. Towata and S. Orimo, Mater. Trans., 2008, 49, 2751–2752 CrossRef CAS.
- D. Y. Kim and G. S. Girolami, Inorg. Chem., 2010, 49, 4942–4948 CrossRef CAS PubMed.
- A. Remhof, Y. G. Yan, D. Rentsch, A. Borgschulte, C. M. Jensen and A. Züttel, J. Mater. Chem. A, 2014, 2, 7244–7249 CAS.
- H. J. Hrostowski and G. C. Pimentel, J. Am. Chem. Soc., 1954, 76, 998–1003 CrossRef CAS.
- J. L. Duncan, D. C. McKean, I. Torto and G. D. Nivellini, J. Mol. Spectrosc., 1981, 85, 16–39 CrossRef CAS.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- H. W. Li, K. Miwa, N. Ohba, T. Fujita, T. Sato, Y. Yan, S. Towata, M. W. Chen and S. Orimo, Nanotechnology, 2009, 20, 1–7 Search PubMed.
- Y. S. Zhang, E. Majzoub, V. Ozolins and C. Wolverton, J. Phys. Chem. C, 2012, 116, 10522–10528 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta00511f |
| ‡ Current affiliation: Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan. |
| § If the degree of materials purity is not mentioned, it was not provided by the supplier. |
| This journal is © The Royal Society of Chemistry 2015 |