
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical and electrocatalytic water oxidation activity of cobalt carbodiimide†
Debora
Ressnig
*a,
Menny
Shalom
a,
Jörg
Patscheider
c,
René
Moré
b,
Fabio
Evangelisti
b,
Markus
Antonietti
a and
Greta R.
Patzke
*b
aMax Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, D-14476 Potsdam, Germany. E-mail: debora.ressnig@mpikg.mpg.de
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: greta.patzke@aci.uzh.ch
cLaboratory for Nanoscale Materials Science, EMPA Dübendorf, CH-8600 Dübendorf, Switzerland
First published on 26th January 2015
Abstract
Cobalt carbodiimide is introduced as a heterogeneous non-oxidic water oxidation catalyst prototype with dual photochemical and electrocatalytic activity in neutral and basic media. CoNCN exhibits higher initial turnover frequencies of (TOF/SBET: 2.1 × 10−1) for visible-light-driven oxygen evolution than cobalt oxide catalysts (TOF/SBET: 3.5 × 10−3) and a 18% higher oxygen yield (Ru-dye sensitized standard setup). Furthermore, CoNCN maintains stable current densities in electrolysis over 20 h, and structural tuning through cationic substitution revealed that mixed (Co, Ni)NCN catalysts with low Ni contents display higher current densities than pristine CoNCN. A wide range of bulk (XAFS/EXAFS, XRD, FTIR) and surface (XPS, EELS, HRTEM) analytical methods together with catalytic parameter variations and reference experiments were performed to confirm the stability of CoNCN under standard operational conditions. The carbodiimide matrix thus offers a straightforward structural alternative to oxide systems and a clear-cut starting point for optimization strategies and for mechanistic studies on the possible role of active carbon or nitrogen sites. This paves the way to metal carbodiimides as a novel catalyst design platform for heterogeneous energy conversion systems.
Introduction
Artificial photosynthesis as a clean, bio-inspired and sustainable route to solar fuels depends on the development of stable and efficient water oxidation catalysts (WOCs). Economic heterogeneous catalysts are essential for the direct conversion of solar energy via water splitting.1–7 Even after decades of research into the complex four-electron-transfer processes of water oxidation, WOC development remains a major, general challenge.8–12 Manifold mechanistic studies have been performed on molecular WOCs with bio-inspired reaction pathways along the lines of nature's OEC of photosystem II.13,14 In comparison, understanding the interfaces, active species and mechanisms associated with most heterogeneous colloidal water oxidation catalysts remains far more demanding,15 because pioneering surface spectroscopic techniques16–18 and operando monitoring approaches19–22 are required. However, detailed mechanistic insight as well as structure–activity relationships for a wide spectrum of materials are indispensable for informed WOC construction. This calls for new structurally clear-cut model systems which facilitate the investigation of catalytic stability and active species.We therefore introduce cobalt carbodiimide (CoNCN) as a new prototype for the investigation of three key WOC design issues. First, we report on the dual photochemical and electrocatalytic activity of CoNCN. Second, we explore the challenging question of the “true catalyst” for cobalt centers in an all-nitrogen/carbon environment. Third, we present clear-cut transition metal tuning options for the robust carbodiimide catalyst matrix.
Traditionally, artificial photosynthesis research has placed special emphasis on oxide-based materials, such as spinel- and perovskite-type WOCs,23 oxynitrides,16,17 powerful electrodeposited catalysts20 and PSII-inspired molecular WOCs.24 Whereas molecular WOCs provide valuable insight to into reaction mechanisms, they are also likely to undergo transformations under operational conditions.25 Heterogeneous WOCs are more robust, but they display a complex interdependence of composition, structure, morphology/surface area, oxidation states and preparative history.2,26 The stability of each WOC type and its possible in situ transformation gives rise to substantial discussions about the true active species. As a consequence, key performance parameters are quite difficult to assign,27 which also renders precise extrapolation of photo- on electrocatalytic activity and vice versa rather difficult.28
Efficient and economic cobalt-containing catalysts perfectly illustrate these complex challenges, given that they currently attract increasing research interest among the multitude of WOC types.29–31 Cluster-based Co–WOCs have launched an ongoing debate about in situ transformations into active oxide species.32,33 On the other hand, in-depth post-catalytic studies on heterogeneous cobalt-containing electrocatalysts, e.g. Co(PO3)2 (ref. 34) and Co-perovskites,23 outlined the productive aspects of restructuring effects. Co3O4 materials are leading heterogeneous catalysts due to their flexible application in either dye sensitized or electrochemical systems.35,36 Only recently first light has been shed on their intermediates under operational water oxidation conditions.37 CoOx materials are furthermore applied as exceptional electrocatalysts or as co-catalysts in photocatalytic conversions with high efficiency in neutral media.38 They are typically electrodeposited from various buffer solutions (e.g. phosphate buffer CoPi) onto electrodes or semiconductor substrates. Latest results underscore the importance of technical processing factors for their performance, such as electrode microstructure and layer thickness, along with innovative approaches for the preparation of mixed amorphous catalysts.39,40 Nevertheless, the mechanisms and local structures of amorphous cobalt oxide-based catalysts keep giving rise to multidisciplinary studies depicting the complexity associated with structure–activity relationships of these materials.20,30,41–45 Studies on the oxidative decomposition of labile molecular Co–WOCs to CoOx species highlight the crucial role of intercalated carbonaceous materials in tuning the catalytic activity.29
This renders alternative WOC types with less variable and demanding structural motifs highly desirable to expand the spectrum of tuning and mechanistic studies. Over the past years, non-oxide WOCs have attracted increasing research attention.46,47 Amongst them, nitrogen and carbon based materials emerge as low-cost and efficient oxide- and metal-free photo- and electrocatalysts with a wide application spectrum including OER, HER, ORR and CO2 reduction processes.48–55 Their mechanistic functionality models involve nitrogen moieties, defects and, most interestingly, positively polarized carbons as active sites. Metal doping or deposition of co-catalysts emerge as the most recent strategies to modify the performance of such materials through synergistic effects.56–59 Moreover, silver cyanamide has shown promising activity as semiconductor photoanode,60 albeit with the pending issue of typically limited photostability of Ag-based catalysts. Most recently, Prussian Blue-type electrocatalysts have been reported as an exceptionally stable alternative to cobalt oxide catalysts.61,62 This combination of non-oxide matrices with catalytically active cobalt centers still leaves plenty of room for discoveries.63–65 and inspired us to further tap into the widely unexplored potential of early transition metal WOCs with nitrogen-based matrices.
We selected CoNCN as an innovative target material with {CoN6} moieties embedded into a NiAs-related matrix. These moieties not only resemble structural units in amorphous CoOx materials but also complement most hitherto known oxide catalyst structure types in highly crystalline form (Fig. 1). In the following, we compare the photo- and electrocatalytic performance of CoNCN. Furthermore, we present coherent trends for the operational stability of the new dual WOC. Finally, we demonstrate that CoNCN electrocatalysts display high optimization potential through Ni doping.
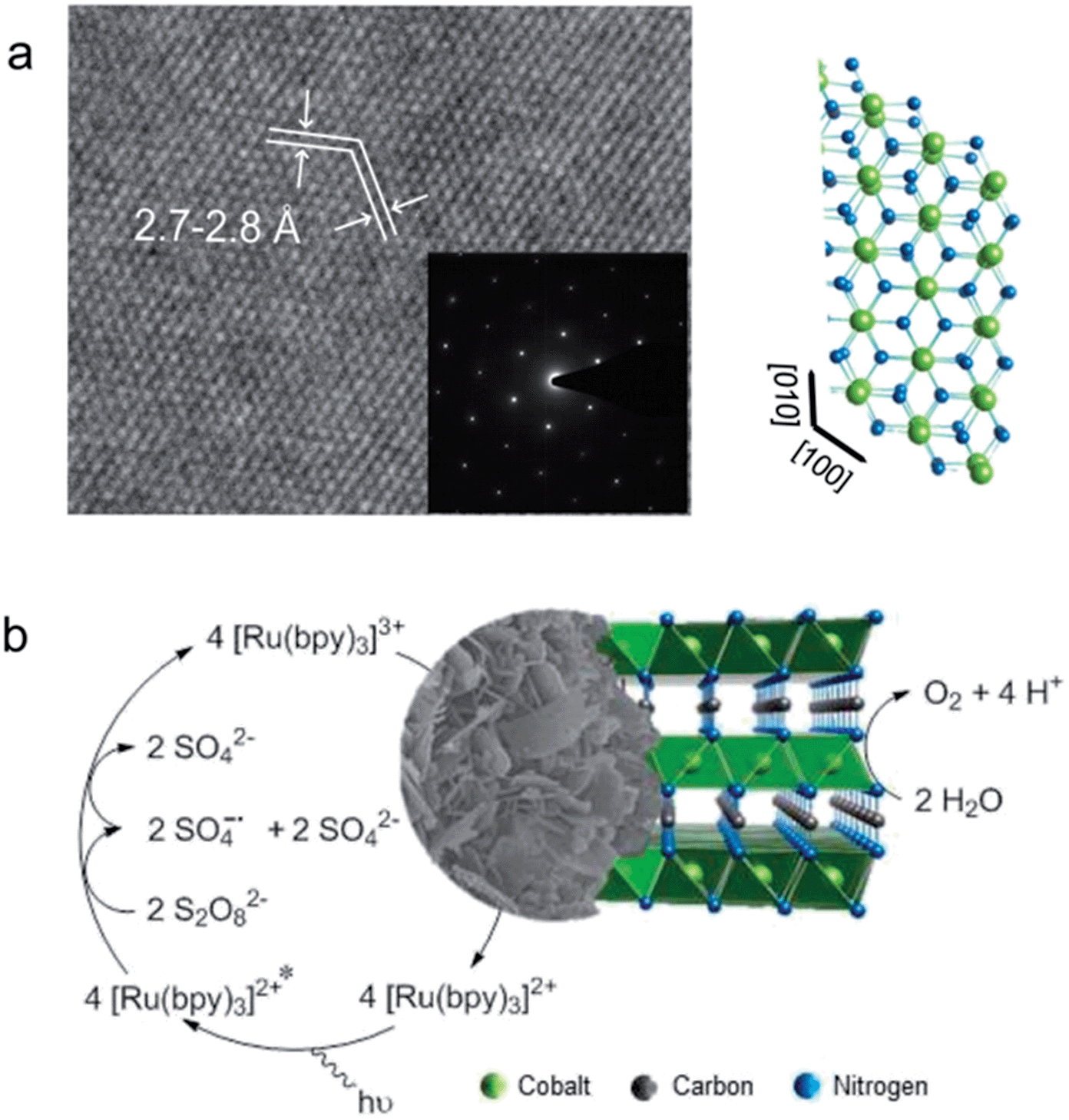 | ||
| Fig. 1 (a) HRTEM and SAED pattern of a CoNCN platelet along with the structural motif. (b) Photocatalytic water oxidation cycle with cobalt carbodiimide. | ||
Results and discussion
Synthetic route and characterization
CoNCN was synthesized via a modified hydrogencyanamide route through dissolution of CoCl2 in molten cyanamide at 75 °C, followed by addition of ammonium carbonate and decomposition/recrystallization in a LiCl/KCl salt melt at 400 °C.66 The advantages of this synthetic protocol include fast precipitation of the precursor, which is favorable for the formation of small particles, and facile scale-up to gram quantities (cf. ESI† for experimental details). Characterization of CoNCN with a wide range of analytical techniques ((HR)TEM, SEM/EDX, PXRD, UV/vis & FT-IR spectroscopy, BET, elemental analyses) verifies the formation of phase pure CoNCN platelets with exposed (002) surfaces (a = 3.02(9) Å, c = 9.37(9) Å, cf.Fig. 1 and S1–S4, Table S1†).Photochemical water oxidation with CoNCN
Standard conditions were defined after optimization runs in various buffer media as follows: 1.5 mM CoNCN in phosphate or carbonate buffer with 1 mM [Ru(bpy)3]2+ and 5 mM Na2S2O8 (Fig. 2 and Table S2†). Reference experiments in the absence of light, catalyst, PS or electron acceptor (Tables S2 and S3†) demonstrate that all components of the catalytic scheme are indispensable for oxygen evolution. No significant induction time was observed for oxygen evolution with the CoNCN–WOC under optimized conditions, whereas a slight delay was observed for catalyst concentrations of 2.5 mM and above, which is likely due to concentration-related shielding, charge accumulation and other secondary effects. These results strongly suggest that CoNCN is the active WOC under optimized working conditions.
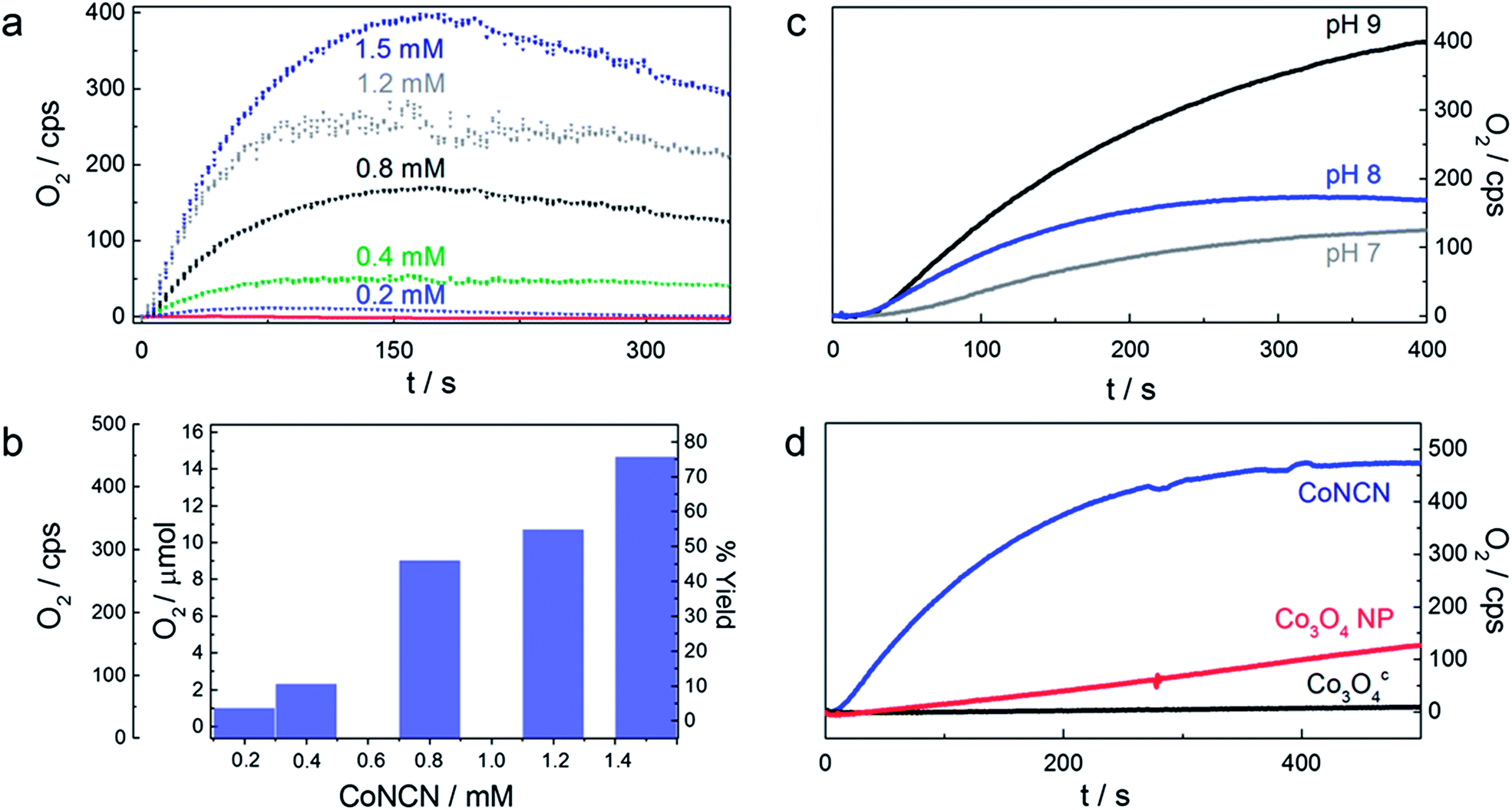 | ||
| Fig. 2 (a) Clark-electrode kinetics of visible-light-driven O2 evolution for different concentrations of CoNCN, (b) corresponding water oxidation activity under standard photocatalytic conditions (470 nm LED; 1 mM [Ru(bpy)3]Cl2, 5 mM Na2S2O8, pH 9 (carbonate buffer)). (c) pH dependent visible-light-driven oxygen evolution activity of CoNCN in phosphate buffer at pH 7 (grey), pH 8 (blue) and pH 9 (black). (d) Oxygen evolution activity of CoNCN (blue) vs. Co3O4 nanoparticles (red) and commercially purchased Co3O4c (black) in carbonate buffered solution at pH 9 (cf.Table 1). | ||
The WOC performance of CoNCN increases with the pH value, and a maximum oxygen yield of 75% (TOF/SBET: 2.1 × 10−1) is obtained in a carbonate buffer at pH 9 (Fig. 2). Interestingly, this trend overcompensates the opposite effect of PS decomposition with increasing pH value. Mechanistic reasons for this behavior might include kinetic effects via a rate-determining step depending on [HO−] or enhanced PS adsorption through electrostatic surface phenomena. Buffer-dependent WOC performance was furthermore found to increase in the order carbonate ≈ phosphate ≫ borate, despite the higher stability of [Ru(bpy)3]2+ in borate compared to phosphate media.68,69
CoNCN exhibits considerably higher normalized activity (TOF/SBET: 2.1 × 10−1) under standard conditions than both nanoscale (TOF/SBET: 3.5 × 10−3) and microscale Co3O4 (TOF/SBET: 4.4 × 10−4; cf.Table 1 and Fig. 2). Most importantly, the lower surface area of CoNCN (14 m2 g−1) compared to the Co3O4 references (nanoparticles: 71 m2 g−1, commercial standard: 56 m2 g−1) suggests that the influence of the structural motif in combination with interface polarity effects outweighs any surface area related effects. This superior performance of CoNCN over Co3O4 as a benchmark oxide was further backed with stability tests as detailed in the following.
| Catalyst | S BET [m2 g−1] | O2 yieldb [%] | TOF SBET−1 [g s−1 m−2] |
|---|---|---|---|
a Standard conditions: 1.5 mM WOC, 1 mM [Ru(bpy)3]2+, 5 mM Na2S2O8, 470 nm, carbonate buffer (pH 9).
b O2 amount relative to initial S2O82− concentration (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
c Commercially purchased material. :1).
c Commercially purchased material.
|
|||
| CoNCN | 14 | 76 | 2.1 × 10−1 |
| Co3O4 NPs | 71 | 56 | 3.5 × 10−3 |
| Co3O4c | 56 | 3 | 4.4 × 10−4 |
CoNCN as a model system for catalytic stability studies
PXRD patterns of recovered CoNCN WOCs do not display notable differences from the pristine material nor any indications for the presence of crystalline cobalt oxide (Fig. S12†). Moreover, neither FT-IR, EDX nor EELS spectra indicated cobalt oxide formation during catalysis as evident from the absence of structural O/OH signals (Fig. S13–S15†). Next, we examined post-catalytic CoNCN with complementary X-ray techniques including HRTEM and XAFS/XANES in combination with XPS for the potential formation of amorphous species. Both pristine and post-catalytic CoNCN display identical lattice fringes in HRTEM measurements that indicate the retention of a high degree of crystallinity in line with the PXRD results (Fig. S16†). The corresponding SAED patterns exclusively display clear diffraction spots from crystalline material without diffuse reflectance (Fig. S17†). All in all, no indications for amorphization, extensive atomic displacements or growth of another catalytic species on the surface of CoNCN during photo driven water oxidation were found.
Comparison of the XANES region for pristine CoNCN and for a sample aged in carbonate buffer shows no significant differences in the respective electronic and local geometric structures (see Fig. S18†), and only minor changes are visible for post-catalytic CoNCN (Fig. 3). The edge position for the post-catalytic sample is shifted by 1.1 eV to higher energies relative to the pristine sample, which may be due to the formation of minor Co3+ amounts during the photocatalytic process. The cobalt valence states of the pristine and post-catalytic samples were determined as 1.95 ± 0.02 and 2.15 ± 0.08, respectively, using a calibration curve based on cobalt reference samples. The EXAFS regions for pristine and post-catalytic CoNCN were fitted against room temperature crystallographic data.7 EXAFS data fits for both samples are of good quality with r-factors of 1.7% and 1.9%, respectively (Fig. 3 and Table S5†). The slight decrease of the interatomic distances for the post-catalytic sample is within the error ranges (Table S5†) associated with an increase of the mean squared displacements with respect to the pristine sample. The interatomic distances for both pristine and post-catalytic samples could be fitted against the same structural model with a rather small difference of the interatomic distances (<0.1 Å), i.e. they are practically congruent with literature values for CoNCN.66 Furthermore, comparison of the edge positions for pristine and post-catalytic CoNCN reveal a rather negligible extent of oxidation under photocatalytic conditions.
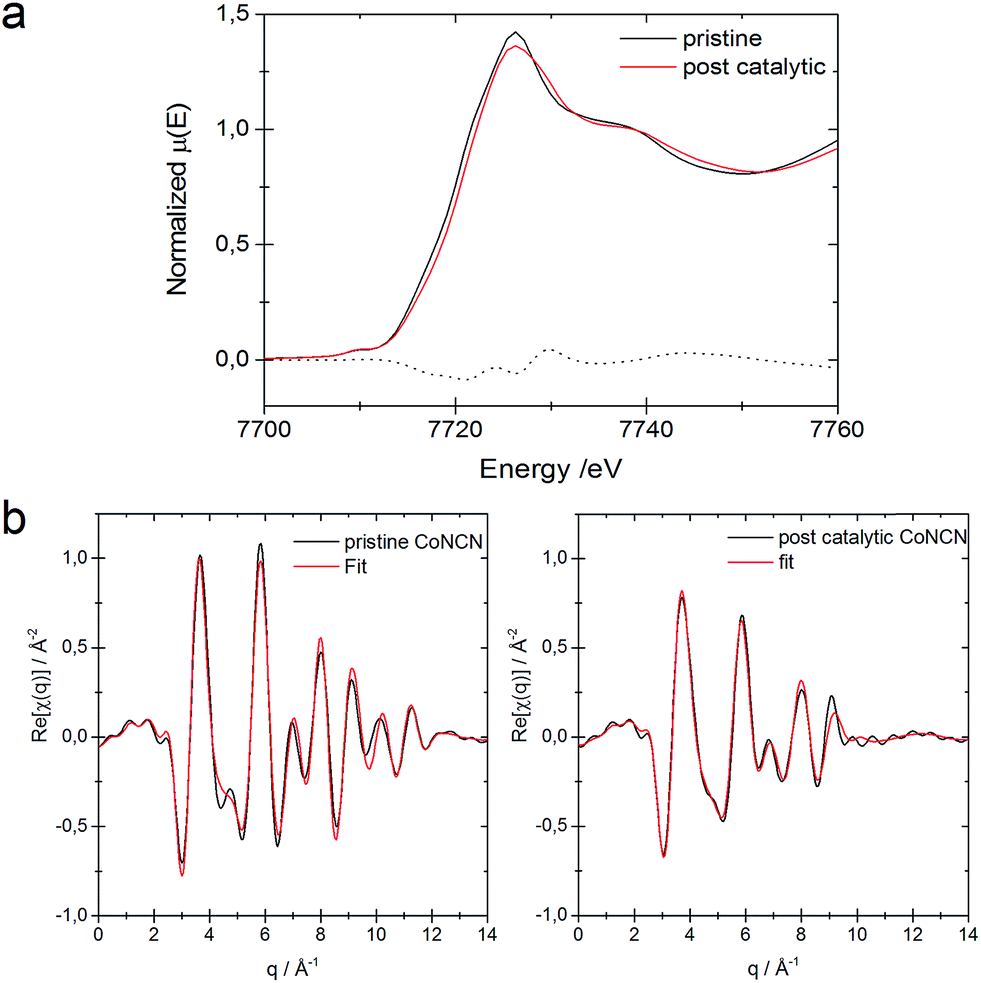 | ||
| Fig. 3 (a) XANES region for pristine and post-catalytic CoNCN and their difference spectrum. (b) EXAFS fit for pristine catalytic CoNCN vs. crystallographic data.66 | ||
In summary, XAFS analyses in their entirety show that CoNCN did not undergo any significant structural changes during photocatalysis.
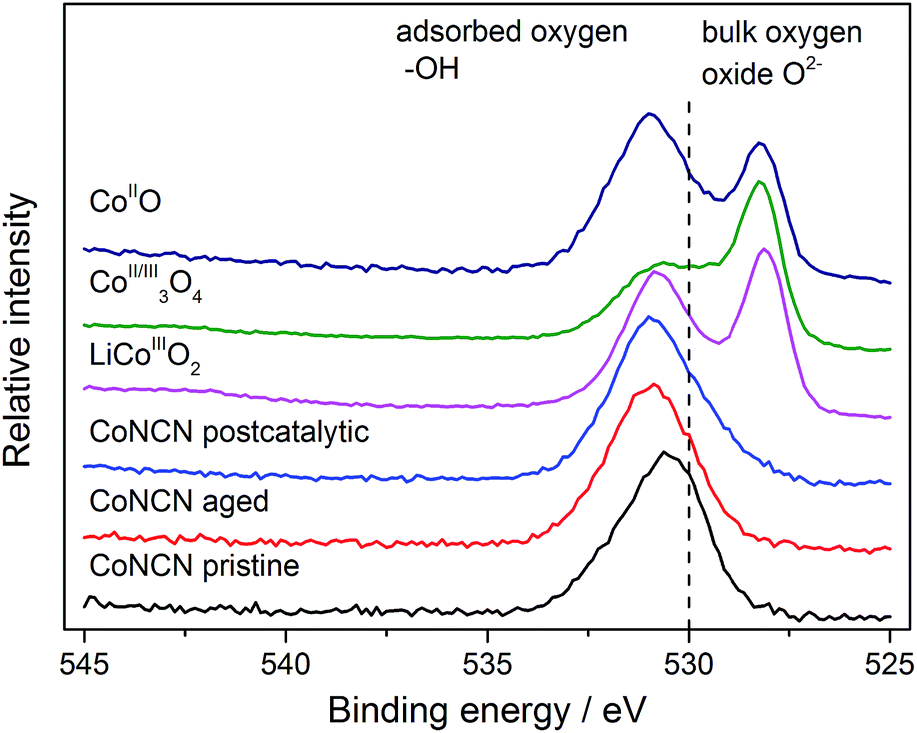 | ||
| Fig. 4 XPS analysis of CoNCN O1s XPS peaks (pristine, aged in carbonate buffer and post-photocatalytic) vs. cobalt oxide references. | ||
Whereas the present set on CoNCN strongly suggests stability under operational photocatalytic conditions, the complete analysis of catalytic species and mechanisms remains a complex multidisciplinary problem that requires follow-up studies in their own right with highly specialized in situ analytical equipment.15,21,73
Electrochemical water oxidation with CoNCN
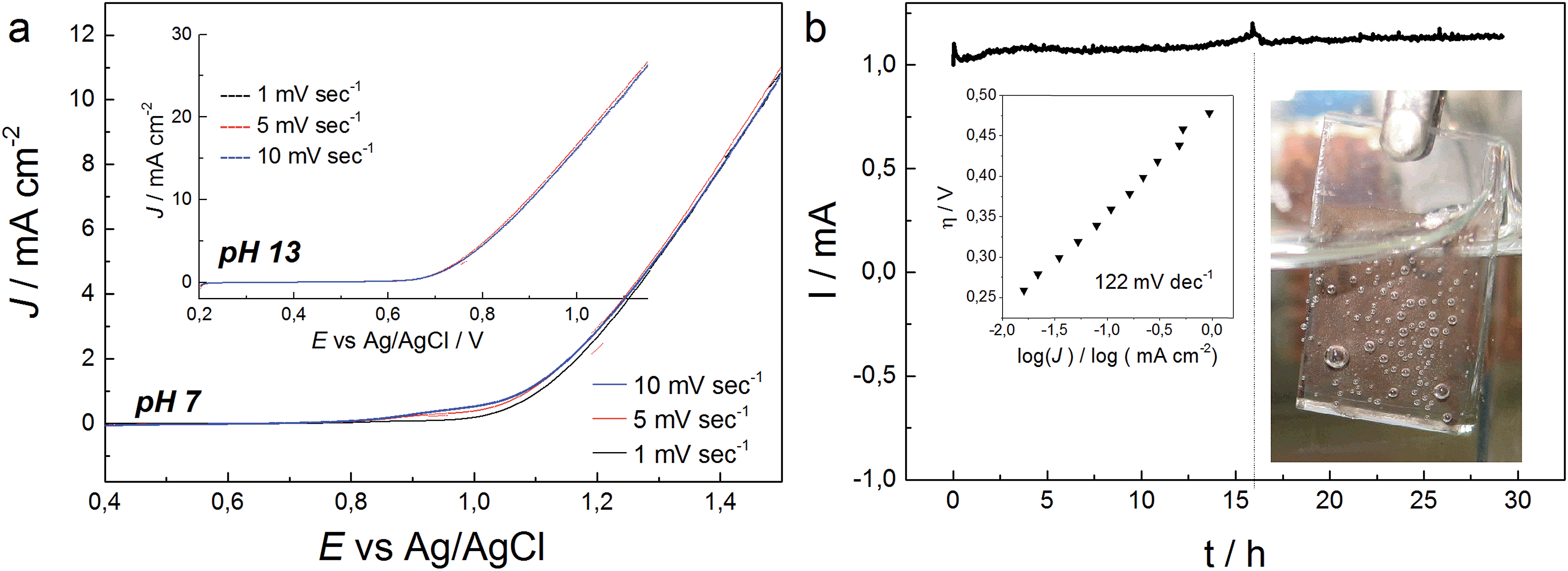 | ||
| Fig. 5 (a) LSV curves of CoNCN, recorded at different scan rates in 0.1 M phosphate buffer at a rotation speed of 1500 rpm (E0 (pH 7) = 0.62 V and E0 (pH 13) = 0.27 V vs Ag/AgCl). (b) Representative chronoamperometry measurement of CoNCN on a FTO electrode at 1.2 V vs. Ag/AgCl in 0.1 M phosphate buffer (E0 (pH 7) = 0.62 V vs. Ag/Ag/Cl). The dotted line indicates mechanical removal of bubbles. The inset shows the Tafel plot with a glassy carbon electrode under the same conditions. | ||
Catalytic current densities of 55 μA cm−2 and 1 mA cm−2 were measured 320 mV and 480 mV above the thermodynamic potential of the OER (E0 (pH 7) = 0.62 V vs. Ag/AgCl) in chronoamperometry measurements (Fig. S21†). The catalytic onset potential is therefore within the reported range for cobalt oxide-based electrocatalysts, such as CoPi (η = 280 mV) or Co(PO3)2 (η = 310 mV) and Co3O4 (η = 434 mV), respectively.34,72,74 However, the current density increase appears delayed as the Tafel slope accounts for around 122 mV dec−1 (Fig. 5).30
Stability of CoNCN under electrocatalytic conditions was tested with XANES and EXAFS measurements on three post-catalytic FTO electrodes after 20 h of electrolysis in different buffer media (Fig. S26–S28†), namely sodium phosphate (NaPi, pH 7), potassium phosphate (KPi, pH 7) and KOH (pH 13). XANES linear combination fits of the first derivatives of the normalized spectra were performed with CoO and LiCoO2 as references. The results revealed that all three materials contain Co3+ amounts in the range of 22–28% after electrolysis (Fig. S27 and Table S6†). EXAFS fits against literature models66 for CoNCN displayed good agreements of experimental and structural data with R values in the range of 1.5–2.7 % (Fig. 6 and Table S7†). Co–N and Co–C bond lengths agree with crystallographic data within the experimental error range, given that data were recorded in fluorescence mode with a rather small k space. All in all, XAS data indicate overall robustness of the CoNCN under electrocatalytic conditions. However, slight surface modifications cannot be excluded with the present data set.
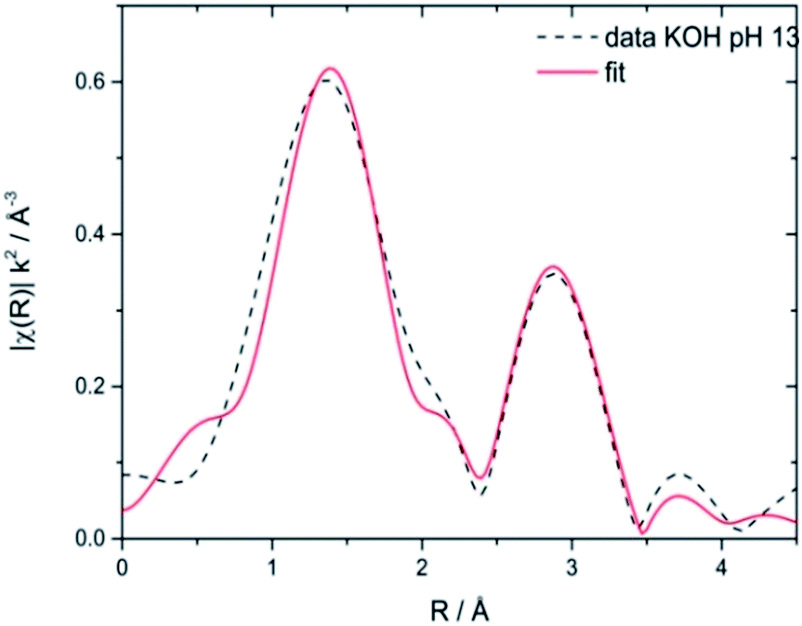 | ||
| Fig. 6 Representative EXAFS spectrum of CoNCN electrocatalyst on a FTO electrode after 20 h of electrolysis in KOH (pH 13) fitted vs. structural reference data.66 | ||
Therefore, FTO electrodes regenerated from NaPi and KPi solutions were investigated with Raman spectroscopy for the presence of secondary oxide phases (Fig. 7; FTO electrodes treated in KOH did not contain sufficient CoNCN amounts). Comparison of electrode Raman spectra with pristine CoNCN, Co3O4 and CoO references clearly show the absence of characteristic oxide vibrations and good agreement of band positions for pristine and post-electrocatalytic CoNCN. As shown in previous studies,34 the formation of significant operational amounts of cobalt oxide and CoPi films would be evident from Raman spectra. Major cobalt leaching and re-deposition processes can thus be excluded. Surface analysis techniques such as XPS are not applicable in the present case, because the particles are covered with Nafion binder. Further analyses of the interplay of cobalt oxidation with the carbodiimide matrix with high-end analytical techniques are now required, beyond the scope of the present work.
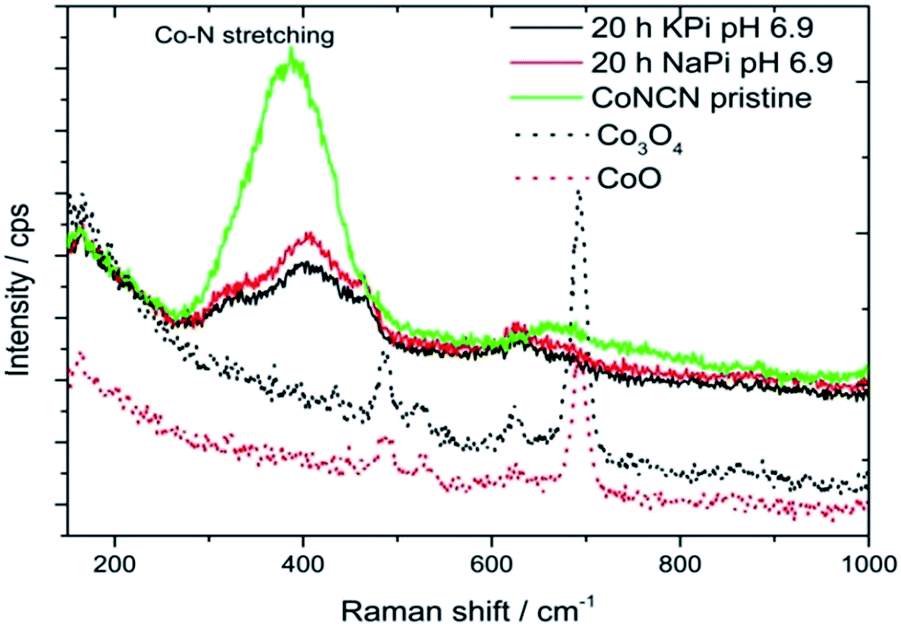 | ||
| Fig. 7 Raman spectra of post-electrocatalytic CoNCN on FTO electrodes after 20 h of electrolysis in KPi and NaPi media (pH 7) vs. reference data for Co3O4 and CoO. | ||
Transition metal tuning of carbodiimide catalysts
We furthermore explored tuning options for the clear-cut carbodiimide matrix, starting with Co/Ni substitution. The successful extension of the above-mentioned synthetic protocol on the formation of Co1−xNixNCN (x = 0–1) solid solutions is evident from the gradual decrease of the lattice constants and from characteristic peaks in the FT-IR spectra among the series (Fig. 7 and S29–S30†). Replacement of Co with Ni leads to a sharp decrease of particle size from microscale platelets for CoNCN to nanoparticles of NiNCN with an average diameter around 21 nm owing to different recrystallization behavior in the salt melt (Fig. S31†). Despite an 8.5-fold higher BET surface area (115 m2 g−1) of NiNCN the main performance trends among the Co1−xNixNCN series point towards lower current densities, except for low substitution levels (LSV data in Fig. S32†). These trends are more pronounced from CV curves recorded in alkaline 0.1 M NaOH solution (pH 13) shown in Fig. 8. The current density increases faster for low degrees of Co2+ substitution so that maximum performance was observed for Co0.9Ni0.1NCN. The negligible activity of pristine NiNCN indicates that the increased activity indeed arises from CoNCN matrix tuning. However, follow-up analyses are now required to explore the combination of structural, electronic and surface factors required to enhance the catalytic performance of CoNCN-based WOCs towards current benchmarks (Fig. 9).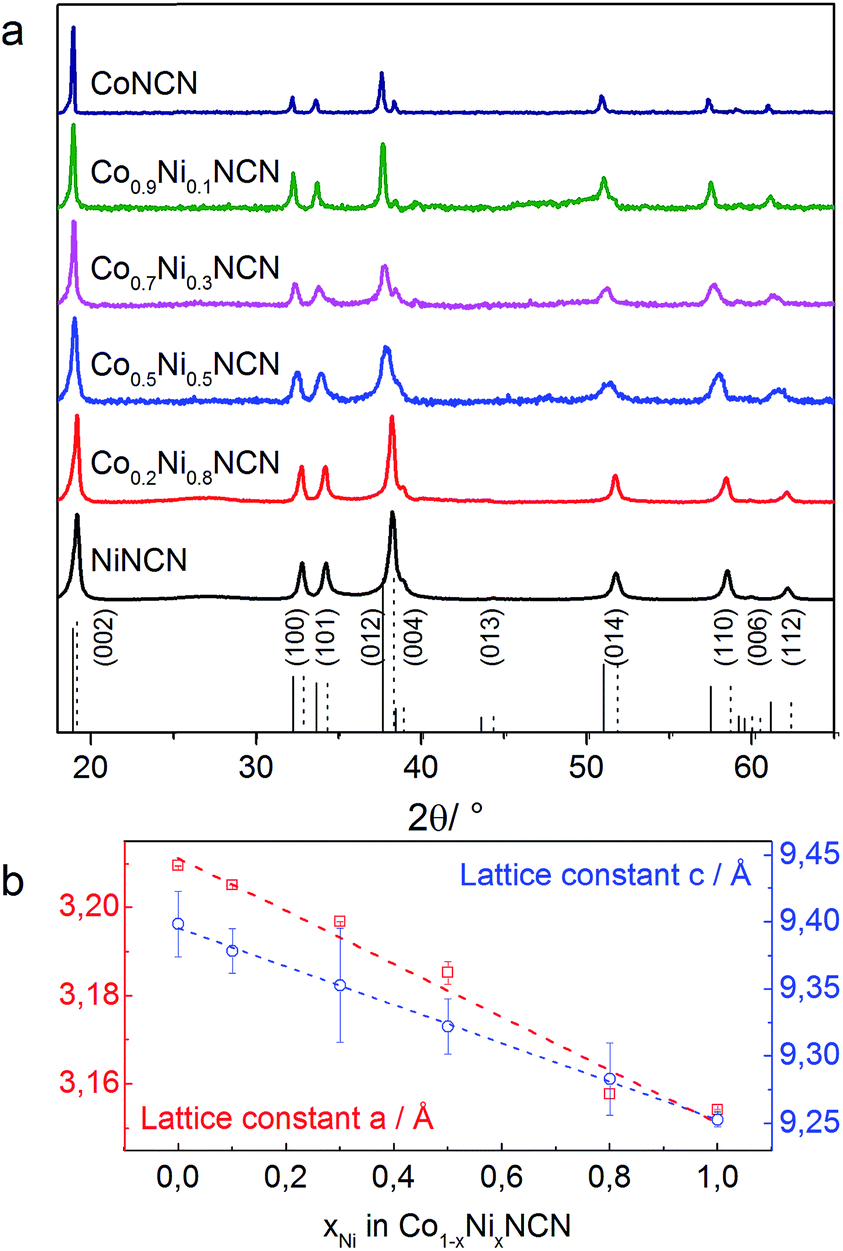 | ||
| Fig. 8 (a) PXRD patterns of Co1−xNixNCN catalysts with different metal ratios and (b) the corresponding Vegard diagram (reference patterns: – CoNCN [ICDD 04-015-1039] and - - - NiNCN [ICDD 04-015-1038]). | ||
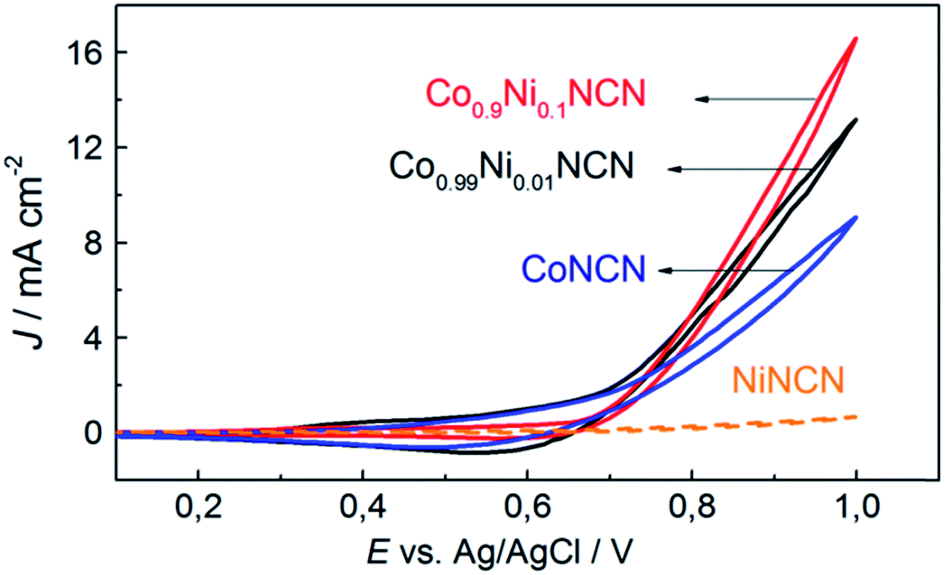 | ||
| Fig. 9 CV curves of Co1−xNixNCN catalysts with different metal ratios (glassy carbon electrodes); scan rate: 25 mV s−1, 0.1 M NaOH (E0 (pH 13) = 0.27 V). | ||
Conclusions
Cobalt carbodiimide CoNCN emerges as a new non-oxidic water oxidation catalyst prototype with dual activity in visible-light-driven and electrocatalytic water oxidation. CoNCN exhibits high photochemical oxygen evolution performance and outperformed appropriate reference samples of the widely used WOC Co3O4. This points to different catalytic reactivity patterns for metal oxides and metal carbodiimides. We thus introduce the carbodiimide matrix as a simple but versatile new system for fundamental studies of structure–activity relationships, catalytic mechanisms and surface effects.A wide range of complementary analytical techniques clearly pointed to stability of CoNCN under photocatalytic conditions and to the absence of secondary catalytic species in solution. Bulk structural changes were excluded with PXRD, XAS and FT-IR spectroscopic techniques. Furthermore, HRTEM and EDX/EELS data did not give any indications of surface alterations. The formation of significant oxide layers during the catalytic process could be clearly excluded with XPS analyses. This sets the stage for new in situ studies into the involved active species and mechanisms, especially with respect to the possible involvement of the anions carbon or nitrogen atoms as Lewis acidic or basic centres.75–77
Moreover, CoNCN exhibits promising electrocatalytic properties, such as stability during long-term water electrolysis and an overpotential in the range of leading Co-based oxide catalysts. Major in situ transformations of CoNCN into secondary oxide electrocatalysts were excluded through XAS and Raman investigations of post-catalytic electrode materials, while cobalt leaching into the aqueous phase was ruled out with ICP-OES analyses. Advanced in-depth studies beyond the detection limits of standard analytical techniques are now required to check the new catalyst for the possible presence of subtle nanoscale heterojunctions.
Next, the tuning potential of the CoNCN host material for electrocatalytic applications was demonstrated through systematic nickel doping, and Co0.9Ni0.1NCN solid solutions displayed the most pronounced increase of current densities. This corresponds well with the optimal Ni-doping range for Co3O4 catalysts, so that design principles for other WOC types can be transferred on the novel oxide-free system.78 The carbodiimide matrix leaves room for further optimization strategies, such addressing the electrocatalytic performance with ternary metal combinations.79,80
Most importantly, CoNCN warrants novelty in structural WOC design on various levels. We present the first immobilization of abundant and powerful catalytic cobalt centers in a non-oxidic WOC matrix with the striking simplicity of the NiAs structure type. This opens up interesting alternatives to the long-standing “magic cube” design paradigm that inspired a long line of work from PSII mimics over layered oxidic WOCs to spinels,81 all of which exhibit a far more complex structural chemistry than carbodiimides. The interconnected multilayer motif of CoNCN provides excellent prerequisites for hole delocalization and rapid diffusion of active species.82 Indeed, the layered hexagonal arrangement of {CoN6} octahedra was associated with higher activity than cubic Co3O4 under representative test conditions. Furthermore, CoNCN translates two key principles of molecular WOCs to heterogeneous catalysts, namely the embedment of Co centers into a stable nitrogen-containing environment83 along with the stabilizing effect of N–Co bonds in trans position to the water binding site.29 CoNCN furthermore stands out through possible new reaction patterns arising from different electronic configurations upon replacing O2− with NCN2−, polarity effects at the catalyst–solution interface, and synergistic effects between metals and anions along with higher anionic flexibility.56
In summary, CoNCN opens up new design avenues for combining assets of homo- and heterogeneous catalysts. CoNCN offers a straightforward and stable solid matrix with highly active and stable all-nitrogen coordination environments that can be further optimized via versatile cationic tuning. The well-defined structure of metal carbodiimide catalysts is a promising and convenient model system to obtain different theoretical and mechanistic insights into the complex multi-parameter challenge of WOC construction. Dual CoNCN catalysts are a creative “melting pot” for merging non-metallic, molecular and solid state WOC features on the way to applied artificial photosynthesis.
Experimental
Synthesis of Co1−xNix(NCNH)2
Co1−xNix(NCNH)2 powders were synthesized by heating a mixture of anhydrous CoCl2/NiCl2 with the target metal ratio (130 mg, 1 mmol) with cyanamide (1.2 g, 30 mmol) to 75 °C in a round bottom flask under nitrogen flow. The melt was stirred until the total amounts of the metal chloride were dissolved and stirring was continued for 5 min. (NH4)2CO3 (196 mg, 2 mmol) was poured into the melt to initiate the reaction by the deprotonation process, followed by 5 min of additional stirring. An excess of cyanamide was removed by washing with H2O prior to solidification of the melt. The precipitate was collected by centrifugation, repeatedly washed with water and dried in vacuo at 50 °C to obtain Co1−xNix(NCNH)2 (typical yield: 91%).Synthesis of Co1−xNix(NCN)
250 mg of Co1−xNix(NCNH)2 precursor powder was mixed with a LiCl–KCl eutectic salt system (6 g, 45–55 wt%) and heated in a heating mantle to 400 °C within 20 min. The temperature was held for 2.5 h, followed by natural cooling of the melt to RT. The obtained salt cake was dissolved in H2O and the product was separated from the solution with a Buchner funnel and washed with H2O. Drying in vacuo at 50 °C affords reddish brown (CoNCN) or yellowish (NiNCN) powders (typical yield: 92%).Photocatalysis
Water oxidation was performed in a 10 mL headspace vial sealed with an aluminum crimp cap with a rubber septum (PTFE). The reaction dispersions were prepared under dark conditions by adding the corresponding amounts of CoNCN catalysts, Na2S2O8 (9.52 mg, 0.04 mmol) and [Ru(bpy)3]Cl2 (5.12 mg, 0.006 mmol) to 8 mL of DI H2O in a glass vessel, which was subsequently sealed. The experiments were performed in different buffer solutions (0.4 M phosphate, 0.1 M carbonate and 0.5 M borate). The solution was degassed by purging with helium (6.0 purity) for 30 min under stirring. Co3O4–NP catalyst samples were synthesized according to literature protocols.16 Commercial Co3O4 catalysts were purchased from Alfa Aesar and used as received.After purging, an initial sample of the headspace (100 μL) was injected into a gas chromatograph as background reference for the GC calibration. Gas chromatograms were recorded using an Agilent Technologies 7820A gas chromatograph with helium as carrier gas and a 3 m × 2 mm packed molecular sieve 13X 80–100 column to separate O2 and N2. The oven was operated isothermally at 80 °C. Analysis of the headspace was performed by taking 100 μL samples with a Hamilton (1825 RN) gas-tight microliter syringe. Gases were detected using a thermal conductivity detector (Varian) operated at 200 °C. Contamination of the headspace by air was constantly quantified by measuring the N2 peak on GC chromatograms. Calibration was performed by injection of known quantities of pure oxygen diluted in the same headspace vial containing the same volume and concentration of buffered solution as used for the measurements.
Oxygen evolution was measured both in solution using an Oxygen Sensor (OX-N) Clark electrode (Unisense). Constant temperature was maintained with a mineral insulated thermosensor (2 mm tip diameter, TP2000, Unisense). After the background calibration of the GC, the Clark-type O2 probe was introduced through the headspace until the signal remained constant, afterwards dipped into the solution and the experiment was started after equilibrium was reached.
Signals were first calibrated using the standard procedure supplied for the applied electrode model. After complete aeration of the water (5 min of vigorous bubbling) in the Unisense calibration chamber CAL300, the gas stream was turned off and the saturation point for the calibration line for atmospheric partial pressure was added. Background calibration was performed with an anoxic solution prepared from sodium ascorbate and NaOH. All experiments were conducted under controlled stirring (500 rpm). Data collection was performed with the SensorTrace software from Unisense and a frequency of 1 data point per sec for both probes (temperature and oxygen). After a constant signal for the O2 sensor was recorded, the catalytic reaction was initiated by exposing the reaction vessel to the light of a 470 nm high flux LED from Rhopoint Components LTD. LED power was determined as 26.1 mW cm−2. Kinetic evaluations were performed for the initial phase of approximately linear time-dependent increase of O2 evolution (0–60 s) to exclude the influence of oxygen diffusion into the headspace at a later stage.
Electrocatalysis
Glassy carbon electrodes were covered with 5 μL of an ultrasonicated suspension (5 mg catalyst, 350 μL EtOH, 9.5 μL Nafion (20% in H2O)) and FTO glass electrodes (∼1 × 1.5 cm) with 20 μL of an ultrasonicated suspension (5 mg catalyst, 700 μL EtOH, 19 μL Nafion (20% in H2O)). Electrochemical measurements were carried out with a Gamry Instruments potentiostat 600 and evaluated with the Gamry EIS 300/Physical Electrochemistry software. The experiments were performed in a standard three-electrode setup (sat. Ag/AgCl reference electrode (Ag/AgCl vs. NHE: +197 mV), platinum wire counter electrode) in 0.1 M of phosphate (pH 7) or KOH (pH 13.1) buffer solutions. CV cycles were recorded with a scan rate of 25 mV s−1 and LSV curves with a scan rate of 10, 5 or 1 mV s−1 at an electrode rotation speed of 1500 rpm. Chronoamperometry scans were performed with an equilibrium time of 60 s at a given potential. The double layer capacitance of CoNCN was determined from cycling at different scan rates in the non-Faradaic region in a potential window of 0.1 V (from 0.0 V to 0.1 V vs Ag/AgCl). The working electrode was held at each potential for 10 s before the measurement. The electrochemical double layer capacitance CDL was determined by applying ic = vCDL, where v is the scan rate and ic the current density at 0.05 mV.Analytical characterization
Powder X-ray diffraction patterns (PXRD) were collected in reflection mode on a Bruker D8 device, equipped with a Kevex fluorescence shield detector (Cu Kα radiation). The Scherrer equation was applied with a form factor of K = 0.9. Scanning electron microscopy (SEM) images were recorded with a JEOL JSM-7500 F instrument. Fourier transform infrared spectroscopy (FT-IR) spectra were collected on a Thermo Scientific Nicolet iS5 spectrometer in ATR mode with a resolution of 4 cm−1. Elemental analysis for nitrogen and carbon was performed with an Elementarvario MICRO cube. Nitrogen sorption isotherms were recorded after degassing at 80 °C for 20 h by using a Quantachrome Quadrasorb SI porosimeter at 77 K. Absorption branch points (0.1 > p/p0 < 0.3) were applied in the BET model to calculate the apparent surface area. Transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDX) analyses were performed on a Philips CM200FEG microscope (200 kV, field emission gun). UV/vis absorbance spectra were recorded on a Varian spectrophotometer equipped with an optical integrating sphere. ICP-OES was measured on a Varian Vista-MPX instrument equipped with a CCD detector. Aqueous calibration solutions of 5, 20 and 50 ppm were freshly prepared from Merck millipore standard solutions (1000 ppm).Cobalt K-edge X-ray absorption spectroscopy was performed at the ESRF Swiss Norwegian Beamline BM01B in transmission mode. CoNCN was measured as solid samples dispersed in cellulose. CoO, Co3O4 and LiCoO2 were used as reference samples for cobalt valence determination. Due to the low concentration of cobalt post-catalytic FTO electrodes were measured in fluorescence mode using the vortex fluorescence detector. Cobalt metal foil was used as calibration standard for all experiments. Energy calibration, background removal and Fourier transformation of XAS spectra were performed with the Athena software and EXAFS fits were performed with the Artemis software.84 Raman spectra were recorded on a Remishaw Ramascope spectrometer equipped with diode laser (785 nm). XPS spectra were recorded with a Quantum 2000 spectrometer from Physical Electronics Inc. using monochromatized Al Kα radiation and an electron as well as an ion neutralizer. The samples were measured as prepared, i.e. no sputter cleaning was performed. No peak shifting (energy scale calibration) was performed.
Acknowledgements
Financial support by the Swiss National Science Foundation (Sinergia Grant no. CRSII2_136205/1), by the University of Zurich (UFSP LightChEC: F. E., R. M. and G. R. P.), and by the Max Planck Society (D. R., M. S. and M. A.) is gratefully acknowledged. We thank Dr Guylhaine Clavel (Fritz Haber Institute) for HRTEM and EELS analyses. We thank Dr Paula Abdala and Dr Herman Emerich (ESRF Grenoble) for beamline support and Adrià Garcia Gil (UZH) for experimental assistance.Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 43, 15729–15735 CrossRef PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- S. Styring, Faraday Discuss., 2012, 155, 357–376 RSC.
- Y. Tachibana, L. Vayssieres and J. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- F. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607–2622 CrossRef CAS.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC.
- J. G. McAlpin, T. A. Stich, W. H. Casey and R. D. Britt, Coord. Chem. Rev., 2012, 256, 2445–2452 CrossRef CAS.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondini, Chem. Sci., 2014, 5, 3591–3597 RSC.
- S. P. Phivilay, A. P. Puretzky, K. Domen and I. E. Wachs, ACS Catal., 2013, 3, 2920–2929 CrossRef CAS.
- M. Matsukawa, R. Ishikawa, T. Hisatomi, Y. Moriya, N. Shibata, J. Kubota, Y. Ikuhara and K. Domen, Nano Lett., 2014, 14, 1038–1041 CrossRef CAS PubMed.
- M. M. Waegele, X. Chen, D. M. Herlihy and T. Cuk, J. Am. Chem. Soc., 2014, 136, 10632–10639 CrossRef CAS PubMed.
- H. Han and H. Frei, J. Phys. Chem. C, 2008, 112, 16156–16159 CAS.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 126, 7297–7300 CrossRef.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CAS.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- C. Zuccaccia, G. Bellachioma, O. Bortolini, A. Bucci, A. Savini and A. Macchioni, Chem.–Eur. J., 2014, 20, 3446–3456 CrossRef CAS PubMed.
- M. Grzelczak, J. Zhang, J. Pfrommer, J. Hartmann, M. Driess, M. Antonietti and X. Wang, ACS Catal., 2013, 3, 383–388 CrossRef CAS.
- B. Klahr and T. Hamann, J. Phys. Chem. C, 2014, 118, 10393–10399 CAS.
- J. M. Thomsen, S. W. Sheehan, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136, 13826–13843 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 79–89 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110–14118 CrossRef CAS PubMed.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- A. Harriman, I. J. Pickering and J. M. Thomas, J. Chem. Soc., Faraday Trans., 1988, 84, 2795–2806 RSC.
- D. Shevchenko, M. F. Anderlund, A. Thapper and S. Styring, Energy Environ. Sci., 2011, 4, 1284–1287 CAS.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- Y. Liu and D. G. Nocera, J. Phys. Chem. C, 2014, 118, 17060–17066 CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- X. L. Hu, S. Piccinin, A. Laio and S. Fabris, ACS Nano, 2012, 12, 10497–10504 Search PubMed.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542–549 CrossRef CAS PubMed.
- M. Richter and D. Schmeißer, Appl. Phys. Lett., 2013, 102, 253904 CrossRef.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- Y.-R. Zheng, M.-R. Gao, Q. Gao, H.-H. Li, J. Xu, Z.-Y. Wu and S.-H. Yu, Small, 2015, 11, 182–188 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- M. Shalom, S. Gimenez, F. Schipper, I. Herraiz-Cardona, J. Bisquert and M. Antonietti, Angew. Chem., Int. Ed., 2014, 53, 3654–3658 CrossRef CAS PubMed.
- T.-P. Fellinger, F. Hasché, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 Search PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 1–7 CrossRef.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Cora, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CAS.
- P. Chen, L.-K. Wang, G. Wang, M.-R. Gao, J. Ge, W.-J. Yuan, Y.-H. Shen, A.-J. Xie and S.-H. Yu, Energy Environ. Sci., 2014, 7, 4095–4103 CAS.
- J. Duan, S. Chen, S. Dai and S. Z. Qiao, Adv. Funct. Mater., 2014, 24, 2072–2078 CrossRef CAS.
- J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378–401 CrossRef CAS.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- M. L. Toebes, T. Alexander Nijhuis, J. Hájek, J. H. Bitter, A. Jos van Dillen, D. Y. Murzin and K. P. de Jong, Chem. Eng. Sci., 2005, 60, 5682–5695 CrossRef CAS.
- W. Zhao, Y. Liu, J. Liu, P. Chen, I.-W. Chen, F. Huang and J. Lin, J. Mater. Chem. A, 2013, 27, 7942–7948 Search PubMed.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2013, 36, 13270–13273 CrossRef PubMed.
- S. Goberna-Ferrón, W. Y. Hernández, B. Rodríguez-García and J. R. Galán-Mascarós, ACS Catal., 2014, 4, 1637–1641 CrossRef.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- J. Pfrommer, M. Lublow, A. Azarpira, C. Göbel, M. Lücke, A. Steigert, M. Pogrezeba, P. W. Menezes, A. Fischer, T. Schnedel-Niedrig and M. Driess, Angew. Chem., Int. Ed., 2014, 53, 5183–5187 CAS.
- A. J. Bloomfield, S. W. Sheehan, S. L. Collom, R. H. Crabtree and P. T. Anastas, New J. Chem., 2014, 38, 1540–1545 RSC.
- M. Krott, X. Liu, B. P. T. Fokwa, M. Speldrich, H. Lueken and R. Dronskowski, Inorg. Chem., 2007, 46, 2204–2207 CrossRef CAS PubMed.
- F. Evangelisti, R. Güttinger, R. Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 50, 18734–18737 CrossRef PubMed.
- M. Hara, C. C. Warasaka, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- H. Lv, J. A. Rudd, P. F. Zhuk, J. Y. Lee, E. C. Constable, C. E. Housecroft, C. L. Hill, D. G. Musaev and Y. V. Geletii, RSC Adv., 2013, 3, 20647–20654 RSC.
- H. Liu and G. R. Patzke, Chem.–Asian J., 2014, 9, 2249–2259 CrossRef CAS PubMed.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787–1793 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 Search PubMed.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- X. Liu, L. Stork, M. Speldrich, H. Lueken and R. Dronskowski, Chem.–Eur. J., 2009, 15, 1558–1561 CrossRef CAS PubMed.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS PubMed.
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material characterizations and addition photo- and electrocatalytic data are available. See DOI: 10.1039/c5ta00369e |
| This journal is © The Royal Society of Chemistry 2015 |