
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous graphene wrapped CoO nanoparticles for highly efficient oxygen evolution†
Yufei
Zhao
ab,
Bing
Sun
a,
Xiaodan
Huang
a,
Hao
Liu
a,
Dawei
Su
a,
Kening
Sun
*b and
Guoxiu
Wang
*a
aCenter for Clean Energy Technology, School of Chemistry and Forensic Science, Faculty of Science, University of Technology, Sydney, Sydney, NSW 2007, Australia. E-mail: Guoxiu.Wang@uts.edu.au
bBeijing Key Laboratory for Chemical Power Source and Green Catalysis, School of Chemical Engineering and Environment, Beijing Institute of Technology, Beijing, 100081, China. E-mail: bitkeningsun@163.com
First published on 28th January 2015
Abstract
The design of highly efficient, robust and earth-abundant electrocatalysts for the oxygen evolution reaction (OER) is a prodigious challenge for the rapid growth of global energy demand. Herein, an active catalyst composed of porous graphene and cobalt oxide (PGE–CoO) has been synthesized, demonstrating high porosity, large specific surface area and fast charge transport kinetics. The catalyst also exhibits excellent electrochemical performance towards OER with a low onset potential and high catalytic current density. The enhanced catalytic activity could be ascribed to porous structure, high electroactive surface area and strong chemical coupling between graphene and CoO nanoparticles. Moreover, this OER catalyst also shows good stability in the alkaline solution. The high performance and strong durability suggest that the porous structured composite is favorable and promising for water splitting.
Introduction
The electrochemical or photoelectrochemical splitting of water is an efficient solution for the current energy demands.1–3 However, water splitting is mainly hindered by the sluggish kinetics of the oxygen evolution reaction (OER).4,5 The major challenge for OER is to develop catalysts with high efficiency, low overpotential, as well as long-term stability. At present, precious metal oxides or complexes of ruthenium (Ru) and iridium (Ir) are the most active catalysts towards OER, achieving high electrochemical performance.6–8 However, the scarcity and prohibitive cost of these noble metals impede them to be used on a large scale. Therefore, it is important to develop new highly active catalysts for oxygen evolution, especially those composed of earth-abundant and inexpensive materials.Recently, electrode materials based on transition metals or their composites, such as cobalt,9–11 nickel12–15 and manganese,16,17 have attracted extensive attention. Among them, cobalt-based materials, such as cobalt oxides (CoO, Co3O4),18–20 substituted cobaltites MxCoyO4 (M = Ni, Fe, or rare-earth cations),21–24 and cobalt phosphate (Co3(PO4)2),25–27 have been investigated due to their high performance. Particularly, cobalt oxides are the most preferable because of facile preparation and high activity. However, further improvement of the performance of cobalt oxides is plagued by the inherently low electrical conductivity and the intense causticity in alkaline solutions. Therefore, the structure and electrical conductivity should be considered when developing catalysts with high catalytic performance and good stability. Recently, cobalt oxides have been studied by depositing on different substrates, such as Au,28 nickel foam,29 or a series of carbon materials.30–33 These substrates can not only improve the conductivity, but also avoid the agglomeration of cobalt oxide particles so that the activity of the composite could be enhanced. Bell and coworkers developed gold-supported cobalt oxide, showing a higher turnover frequency of 40 times compared to that of the bulk one.28 Chen's group reported 3D crumpled graphene–cobalt oxide nanohybrids as high performance bi-functional electrocatalysts.33 These results have illustrated that a suitable substrate could efficiently enhance catalytic activity.
Porous carbon materials have garnered extensive interest because of their attractive properties such as ordered pores, large pore volume and good conductivity.34,35 The porous structure could efficiently prevent graphene sheets from restacking and further provide a significantly enhanced specific surface area.36 In addition, the porous structure, especially mesoporous and macroporous structures, can facilitate electrolyte diffusion.1,37 These properties make porous graphene an ideal matrix for improving the catalytic activity of embedded electrocatalysts.
Herein, we developed an active catalyst, CoO nanoparticles wrapped by porous graphene sheets. The porous graphene was prepared by using 1D silica nanorods as the template. The results indicate that ultrafine CoO nanoparticles were uniformly dispersed and wrapped by porous graphene, which could efficiently avoid the corrosion during an electrochemical test. The homogeneous distribution of particles and porous structure of graphene enable the efficient utilization of catalysts, which could be used as an active non-precious metal material for oxygen evolution.
Experimental
Material synthesis
Structural characterization
The morphology of the as-prepared materials was characterized by field emission scanning electron microscopy (FESEM, Zeiss Supra 55VP) and transmission electron microscopy (TEM, Model JEM-2011, JEOL). Element mapping was performed with a Zeiss Evo SEM. X-ray diffraction (XRD) patterns were recorded by a Siemens D5000 using Cu Kα radiation with a scanning step of 0.02° per second. X-Ray Photoelectron Spectroscopy (XPS) was performed on an ESCALAB250Xi (Thermo Scientific, UK) equipped with mono-chromated Al K alpha (energy: 1486.68 eV). The Brunauer–Emmett–Teller (BET) surface area of the obtained materials was calculated by experimental points at a relative pressure of P/P0 = 0.05–0.25. The percentage of CoO nanoparticles in a PGE–CoO composite was analyzed using a TGA/differential thermal analysis (DTA) analyzer (TA Instruments, SDT 2960 module, New Castle, DE, USA) at a heating rate of 10 °C min−1 from room temperature to 800 °C in an air atmosphere.Electrochemical measurements
All electrochemical measurements were performed in a standard three electrode glass cell on an electrochemical workstation (CHI 660E) with platinum wire as the counter electrode and Ag/AgCl (1 M KCl) as the reference electrode. The potentials reported here were quoted against the reversible hydrogen electrode (RHE) according to the Nernst equation ERHE = EAg/AgCl + 0.059pH + 0.2224. The working electrode was prepared by depositing the target materials on the glassy carbon (GC) electrode with a diameter of 3 mm. 4 mg of the material was dispersed in 1 ml solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v water/isopropanol) by ultrasonication, followed by addition of 80 μl Nafion (5 wt%) to obtain a homogeneous ink. The catalyst ink (10 μl) was loaded onto the GC electrode and then dried at room temperature. Linear sweep voltammetry (LSV) was conducted in a KOH solution (selected concentration) at a scan rate of 10 mV s−1. The polarization curves were all corrected by 95% iR compensation and turnover frequency (TOF) was calculated by the equation of TOF = nO2/nCo= (Q/4F)/nCo (F is the faraday constant, 96485 C mol−1). Electrical impedance spectroscopy (EIS) was recorded under the following conditions: ac voltage amplitude of 5 mV, frequency ranging from 106 to 0.01 Hz, and open circuit or selected overpotential. Cyclic voltammetry measurements were conducted from −0.12 to −0.04 V (vs. Ag/AgCl) without faradaic processes at different scan rates (1 to 20 mV s−1).
:1 v/v water/isopropanol) by ultrasonication, followed by addition of 80 μl Nafion (5 wt%) to obtain a homogeneous ink. The catalyst ink (10 μl) was loaded onto the GC electrode and then dried at room temperature. Linear sweep voltammetry (LSV) was conducted in a KOH solution (selected concentration) at a scan rate of 10 mV s−1. The polarization curves were all corrected by 95% iR compensation and turnover frequency (TOF) was calculated by the equation of TOF = nO2/nCo= (Q/4F)/nCo (F is the faraday constant, 96485 C mol−1). Electrical impedance spectroscopy (EIS) was recorded under the following conditions: ac voltage amplitude of 5 mV, frequency ranging from 106 to 0.01 Hz, and open circuit or selected overpotential. Cyclic voltammetry measurements were conducted from −0.12 to −0.04 V (vs. Ag/AgCl) without faradaic processes at different scan rates (1 to 20 mV s−1).
Results and discussion
Graphene with a highly porous structure was synthesized by a hard template method, which is effective in achieving porous graphene with controlled pore size. Scheme 1 displays the schematic illustration of the synthesis route to the PGE–CoO hybrid. The 1D silica nanorods with a size about 200 nm wide and 400 nm long were first prepared by the well-known Stöber method (see Fig. S1†) and then functionalized with surface methyl groups. Subsequently, GO/silica composites were formed by mixing silica nanorods and GO nanosheet suspension together. The as-prepared composites were then calcinated in the inert atmosphere to reduce GO into PGE. The final product PGE–CoO was obtained by heating the mixture of PGE and cobalt precursor.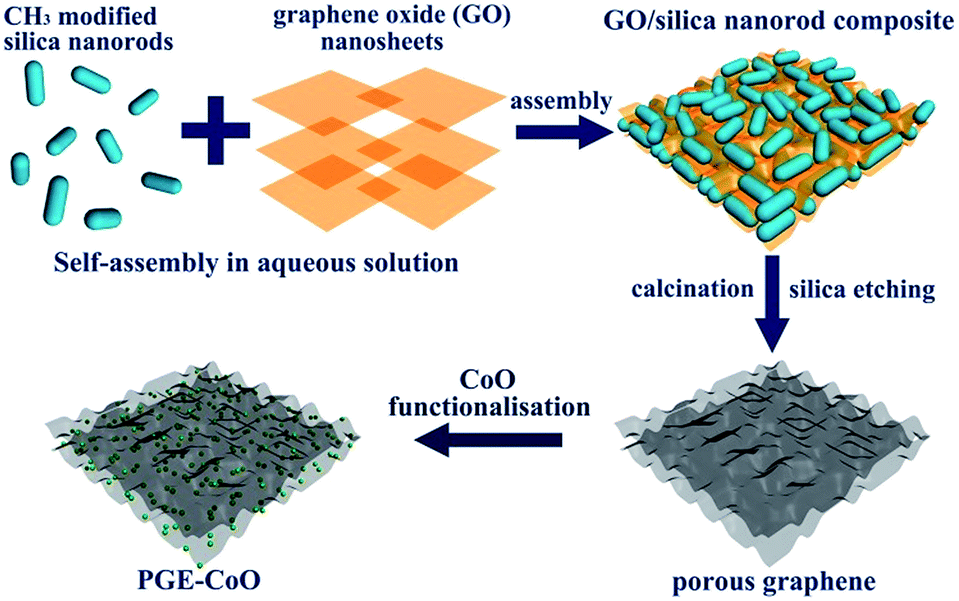 | ||
| Scheme 1 Schematic illustration of the synthesis procedure of PGE–CoO materials. | ||
The SEM image of PGE in Fig. 1a depicts the highly porous architecture with nanorod-shaped pores. The structure and size of the pore are consistent with the particle shape of the silica nanorod templates. The BET results in Fig. S2a† reveal that PGE has a surface area of 417 m2 g−1 and a large pore volume of 1.69 cm3 g−1. The low magnification SEM image of a PGE–CoO composite in Fig. S3† shows that the material maintains the structure of porous graphene. Fig. 1b shows the high magnification SEM image of a PGE–CoO composite. PGE and CoO particles are co-assembled into a well-organized hybrid. Moreover, highly distributed and ultrafine CoO nanoparticles were wrapped by porous graphene, which could efficiently avoid aggregation and corrosion of CoO particles. TEM images in Fig. 1c and d not only show the uniform distribution and good crystalline structure of CoO nanoparticles, but also confirm the wrapped structure. The uniform distribution of CoO particles on porous graphene was also proved by element mapping, which shows homogeneous distribution of C, Co, and O elements inside the PGE–CoO composite (see Fig. S4†). The SEM images of pure CoO and GE–CoO are shown in Fig. S5.† The pure CoO particles highly aggregated together, while GE–CoO displayed a relatively uniform distribution of CoO particles on the graphene surface. The nitrogen sorption isotherm of the PGE–CoO hybrid shows a surface area of 254 m2 g−1 and a pore volume of 1.35 cm3 g−1, which are much higher than those of GE–CoO (32 m2 g−1, 0.14 cm3 g−1) (see Fig. S2b and c†).
 | ||
| Fig. 1 The characterization of pure PGE and PGE–CoO hybrid: (a) SEM image of PGE, (b) high magnification SEM image of PGE–CoO, and (c and d) TEM images of PGE–CoO. | ||
Fig. 2a shows the wide-angle XRD patterns of the PGE and PGE–CoO composite. The diffraction peak around 25° could be assigned to graphene (002) for both PGE and PGE–CoO. After loading CoO nanoparticles into PGE, two broad weak diffraction peaks appeared in the XRD pattern, which can be indexed to the (111) and (200) crystal planes of the cubic CoO (PDF no. 43-1004). XPS measurements were carried out on PGE–CoO to further investigate the composition and determine the surface electronic state. The high-resolution Co 2p spectrum exhibits two prominent peaks at 797.6 and 781.4 eV, corresponding to the Co 2p1/2 and Co 2p3/2 spin–orbit peaks of CoO (Fig. 2b). The peak O at 531.5 eV corresponds to the dominant O 1s feature in CoO (Fig. 2c).33,40 The TGA curve of PGE–CoO is presented in Fig. 2d, from which the content of CoO can be determined to be 16.8 wt%.
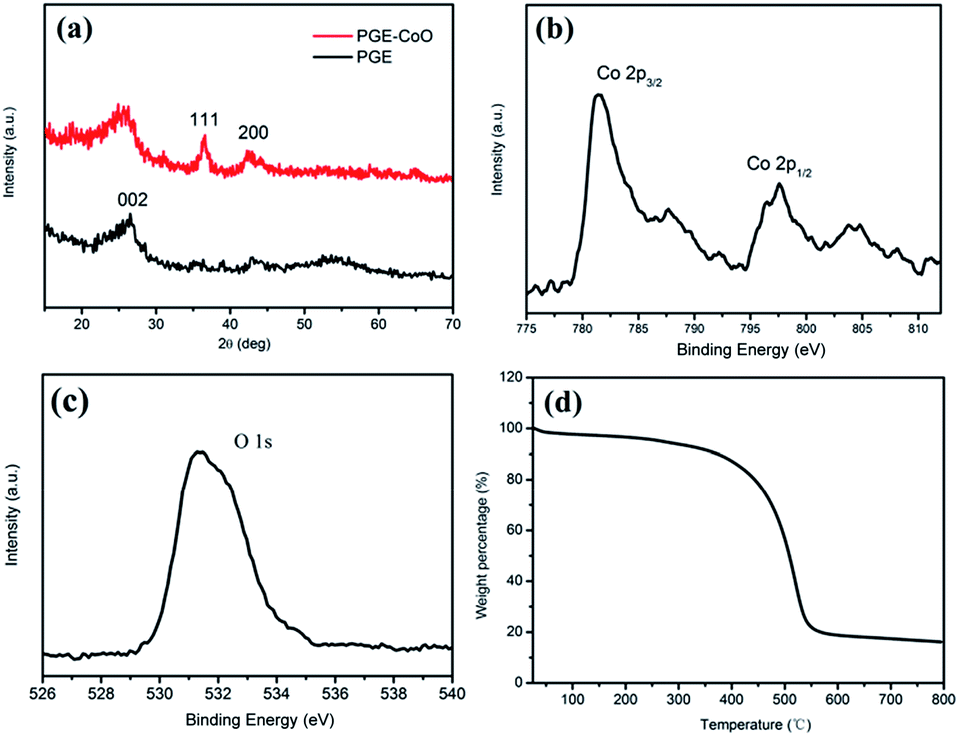 | ||
| Fig. 2 (a) XRD patterns of PGE–CoO and PGE, high-resolution XPS spectra of (b) Co 2p and (c) O 1s, and (d) thermogravimetric analysis in air of PGE–CoO. | ||
The OER catalytic properties were investigated in a three-electrode system in a 0.1 M KOH solution by LSVs. Comparative studies were performed on GE–CoO, CoO and PGE with the same loading. The PGE–CoO hybrid shows a high activity with a small onset potential of 504 mV (vs. Ag/AgCl, determined by Chen's method, Fig. 3a),41 which is more negative than that of other samples, GE–CoO (560 mV) and CoO (570 mV). Moreover, the high activities can also be derived by the comparison of current densities of the samples, especially for the current density of 10 mA cm−2. The PGE–CoO composite achieved a current density of j = 10 mA cm−2 at the overpotential of 348 mV, which is even comparable to those of previously reported noble metal catalysts,7,8 and higher than those of many other Co-based materials (366 mV for NG-CoSe2 and 390 mV for Co3O4/CNT) under the same conditions.30,42 In contrast, both CoO (510 mV) and GE–CoO (438 mV) exhibit much lower OER activities in terms of the same current density.
 | ||
| Fig. 3 Electrochemical performance of the OER catalysts: (a) polarization curves for PGE–CoO, GE–CoO, CoO and PGE on GC electrodes in 0.1 M KOH. (b) Tafel curves of PGE–CoO, GE–CoO and CoO in 0.1 M KOH. (c) Electrochemical capacitance measurements: cyclic voltammograms (CV) were performed in a 0.1 M KOH solution in a potential window without faradaic processes. (d) Scan rate dependence of the average capacitive currents at −0.08 V vs. Ag/AgCl for PGE–CoO and GE–CoO. | ||
TOF could be calculated from the proportion of the CoO in the hybrid obtained from the TGA result and the current density. We achieved a high TOF of 1.37 s−1 referring to per Co atom for a PGE–CoO hybrid at the overpotential of 400 mV (GE–CoO: 0.38 s−1 and CoO: 0.045 s−1) in 0.1 M KOH. This value is the lower limit for turnover frequency, assuming that deposited materials were all involved in the electrochemical reaction. The TOF of PGE–CoO is also much higher than those of other previously reported Co based materials.19,20
The Tafel plots were derived from the LSVs, which are usually used to evaluate the efficiency of the catalytic reaction. The linear regions were fitted to the Tafel equation (η = blog(j/j0), where η is the overpotential, b is the Tafel slope, j is the current density, and j0 is the exchange current density). The PGE–CoO composite exhibits a Tafel slope of 79 mV dec−1 (Fig. 3b), which is much smaller than those of GE–CoO (192 mV dec−1) and CoO (354 mV dec−1). The small Tafel slope and high current density of the PGE–CoO catalyst could be ascribed to the fast charge transport kinetics of the porous graphene and the strong interaction between PGE and CoO particles. This theory can also be confirmed by the EIS results under the conditions of an open circuit (see Fig. S6†). The samples displayed similar trends with both the semi-circles and the slopes. This indicated the similar mass transport properties and the reaction mechanism. However, the PGE–CoO hybrid shows much lower impedance compared to GE–CoO and CoO, which contributes to the high catalytic activities.
The electrochemically active surface areas for PGE–CoO and GE–CoO were estimated from the electrochemical double-layer capacitance of the catalytic surface.43,44 Representative plots for the determination of PGE–CoO and GE–CoO surface areas are shown in Fig. 3c and d and S7,† respectively. Cyclic voltammetry measurements were conducted in a region of −0.12 to −0.04 V (vs. Ag/AgCl), where the currents are mainly attributed to the charging of the double layer. The capacitance of PGE–CoO is 14.29 mF cm−2, while the capacitance of GE–CoO is only 1.62 mF cm−2. The measured active surface area is not an absolute value; however, it could serve as a guide for the comparison of the surface roughness of similar materials. The results are in good agreement with the BET results (PGE–CoO: 254 m2 g−1 and GE–CoO: 32 m2 g−1). The larger specific surface areas and active surface areas play an important role in the high performance of PGE–CoO towards OER.
EIS at various overpotentials was measured to further investigate the interface reaction and electrode kinetics of the PGE–CoO hybrid towards OER in a 0.1 M KOH solution. The representative Nyquist and Bode plots are presented in Fig. 4. The analogous plot profiles at different overpotentials suggest the similar electrochemical mechanism towards OER. The PGE–CoO hybrid electrode displays two semicircles in the Nyquist plots at the selected overpotentials, which reveals the presence of two time constants: high-frequency time constant and low-frequency time constant.45,46 The semicircle at high-frequency is attributed to the porous structure of the modified electrode and it displays constant properties over the different overpotentials; while the other one at low-frequency is related to the process of electrochemical reaction at the interface. The Bode plots also display the same trend of two time constants. The superior electrocatalytic performance of the PGE–CoO hybrid could be explained by the observation of the lower charge transfer resistance (Rct) and higher interfacial capacitances during the reaction process (derived from the fitted data of low frequency) (see Fig. S8†). Rct is related to the kinetics of electrocatalysis, and a lower value represents a fast reaction rate, which is strongly reliant on the overpotential. In this system, Rct decreased from 1145 Ω at 500 mV (vs. Ag/AgCl) to 54.7 Ω at 600 mV. Furthermore, PGE–CoO displayed a high constant phase element (CPE) during the test, varying from 13.25 mF cm−2 to 6.97 mF cm−2 and the decreased value of CPE may be ascribed to the bubbles blocking the active sites of the electrode materials during the test. The low charge transfer resistance and high interfacial capacitances during the test are mainly attributed to the superior conductivity of porous graphene and large specific surface area, which further illustrated the high electrocatalytic performance of the PGE–CoO hybrid towards OER.
 | ||
| Fig. 4 (a) Nyquist and (b) Bode plots for the PGE–CoO hybrid modified GC electrode recorded at various OER overpotentials in 0.1 M KOH. | ||
Besides the high catalytic activities, the good stability towards OER is also important for an energy conversion system. The stability of the materials was measured for 1000 cycles. The PGE–CoO hybrid showed excellent durability in a 0.1 M alkaline solution. Even after long time cycles, the PGE–CoO electrocatalyst still retained a polarization curve similar to that in the initial cycle (Fig. 5), which should be ascribed to the unique wrapped structure and synergistic effects between CoO and flexible porous graphene. However, the stability of a GE–CoO modified GC electrode was not as good as that of the PGE–CoO. After 1000 cycles, the anodic current density of the GE–CoO electrode was reduced by about 20% (from 21.3 mA cm−2 to 16.9 mA cm−2 at 0.8 V, vs. Ag/AgCl). In addition, the ex situ SEM also confirmed that the morphology of PGE–CoO is well maintained; however, the particles of GE–CoO all aggregated together after the stability test (see Fig. S9†). This further demonstrates the advantage of the wrapped structure of the PGE–CoO composite, in which CoO particles are well confined by the porous graphene, which could not only prevent aggregation, but also protect CoO particles from corrosion, and therefore enhance the stability.
 | ||
| Fig. 5 The stability of PGE–CoO and GE–CoO modified electrodes before and after LSV testing for 1000 cycles. | ||
The excellent catalytic activity and durability indicate that PGE–CoO is an efficient OER catalyst, which could be attributed to the porous, wrapped structure and the strong chemical and electronic coupling between graphene and CoO. Normally, there are three intermediate steps in the oxygen evolution process. These include the adsorption of water onto the electrode surface, splitting of water into molecular oxygen and oxygen evolution. The porous structure of graphene can provide large specific surface areas for water adsorption and also play an important role in enabling the cobalt precursor to form the deposition of the active center (CoO particles). The porous structure could efficiently suppress the agglomeration of CoO particles and further expose many more active sites to oxidise OH− to oxygen, which could accelerate the reaction process. This study also illustrates that electrolyte concentration (OH−) is also an important parameter for electrochemical reactions. Electrochemical catalysis using the PGE–CoO catalyst was conducted in a series of KOH solutions with different pH. The results are presented in Fig. 6. It was found that the oxidative catalytic activity gradually decreased with the lowering of alkaline concentration (from pH = 14, 224.89 mA cm−2 to pH = 12, 14.89 mA cm−2 at 0.7 V, vs. Ag/AgCl). Moreover, the high conductivity of graphene can offer fast charge transport between the material and the electrode. The porous structure plays an important role in the access by water to “inner” catalytic layers in the electrocatalytic reaction. The fast diffusional mass transport to the particles further confirms the efficient utilization of the catalyst. The wrapped structure is also attractive because it could not only enhance their interface contact, but also efficiently avoid the corrosion of CoO particles during the test and further enhance the stability. Finally, the synergetic effect between PGE and CoO is also an important factor for the enhanced catalytic activity and high durability.
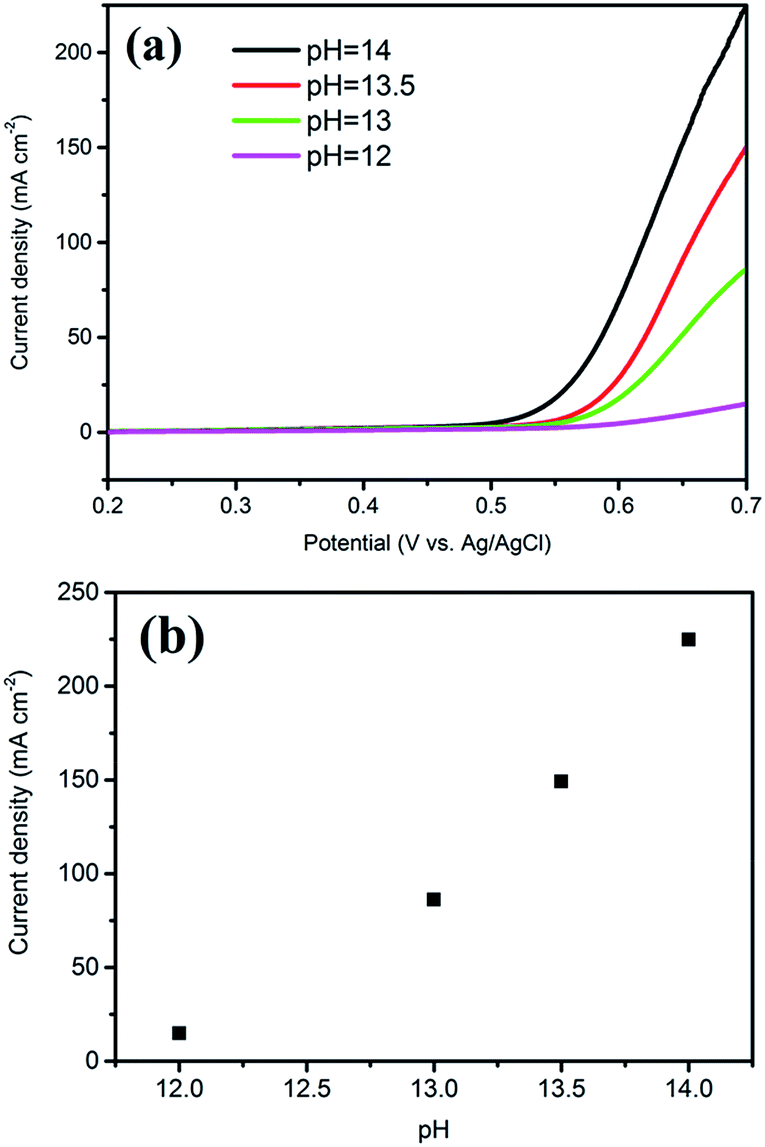 | ||
| Fig. 6 (a) Polarization curves for PGE–CoO at various KOH concentrations. (b) Current densities at 0.7 V vs. Ag/AgCl for PGE–CoO in solutions with various KOH concentrations. | ||
Conclusions
In conclusion, we devised a noble-metal free electrocatalytic material for efficient oxygen evolution. The material consists of a well-organized structure, in which the CoO nanoparticles are well wrapped by porous graphene. The highly porous structure and the excellent chemical and electronic coupling within the composite material lead to the superior OER activity with a low overpotential and high catalytic current density. Furthermore, PGE–CoO also exhibited good stability in alkaline solutions, indicating that the wrapped structure could efficiently prevent the corrosion of CoO nanoparticles. PGE–CoO could be a promising electrocatalyst for the oxygen evolution reaction.Acknowledgements
This project is financially supported by the Australian Research Council (ARC) through the ARC Discovery project (DP1093855), ARC Future Fellowship project (FT110100800), ARC DECRA project (DE140100619), Ministry of Science and Technology (2012DFR40240) and partially supported from the China Scholarship Council (CSC, no. 201306030039).Notes and references
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- K. S. Joya, J. L. Valles-Pardo, Y. F. Joya, T. Eisenmayer, B. Thomas, F. Buda and H. J. M. de Groot, ChemPlusChem, 2013, 78, 35–47 CrossRef CAS.
- Y. F. Zhao, Y. X. Zhang, Z. Y. Yang, Y. M. Yan and K. N. Sun, Sci. Technol. Adv. Mater., 2013, 14, 043501 CrossRef.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed.
- T. Zidki, L. H. Zhang, V. Shafirovich and S. V. Lymar, J. Am. Chem. Soc., 2012, 134, 14275–14278 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516–4521 CrossRef CAS PubMed.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- S. Chen, J. J. Duan, J. R. Ran, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2013, 6, 3693–3699 CAS.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- W. J. Zhou, X. J. Wu, X. H. Cao, X. Huang, C. L. Tan, J. Tian, H. Liu, J. Y. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- Y. Gorlin, C. J. Chung, J. D. Benck, D. Nordlund, L. Seitz, T. C. Weng, D. Sokaras, B. M. Clemens and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 4920–4926 CrossRef CAS PubMed.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400–2408 CAS.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- H. Tuysuz, Y. J. Hwang, S. B. Khan, A. M. Asiri and P. D. Yang, Nano Res., 2013, 6, 47–54 CrossRef.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CAS.
- D. D. Wang, X. Chen, D. G. Evans and W. S. Yang, Nanoscale, 2013, 5, 5312–5315 RSC.
- X. J. Liu, Z. Chang, L. Luo, T. H. Xu, X. D. Lei, J. F. Liu and X. M. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- Y. G. Li, P. Hasin and Y. Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS PubMed.
- C. Jin, F. L. Lu, X. C. Cao, Z. R. Yang and R. Z. Yang, J. Mater. Chem. A, 2013, 1, 12170–12177 CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- G. L. Wang, D. X. Cao, C. L. Yin, Y. Y. Gao, J. L. Yin and L. Cheng, Chem. Mater., 2009, 21, 5112–5118 CrossRef CAS.
- X. Y. Lu and C. Zhao, J. Mater. Chem. A, 2013, 1, 12053–12059 CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- J. Wu, Y. Xue, X. Yan, W. S. Yan, Q. M. Cheng and Y. Xie, Nano Res., 2012, 5, 521–530 CrossRef CAS.
- S. Mao, Z. H. Wen, T. Z. Huang, Y. Hou and J. H. Chen, Energy Environ. Sci., 2014, 7, 609–616 CAS.
- C. D. Liang, K. L. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- D. Feng, Y. Y. Lv, Z. X. Wu, Y. Q. Dou, L. Han, Z. K. Sun, Y. Y. Xia, G. F. Zheng and D. Y. Zhao, J. Am. Chem. Soc., 2011, 133, 15148–15156 CrossRef CAS PubMed.
- X. C. Dong, H. Xu, X. W. Wang, Y. X. Huang, M. B. Chan-Park, H. Zhang, L. H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- L. Liao, J. Zhu, X. J. Bian, L. N. Zhu, M. D. Scanlon, H. H. Girault and B. H. Liu, Adv. Funct. Mater., 2013, 23, 5326–5333 CrossRef CAS.
- J. Zhu, J. W. Tang, L. Z. Zhao, X. F. Zhou, Y. H. Wang and C. Z. Yu, Small, 2010, 6, 276–282 CrossRef CAS PubMed.
- B. Sun, H. Liu, P. Munroe, H. Ahn and G. X. Wang, Nano Res., 2012, 5, 460–469 CrossRef CAS PubMed.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, J. Mater. Chem., 2012, 22, 13826–13831 RSC.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. M. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- M. R. Gao, X. Cao, Q. Gao, Y. F. Xu, Y. R. Zheng, J. Jiang and S. H. Yu, ACS Nano, 2014, 8, 3970–3978 CrossRef CAS PubMed.
- D. S. Kong, H. T. Wang, Z. Y. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed.
- C. C. L. McCrory, S. H. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- L. Liao, S. N. Wang, J. J. Xiao, X. J. Bian, Y. H. Zhang, M. D. Scanlon, X. L. Hu, Y. Tang, B. H. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 CAS.
- R. L. Doyle and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 5224–5237 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image of silica nanorod templates, low magnification SEM image of PGE–CoO, SEM images of pure CoO and nonporous GE–CoO, SEM images of PGE–CoO and GE–CoO after the stability test, nitrogen adsorption/desorption isotherm of PGE, PGE–CoO and GE–CoO, element mapping of PGE–CoO, Nyquist plots of the PGE–CoO, GE–CoO and CoO modified electrodes, cyclic voltammograms of PGE–CoO and GE–CoO, and equivalent electrical circuit. See DOI: 10.1039/c5ta00158g |
| This journal is © The Royal Society of Chemistry 2015 |