
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A spiro-bifluorene based 3D electron acceptor with dicyanovinylene substitution for solution-processed non-fullerene organic solar cells†
Debin
Xia
,
Dominik
Gehrig
,
Xin
Guo
 ,
Martin
Baumgarten
*,
Frédéric
Laquai
* and
Klaus
Müllen
*
,
Martin
Baumgarten
*,
Frédéric
Laquai
* and
Klaus
Müllen
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: baumgart@mpip-mainz.mpg.de; laquai@mpip-mainz.mpg.de; muellen@mpip-mainz.mpg.de
First published on 20th April 2015
Abstract
A novel electron acceptor, namely 2,2′-(12H,12′H-10,10′-spirobi[indeno[2,1-b]fluorene]-12,12′-diylidene)dimalononitrile (4CN-spiro), exhibiting a three-dimensional molecular structure was synthesized and its thermal, photophysical, electrochemical, crystal, and photovoltaic properties were investigated. The novel acceptor exhibits excellent thermal stability with a decomposition temperature of 460 °C, an absorption extending to 600 nm, and a LUMO level of −3.63 eV. Solution processed bulk-heterojunction (BHJ) organic solar cells were fabricated using 4CN-spiro as an acceptor and polythieno[3,4-b]-thiophene-co-benzodithiophene (PTB7) as a donor polymer. The effect of the donor-to-acceptor ratio and processing conditions on the device performance was investigated. A device processed from tetrachloroethane with a donor to acceptor weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 yielded a power conversion efficiency (PCE) of 0.80%.
:1 yielded a power conversion efficiency (PCE) of 0.80%.
Introduction
Organic photovoltaic (OPV) devices that can be fabricated by different techniques, such as vacuum deposition,1–3 solution processing and roll-to-roll printing,4,5 have received a great deal of attention. Remarkable progress has been made on solution-processed, bulk-heterojunction (BHJ) devices,6–13 and the reported power conversion efficiency (PCE) has recently exceeded 10% in single junction cells.14 Undoubtedly, fullerene-based electron acceptors have played a vital role in achieving high PCEs owing to their high electron mobility, strong electron affinity and three-dimensional (3D) electron transport.15–18 However, fullerene acceptors have a few obvious disadvantages, such as low absorption in the visible region, limited energy-level variability, as well as tough synthesis and purification, which limit further improvements of fullerene-based OPV devices and their commercial applications.In the ongoing search for non-fullerene acceptors, special attention has been paid to small molecule acceptors.19–30 Compared to one-dimensional (1D) and two-dimensional (2D) analogues, 3D acceptors have attracted much more scientific interest because of their unique advantages, specifically: the strong self-aggregation propensity of acceptors31 might be prevented by the 3D arrangement, e.g. perylenediimide substituted spiro-bifluorene, thiophene or triphenylamine,32–34 which could avoid undesirable large scale phase separation from the donor. Furthermore, 3D isotropic electron transport pathways as exhibited by fullerene derivatives are formed in BHJ OPVs, enhancing the exciton diffusion/separation efficiencies and the PCE of the devices.35,36
Non-fullerene 3D electron-withdrawing materials have shown good performance in OPVs including diketopyrrolopyrrole,37 benzothiadiazole,38 perylenediimide,32–35,39 naphthalenediimide and bifluorenylidene derivatives used as acceptors.40–42 However, to our knowledge, there have been no reports on dicyanovinylene-substituted 3D acceptors for OPVs, even though dicyanovinylene derivatives have already proven their potential as n-type organic semiconductors.43–46
Herein, we report the synthesis and characterization of a novel 3D small-molecule acceptor 2,2′-(12H,12′H-10,10′-spirobi[indeno[2,1-b]fluorene]-12,12′-diylidene)dimalononitrile (4CN-spiro) (Scheme 1). We demonstrate its potential as a non-fullerene electron acceptor in BHJ solar cells. Using a simple spin-coating fabrication technique, with polythieno[3,4-b]-thiophene-co-benzodithiophene (PTB7)47 as an electron donor, the best device efficiency was obtained when processed from tetrachloroethane, while further solvent additives like diiodooctane did not improve the performance further.
 | ||
| Scheme 1 Synthetic route towards 4CN-spiro. | ||
Results and discussion
Synthesis and characterization
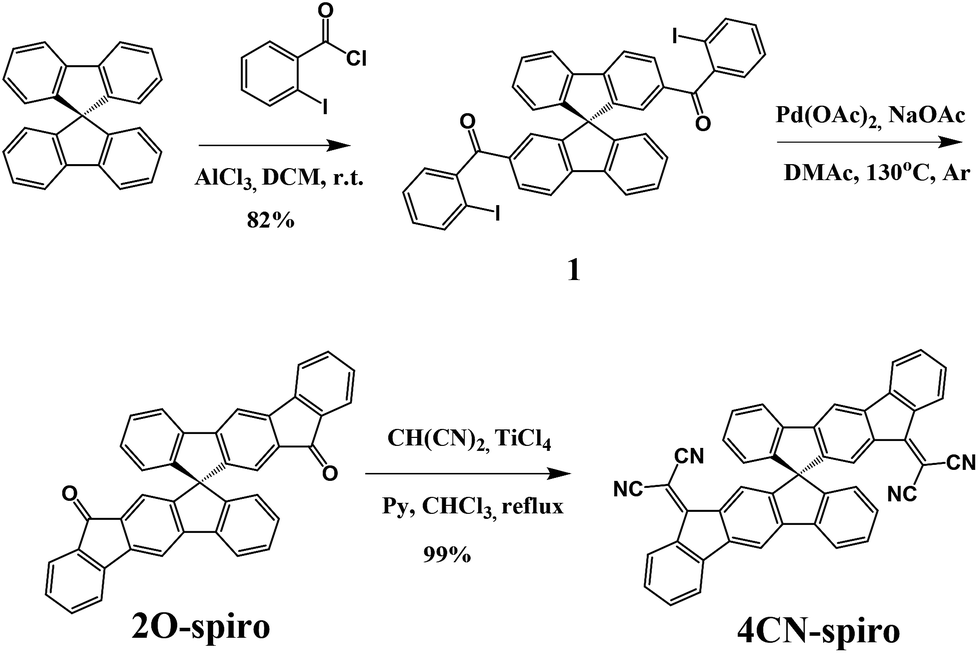
The synthetic route towards 2,2′-(12H,12′H-10,10′-spirobi[indeno[2,1-b]fluorene]-12,12′-diylidene)dimalononitrile (4CN-spiro) is shown in Scheme 1. 9,9-Spiro-bifluorene was acylated by 2-iodobenzoyl chloride to give 9,9′-spirobi[fluorene]-2,2′-diylbis((2-iodophenyl)methadone) (1) in 82% yield. The intermediate compound 12H,12′H-10,10′-spirobi[indeno[2,1-b]fluorene]-12,12′-dione (2O-spiro) was synthesized by a cyclization reaction using palladium acetate as a catalyst and sodium acetate as a base. Finally, the target product 4CN-spiro was obtained in 99% yield by the Knoevenagel condensation of 2O-spiro with the Lehnert reagent48,49 (TiCl4, malononitrile, pyridine). 2O-spiro and 4CN-spiro were fully characterized by FD-Mass, HRMS, 1H NMR, and 13C NMR. The molecular structures were further confirmed by single crystal X-ray analysis (see Crystallography section below). Thermogravimetric analysis (TGA) of 2O-spiro and 4CN-spiro revealed excellent thermal stability, with 5% weight loss occurring at 423 °C and 460 °C, respectively (Fig. 1a).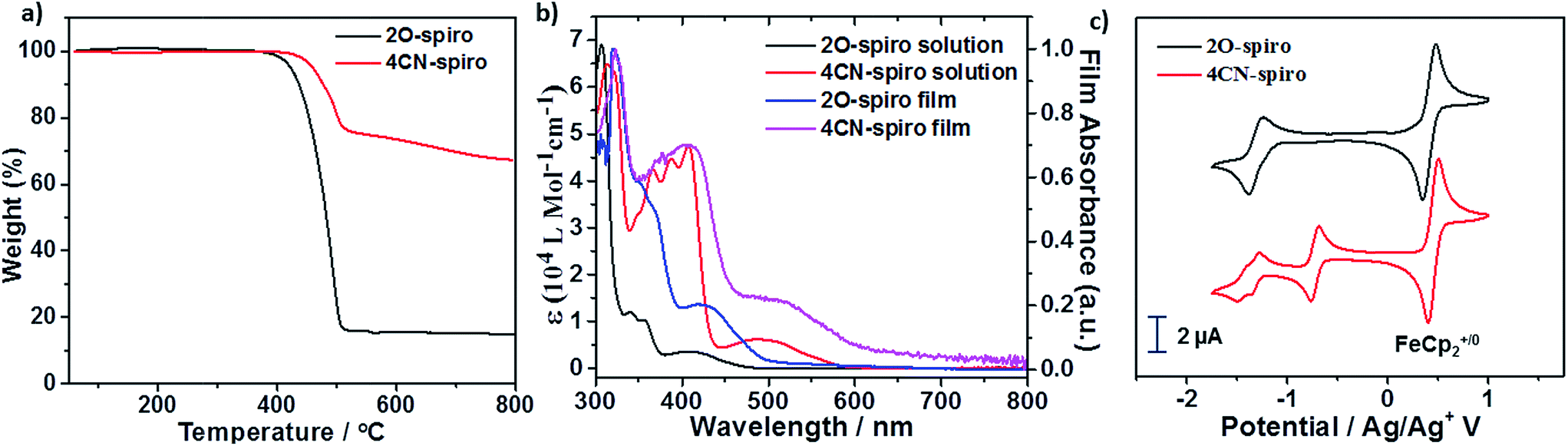 | ||
| Fig. 1 Thermal, photophysical and electrochemical properties of 2O-spiro and 4CN-spiro. (a) TGA curves; (b) UV-vis absorption spectra in dichloromethane solution and in thin film; (c) cyclic voltammograms in dichloromethane/0.1 M nBu4NPF6 at 100 mV s−1. | ||
Optical and electrochemical properties
Fig. 1b shows the absorption spectra of 2O-spiro and 4CN-spiro in dichloromethane solution and in thin solid films. The absorption bands of 2O-spiro and 4CN-spiro in the thin film spectra are only slightly red-shifted compared to those in solution. This is different from the previously reported 3D star-shaped materials,34,37 whose absorption bands are significantly red-shifted in the solid state, which might be due to the different electron donating and electron withdrawing groups (the push–pull effect). The optical gap (Eg) estimated from the absorption edge of the solution spectrum is 2.56 eV for 2O-spiro and 2.08 eV for 4CN-spiro, respectively.The electrochemical properties of 2O-spiro and 4CN-spiro were investigated by cyclic voltammetry (CV) in dichloromethane solution with 0.1 M nBu4NPF6 as the supporting electrolyte. As shown in Fig. 1c, both 2O-spiro and 4CN-spiro exhibit reversible reduction waves. No oxidation waves could be observed in the measured potential range. The half-wave potential of 2O-spiro is −1.29 V. Upon dicyanovinylene functionalization, the reduction potentials of 4CN-spiro shift to more positive values with the E1/2 potentials at −0.72, −1.31 and −1.44 V, respectively. The overlapping second and third reduction waves of 4CN-spiro with a lower associated current are similar to those of dicyanovinylene-functionalized (bis)indenofluorenes.43 The LUMO energy levels of 2O-spiro and 4CN-spiro estimated from the equation ELUMO = −[Ered1/2 − EFc1/2 + 4.8] eV are −3.09 eV and −3.63 eV, respectively. The strong electron-withdrawing character of dicyanovinylene causes a low LUMO level of 4CN-spiro, which guarantees sufficient driving force for exciton dissociation in OPV devices. The HOMO energies of 2O-spiro and 4CN-spiro are virtually identical to the value of −5.80 eV, calculated from the optical gap according to the equation HOMO = LUMO − Eg.
Crystallography
Single crystals of the new compounds 2O-spiro and 4CN-spiro were grown by slow evaporation of the solvent mixture dichloromethane–hexane, and pure dichloromethane, respectively. The crystal structure determined by X-ray diffraction is presented in Fig. 2. The functionalized 9,9-spiro-bifluorene consists of two identical fluorene π-systems, which are perpendicular to each other via a common sp3-hybridized carbon atom. Crystal packing of the two molecules is dominated by π–π stacking interactions. The π-stacking of 2O-spiro obviously extends into three dimensions in the single crystal, as it can clearly be seen from the packing structure (Fig. 2a). Three π-stacking axes are almost perpendicular to each other. To the best of our knowledge, this is the first example of an organic semiconductor that can adopt a 3D isotropic π-stacking. In the case of the acceptor 4CN-spiro, π-stacking with interplanar distances of ca. 3.48 Å along the a axis and 3.40 Å along the b axis (Fig. 2b) was observed. Therefore, the packing geometry of the two compounds indicates the possibility of π–π interactions, implying that isotropic electron transport pathways as in fullerene derivatives could potentially be formed in donor–acceptor BHJ solar cells.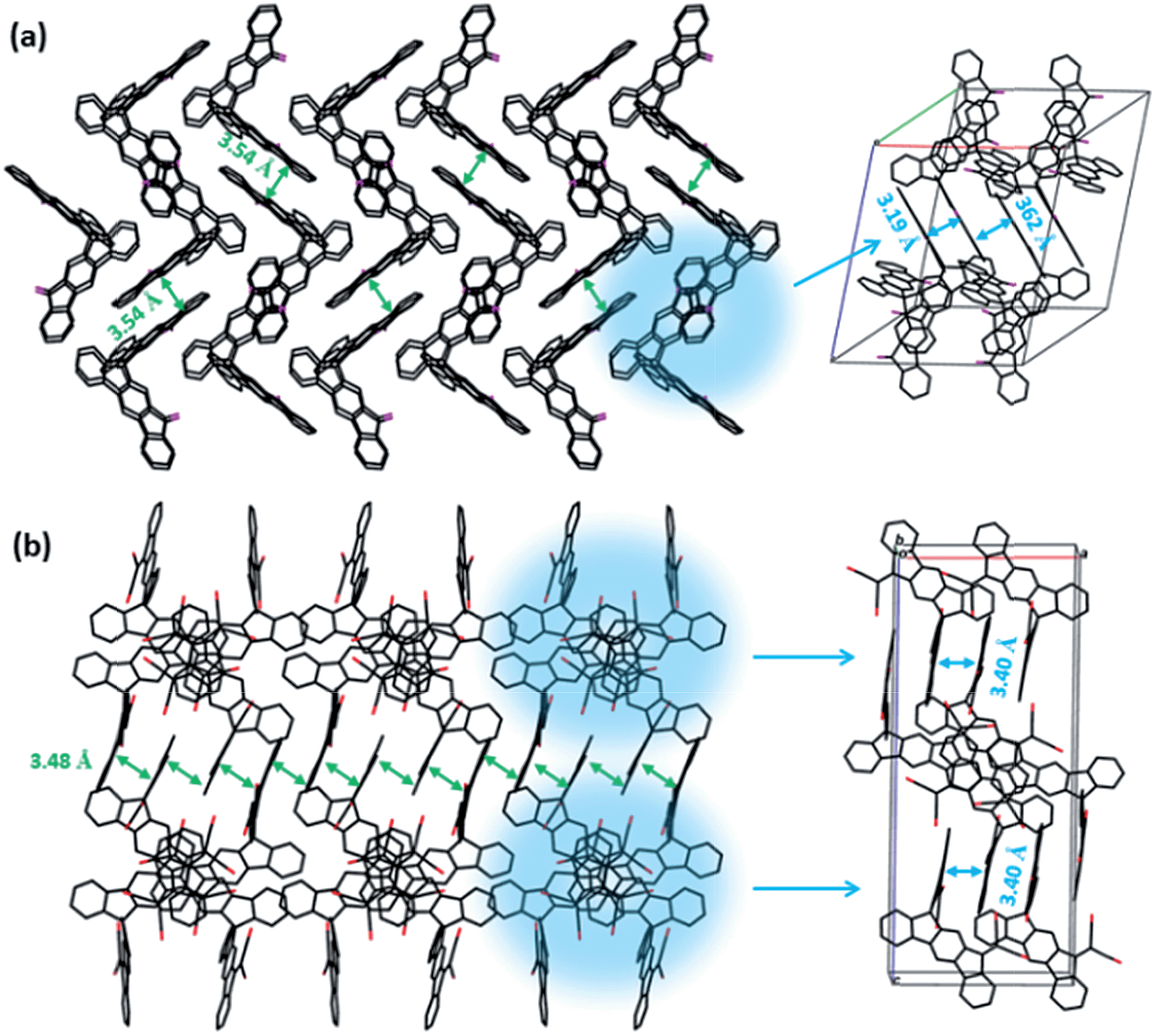 | ||
| Fig. 2 Crystal packing diagrams of 2O-spiro (a) and 4CN-spiro (b) projected along the a axis. C, black; O, purple; N, red. | ||
Photovoltaic properties
To demonstrate the potential application of 4CN-spiro as an acceptor in photovoltaic devices, it was blended with PTB7 as the electron donor polymer, whose absorption band is more red-shifted than that of P3HT and partially complementary to the absorption of 4CN-spiro. BHJ OPV cells of the structure ITO/PEDOT:PSS/PTB7:4CN-spiro/Ca/Al were prepared. In addition, the effects of varying processing solvents and blend composition were investigated. Table 1 summarizes the obtained open-circuit voltage (Voc), short-circuit current density (Jsc), fill factor (FF), and PCE of the devices.| Blend ratio (A/D) | Solvent | V oc [V] | J SC [mA cm−2] | FF | PCE [%] |
|---|---|---|---|---|---|
| a TCE: tetrachloroethane and DCB: dichlorobenzene. | |||||
| 1:2 |
TCE | 0.87 | 1.13 | 0.49 | 0.64 |
| 1:1 |
TCE | 0.89 | 1.41 | 0.48 | 0.80 |
| 1:1 |
TCE/DCB | 0.89 | 0.22 | 0.19 | 0.05 |
| 1:1 |
DCB | 0.09 | 0.10 | 0.09 | 0.01 |
| 1:1 |
TCE/3%DIO | 0.42 | 0.03 | 0.20 | 0.00 |
The device prepared from tetrachloroethane (TCE)with a donor–acceptor weight ratio of 1:2 exhibited an open-circuit voltage of 0.87 V, a short-circuit current density of 1.13 mA cm−2, a fill factor of 0.49, and a power conversion efficiency of 0.64% (Fig. 3). Encouragingly, the blend at a donor–acceptor weight ratio of 1:1 demonstrated an increased PCE of up to 0.80% with a Voc, Jsc and FF of 0.89 V, 1.41 mA cm−2 and 0.48, respectively. We note that TCE is a rather uncommon solvent in photovoltaic device preparation, however, TCE is a good solvent for the acceptors used in the present study. AFM height images show a homogeneous surface structure of the blend with demixing on the length scale of exciton diffusion, which ensures efficient exciton quenching and dissociation (Fig. 4). We have also tried further device optimization including the use of solvent additives. In fact, a small amount of 1,8-diiodooctane (DIO) in a volume ratio of 3% was used as a solvent additive to improve the photovoltaic performance in PTB7:PCBM devices.47 However, in the present case the device prepared with the additive exhibited no response to light at all, even though the AFM images indicated a rather uniform surface structure (Fig. S2†). This implies that other processes such as geminate recombination of interfacial charge-transfer (CT) states or insufficient charge carrier transport due to a lack of charge carrier percolation pathways limit the photovoltaic performance. Furthermore, we have also investigated the device performance upon using DCB as a solvent or a mixture of DCB and TCE. However, in these cases, the AFM images indicated the formation of large crystallites as high RMS values were observed. This is detrimental to the device efficiency (Fig. S2†) as demonstrated also by the low PCE value. We note that even for the optimized device only a moderate short circuit current and fill factor were obtained compared to devices that use fullerene as an acceptor. The latter is a consequence of the pronounced bias dependence of the current density, which does not even saturate at high negative bias, as previously also observed by us for polymer–PDI blends.50 To better understand the origin of the former, that is, the moderate short circuit current of the PTB7:4CN-spiro blends spun from C2D2Cl4, transient absorption (TA) spectroscopy was performed on the picosecond to nanosecond timescale. The transient absorption spectra shown in Fig. 5a are dominated by a positive feature in the spectral region from 540 to 830 nm peaking at 685 nm, which we assigned to a combination of the ground-state bleaching (GSB) of PTB7, as it coincides with the ground state absorption of the polymer and stimulated emission (SE) of PTB7 singlet excitons. The observation of SE points towards inefficient exciton quenching in these blends, in part explaining the lower device performance compared to PTB7:fullerene blends. Fig. 5b shows the decay dynamics of the GSB at various excitation intensities. Clearly, the signal decay is independent of the excitation intensity pointing towards geminate recombination of charges that do not manage to entirely dissociate into free charges. We note that a substantial fraction of >80% of the initially created excited states decays on the sub-ns timescale and consequently does not contribute to the photocurrent of the device. Thus, we conclude that the devices are limited by both inefficient exciton quenching as well as pronounced geminate recombination of bound charges on the sub-ns timescale.
 | ||
| Fig. 3 Current density–voltage (J–V) curve of the OPV cell processed from tetrachloroethane with a donor–acceptor weight ratio of 1:1. | ||
 | ||
| Fig. 4 AFM height images of a PTB7:4CN-spiro (1:1, w/w) blend processed from tetrachloroethane. | ||
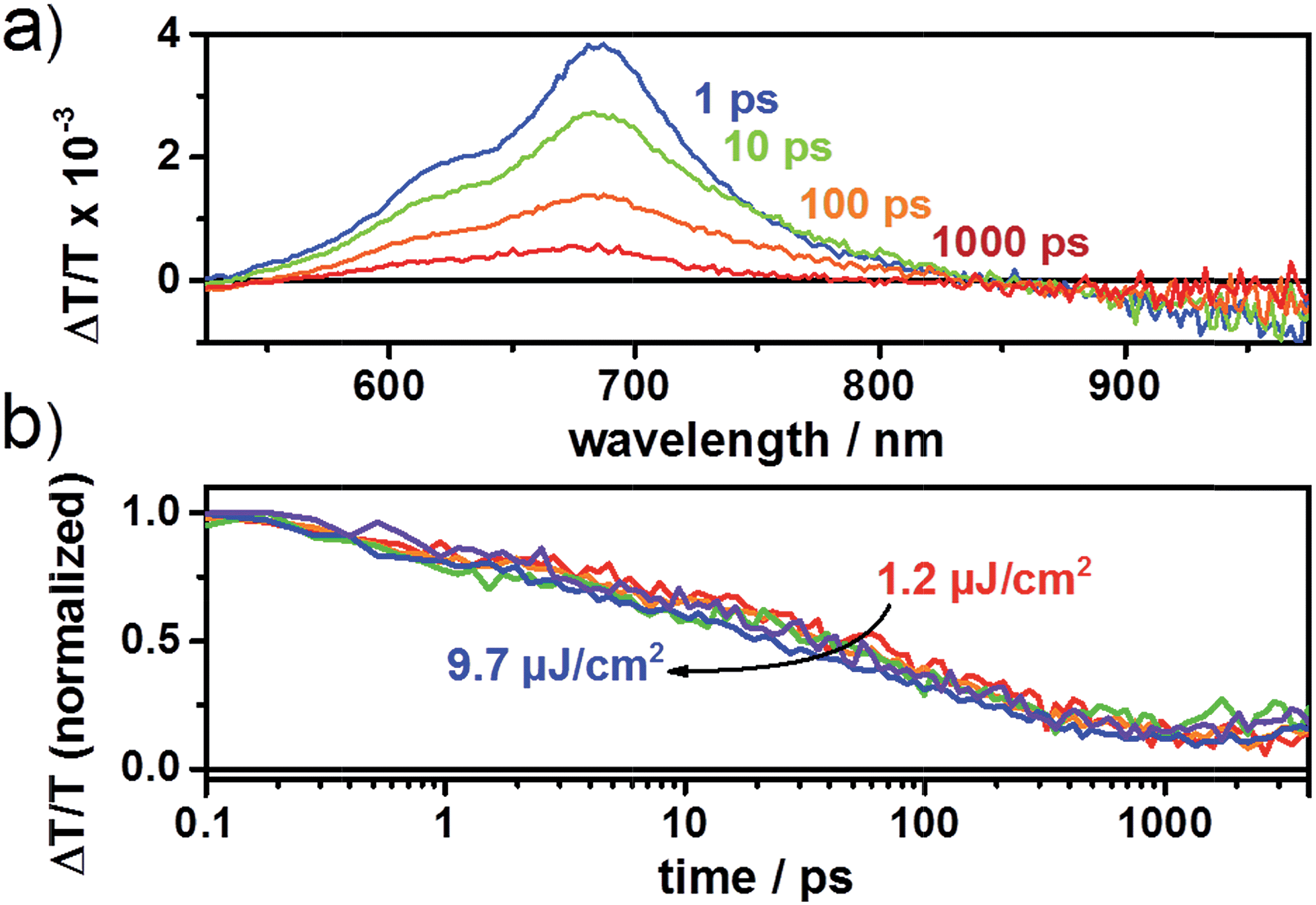 | ||
| Fig. 5 (a) ps–ns transient absorption spectra at different time delays after photoexcitation at 650 nm with a fluence of 6.0 μJ cm−2 and (b) ground-state bleaching (680–700 nm) dynamics at various excitation densities. | ||
Conclusions
In conclusion, a novel 3D acceptor 4CN-spiro containing spiro-bifluorene as the core was synthesized and fully characterized. As revealed by single-crystal analysis, the new compound presents the possibility of isotropic charge transport, which is similar to the situation in fullerene derivatives. The solar cells based on PTB7:4CN-spiro processed from tetrachloroethane yielded the highest PCE of 0.80%. Our results demonstrated for the first time that dicyanovinylene substituted 4CN-spiro could be an alternative to fullerene-based acceptors. However, we also observed that the device performance is limited by incomplete exciton quenching and fast geminate recombination on the sub-ns timescale. Further experiments are required to determine whether the bias dependence of the photocurrent is caused by field-dependent charge generation or limited by a low charge carrier mobility leading to a competition of charge extraction and non-geminate recombination or a combination of both processes. Finally, future work will aim towards new acceptor structures to improve charge separation and to overcome the limits set by geminate recombination and the bias dependence observed in the present study.Experimental section
Synthesis
Materials and characterization
1H NMR and 13C NMR spectra were recorded in deuterated solvents such as C2D2Cl4, using a Bruker DPX 500 spectrometer, with the solvent proton or carbon signal as an internal standard. FD mass spectra were recorded with a VG-Instrument ZAB 2-SE-FDP. High resolution mass spectra (HRMS) were recorded by the Microanalytical Laboratory of Johannes Gutenberg-University, Mainz. UV-vis absorption spectra were recorded at room temperature using a Perkin Elmer Lambda 900 spectrophotometer. Fluorescence spectra were recorded on a SPEX-Fluorolog II (212) spectrometer. CV measurements were carried out on a computer-controlled GSTAT12 in a three-electrode cell in a DCM solution of Bu4NPF6 (0.1 M) with a scan rate of 100 mV s−1 at room temperature, with a glassy carbon disc as the working electrode, a Pt wire as the counter electrode, and an Ag/AgCl electrode as the reference electrode. Thermogravimetric analysis (TGA) was carried out on a Mettler 500 at a heating rate of 10 °C min−1 under nitrogen flow. All reagents and starting materials were obtained from commercial suppliers and used without further purification. Column chromatography was performed on silica gel 60 (Macherey-Nagel, Si60) with dichloromethane, hexane, ethyl acetate or tetrahydrofuran (Sigma-Aldrich). All reported yields are isolated yields.Solar cell preparation and measurement
Solar cells were fabricated on patterned ITO-coated glass substrates (Präzisions Glas & Optik GmbH, Germany). Cleaning included successive ultrasonication in detergent, acetone and isopropanol. Furthermore, the samples were treated with an argon plasma before spincoating an ∼40 nm thick poly(3,4-ethylene-dioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) (Clevios P VP al 4083, H.C. Stark) layer. The substrates were heated to 120 °C for 30 min in a nitrogen-filled glovebox. The active layer was deposited via spin-coating a 1:1 mixture of the donor poly(3-fluoro-2-[(2ethylhexyl)carbonyl]-thieno[3,4-b]thiophenediyl) (PTB7) and the acceptor (4CN-spiro) at 800 rpm. The total weight concentration was 15 mg mL−1 in 1,1,2,2-tetrachloroethane, 20 mg mL−1 in a 1,1,2,2-tetrachloroethane and dichlorobenzene (DCB) mixture (1:1 by volume), 25 mg mL−1 in DCB and 15 mg mL−1 in 1,1,2,2-tetrachloroethane containing 3 vol% 1,8-diiodooctane. Subsequent to the active layer deposition, a bilayer of 5 nm calcium and 100 nm aluminum was evaporated through a shadow mask.
I–V characteristics were obtained under illumination with a solar simulator (K.H. Steuernagel Lichttechnik GmbH, Germany) using a 575 W metal halide lamp in combination with a filter system to create a spectrum according to AM1.5G conditions. Yet, the intensity was 70 mW cm−2. Current–voltage curves were measured with a Keithley 236 Source Measure Unit (SMU) within a glovebox. The light intensity was measured with a calibrated silicon photodiode.
AFM images were taken with a Dimension Icon FS with ScanAsyst using an Olympus OMCL-AC 240TS-W2 Cantilever Type at F0 = 70 kHz in non-contact mode.
Transient absorption spectroscopy was described previously by our group. Transient absorption (TA) measurements were performed with a home-built pump–probe setup. To measure in the time range of 1–4 ns with a resolution of ∼100 fs, the output of a commercial titanium:sapphire amplifier (Coherent LIBRA-HE, 3.5 mJ, 1 kHz, 100 fs) was split into two beams that pumped two independent commercial optical parametric amplifiers (Coherent OPerA Solo). One optical parametric amplifier (OPA) was used to generate the tunable excitation pulses in the visible range, while the second OPA was used to generate the seed beam for white-light generation. For measurements in the spectral range between 550 and 1100 nm, a 1300 nm seed of a few μJ was focused into a c-cut 3 mm thick sapphire window for white-light generation. The variable delay of up to 4 ns between the pump and the probe was introduced by a broadband retroreflector mounted on a mechanical delay stage. Mostly reflective elements were used to guide the probe beam to the sample to minimize chirp. The excitation pulse was chopped at 500 Hz, while the white-light pulses were dispersed onto a linear silicon photodiode array, which was read out at 1 kHz by home-built electronics. Adjacent diode readings corresponding to the transmission of the sample after an excitation pulse and without an excitation pulse were used to calculate ΔT/T.
Acknowledgements
This work was financially supported by the Transregio TR49. We acknowledge Dr. Dieter Schollmeyer in Johannes Gutenberg-University for crystal structure analysis. Dr. Frédéric Laquai thanks the Max Planck Society for funding the Max Planck Research Group of Organic Optoelectronics. FL and KM thank the Deutsche Forschungsgemeinschaft (DFG) for funding in the framework of the priority program SPP1355 “Elementary Processes in Organic Photovoltaics”. D.G. acknowledges a Kekulé scholarship of the Fonds der Chemischen Industrie (FCI).Notes and references
- R. Fitzner, C. Elschner, M. Weil, C. Uhrich, C. Körner, M. Riede, K. Leo, M. Pfeiffer, E. Reinold, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2012, 24, 675–680 CrossRef CAS PubMed
.
- A. Mishra, D. Popovic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2014, 26, 7217–7223 CrossRef CAS PubMed
- R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, H. Ziehlke, C. Körner, K. Leo, M. Riede, M. Weil, O. Tsaryova, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, Adv. Funct. Mater., 2011, 21, 897–910 CrossRef CAS PubMed
- T. R. Andersen, H. F. Dam, M. Hosel, M. Helgesen, J. E. Carle, T. T. Larsen-Olsen, S. A. Gevorgyan, J. W. Andreasen, J. Adams, N. Li, F. Machui, G. D. Spyropoulos, T. Ameri, N. Lemaitre, M. Legros, A. Scheel, D. Gaiser, K. Kreul, S. Berny, O. R. Lozman, S. Nordman, M. Valimaki, M. Vilkman, R. R. Sondergaard, M. Jorgensen, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 2925–2933 CAS
- G. D. Spyropoulos, P. Kubis, N. Li, D. Baran, L. Lucera, M. Salvador, T. Ameri, M. M. Voigt, F. C. Krebs and C. J. Brabec, Energy Environ. Sci., 2014, 7, 3284–3290 CAS
- A. K. K. Kyaw, D. H. Wang, V. Gupta, J. Zhang, S. Chand, G. C. Bazan and A. J. Heeger, Adv. Mater., 2013, 25, 2397–2402 CrossRef CAS PubMed
- J. Zhou, X. Wan, Y. Liu, Y. Zuo, Z. Li, G. He, G. Long, W. Ni, C. Li, X. Su and Y. Chen, J. Am. Chem. Soc., 2012, 134, 16345–16351 CrossRef CAS PubMed
- Y. Zhang, X.-D. Dang, C. Kim and T.-Q. Nguyen, Adv. Energy Mater., 2011, 1, 610–617 CrossRef CAS PubMed
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. El Labban, G. O. Ngongang Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608–9618 CrossRef CAS PubMed
- J. E. Coughlin, Z. B. Henson, G. C. Welch and G. C. Bazan, Acc. Chem. Res., 2013, 47, 257–270 CrossRef PubMed
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed
- C. D. Wessendorf, G. L. Schulz, A. Mishra, P. Kar, I. Ata, M. Weidelener, M. Urdanpilleta, J. Hanisch, E. Mena-Osteritz, M. Lindén, E. Ahlswede and P. Bäuerle, Adv. Energy Mater., 2014, 4, 1400266 Search PubMed
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 593–597 CrossRef CAS
- F. G. Brunetti, R. Kumar and F. Wudl, J. Mater. Chem., 2010, 20, 2934–2948 RSC
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. Van Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208–212 CrossRef CAS PubMed
- G. Zhao, Y. He and Y. Li, Adv. Mater., 2010, 22, 4355–4358 CrossRef CAS PubMed
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC
- L. Chen, L. Huang, D. Yang, S. Ma, X. Zhou, J. Zhang, G. Tu and C. Li, J. Mater. Chem. A, 2014, 2, 2657–2662 CAS
- Y. Zhou, L. Ding, K. Shi, Y.-Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957–961 CrossRef CAS PubMed
- Y.-Q. Zheng, Y.-Z. Dai, Y. Zhou, J.-Y. Wang and J. Pei, Chem. Commun., 2014, 50, 1591–1594 RSC
- Y. Zhou, Y.-Z. Dai, Y.-Q. Zheng, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Commun., 2013, 49, 5802–5804 RSC
- Y. Lin, Y. Li and X. Zhan, Adv. Energy Mater., 2013, 3, 724–728 CrossRef CAS PubMed
- A. Sharenko, D. Gehrig, F. Laquai and T.-Q. Nguyen, Chem. Mater., 2014, 26, 4109–4118 CrossRef CAS
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC
- P. Sonar, J. P. Fong Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558–1574 CAS
- A. a. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201–1213 CAS
- J. E. Anthony, Chem. Mater., 2010, 23, 583–590 CrossRef
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 Search PubMed
- V. Kamm, G. Battagliarin, I. A. Howard, W. Pisula, A. Mavrinskiy, C. Li, K. Müllen and F. Laquai, Adv. Energy Mater., 2011, 1, 297–302 CrossRef CAS PubMed
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed
- Y. Ie, T. Sakurai, S. Jinnai, M. Karakawa, K. Okuda, S. Mori and Y. Aso, Chem. Commun., 2013, 49, 8386–8388 RSC
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435–441 CAS
- Y. Lin, P. Cheng, Y. Li and X. Zhan, Chem. Commun., 2012, 48, 4773–4775 RSC
- Y. Lin, H. Wang, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2013, 1, 14627–14632 CAS
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K. Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed
- F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2010, 49, 532–536 CrossRef CAS PubMed
- H. U. Kim, J.-H. Kim, H. Suh, J. Kwak, D. Kim, A. C. Grimsdale, S. C. Yoon and D.-H. Hwang, Chem. Commun., 2013, 49, 10950–10952 RSC
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272–2277 CrossRef CAS PubMed
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed
- X. Shi, J. Chang and C. Chi, Chem. Commun., 2013, 49, 7135–7137 RSC
- H. Tian, Y. Deng, F. Pan, L. Huang, D. Yan, Y. Geng and F. Wang, J. Mater. Chem., 2010, 20, 7998–8004 RSC
- Q. Wu, S. Ren, M. Wang, X. Qiao, H. Li, X. Gao, X. Yang and D. Zhu, Adv. Funct. Mater., 2013, 23, 2277–2284 CrossRef CAS PubMed
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed
- W. Lehnert, Tetrahedron Lett., 1970, 11, 4723–4724 CrossRef
- W. Lehnert, Synthesis, 1974, 1974, 667–669 CrossRef
- D. W. Gehrig, S. Roland, I. A. Howard, V. Kamm, H. Mangold, D. Neher and F. Laquai, J. Phys. Chem. C, 2014, 118, 20077–20085 CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1035931 and 1035932. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ta00108k |
| This journal is © The Royal Society of Chemistry 2015 |