
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced triplet-state electron transfer of platinum porphyrin: a one-step direct method for sensing iodide with an unprecedented detection limit†
Dilshad
Masih
,
Shawkat M.
Aly‡
,
Erkki
Alarousu
and
Omar F.
Mohammed
*
Solar and Photovoltaics Engineering Research Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: omar.abdelsaboor@kaust.edu.sa
First published on 5th February 2015
Abstract
Here, we report for the first time a one-step direct method for sensing halides in aqueous solution using phosphorescence quenching of platinum-cationic porphyrin. This method offers an easy, rapid, environmentally friendly, ultra-sensitive (with a previously unattained detection limit of 1 × 10−12 M) and economical method for the determination of iodide. To fully understand the reaction mechanism responsible for the phosphorescence quenching process, we have employed cutting-edge time-resolved laser spectroscopy with broadband capabilities.
Anions play a pivotal role in biological, chemical and environmental processes, and they have begun to receive increasing attention.1–3 Among anions, iodide is of special note not only because of its physiological importance4,5 in controlling metabolic activities but also as an essential factor in energy conversion processes.6–11 Both deficiency and excess of iodide can result in malfunctions and disorders in the human body.12–14 In addition to its environmental and biological importance, iodide is a key component of a redox couple often used in solar-energy-harvesting systems.15 Various methods can be used for the determination of iodide,16 including photoluminescence,17–32 colorimetric detection,33–37 time-resolved absorption techniques,38 mass spectrometry,39–41 chromatography,42 Raman scattering43,44 and electrochemical profiling.45,46 However, because of its large size and weakly basic nature, the binding capacity of iodide is the weakest among the halide ions. Therefore, the development of a simple, rapid, direct and economical method for the determination of iodide is still under investigation.
Researchers have recently demonstrated great interest in the development of photoluminescence-based methods for the highly sensitive, rapid and selective determination of iodide.17–30 However, the complex preparation of the fluorophores, their low hydrophilicity, and the fact that the multistep determination of iodide usually involves a toxic reagent (Hg) have hampered the widespread application of photoluminescence-based methods. In this study, we report a 5,10,15,20-tetra(1-methyl-4-pyridino)-porphyrin Pt(II) tetrachloride (Pt(II)TMPyP)-based photoluminescence sensor for the ultra-sensitive, easy, rapid, environmentally friendly and economical determination of iodide. This method represents a one-step direct technique for monitoring iodide in an aqueous phase with a pico-molar (pmol) detection limit. In the presence of iodide, the quenching of the photoluminescence of Pt(II)TMPyP can be directly monitored upon irradiation with visible light. This method does not require complex preparatory steps or an additional species such as Hg, which is usually used for the activation of fluorophores as a fluorescence turn-on signal.20–26 The extreme toxicity and special handling and disposal requirements of Hg pose great concerns and limit the use of sensors activated with Hg. In this study, we not only provide a simple method with a previously unattained (1 × 10−12 M) detection limit in aqueous solution but also propose a detailed mechanism for photoluminescence quenching using cutting-edge ultrafast laser spectroscopy with broadband capabilities. Interestingly, a control experiment using Zn(II)TMPyP clearly indicated that the Pt metal center is the key element for the quenching and the extremely low detection limit.
Both of the investigated porphyrins, Pt(II)TMPyP and Zn(II)TMPyP, were supplied by Frontier Scientific. High-purity halide salts of NH4F, NaCl, KBr and KI were purchased from Sigma-Aldrich. Porphyrins and halide salts were completely dissolved in Milli-Q water, and all spectral measurements were performed without any additions. Absorption spectra were recorded on a Cary 6000i UV-Vis-NIR spectrophotometer (Varian, Inc.). Steady-state photoluminescence spectra were recorded with a Jobin-Yvon-Horiba Fluoromax-4 spectrofluorometer. We utilized femto- and nanosecond transient absorption spectroscopy to probe the photophysical processes that occur upon photoexcitation of Pt(II)TMPyP with and without iodide. The experimental setup is detailed elsewhere.47–49
Fig. 1 displays the steady-state absorption and photoluminescence spectra of Pt(II)TMPyP alone and with various halide ions, i.e., chloride, bromide and iodide. Three milliliters of 12.8 μM Pt(II)TMPyP aqueous solution was added to a quartz cuvette with a 1 cm path length, and an aqueous solution of a halide ion was then added. Samples were excited at 512 nm, and the aperture size for both the entrance and exit slits was kept the same for all experiments. The obtained emission agrees in shape and position with previously reported Pt(II) porphyrin emission which is mainly due to the heavy atom effect of Pt(II) as the central atom leading to efficient intersystem crossing (ISC).50,51 As evident from Fig. 1 (left panel), the change in peak intensity and position in the absorption spectrum of Pt(II)TMPyP after halide addition was negligible. In contrast, a significant intensity quenching in the photoluminescence spectrum upon halide ion addition is clearly evident in the figure. The quenching magnitude for the different halides decreased in the order iodide > bromide > chloride. The addition of 1.2 μmol of chloride led to approximately 28% quenching of the photoluminescence of Pt(II)TMPyP, and the extent of quenching increased down the halogen group, with almost 75 and 90% quenching observed after the addition of similar amounts of bromide (1.05 μmol) and iodide (1.07 μmol), respectively. It is worth mentioning that fluoride did not show any significant tendency to interact with Pt(II)TMPyP (see Fig. S10†).
 | ||
| Fig. 1 Absorption (left) and photoluminescence (right, λex = 512 nm) spectra of Pt(II)TMPyP before and after the addition of chloride, bromide and iodide in the aqueous phase. | ||
The literature contains only a few reports on the ultra-trace, low-pmol-level detection of iodide. Wygladacz et al. reported a fluorescent microsphere fiber optic microsensor array for the low-pmol detection of iodide.29 However, the microsensor exhibited the lowest limit of detection of 3 pmol only at pH 3.5. Recently, Dasary et al. reported a surface-enhanced Raman scattering probe for ultra-sensitive detection.43 An iodide detection limit of 30 pmol was demonstrated on the basis of indirect Raman intensity originating from the desorption of rhodamine from gold nanoparticles.
The detection limit was determined from our photoluminescence quenching results using the 3σ IUPAC method, where the detection limit = 3σ/slope.20,24,26,33,36 A calibration curve for photoluminescence intensity against iodide concentration was first constructed (Fig. S1†). The slope value was taken from the curve, and the standard deviation σ was calculated for the photoluminescence intensity of the blank Pt(II)TMPyP solution in the absence of iodide. Interestingly, a never-before-attained detection limit of 1 pmol was obtained.
The photoluminescence quenching upon the addition of halides indicates the presence of a photoinduced electron transfer event between the halide donor and the Pt(II)TMPyP acceptor. Stern–Volmer plots for all of the halide ions show a deviation from linearity with Pt(II)TMPyP (Fig. S2 and S3†), indicating that a static reaction is involved in the quenching process. The observed negative deviation from linearity in Stern–Volmer plots at very high halide concentration can be attributed to the formation of a luminescent exciplex between the negatively charged halide and the poor electron density on the Pt metallic center (explained below in transient measurements).52 From the relationship KSV = kqτ°,53 where KSV is the Stern–Volmer constant, kq is the bimolecular quenching rate constant and τ° is the phosphorescence lifetime of Pt(II)TMPyP (1.03 μs), we calculated kq ≈ 7.8 × 1012 M−1 s−1. This quenching rate far exceeds the diffusion-controlled limit (the estimated rate for iodide diffusion for an aqueous sample under the current experimental conditions is ∼3 × 1010 M−1 s−1, as determined using the Stokes–Einstein equation),54 establishing the fact that quenching is due to the static interaction between the donor–acceptor components. To confirm the static quenching, the phosphorescence lifetimes of the free Pt(II)TMPyP and in the presence of different iodide concentrations giving 30, 50, and 70% quenching were measured; the results are displayed in Fig. S4.† The time decay curves collected for the four solutions exhibited the same lifetime, confirming the static nature of the quenching mechanism.53 Further confirmation of the static nature of the quenching mechanism is indicated by the similarity of the Stern–Volmer quenching constants for samples in water and methanol (see Fig. S5†). Despite the different viscosities of water and methanol, the KSV values extracted for the iodide interaction with Pt(II)TMPyP in methanol is 8.6 μM−1 and that in water is 8.01 μM−1. The similarity between these values suggests that quenching does not depend on solvent viscosity, which favors a static-interaction explanation.
Similar to the Pt(II)TMPyP and iodide system, 1.07 μmol of iodide was sequentially added to 3 mL of 12.7 μM Zn(II)TMPyP. The steady-state and excited-state absorptions remained essentially the same before and after the iodide addition (Fig. S6 and S7†). For the photoluminescence measurements of Zn(II)TMPyP with the addition of 1.07 μmol iodide, in contrast to the 90% photoluminescence quenching observed in the iodide–Pt(II)TMPyP mixture, only approximately 7% quenching was observed (Fig. S6†). This quenching behavior demonstrates that the change in the photoluminescence was specific to the metal center in the cavity of the porphyrin and pertained to its emissive state, as discussed below.
Transient absorption (TA) spectroscopy is an essential method for investigating photoinduced excited-state interactions.55–57 In the present work, the excited-state interaction of halides with Pt(II)TMPyP was examined using nanosecond (ns) and femtosecond (fs) TA spectroscopy. The recorded ns-TA spectra of aqueous solutions of free Pt(II)TMPyP and its mixtures with chloride, bromide, and iodide collected after 350 nm photoexcitation are shown in Fig. 2.
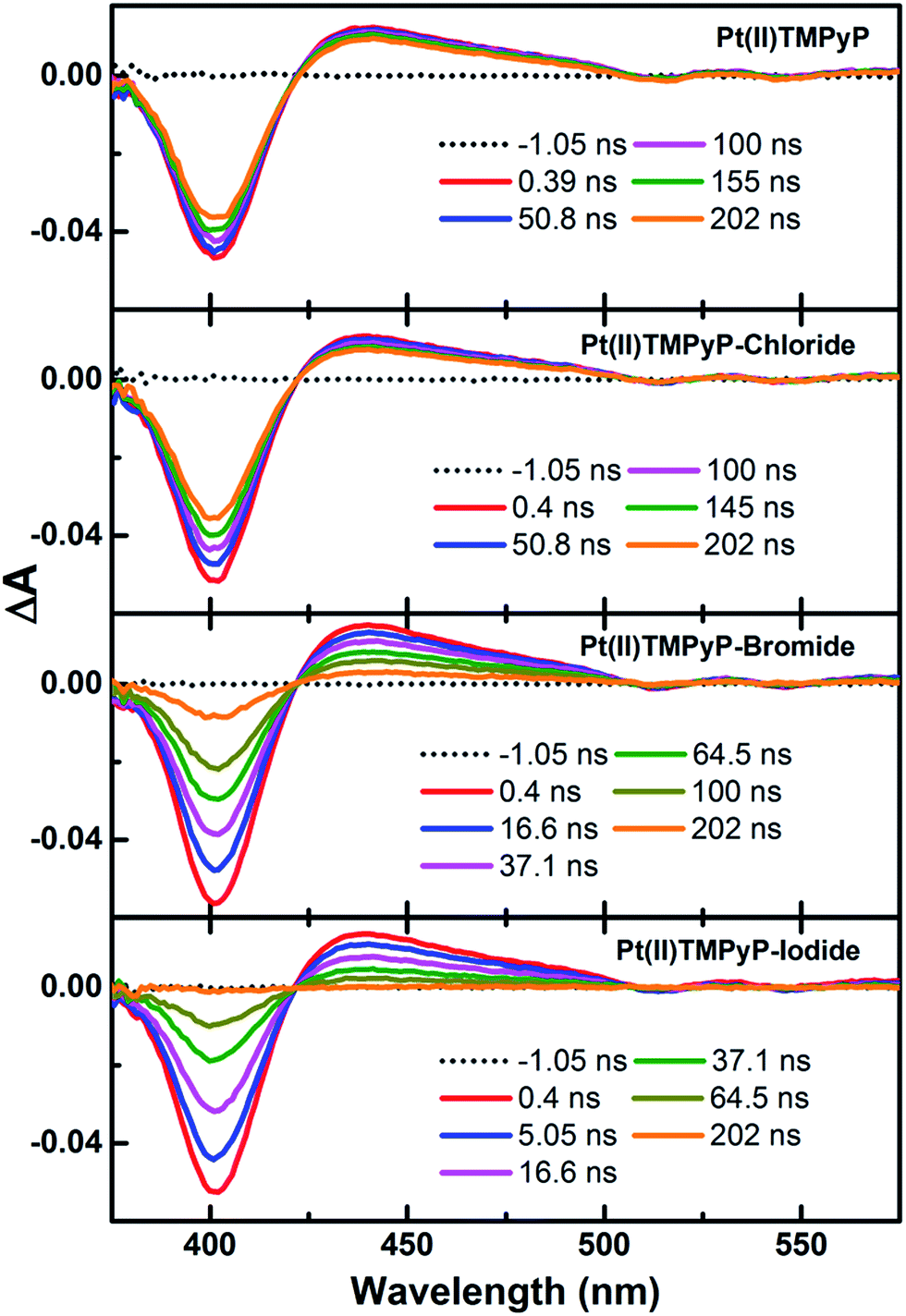 | ||
| Fig. 2 Transient absorption spectra of Pt(II)TMPyP before and after successive additions of chloride, bromide and iodide in the aqueous phase (λex = 350 nm; probing range = 350–900 nm), excitation energy density of 2.17 mJ cm−2. | ||
The ns-TA spectra of Pt(II)TMPyP with and without halides show ground-state bleaching at ∼400 nm and excited-state T1–Tn absorption over the range of 420–600 nm. These spectral features are consistent with the reported excited-state triplet absorption of porphyrin molecules.58,59 Members of this class of porphyrins in which Pt is the central atom are known to exhibit a very rapid and efficient singlet-to-triplet (S1 → T1) intersystem crossing associated with strong spin–orbit coupling.60 The TA spectra of Pt(II)TMPyP show a change over a time window of up to 4 μs, which is in agreement with the reported triplet lifetime of aerated solutions (∼1 μs).58 The TA spectra recorded in the presence of halides demonstrate a significant shortening of the time window. Moreover, the time window over which TA spectra are detected shows a further shortening in the same order of chloride > bromide > iodide. The kinetic traces collected from the TA spectra at both the ground-state bleach recovery and the excited-state absorption decay are shown in Fig. 3.
 | ||
| Fig. 3 Kinetic traces of Pt(II)TMPyP with and without the successive addition of chloride, bromide and iodide in the aqueous phase. Monitoring wavelengths are 443 nm for excited-state absorption and 401 nm for ground-state bleach. | ||
All kinetic traces both for ground-state bleach and excited-state decay are fitted to single exponent fitting curves as shown in the figure. The ground-state bleach recovery extracted from the TA spectra of Pt(II)TMPyP in the presence of halides decreases in the order of Pt(II)TMPyP (1.03 μs) > Pt(II)TMPyP-chloride (0.56 μs) > Pt(II)TMPyP-bromide (0.09 μs) > Pt(II)TMPyP-iodide (0.04 μs). This overall fast deactivation of the triplet signal implies the presence of an extra process involved in the deactivation of the excited state when halides are present compared to the case of free Pt(II) porphyrin. Notably, the ring reduction potential of Pt(II) porphyrin are in the range of −1.39 to −1.3 V,61 whereas the oxidation potentials of the halides (X−/X) are 1.36, 1.087, and 0.535 V for chloride, bromide, and iodide, respectively.62 The rapid changes in the TA spectra, the redox properties and the lack of any possibility for energy transfer suggests a photoinduced electron transfer from the halides to the Pt(II)TMPyP*. In general, a peripheral substituent at the meso position on a porphyrin macrocycle can significantly change its electrochemical potential.63
In this regime, the electron-accepting positively charged pyridinium units (CH3–N+) on the meso positions of the porphyrin will extract the electron density from the porphyrin macrocycle via intramolecular charge transfer (ICT).64 If we consider the ICT in conjunction with Pt(II) as the central atom in the TMPyP core, Pt-to-porphyrin back electron donation (d → eg(π*))50,65 produces a region of poor electron density on the Pt metallic center. The generation of this region is expected to facilitate the axial electron transfer interaction with the Pt central atom. Hence, upon photoexcitation, an attractive center is anticipated to be formed around the Pt in the center of Pt(II)TMPyP, which, in turn, facilitates the attraction of the negatively charged halides, triggering electron transfer. Notably, the strong spectral overlap between the porphyrin anion radical, which is reported to occur at 470 nm, and T1–Tn TA and the very low concentration of halide used in these experiments make monitoring of the anion radical absorption difficult.66
To confirm the importance of Pt as the central atom in the suggested mechanism, a control experiment was carried out using Zn(II)TMPyP with iodide; in this experiment, the TA did not exhibit any change (see Fig. S7†), confirming that the interaction occurs with the Pt at the porphyrin macrocycle center. Further confirmation of the fast photoinduced electron transfer was provided by the fs-TA in Fig. S8 and S9.†
In the case of Pt(II)TMPyP, the triplet–triplet absorption developed within 120 fs, which was the temporal resolution of our experiment. This result is in good agreement with the efficient and fast ISC.50,51 In contrast, the fs-TA for Pt(II)TMPyP in the presence of iodide (see Fig. S9†) exhibited a rapid change in the ground-state bleach, indicating the rapid deactivation of the excited state because of the photoinduced electron transfer between iodide and Pt(II)TMPyP, as simplified in Fig. 4. Taking advantage of this Pt(II)TMPyP-iodide system, further research is going on towards the development of porphyrin-based sensors for other crucial anions like sulfide and cyanide.
 | ||
| Fig. 4 Illustration showing luminescence quenching of Pt(II)TMPyP due to photoinduced reduction by iodide in the aqueous phase. | ||
In summary, many halogen containing compounds have come into everyday use in the fields of chemistry, biology, medicine, plastics, food and even photography. Many techniques for the detection of these halogen compounds and ions have been developed not only to reduce the complexity and cost of the analysis but also to improve the detection sensitivity. In these techniques various types of materials namely polymers, porphyrins, nano-particles/clusters, quantum dots, DNA logic gates, and composites have been tried as a photoluminescence sensor for the detection of halides.17–32 The requirement of controlled shape and size makes nano-sized materials a less preferred choice as a sensor. Furthermore, deterioration and modification of nano-sized materials also question their application. Herein, we report for the first time the photoinduced triplet-state electron transfer of Pt(II)TMPyP as an easy, rapid, environmentally friendly, ultra-sensitive (a never-before-attained detection limit of 1 × 10−12 M) and economical method for the determination of iodide in the aqueous phase. Pt(II) porphyrin phosphorescence was observed to be quenched to different magnitudes through the use of different halides. The efficiency of quenching was experimentally demonstrated to increase in the order chloride < bromide < iodide. The very low concentration range over which the Pt(II) porphyrin exhibit quenching and the simplicity of the measurements constitute the basis for a very safe and efficient halide sensor in the analytical market.
Acknowledgements
Shawkat M. Aly is grateful for the post-doctoral fellowship provided by the Saudi Basic Industries Corporation (SABIC). This work was supported by the King Abdullah University of Science and Technology.Notes and references
- J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, UK, 2006 Search PubMed.
- V. Amendola, L. Fabbrizzi, M. Licchelli and A. Taglietti, Anion Coordination Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- J. W. A. Smit, J. P. Schroder-Van der Elst, M. Karperien, I. Que, J. A. Romijn and D. Van der Heide, Exp. Clin. Endocrinol. Diabetes, 2001, 109, 52–55 CrossRef CAS PubMed.
- C. G. Brown, R. F. Harland, I. R. Major and C. K. Atterwill, Food Chem. Toxicol., 1987, 25, 787–794 CrossRef CAS.
- C. Nasr, S. Hotchandani and P. V. Kamat, J. Phys. Chem. B, 1998, 102, 4944–4951 CrossRef CAS.
- K. G. Stamplecoskie, J. S. Manser and P. V. Kamat, Energy Environ. Sci., 2015, 8, 208–215 Search PubMed.
- B. H. Meekins and P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 2304–2310 CrossRef CAS.
- A. J. Nozik and J. Miller, Chem. Rev., 2010, 110, 6443–6445 CrossRef CAS PubMed.
- A. J. Nozik, M. C. Beard, J. Johnson, M. C. Hanna, J. M. Luther, A. Midgett, O. Semonin and J. Michel, Prepr. Symp. – Am. Chem. Soc., Div. Fuel Chem., 2012, 57, 14 CAS.
- M. Law, J. M. Luther, M. C. Beard, S. Choi and A. J. Nozik, Photovoltaic Specialists Conference, 2009 34th IEEE, 002068–002073 Search PubMed.
- V. R. Preedy, G. N. Burrow and R. R. Watson, Comprehensive Handbook of Iodine: Nutritional, Biochemical, Pathological, and Therapeutic Aspects, Academic Press, New York, 2009 Search PubMed.
- E. Nystrom, G. E. B. Berg, S. K. G. Jansson, O. Torring and S. V. Valdemarsson, Thyroid Disease in Adults, Berlin, Germany, 2011 Search PubMed.
- S. Venturi, Curr. Chem. Biol., 2011, 5, 155–162 CAS.
- R. Abe, K. Shinmei, N. Koumura, K. Hara and B. Ohtani, J. Am. Chem. Soc., 2013, 135, 16872–16884 CrossRef CAS PubMed.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS.
- J. M. Fang, P. F. Gao, X. L. Hu and Y. F. Li, RSC Adv., 2014, 4, 37349–37352 RSC.
- Y. Xiao, Y. Zhang, H. Huang, Y. Zhang, B. Du, F. Chen, Q. Zheng, X. He and K. Wang, Talanta, 2015, 131, 678–683 CrossRef CAS PubMed.
- M. M. Kruk, Y. B. Ivanova, V. B. Sheinin, A. S. Starukhin, N. Z. Mamardashvili and O. I. Koifman, Makrogeterotsikly, 2008, 1, 50–58 CAS.
- L. Wang, G. Fang, D. Ye and D. Cao, Sens. Actuators, B, 2014, 195, 572–580 CrossRef CAS PubMed.
- J. Liu, Q. Lin, Y.-M. Zhang and T.-B. Wei, Sens. Actuators, B, 2014, 196, 619–623 CrossRef CAS PubMed.
- S. Nabavi and N. Alizadeh, Sens. Actuators, B, 2014, 200, 76–82 CrossRef CAS PubMed.
- S. Chen, P. Wang, C. Jia, Q. Lin and W. Yuan, Spectrochim. Acta, Part A, 2014, 133, 223–228 CrossRef CAS PubMed.
- S. Hussain, S. De and P. K. Iyer, ACS Appl. Mater. Interfaces, 2013, 5, 2234–2240 CAS.
- F. Du, F. Zeng, Y. Ming and S. Wu, Microchim. Acta, 2013, 180, 453–460 CrossRef CAS.
- X. Wu, J. Chen and J. X. Zhao, Analyst, 2013, 138, 5281–5287 RSC.
- S.-C. Wei, P.-H. Hsu, Y.-F. Lee, Y.-W. Lin and C.-C. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2652–2658 CAS.
- N. Singh and D. O. Jang, Org. Lett., 2007, 9, 1991–1994 CrossRef CAS PubMed.
- K. Wygladacz and E. Bakker, Analyst, 2007, 132, 268–272 RSC.
- H. Zhang, Y. Li, X. Liu, P. Liu, Y. Wang, T. An, H. Yang, D. Jing and H. Zhao, Environ. Sci. Technol. Lett., 2013, 1, 87–91 CrossRef.
- M. S. Mehata and H. B. Tripathi, J. Lumin., 2002, 99, 47–52 CrossRef CAS.
- E. Nyarko, N. Hanada, A. Habib and M. Tabata, Inorg. Chim. Acta, 2004, 357, 739–745 CrossRef CAS PubMed.
- B. Basumatary, M. Ayoub Kaloo, V. Kumar Singh, R. Mishra, M. Murugavel and J. Sankar, RSC Adv., 2014, 4, 28417–28420 RSC.
- A. Nayal, A. Kumar, R. K. Chhatra and P. S. Pandey, RSC Adv., 2014, 4, 39866–39869 RSC.
- X. Hou, S. Chen, J. Tang, Y. Xiong and Y. Long, Anal. Chim. Acta, 2014, 825, 57–62 CrossRef CAS PubMed.
- G. Zhou, C. Zhao, C. Pan and F. Li, Anal. Methods, 2013, 5, 2188–2192 RSC.
- X.-H. Yang, J. Ling, J. Peng, Q.-E. Cao, Z.-T. Ding and L.-C. Bian, Anal. Chim. Acta, 2013, 798, 74–81 CrossRef CAS PubMed.
- M. E. Vaida, R. Tchitnga and T. M. Bernhardt, Beilstein J. Nanotechnol., 2011, 2, 618–627 CrossRef CAS PubMed.
- W. Brüchert, A. Helfrich, N. Zinn, T. Klimach, M. Breckheimer, H. Chen, S. Lai, T. Hoffmann and J. Bettmer, Anal. Chem., 2007, 79, 1714–1719 CrossRef PubMed.
- S. Mishra, V. Singh, A. Jain and K. K. Verma, Analyst, 2000, 125, 459–464 RSC.
- I. A. Carasel, C. R. Yamnitz, R. K. Winter and G. W. Gokel, J. Org. Chem., 2010, 75, 8112–8116 CrossRef CAS PubMed.
- Y. Miura, M. Hatakeyama, T. Hosino and P. R. Haddad, J. Chromatogr. A, 2002, 956, 77–84 CrossRef CAS.
- S. S. R. Dasary, P. Chandra Ray, A. K. Singh, T. Arbneshi, H. Yu and D. Senapati, Analyst, 2013, 138, 1195–1203 RSC.
- P. Pienpinijtham, X. X. Han, S. Ekgasit and Y. Ozaki, Anal. Chem., 2011, 83, 3655–3662 CrossRef CAS PubMed.
- H. S. Toh, K. Tschulik, C. Batchelor-McAuley and R. G. Compton, Analyst, 2014, 139, 3986–3990 RSC.
- F. C. Pereira, L. M. Moretto, M. De Leo, M. V. Boldrin Zanoni and P. Ugo, Anal. Chim. Acta, 2006, 575, 16–24 CrossRef CAS PubMed.
- A. O. El-Ballouli, E. Alarousu, M. Bernardi, S. M. Aly, A. P. Lagrow, O. M. Bakr and O. F. Mohammed, J. Am. Chem. Soc., 2014, 136, 6952–6959 CrossRef CAS PubMed.
- A. O. El-Ballouli, E. Alarousu, A. Usman, J. Pan, O. M. Bakr and O. F. Mohammed, ACS Photonics, 2014, 1, 285–292 CrossRef CAS.
- J. Sun, W. Yu, A. Usman, T. T. Isimjan, S. Dgobbo, E. Alarousu, K. Takanabe and O. F. Mohammed, J. Phys. Chem. Lett., 2014, 5, 659–665 CrossRef CAS.
- F. Nifiatis, W. Su, J. E. Haley, J. E. Slagle and T. M. Cooper, J. Phys. Chem. A, 2011, 115, 13764–13772 CrossRef CAS PubMed.
- D. Eastwood and M. Gouterman, J. Mol. Spectrosc., 1970, 35, 359–375 CrossRef CAS.
- T. Htun, J. Fluoresc., 2004, 14, 217–222 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Singapore, 3rd edn, 2006 Search PubMed.
- C. P. Ponce, R. P. Steer and M. F. Paige, Photochem. Photobiol. Sci., 2013, 12, 1079–1085 CAS.
- B. Lang, S. Mosquera-Vazquez, D. Lovy, P. Sherin, V. Markovic and E. Vauthey, Rev. Sci. Instrum., 2013, 84, 073107 CrossRef PubMed.
- E. Vauthey, J. Photochem. Photobiol., A, 2006, 179, 1–12 CrossRef CAS PubMed.
- A. Rosspeintner, B. Lang and E. Vauthey, Annu. Rev. Phys. Chem., 2013, 64, 247–271 CrossRef CAS PubMed.
- P. M. Keane and J. M. Kelly, Photochem. Photobiol. Sci., 2011, 10, 1578–1586 CAS.
- T. Kobayashi, K. D. Straub and P. M. Rentzepis, Photochem. Photobiol., 1979, 29, 925–931 CrossRef CAS PubMed.
- T. Mani, D. M. Niedzwiedzki and S. A. Vinogradov, J. Phys. Chem. A, 2012, 116, 3598–3610 CrossRef CAS PubMed.
- Z. Ou, P. Chen and K. M. Kadish, Dalton Trans., 2010, 39, 11272–11276 RSC.
- A. Jammoul, S. Dumas, B. D'Anna and C. George, Atmos. Chem. Phys., 2009, 9, 4229–4237 CrossRef CAS.
- O. S. Finikova, P. Chen, Z. Ou, K. M. Kadish and S. A. Vinogradov, J. Photochem. Photobiol., A, 2008, 198, 75–84 CrossRef CAS PubMed.
- F. J. Vergeldt, R. B. M. Koehorst, A. Vanhoek and T. J. Schaafsma, J. Phys. Chem., 1995, 99, 4397–4405 CrossRef CAS.
- V. A. Galievsky, V. S. Chirvony, S. G. Kruglik, V. V. Ermolenkov, V. A. Orlovich, C. Otto, P. Mojzes and P.-Y. Turpin, J. Phys. Chem., 1996, 100, 12649–12659 CrossRef CAS.
- T. Strozik, M. Wolszczak and M. Hilczer, Radiat. Phys. Chem., 2013, 91, 156–165 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculation of the iodide detection limit, steady-state absorption and emission, Stern–Volmer plots, and ns-TA and fs-TA. See DOI: 10.1039/c4ta07033j |
| ‡ On leave from Chemistry Department, Assiut University, Egypt. |
| This journal is © The Royal Society of Chemistry 2015 |