
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of micro/mesoporosity in non-hydrolytic hybrid silicophosphate xerogels†‡
Ales
Styskalik
ab,
David
Skoda
ab,
Zdenek
Moravec
a,
Michal
Babiak
ab,
Craig E.
Barnes
c and
Jiri
Pinkas
 *ab
*ab
aMasaryk University, Department of Chemistry, Kotlarska 2, CZ-61137 Brno, Czech Republic. E-mail: jpinkas@chemi.muni.cz; Fax: +420 549492443; Tel: +420 549496493
bMasaryk University, CEITEC MU, Kamenice 5, CZ-62500 Brno, Czech Republic
cUniversity of Tennessee, Department of Chemistry, Knoxville, TN 37996-1600, USA
First published on 24th February 2015
Abstract
Non-hydrolytic sol–gel reactions of acetoxysilanes with trimethylsilyl esters of phosphoric and phosphonic acids produce cross-linked matrices containing homogeneous dispersions of silicon and phosphoryl groups connected together by networks of Si–O–P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) linkages. These polycondensation reactions proceed cleanly and under mild conditions for a wide variety of precursor silanes RnSi(OAc)4−n (R = alkyl, aryl; n = 1, 2) and phosphoryl compounds RP(O)(OSiMe3)2 (R = alkyl, aryl) to provide hybrid xerogels, the final properties of which are a sensitive function of the organic substituents and the Si
O) linkages. These polycondensation reactions proceed cleanly and under mild conditions for a wide variety of precursor silanes RnSi(OAc)4−n (R = alkyl, aryl; n = 1, 2) and phosphoryl compounds RP(O)(OSiMe3)2 (R = alkyl, aryl) to provide hybrid xerogels, the final properties of which are a sensitive function of the organic substituents and the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio of the precursors. The reactions of bridged acetoxysilanes (AcO)3Si–X–Si(OAc)3 and phosphoryl reagents (Me3SiO)2P(O)–X–P(O)(OSiMe3)2 have also been investigated and found to produce gels that exhibit large surface areas (up to 700 m2 g−1). The presence of SiO6 structural units in bridged-phosphoryl xerogels is related to their microporosity while the absence of such moieties in bridged-acetoxysilane networks is congruent with significant mesoporosity. Several important parameters are identified which can be used to tailor the properties of these hybrid matrices such that gels with specific polarity, porosity and surface area can be targeted at the time of synthesis.
:P ratio of the precursors. The reactions of bridged acetoxysilanes (AcO)3Si–X–Si(OAc)3 and phosphoryl reagents (Me3SiO)2P(O)–X–P(O)(OSiMe3)2 have also been investigated and found to produce gels that exhibit large surface areas (up to 700 m2 g−1). The presence of SiO6 structural units in bridged-phosphoryl xerogels is related to their microporosity while the absence of such moieties in bridged-acetoxysilane networks is congruent with significant mesoporosity. Several important parameters are identified which can be used to tailor the properties of these hybrid matrices such that gels with specific polarity, porosity and surface area can be targeted at the time of synthesis.
1. Introduction
Sol–gel syntheses of organic–inorganic hybrid materials with covalently incorporated organic functionalities in silicate matrices have been studied with increasing interest for the application in and also in providing an extensive tool-box for the controlled fabrication of xerogels, nanoparticles, thin films and other advanced materials.1–8 The refined procedures allow tailoring textural,9,10 optical,11 mechanical,12,13 catalytic,14,15 sensor,16 and electrical properties.17,18 Synthetic strategies rely, for example, on the copolymerization of metal alkoxide/oxide clusters functionalized with polymerizable groups with suitable co-monomers,19–21 or on the controlled radical polymerization of polyhedral oligomeric silsesquioxanes (POSS).22 Hybrid composites based on POSS precursors have reached the industrial scale of applications.23–25 Furthermore, hydrolysis and condensation of alkyl- or aryltrialkoxysilanes provide single-phase organosilicagels composed of polysilsequioxane networks (so-called T-resins).26 Despite the high number of reactive groups in these trifunctional organosilanes RSiX3, their sol–gel polymerization products are incompletely condensed nonporous T-resins or the molecular polyhedral silsesquioxanes. However, network materials with high surface areas can be formed from RSiX3 by co-condensation with monomers that contain a higher level of functionality, such as tetraethoxysilane (TEOS) or tetramethoxysilane (TMOS).Two interconnected phenomena – surface area decrease and pore size increase – were observed in the hydrolytic sol–gel synthesis of silicagels from TEOS and trimethylethoxysilane, the latter of which was used as a condensation inhibitor.27 Similar results were observed in the hydrolytic sol–gel co-polymerization of TEOS with alkyltrimethoxysilane RSi(OMe)3.28 In general, the bulkier the organic group R, the higher the decrease of surface area. Moreover, the different reactivities of Si(OR)4 and RSi(OR)3 in hydrolysis–condensation reactions at various pHs may preclude homogeneous incorporation and even lead to phase separation.29
Changing the precursor structure to bridged trialkoxysilanes (RO)3Si–X–Si(OR)3 gives rise to functional materials with controllable self-assembling properties30–33 and mesoporous architectures.34 By introducing hydrocarbon spacers X into the silicate network, properties such as hydrophobicity, porosity, UV-visible absorption and fluorescence, can be significantly modified. The morphology of a material can be controlled at the molecular level by carefully designing suitable spacer groups that influence textural parameters. For example, an average pore diameter increases with longer alkylene spacers.35
Examples of non-hydrolytic preparations of hybrid xerogel materials in the present literature are much less common. Hay et al.36 and Bourget et al.37 used alkyhalogenide elimination from RSiCl3 and RSi(OEt)3 for the synthesis of organosilsesquioxane gels that were, however, nonporous. Non-hydrolytic condensations usually show better results in terms of homogeneity in mixed oxides, such as hybrid titania–silica xerogels also possessing high surface areas.38–42 In general, it is not possible to predict how the precursor binding capacity and size of organic groups will influence the pore size and surface area in various final gel matrices and to discern some general trends. The preparation of homogeneous mixed oxides with controllable surface areas and pore size distribution through the introduction of organic groups still presents a challenge.
Hybrid silicophosphates are studied as promising materials for proton conducting membranes of fuel cells, gas sensors, and optical applications.43–48 The synthesis of hybrid materials with an Si–O–P skeleton was accomplished by non-hydrolytic polycondensation induced by heating RP(O)Cl2 (R = Me or Ph) with TEOS, or by their reaction with SiCl4 in tBuOH.49 Recently, preparation of low melting silicophosphate glasses has been described utilizing non-hydrolytic polycondensation with HCl elimination from anhydrous, solid H3PO4 or RP(O)(OH)2 and alkyl-, aryl-, and vinylchlorosilanes, RnSiCl4−n (n = 1, 2), without employing any solvent.50–53 However, the homocondensation and phase separation cannot be prevented in these systems. So far no porous hybrid organic–inorganic materials with an Si–O–P skeleton have been described.
We recently described a new non-hydrolytic ester elimination route to microporous silicophosphate xerogels with a purely inorganic framework by condensing silicon acetate, Si(OAc)4, and tris(trimethylsilyl)phosphate, OP(OSiMe3)3 (TTP).54 Optimization of reaction conditions leads to products with a high degree of condensation and large specific surface areas, while maintaining a homogeneous distribution of the components (eqn (1)).
| 3Si(OAc)4 + 4OP(OSiMe3)3 → Si3P4O16 + 12CH3C(O)OSiMe3 | (1) |
2. Experimental
2.1 General
All reactions and manipulations of the materials described here were performed in a dry N2 atmosphere using Schlenk techniques or in dry nitrogen gloveboxes. Sources, procedures and characterization data for all solvents and reagents used in these investigations are summarized in the ESI† of this article.2.2 General procedure for non-hydrolytic sol–gel reactions
The parent unmodified silicophosphate xerogel was prepared under conditions optimized to give the maximum surface area.54 In a typical reaction, OP(OSiMe3)3 (TTP) was added dropwise to a stirred solution of Si(OAc)4 (the molar ratio 4:3) in toluene to obtain a clear colorless gel (eqn (1)). The gel was aged at 80 °C for one week and then dried under vacuum for two days providing an opaque xerogel (SiP). Characterization and spectral analysis data of SiP were the same as reported previously.54 Hybrid xerogels were synthesized in the same manner under the same conditions (toluene, concentration, and 80 °C) to estimate the influence of organic groups covalently bound to silicon and phosphorus (Sections 1, 2, and 3). Detailed experimental procedures for all the reactions of alkyl- and arylacetoxysilanes and alkylphosphonic acid esters can be found in the ESI† along with the characterization data for the obtained xerogels. Samples are denoted by abbreviations that point to the particular employed precursors as summarized in Table 1. For example, the abbreviation SiP denotes the reaction of 3Si(OAc)4 + 4OP(OSiMe3)3; MeSiP: MeSi(OAc)3 + OP(OSiMe3)3; SiP-MeSiX: Si(OAc)4 + nMeSi(OAc)3 + (1.33 + n) OP(OSiMe3)3 (n = 0–1); SiC1SiP: (AcO)3Si–CH2–Si(OAc)3 + 2OP(OSiMe3)3.
| Abbreviation | Precursor | Abbreviation | Precursor |
|---|---|---|---|
| Si | Si(OAc)4 | P | OP(OSiMe3)3 |
| MeSi | MeSi(OAc)3 | SiCnSi | (AcO)3Si–(CH2)n–Si(OAc)3 |
| Me2Si | Me2Si(OAc)2 | HexP | C6H11P(O)(OSiMe3)2 |
| PhSi | PhSi(OAc)3 | PhP | PhP(O)(OSiMe3)2 |
| Ph2Si | Ph2Si(OAc)2 | PC2P | (Me3SiO)2P(O)–(CH2)2–P(O)(OSiMe3)2 |
| BuSi | t BuSi(OAc)3 | PC6P | (Me3SiO)2P(O)–C6H4–P(O)(OSiMe3)2 |
| BuOSi | t BuOSi(OAc)3 |
The organic byproduct of polycondensation was identified as trimethylsilylacetate by 1H, 13C NMR, and GC-MS.54 NMR tube reactions between Si and P precursors were carried out to follow the reaction progress by 31P NMR taken at 1, 24, and 168 h after the mixing of precursors in C6D6.
Tables 2–4 summarize the data for three different types of hybrid silicophosphate network solids prepared from (1) mono- and dialkyl/arylacetoxysilanes and alkyl/arylphosphonic acid esters, (2) co-condensation of Si(OAc)4 and OP(OSiMe3)3 partially substituted with RnSi(OAc)4−n (n = 1, 2; R = Me, Ph) and PhP(O)(OSiMe3)2, and (3) bridged acetoxysilanes and phosphonic acid esters. Detailed experimental procedures and characterization data (NMR, IR spectra) are provided in the ESI.†
| Sample | n Si [mmol] | n P [mmol] | DC [%] | ML [%] | MLcalc [%] |
|---|---|---|---|---|---|
| SiP | 1.0 | 1.33 | 85.7 | 29.4 | 32.8 |
| MeSiP | 1.0 | 1.0 | 87.1 | 32.1 | 35.3 |
| Me2SiP | 1.0 | 0.67 | 89.4 | — | — |
| PhSiP | 1.0 | 1.0 | 63.6 | — | — |
| Ph2SiP | 1.0 | 0.7 | 61.9 | — | — |
| BuSiP | 1.0 | 1.1 | 69.3 | — | — |
| SiHexP | 1.0 | 2.0 | 100 | 65.0 | 42.8 |
| SiPhP | 1.0 | 2.0 | 58.1 | 63.5 | 64.1 |
| Sample | x | DC [%] | ML [%] | MLcalc [%] |
|---|---|---|---|---|
| a Amount of added organosilane/phosphonate components according to eqn (4)–(6). | ||||
| SiP | — | 85.7 | 29.4 | 32.8 |
| SiP-MeSi1 | 0.19 | 79.2 | 35.3 | 41.4 |
| SiP-MeSi2 | 0.29 | 80.7 | 31.6 | 39.7 |
| SiP-MeSi3 | 0.37 | 82.0 | 30.6 | 38.0 |
| SiP-MeSi4 | 0.51 | 83.8 | 29.1 | 35.6 |
| SiP-MeSi5 | 1.24 | 91.0 | 31.5 | 24.1 |
| SiP-Me2Si1 | 0.08 | 74.8 | 26.6 | 46.0 |
| SiP-Me2Si2 | 0.17 | 85.3 | 29.9 | 33.5 |
| SiP-Me2Si3 | 0.21 | 88.2 | 36.5 | 27.1 |
| SiP-Me2Si4 | 0.28 | 84.9 | 29.1 | 34.0 |
| SiP-Me2Si5 | 0.37 | 87.5 | 28.6 | — |
| SiP-Me2Si6 | 0.44 | 83.1 | 30.2 | 36.4 |
| SiP-PhSi1 | 0.53 | 83.7 | 34.6 | 39.2 |
| SiP-PhSi2 | 0.97 | 81.0 | 34.9 | 44.7 |
| SiP-Ph2Si1 | 0.11 | 81.0 | 33.6 | 41.5 |
| SiP-Ph2Si2 | 0.32 | 80.3 | 38.0 | 45.3 |
| SiP-PhP1 | 0.35 | 83.0 | 28.2 | 39.6 |
| SiP-PhP2 | 1.49 | 87.3 | 47.3 | 41.2 |
| Sample | DC [%] | ML [%] | MLcalc [%] |
|---|---|---|---|
| SiC1SiP | 85.6 | 27.2 | 29.3 |
| SiC2SiP | 78.1 | 35.3 | 27.4 |
| SiC3SiP | 88.5 | 36.8 | 31.3 |
| SiC6SiP | — | 45.9 | — |
| SiPC2P | 83.6 | 31.9 | 32.9 |
| SiPC6P | 77.0 | 33.9 | 47.7 |
3. Results and discussion
3.1 Mono- and dialkyl/arylacetoxysilanes and alkyl/arylphosphonic acid esters
A set of sol–gel reactions (80 °C, 168 h, and toluene or 100–130 °C, 2 h, and no solvent) and NMR tube reactions (r.t., C6D6) based on non-hydrolytic polycondensation of acetoxysilane and trimethylsilyl ester moieties was performed in order to evaluate the propensity of acetoxysilanes and phosphonic acid esters to condense and form gels as illustrated in Scheme 1. A 3-dimensionally cross-linked matrix of silicon centers and phosphoryl groups is formed. As the matrix grows and becomes more rigid, some acetate groups on silicon and trimethylsiloxy groups on phosphorus are presumed to become spatially isolated with one another within the matrix and are retained, thus reducing the average degree of condensation from 100%. The value of y then represents the number of uncondensed OAc and SiMe3 groups.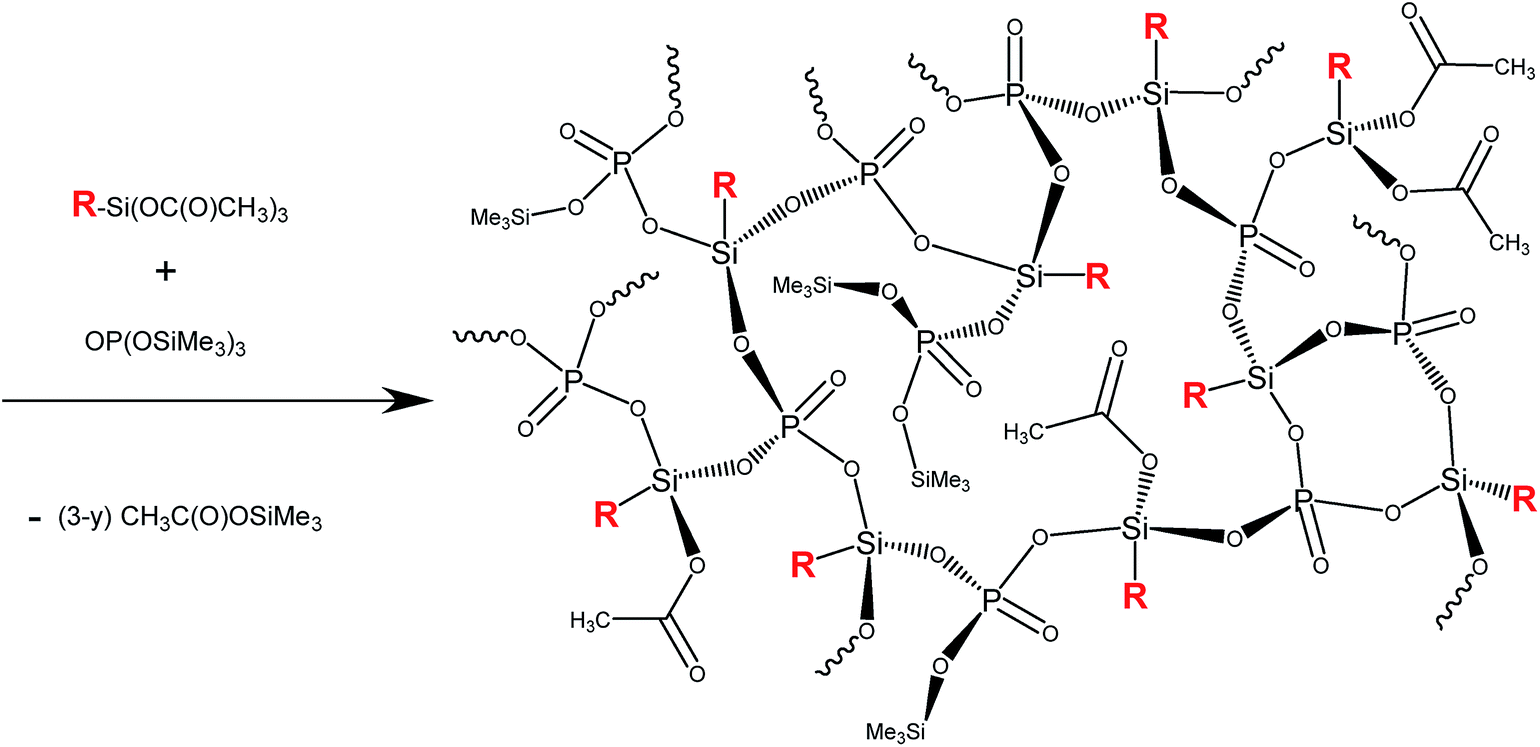 | ||
| Scheme 1 Polycondensation of alkyl/arylacetoxysilanes and OP(OSiMe3)3, R = methyl, phenyl, or tbutyl. | ||
The extent of condensation, followed by 31P NMR (Fig. 1S†), allowed us to distinguish between the formation of molecular products, oligomers and polymers as summarized in Table 2S.† The condensation between Si and P precursors was confirmed by the formation of new NMR signals upfield from the starting esters. Signals of molecular and oligomeric condensation products then usually diminished and a broad signal of polymeric silicophosphates was observed (Fig. 1S†). No signals at a lower field from the starting phosphorus esters were observed attesting to the absence of ligand exchange and the consequent homocondensation.
In all reactions, the yield of the product as well as the mass of the starting precursors were precisely weighed to allow gravimetric estimation of the degree of condensation, DC, (Table 2, eqn (2)). Degree of condensation as defined by eqn (2) represents the relative difference between the maximum theoretical loss of Me3SiOAc in comparison to what is experimentally observed. This difference also defines the number of acetoxy groups on silicon and trimethylsilyl ester groups on phosphorus that are left in the matrix. ntotal is the number of moles of organic groups in the starting materials and nresidual is the molar amount of residual organic groups in the xerogel computed from the difference of the theoretical and the experimental yield. The condensation reactions were never quantitative, therefore DC was used for calculating the theoretical mass loss for thermogravimetric experiments (MLcalc) assuming the removal of residual OAc and SiMe3 groups upon heating.
| DC = 100(ntotal − nresidual)/ntotal | (2) |
The stability of Si–C and P–C bonds in these reactions was confirmed by the IR and solid-state NMR studies of the products (see below) as there is no evidence of Si–C or P–C bond rupture. One can expect that the ability of these precursors to form porous xerogels would be lower in comparison with the parent reaction of Si(OAc)4 and TTP in analogy to trifunctional aryltrialkoxysilanes (e.g. PhSi(OEt)3) that in classical hydrolytic sol–gel reactions preferentially form oligosilsesquioxanes rather than gels. At high monomer concentrations, however, gels with low specific surface areas can be prepared by heating or with the addition of a strong base.55
Covalently bound organic groups caused significant changes in the morphological properties of products in comparison with the parent SiP xerogel (Table 2S†). Only systems with MeSi(OAc)3, C6H11P(O)(OSiMe3)2 and PhP(O)(OSiMe3)2 formed gels, which were nonporous. Once the organic group attached to Si is too bulky (phenyl or tbutyl) or the number of alkyl/aryl substituents is increased (dimethyldiacetoxysilane instead of methyltriacetoxysilane), the ability to form gels is completely lost and the reactions yield oligomeric mixtures. The glue-like products were heated at 250 °C in an oven under nitrogen to promote further condensation of the acetoxy- and trimethylsiloxy-groups. This resulted in the formation of insoluble, polymeric and hard non-porous (BET) glasses similar to low-melting glasses obtained by solventless condensation between alkyl/arylchlorosilanes and H3PO4 (100%).50–53
The reaction of TTP and diphenyldiacetoxysilane (Table 2S,†eqn (3)) produced a mixture of molecular products. One of them crystallized out of the reaction mixture and was isolated. This compound has been spectroscopically characterized and the molecular structure was determined by the single crystal X-ray diffraction analysis (Fig. 1). In this molecular product, condensation stops when a Si2P2O4 ring is formed according to eqn (3). Relevant metric data are gathered in Tables 3S and 4S in the ESI.† The core of the structure is a centrosymmetric Si2P2O4 ring in a chair conformation similar to what is observed in [{tBu2Si{O2P(O)Ph})2].56
| 2Ph2Si(OAc)2 + 2OP(OSiMe3)3 → [(Ph2Si{O2P(O)OSiMe3})2] + 4Me3SiOAc | (3) |
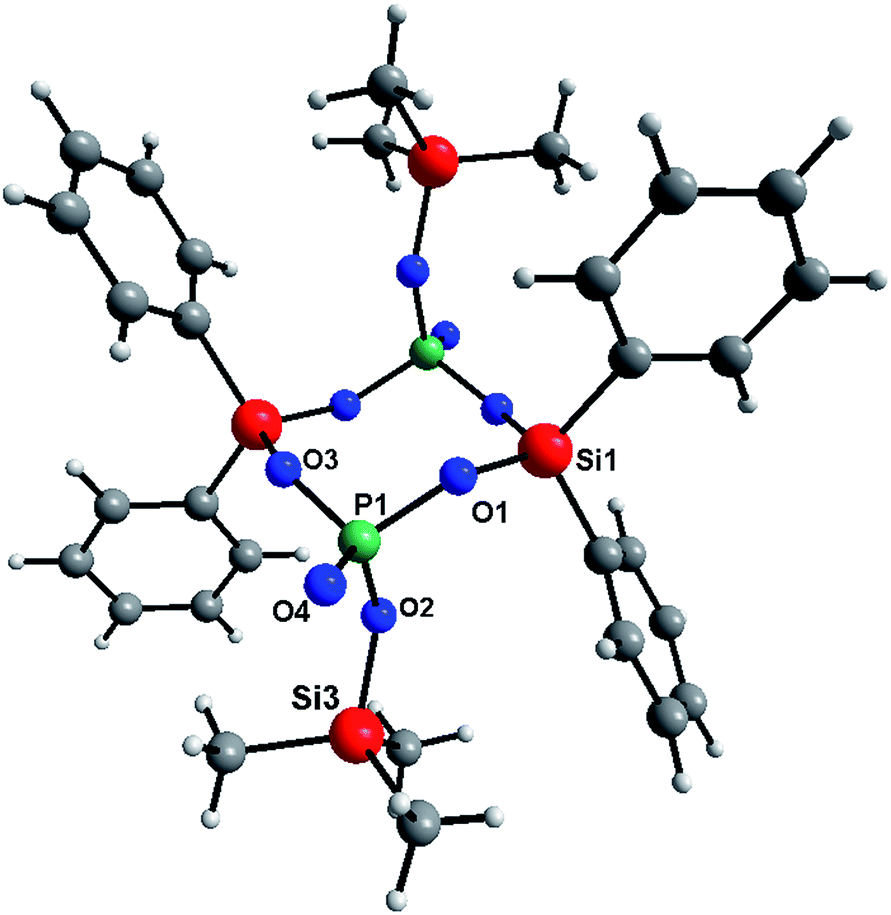 | ||
| Fig. 1 Molecular structure of [(Ph2Si{O2P(O)OSiMe3})2]. | ||
From the presented results, it can be concluded that the organic groups attached to Si have a stronger effect on the polycondensation reaction than the ones bound to the P center, e.g. the condensation reaction between phenyltriacetoxysilane and TTP stopped at the stage of oligomers, while replacing one reactive Me3SiO-group on P in TTP with a phenyl group resulted in the formation of a polymer (Table 2S†). Understanding the reason for this different behavior would potentially allow one to design gels with specific properties. This hypothesis was investigated with the help of IR and solid-state NMR spectroscopic studies of the prepared xerogels.
The IR spectra of SiP xerogel54 display important vibration bands at 1771, 1721, and 1541 cm−1 which are assigned to the νas(COO) stretches of acetates; the band at 1541 cm−1 arises from the bidentate bridging form (Fig. 2).57,58 Strong CH3 symmetric bending and rocking vibrations at 1259 and 854/764 cm−1 are characteristic of the SiMe3 groups.59 A medium intensity shoulder at 1300 cm−1 represents the PO terminal bonds60,61 and finally a strong composite band at 1129 cm−1 serves as a benchmark for the predominant formation of Si–O–P bonds.62,63 The IR spectra of hybrid xerogels with organic groups bound to silicon (MeSiP to BuSiP, Table 2) feature characteristic bands for residual organic groups (1771 and 1721 cm−1 for acetates; 1259 and 854/764 cm−1 for Me3Si groups), but they differ in certain features (Fig. 2): (i) the characteristic vibrations of CH3–SiO3 groups (ρCH3Si 815 and 732 cm−1) are present in MeSiP (that are separate from the absorption bands of the residual Me3SiOP groups), (ii) vibrations of aromatic ν CarH at 3052 cm−1 feature in the PhSiP and Ph2SiP spectra (Fig. 2S†), (iii) the vibration band of bidentate acetate at 1540 cm−1 is missing in all xerogels containing Si–C bonds, and (iv) the strong composite band, which appears in the SiP sample between 900 and 1350 cm−1, is now significantly sharper and is located from 900 to 1200 cm−1. These observations indicate that acetate and the PO groups do not coordinate with silicon and do not form bridges in the hybrid gels (Scheme 1). The absence of these binding modes was additionally corroborated by solid-state NMR (see below). Finally, the changes in the internal structure of these gels correlate with important structural and morphological properties of the hybrid gels.
 | ||
| Fig. 2 IR spectra of a hybrid silicophosphate xerogels with CH3–SiO3 groups (MeSiP, the top) and the parent silicophosphate (SiP, the bottom). | ||
Further evidence for the structural changes deduced from IR data was found in the solid-state NMR spectra of the hybrid silicophosphate xerogels. First, the broad 31P signal of P(OSi)4 moieties observed in the parent SiP xerogels shifted downfield by 10 ppm to −37 ppm indicating the loss of the coordinative PO→Si bonds (Chart 1B). Therefore the signal was assigned to the OP(OSi)3 groups (Chart 1A). No other signal was observed in the 31P MAS NMR spectra (Fig. 3S†). The 13C signals of the bridging acetates (in SiP at 16.3 and 190.6 ppm)54 disappeared and only resonances belonging to unidentate acetates (at 22.7 and 169.7 ppm), CH3–SiO3 groups (1.1 ppm) and residual Me3SiOP (−4.1 ppm) groups were present (Fig. 4S†). As a consequence of the missing PO→Si and CO→Si bridges, signals for six-coordinate silicon species were absent in the 29Si CPMAS NMR spectra which now featured only signals of CH3–SiO3 groups (−61 ppm, T3) and residual Me3SiOP (26 ppm, M) (Fig. 3). All the above described phenomena were also observed in solution NMR spectra of oligomeric samples Me2SiP, PhSiP and BuSiP. In other words, the number of the maximum possible Si–O–P connections available to form a network decreased from 6 (Chart 1B) to 3 (Chart 1A) in the case of silicon and from 4 (Chart 1B) to 3 (Chart 1A) for phosphorus, significantly influencing the structure (oligomers vs. xerogels, low cross-linking) and specific surface area of xerogels (Table 2S†).
 | ||
| Chart 1 Coordination environments of Si atoms in silicophosphate xerogels. | ||
 | ||
| Fig. 3 29Si CPMAS NMR spectra of hybrid xerogels with (CH3)3C–O–SiO3 groups (BuOSiP), C6H11–P(O)O2 groups (SiHexP), CH3–SiO3 groups (MeSiP) and the parent silicophosphate sample (SiP). Asterisks denote spinning side bands. | ||
On the other hand, the structures of xerogels synthesized from RP(O)(OSiMe3)2, R = Ph (SiPhP) or C6H11 (SiHexP), and Si(OAc)4 are similar to the parent SiP material. According to 31P MAS NMR, terminal PO bonds are converted to the PO→Si moieties (Chart 1C and D, Fig. 3S†).
Moreover, some CH3COO groups become bidentate (as confirmed by IR and 13C MAS NMR spectra, Fig. 4S†) by coordinating to Si, and thus there are two types of six-coordinate silicon atoms as confirmed by the 29Si MAS NMR spectra of SiPhP and SiHexP xerogels (Fig. 3). No evidence of a P–C bond rupture was observed. However, the resulting xerogels were non-porous.
The question why RnSi(OAc)4−n precursors do not achieve hexacoordination around silicon was puzzling. It can be seen from Table 2S,† that steric effects play a significant role during condensation reactions. While MeSi(OAc)3 yields a gel, PhSi(OAc)3 produces a mixture of oligomers. Similarly Me2Si(OAc)2 with OP(OSiMe3)3 afforded oligomeric species, while Ph2Si(OAc)2 under the same conditions yielded a mixture of molecular products. Considering these facts, there is a question, if the methyl group in MeSi(OAc)3 causes such a steric hindrance at silicon that the hexacoordination is not possible, or if there are any other effects, which prevent silicon from becoming hypercoordinated. Furthermore the electron donor properties of methyl groups must be considered. Electron rich methyl groups could reduce the positive charge on silicon and lower its Lewis acidity.
In order to distinguish electronic versus steric effects, the condensation employing tBuOSi(OAc)3 and OP(OSiMe3)3 was investigated (BuOSiP). In tbutoxytriacetoxysilane, only OAc groups undergo condensation, the electronic environment of Si closely resembles that in silicon tetraacetate (i.e. SiO4), and the bulky tBuO-groups will cause a strong steric hindrance of the silicon center and possibly suppress hexacoordination. Under standard reaction conditions, polycondensation was rather slow in comparison with Si(OAc)4 and did not afford any gel. While unreacted starting TTP was observed together with trimethylsilylacetate among volatile byproducts, neither tbutylacetate nor tbutoxytrimethylsilane were identified by GC-MS which excluded any elimination of tBuO-groups. Six-coordinate Si(OP)6 was, however, clearly evidenced by a resonance at −215 ppm in the 29Si CPMAS NMR spectrum (Fig. 3) of the isolated solid product. Interestingly, the acetates maintained their unidentate nature since no other signal, such as that of the Si(OP)4(OC)2 species at −195 ppm,54 appeared in the spectrum. These results were supported by 13C CPMAS NMR and IR spectroscopy showing the absence of bridging acetates (ESI†). To conclude, the bulky tBuO-groups slowed condensation reactions but did not affect the ability of silicon atoms to acquire hexacoordination. This property of silicon is therefore controlled by its Lewis acidity.
3.2 Co-condensation of Si(OAc)4 and OP(OSiMe3)3 partially substituted with RnSi(OAc)4−n (n = 1, 2; R = Me, Ph) and PhP(O)(OSiMe3)2
We subsequently turned our attention to systems in which substituents were added to both silicon (RnSi(OAc)4−n (n = 1, 2)) and phosphorus (PhP(O)(OSiMe3)2), which were expected to yield products with high surface areas.1,55,64,65 It is well known that in hydrolytic sol–gel reactions the differences in reactivity at various pHs for Si(OR)4 and RSi(OR)3 may preclude homogeneous incorporation and frequently lead to a precipitous loss of surface areas in the resulting products.29 We have also investigated the effect of partially substituting Si(OAc)4 with alkyl- and arylacetoxysilanes (Tables 3 and 6S†) according to eqn (4) and (5) and also OP(OSiMe3)3 with PhP(O)(OSiMe3)2 (eqn (6)). Hybrid networks containing a homogeneous dispersion of organic groups within the xerogel were expected as observed in Section 1.| (3 − x)Si(OAc)4 + 4/3xRSi(OAc)3 + 4OP(OSiMe3)3 → (1 − x/3)Si3P4O16·(4/3x)RSiPO4 + 12AcOSiMe3 | (4) |
| (3 − x)Si(OAc)4 + 2xR2Si(OAc)2 + 4OP(OSiMe3)3 → (1 − x/3)Si3P4O16·(2x/3)R6Si3P2O8 + 12AcOSiMe3 | (5) |
| 3Si(OAc)4 + (4 − x)OP(OSiMe3)3 + 3/2xPhP(O)(OSiMe3)2 → (1 − x/4)Si3P4O16·(3/4x)SiPh2P2O6 + 12AcOSiMe3 | (6) |
The structure of the resulting xerogels was strongly related to the parent SiP samples. The IR spectra of hybrid xerogels showed almost no changes with respect to the parent SiP gel and featured all characteristic vibrations described earlier. The 13C CPMAS NMR spectra displayed the presence of organic groups covalently bound to silicon (methyl or phenyl) consistent with the incorporation of the alkyl- or arylsilane components into the gel products. Finally, the 29Si CPMAS NMR spectra showed one additional resonance representing the particular MeSiO3/Me2SiO2/PhSiO3/Ph2SiO2 groups in addition to the signals of the residual Me3SiOP and SiO4 and two types of SiO6 moieties for each product (Fig. 5S–7S†).
All of the gel products listed in Table 6S† exhibited significant surface areas (isotherms shown in the ESI, Fig. 8S–14S†). The surface area decreased substantially with the increasing amount of organic groups (Table 6S,†Fig. 4) in agreement with the similar reports on the hydrolytic sol–gel reactions.28 Two effects can be discerned: (i) the bulkier the organic group, the steeper the decrease of the surface area (e.g. dimethyldiacetoxysilane vs. diphenyldiacetoxysilane); (ii) the lower the number of potential cross-link bonds, the steeper the decrease of surface areas in xerogels with low contents of the alkyl/arylacetoxysilane additive (e.g. phenyltriacetoxysilane vs. diphenyldiacetoxysilane). In the xerogels with high contents of dimethyldiacetoxysilane, there is a significant increase of the surface area (from 356 m2 g−1 at 13 mol% of Me2Si(OAc)2 to 547 m2 g−1 at 25 mol%). However, the volatile organic byproducts contained unreacted dimethyldiacetoxysilane which apparently does not enter the network structure of the gel but acts only as a template. It is eliminated from the pores after the reaction during drying in vacuo, leading to progressively higher surface areas. A similar phenomenon was observed in the case of nonvolatile diphenyldiacetoxysilane, where the surface area of products with a high molar ratio of phenyl groups increased significantly, but only after washing the xerogel with toluene. The washings contained diphenyldiacetoxysilane, [(Ph2Si{O2P(O)OSiMe3})2], and other molecular condensation products with TTP. In contrast, hybrid silicophosphates synthesized from methyltriacetoxysilane and phenyltriacetoxysilane contained no unreacted alkyl/arylacetoxysilanes or some molecular condensation products occluded in the xerogels. Fig. 5 illustrates the dependence of the average pore size on the content of alkyl/arylacetoxysilanes. Interestingly, the average pore size increases as a function of the size of organic groups and the number of bonds silicon can form with phosphorus in these gels. This observation is important, because the SiP xerogel has an average pore size just on the boundary between micropores and mesopores (2.0 nm) and is mostly microporous (ca. 70%), while the hybrid organic–inorganic materials with 20% of RnSi(OAc)4−n are mostly mesoporous (0% microporosity for SiP-MeSi4, SiP-PhSi1 and SiP-Ph2Si2). This phenomenon is clearly seen in Fig. 6, where a comparison of DFT pore size distributions of SiP and SiP-Ph2Si2 samples is shown. The general trends allow us to control the surface area and pore sizes in these hybrid xerogels.
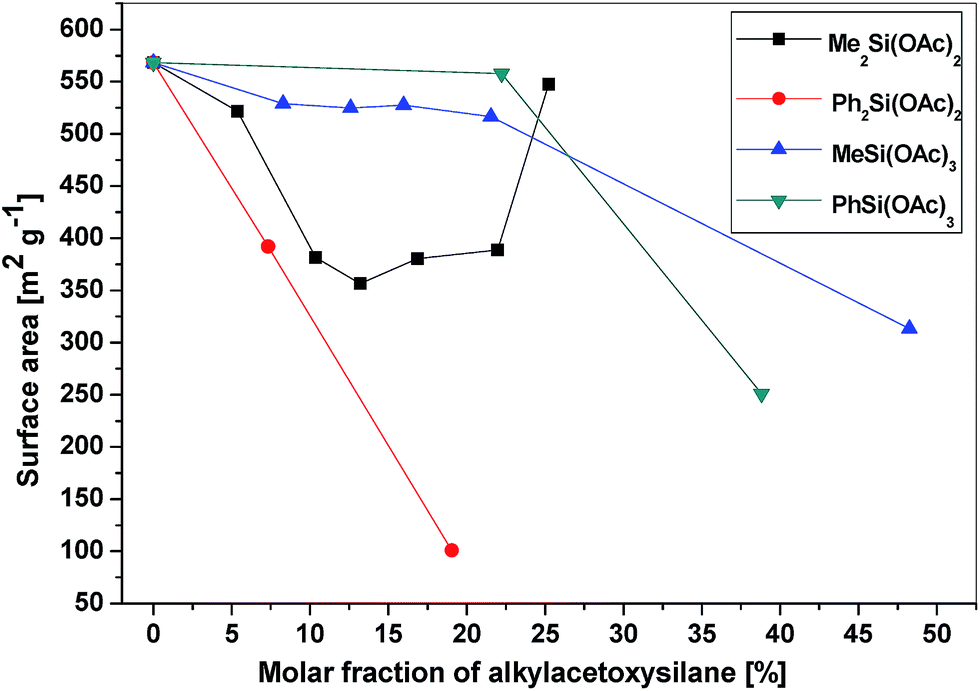 | ||
| Fig. 4 Dependence of the surface area of the hybrid xerogels on the molar fraction of alkyl/arylacetoxysilanes. | ||
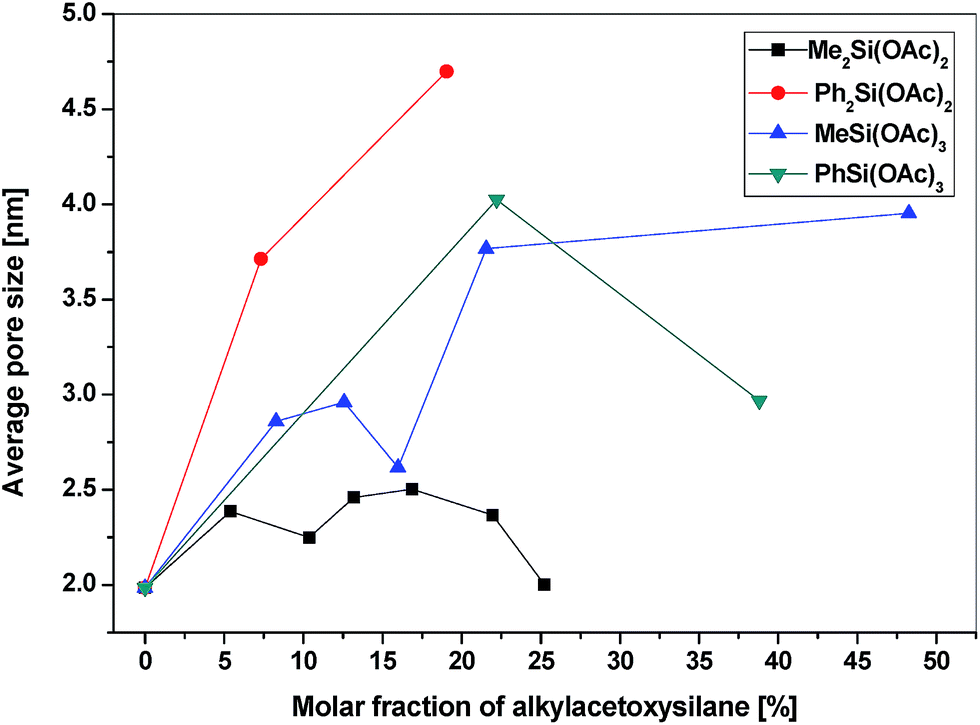 | ||
| Fig. 5 Dependence of the average pore size of the hybrid xerogels on the molar fraction of alkyl/arylacetoxysilanes. | ||
 | ||
| Fig. 6 Pore size distributions of SiP and SiP-Ph2Si2 xerogels (DFT Kernel used: N2 at 77 K on silica, a cylindrical pore, and NSDFT equilibrium model). | ||
To compare the effects of organic groups bound to Si and P, the bis(trimethylsilyl)phenyl phosphonate was introduced into the xerogel in amounts of 13 and 47 mol% replacing TTP (Table 6S:†SiP-PhP1 and SiP-PhP2). As in the case of PhSi(OAc)3 and other RnSi(OAc)4−n (n = 1, 2) precursors, the specific surface area decreased in a similar manner, but the mean pore size stayed small around 2.0 nm and the sample displayed 70% microporosity similar to the unsubstituted SiP sample (Table 6S†). Thus, in contrast to alkyl/arylacetoxysilanes, the phosphonate influenced only the surface area, but did not play a significant role in influencing the mean pore size and micro/mesoporosity of the xerogels. These observations indicate that the mesoporosity was introduced by adding RSiO3 and R2SiO2 units of alkyl/arylacetoxysilanes, while the microporosity is strongly related to the presence of SiO6 groups, which are formed from TTP as well as from phosphonates (Chart 1B and C).
3.3 Bridged acetoxysilanes and phosphonic acid esters
Bis(trialkoxysilyl)alkanes with flexible alkylene spacers are prone to intra- or intermolecular cyclization which hinders the network formation and increases gelation time.66 While rigid spacer groups produce porous xerogels, flexible ones with a hydrocarbon chain longer than six carbons generally cause pore collapse. Also higher DC produces a less compliant network which is in turn more porous.To increase the surface area of xerogels we tested, as the sources of silicon and phosphorus, various bridged bifunctional molecules (Chart 2). An advantage of these bridged precursors is that they are not able to self-condense and form cyclic molecules66 that would impede the gelation process. Homocondensations resulting in acetic acid anhydride or hexamethyldisiloxane elimination are not feasible under reaction conditions used here. All these precursors afforded rigid, colorless (milky) and transparent gels within a short reaction period. They formed two completely different classes of xerogels: (i) hybrid silicophosphates with bridging groups between Si atoms and (ii) hybrid silicophosphates with bridging groups between P atoms. These two classes of xerogels differ in structural and textural properties as will be shown below.
 | ||
| Chart 2 Precursors for hybrid silicophosphate xerogels with bridging organic groups. | ||
From the results presented above, one can expect that the network of xerogels based on (AcO)3Si(CH2)nSi(OAc)3, n = 1–3, 6, will be built from tetrahedral (CH2)nSiO3 and OPO3 units as in the MeSiP and PhSiP samples (Scheme 1 and Chart 1A). The 29Si CPMAS NMR spectra, in fact, displayed only two resonances at −64.5 ppm ((CH2)nSiO3) and 25.6 ppm (residual Me3SiOP) with no evidence for the SiO6 centers and no evidence of Si–C bond cleavage. Also the 31P and 13C MAS NMR and IR spectra were similar to MeSiP supporting their structural similarity (Fig. 15S–17S†). However, the introduction of the bridging molecules with six acetate groups provided an abundant cross-linking, so that the surface area increased significantly in comparison with the MeSiP sample (Table 5).
| Sample | Si source | P source | SA [m2 g−1] | V total [cm3 g−1] | D [nm] | MPb [%] |
|---|---|---|---|---|---|---|
| a Average pore diameters (4Vtotal/SA). b Determined by the t-plot. | ||||||
| SiCSiP | (AcO)3SiCH2Si(OAc)3 | OP(OSiMe3)3 | 505 | 0.462 | 3.7 | 0 |
| SiC2SiP | (AcO)3Si(CH2)2Si(OAc)3 | OP(OSiMe3)3 | 703 | 1.559 | 9.0 | 0 |
| SiC3SiP | (AcO)3Si(CH2)3Si(OAc)3 | OP(OSiMe3)3 | 369 | 1.091 | 11.9 | 0 |
| SiC6SiP | (AcO)3Si(CH2)6Si(OAc)3 | OP(OSiMe3)3 | 385 | 0.753 | 7.5 | 0 |
| SiPC2P | Si(OAc)4 | (Me3SiO)2P(O)CH2CH2P(O)(OSiMe3)2 | 553 | 0.296 | 2.2 | 51 |
| SiPC6P | Si(OAc)4 | (Me3SiO)2P(O)C6H4P(O)(OSiMe3)2 | 617 | 0.328 | 2.1 | 52 |
The surface area of the xerogels ranges from 369 to 703 m2 g−1 and reaches its maximum for the 1,2-ethanediyl bridging unit. Longer spacers exhibit lower surface areas (Table 5). All the samples with bridging units between Si atoms are mesoporous, in contrast to the SiP sample, with the mean pore sizes ranging from 3.7 to 11.9 nm (Fig. 21S†). These observations support our suggestion that the microporosity in silicophosphate xerogels is dependent on the presence of the SiO6 units while mesoporous materials can be obtained by restricting the Si coordination to 4. The average pore size increases with the increasing length of the aliphatic chain for n = 1–3 and then drops down for n = 6 probably because of the non-rigidity of the bridging unit (Table 5). These results are similar to those observed by Oviatt et al.,35 where average pore sizes increased from 4.09 nm to 19.7 nm and the surface area simultaneously decreased from 684 to 88 m2 g−1 on going from bis(triethoxysilyl)butane to bis(triethoxysilyl)decane in the case of base-catalyzed hydrolytic sol–gel condensations.
Completely different structural and textural properties were displayed by the hybrid silicophosphates with the bridging group between P atoms. These materials were strongly related to SiP, SiP-PhP1, and SiP-PhP2 samples. The bridging organic groups bound to phosphorus did not hinder the formation of hexacoordinated silicon (Chart 1C) and both MAS NMR and IR spectra closely resembled the SiP samples including the evidence for SiO6 groups, while no P–C bond rupture was observed (Fig. 15S–17S†). These xerogels reached the maximum surface area of 617 m2 g−1 and their average pore sizes were on the boundary between micro- and mesoporosity (2.2 and 2.1 nm), similar to the SiP samples. The content of the micropore surface area was around 50% (Table 5). The textural diversity caused by the location of the organic spacer (between the Si vs. P atoms) is clearly seen in the comparison of adsorption–desorption isotherms of the corresponding xerogels (Fig. 7, Fig. 18S–20S†). The SiPC6P sample did not show any hysteresis and the main gas adsorption proceeded at low relative pressures similarly as in the SiP sample.54 In contrast, the isotherm of the SiC2SiP sample was of the type IV and displayed hysteresis H3 in medium relative pressures corroborating the mesoporosity of these xerogels.
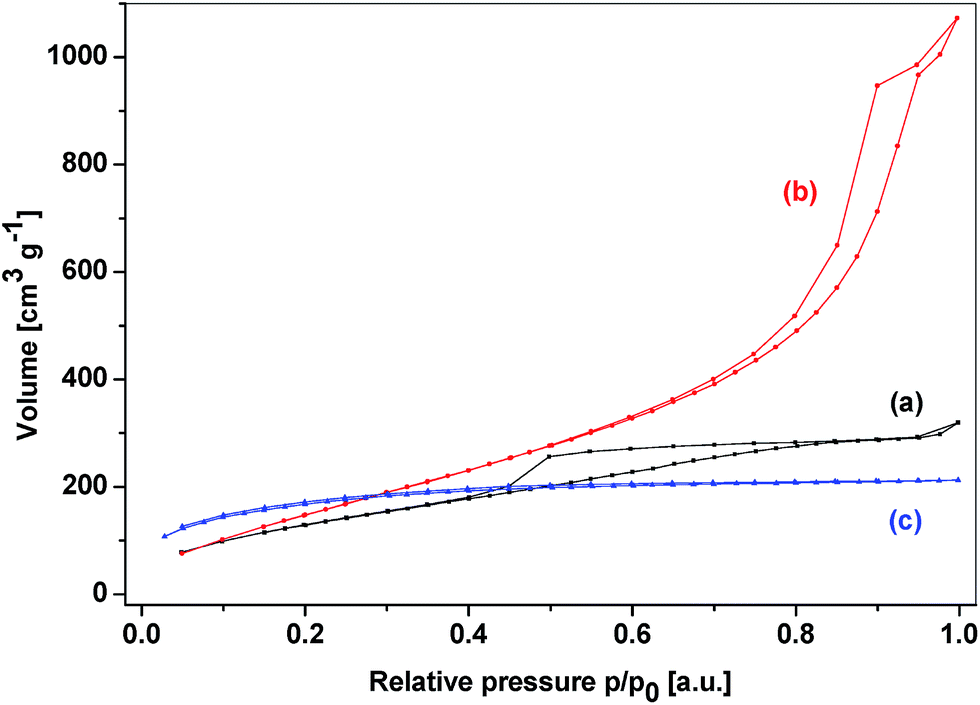 | ||
| Fig. 7 Adsorption–desorption isotherms of xerogels (a) SiC1SiP, (b) SiC2SiP and (c) SiPC6P. | ||
3.4 Thermal properties of the hybrid silicophosphate xerogels
The elimination and oxidation of organic groups and crystallization of silicon phosphates from the SiP sample upon heating in air were described previously.54 The experimental mass losses (ML) for most of the hybrid silicophosphates are in reasonable agreement with the calculated ones (MLcalc) from DC (Tables 2–4). Three exothermic weight loss steps were observed, identically as in the SiP xerogel. They appeared for all the samples in these ranges: from 50 to 250 °C, from 500 to 600 °C and between 900 and 990 °C. Samples with covalently bound phenyl groups showed one more mass loss between 300 and 500 °C apparently caused by the degradation of aromatic rings.The crystallization of Si5P6O25 from the SiP xerogel was observed as early as at 150 °C and was accompanied by a decrease in the surface area. Also the hybrid samples were heated in air in order to describe the textural properties of heated xerogels. All the samples displayed the breakdown of the porous structure and rapid decrease of the surface area upon heating to 200 °C for 4 h. The highest surface area after calcination was only 20 m2 g−1 for the sample SiP-MeSi3 (the parent xerogel 528 m2 g−1). The hybrid samples after TG analysis were amorphous, only some of them contained small amounts of crystalline silicon orthophosphate (PDF 40-457) or cubic silicon diphosphate (PDF 12-1321).
4. Conclusions
This work extends our previous efforts to synthesize homogenous silicophosphates which were prepared by a non-hydrolytic sol–gel polycondensation reaction based on acetic acid ester elimination.54 While the reactions of silicon tetraacetate and tris(trimethylsilyl)phosphate provided microporous gels with SiO6 structural units, the introduction of organic groups in the form of different alkyl/arylacetoxysilanes induced structural and textural changes leading to nonporous hybrid silicophosphate glasses. Such a drastic change of properties was ascribed to the lowering of the cross-linking capacity of silicon. Moreover, the organo-substituted silicon centers were shown not to be sufficiently Lewis acidic to acquire hexacoordination while steric hindrance did not play a major role in preventing the formation of SiO6 moieties. The reaction of OP(OSiMe3)3 with Ph2Si(OAc)2 afforded only molecular products of condensation in contrast to other alkyl/arylacetoxysilanes. One of them, [(Ph2Si{O2P(O)OSiMe3})2], was isolated and its structure was determined by single crystal X-ray diffraction analysis.Partially substituting Si(OAc)4 with RnSi(OAc)4−n (n = 1, 2; R = Me, Ph) and varying the nature of the organic group R allowed us to tune the surface area in the range from 101 to 568 m2 g−1 and average pore sizes from 2.0 to 4.7 nm in resulting xerogels. R2Si(OAc)2 and acetoxysilanes with bulky organic groups caused a bigger decrease of the surface area and average pore size increase in comparison to RSi(OAc)3 and acetoxysilanes with small substituents. However the products were still significantly microporous.
Finally, the introduction of bridging organic groups provided an additional number of cross-linking possibilities which was reflected in the increase of the surface area of the xerogels. The organic spacer between silicon atoms caused the disappearance of the SiO6 units similar to alkyl/arylacetoxysilanes. This structural change was accompanied by a change in textural properties – these samples were completely mesoporous with a highest surface area of 703 m2 g−1 and an average pore size of 9.0 nm for the C2H4 bridge. The length of the bridging unit of (AcO)3Si(CH2)nSi(OAc)3 significantly influenced the pore size distribution – the average pore size increased with the increasing number of carbon atoms in the organic spacer for n = 1 to 3. In the case of the substitution of TTP by (Me3SiO)2P(O)CH2CH2P(O)(OSiMe3)2 and (Me3SiO)2P(O)C6H4P(O)(OSiMe3)2, the structures of the xerogels were still predominantly built up of the SiO6 units and displayed a significant microporosity reaching the highest surface area at 617 m2 g−1 with an average pore size of 2.1 nm in the case of the phenylene bridge. These results point to a significant difference in the properties of xerogels built from units with spacer groups between silicon atoms in contrast to the spacers between phosphorus atoms.
Acknowledgements
The authors thank the project CEITEC – Central European Institute of Technology CZ.1.05/1.1.00/02.0068, KONTAKT II LH11028, GACR GAP207/11/0555 and Single Crystal X-Ray Diffraction Core Facility for financial assistance. J.P. thanks the Fulbright Foundation for a scholarship. A.S. thanks the Brno City Municipality for Brno a Ph.D. Talent Scholarship.References
- D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS.
- G. Cerveau and R. J. P. Corriu, Coord. Chem. Rev., 1998, 178–180(2), 1051–1071 CrossRef CAS.
- C. Sanchez, F. Ribot and B. Lebeau, J. Mater. Chem., 1999, 9, 35–44 RSC.
- R. J. P. Corriu, Angew. Chem., Int. Ed., 2000, 39, 1376–1398 CrossRef CAS.
- G. Kickelbick, Hybrid Materials: Synthesis, Characterization, and Applications, John Wiley & Sons, 2007 Search PubMed.
- C. Sanchez, L. Rozes, F. Ribot, C. Laberty-Robert, D. Grosso, C. Sassoye, C. Boissiere and L. Nicole, C. R. Chim., 2010, 13, 3–39 CrossRef CAS PubMed.
- C. Sanchez, K. J. Shea and S. Kitagawa, Chem. Soc. Rev., 2011, 40, 471–472 RSC.
- D. A. Loy, K. A. Obrey-DeFriend, K. V. Wilson Jr, M. Minke, B. M. Baugher, C. R. Baugher, D. A. Schneider, G. M. Jamison and K. J. Shea, J. Non-Cryst. Solids, 2013, 362, 82–94 CrossRef CAS PubMed.
- G. Cerveau, R. J. P. Corriu and E. Framery, Polyhedron, 2000, 19, 307–313 CrossRef CAS.
- W. Chaikittisilp, M. Kubo, T. Moteki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2011, 133, 13832–13835 CrossRef CAS PubMed.
- C. Sanchez, B. Lebeau, F. Chaput and J.-P. Boilot, Adv. Mater., 2003, 15, 1969–1994 CrossRef CAS.
- Y. Tokudome, T. Sato, M. Hidaka and M. Takahashi, J. Sol-Gel Sci. Technol., 2013, 65, 318–323 CrossRef CAS.
- H. Dong, R. F. Reidy and J. D. Brennan, Chem. Mater., 2005, 17, 6012–6017 CrossRef CAS.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS PubMed.
- M. Sebah, S. P. Maddala, P. Haycock, A. Sullivan, H. Toms and J. Wilson, J. Mol. Catal. A: Chem., 2013, 374–375, 59–65 CrossRef CAS PubMed.
- N. A. Carrington and Z.-L. Xue, Acc. Chem. Res., 2007, 40, 343–350 CrossRef CAS PubMed.
- L. Brigo, V. Auzelyte, K. A. Lister, J. Brugger and G. Brusatin, Nanotechnology, 2012, 23, 325302 CrossRef CAS PubMed.
- F. Eckstorff, Y. Zhu, R. Maurer, T. E. Müller, S. Scholz and J. A. Lercher, Polymer, 2011, 52, 2492–2498 CrossRef CAS PubMed.
- U. Schubert, N. Huesing and A. Lorenz, Chem. Mater., 1995, 7, 2010–2027 CrossRef CAS.
- C. Sanchez, C. Boissiere, S. Cassaignon, C. Chaneac, O. Durupthy, M. Faustini, D. Grosso, C. Laberty-Robert, L. Nicole, D. Portehault, F. Ribot, L. Rozes and C. Sassoye, Chem. Mater., 2014, 26, 221–238 CrossRef CAS.
- G. Fornasieri, L. Rozes, S. Le Calvé, B. Alonso, D. Massiot, M. N. Rager, M. Evain, K. Boubekeur and C. Sanchez, J. Am. Chem. Soc., 2005, 127, 4869–4878 CrossRef CAS PubMed.
- J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436–3448 CrossRef CAS.
- G. Li, L. Wang, H. Ni and C. U. Pittman, J. Inorg. Organomet. Polym., 2001, 11, 123–154 CrossRef CAS.
- J. D. Lichtenhan, Comments Inorg. Chem., 1995, 17, 115–130 CrossRef CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- R. Corriu, Polyhedron, 1998, 17, 925–934 CrossRef CAS.
- S. Lin, C. Ionescu, E. M. Valliant, J. V. Hanna, M. E. Smith and J. R. Jones, J. Mater. Chem., 2010, 20, 1489 RSC.
- R. Al-Oweini, Appl. Surf. Sci., 2010, 257, 276 CrossRef CAS PubMed.
- K. Nakanishi and K. Kanamori, J. Mater. Chem., 2005, 15, 3776–3786 RSC.
- B. Boury, R. J. P. Corriu, V. Le Strat, P. Delord and M. Nobili, Angew. Chem., Int. Ed., 1999, 38, 3172–3175 CrossRef CAS.
- G. Creff, G. Arrachart, P. Hermet, H. Wadepohl, R. Almairac, D. Maurin, J.-L. Sauvajol, C. Carcel, J. J. E. Moreau, P. Dieudonné, M. W. C. Man and J.-L. Bantignies, Phys. Chem. Chem. Phys., 2012, 14, 5672–5679 RSC.
- G. Creff, B. P. Pichon, C. Blanc, D. Maurin, J.-L. Sauvajol, C. Carcel, J. J. E. Moreau, P. Roy, J. R. Bartlett, M. Wong Chi Man and J.-L. Bantignies, Langmuir, 2013, 29, 5581–5588 CrossRef CAS PubMed.
- B. Boury and R. J. P. Corriu, Chem. Commun., 2002, 795–802 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- H. W. Oviatt, K. J. Shea and J. H. Small, Chem. Mater., 1993, 5, 943–950 CrossRef CAS.
- J. N. Hay, D. Porter and H. M. Raval, J. Mater. Chem., 2000, 10, 1811–1818 RSC.
- L. Bourget, D. Leclercq and A. Vioux, J. Sol-Gel Sci. Technol., 1999, 14, 137–147 CrossRef CAS.
- A. Styskalik, D. Skoda, J. Pinkas and S. Mathur, J. Sol-Gel Sci. Technol., 2012, 63, 463–472 CrossRef CAS PubMed.
- O. Lorret, V. Lafond, P. H. Mutin and A. Vioux, Chem. Mater., 2006, 18, 4707–4709 CrossRef CAS.
- D. P. Debecker and P. H. Mutin, Chem. Soc. Rev., 2012, 41, 3624–3650 RSC.
- L. Crouzet, D. Leclercq, P. Mutin and A. Vioux, J. Sol-Gel Sci. Technol., 2003, 26, 335–338 CrossRef CAS.
- P. H. Mutin and A. Vioux, J. Mater. Chem. A, 2013, 1, 11504–11512 CAS.
- M. Nogami, H. Matsushita, Y. Goto and T. Kasuga, Adv. Mater., 2000, 12, 1370–1372 CrossRef CAS.
- A. Matsuda, T. Kanzaki, K. Tadanaga, M. Tatsumisago and T. Minami, Solid State Ionics, 2002, 154&155, 687–692 Search PubMed.
- M. Mika, M. Paidar, B. Klapste, M. Masinova, K. Bouzek and J. Vondrak, J. Phys. Chem. Solids, 2007, 68, 775–779 CrossRef CAS PubMed.
- J.-M. Lim, J.-H. Won, H.-J. Lee, Y. T. Hong, M.-S. Lee, C. H. Kod and S.-Y. Lee, J. Mater. Chem., 2012, 22, 18550–18557 RSC.
- M. Takahashi, M. Suzuki, Y. Miyagawa, R. Ihara, Y. Tokuda, T. Yoko, T. Nemoto and S. Isoda, J. Sol-Gel Sci. Technol., 2010, 54, 319–324 CrossRef CAS PubMed.
- I. N. Tsvetkova, O. A. Shilova, M. G. Voronkov, Y. P. Gomza and K. M. Sukhoy, Glass Phys. Chem., 2011, 34, 68–76 CrossRef.
- R. J. P. Corriu, D. Leclercq, P. H. Mutin, L. Sarlin and A. Vioux, J. Mater. Chem., 1998, 8, 1827–1833 RSC.
- M. Mizuno, M. Takahashi, Y. Tokuda and T. Yoko, Chem. Mater., 2006, 18, 2075–2080 CrossRef CAS.
- Y. Tokuda, Y. Tanaka, M. Takahashi, R. Ihara and T. Yoko, J. Ceram. Soc. Jpn., 2009, 117, 842–846 CrossRef CAS.
- H. Niida, Y. Tokuda, M. Takahashi, T. Uchino and T. Yoko, J. Non-Cryst. Solids, 2002, 311, 145–153 CrossRef CAS.
- M. Mizuno, M. Takahashi, Y. Tokuda and T. Yoko, J. Sol-Gel Sci. Technol., 2007, 44, 47–52 CrossRef CAS PubMed.
- A. Styskalik, D. Skoda, Z. Moravec, J. G. Abbott, C. E. Barnes and J. Pinkas, Microporous Mesoporous Mater., 2014, 197, 204–212 CrossRef CAS PubMed.
- D. W. Schaefer, G. Beaucage, D. A. Loy, K. J. Shea and J. S. Lin, Chem. Mater., 2004, 16, 1402–1410 CrossRef CAS.
- Y. Vaugeois, R. D. Jaeger, J. Levalois-Mitjaville, A. Mazzah, M. Wörle and H. Grützmacher, New J. Chem., 1998, 22, 783–785 RSC.
- N. W. Alcock, V. M. Tracy and T. C. Waddington, J. Chem. Soc., Dalton Trans., 1976, 2243 RSC.
- B. P. Straughan, W. Moore and R. McLaughlin, Spectrochim. Acta, Part A, 1986, 42, 451–456 CrossRef.
- Y. Song, Y. Huang, E. A. Havenga and I. S. Butler, Vib. Spectrosc., 2001, 27, 127–134 CrossRef CAS.
- V. G. Plotnichenko, V. O. Sokolov, V. V. Koltashev and E. M. Dianov, J. Non-Cryst. Solids, 2002, 306, 209–226 CrossRef CAS.
- E. M. Dianov, V. V. Koltashev, V. G. Plotnichenko, V. O. Sokolov and V. B. Sulimov, J. Non-Cryst. Solids, 1999, 249, 29–40 CrossRef CAS.
- N. Shibata, M. Horigudhi and T. Edahiro, J. Non-Cryst. Solids, 1981, 45, 115–126 CrossRef CAS.
- J. Wong, J. Non-Cryst. Solids, 1976, 20, 83–100 CrossRef CAS.
- K. J. Shea, D. A. Loy and O. Webster, J. Am. Chem. Soc., 1992, 114, 6700–6710 CrossRef CAS.
- R. J. P. Corriu, J. J. E. Moreau, P. Thepot and M. W. C. Man, Chem. Mater., 1992, 4, 1217–1224 CrossRef CAS.
- K. J. Shea and D. A. Loy, Acc. Chem. Res., 2001, 34, 707–716 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1038898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ta06823h |
| ‡ Dedicated to Professor H. W. Roesky on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |