
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improvement in flexibility and volumetric performance for supercapacitor application and the effect of Ni–Fe ratio on electrode behaviour†
Liuyang
Zhang
and
Hao
Gong
*
Department of Materials Science and Engineering, National University of Singapore, Singapore 117576, Singapore. E-mail: msegongh@nus.edu.sg; Tel: +65 6516 4632
First published on 25th February 2015
Abstract
To increase the flexibility, volumetric capacitance and energy density of a supercapacitor, an economical but effective method of rolling a foam current collector before the direct growth of electrode materials on it is proposed and studied for the first time. By using this novel economic approach, volumetric capacitance and energy density are demonstrated to increase almost 3 times, even though they have not been optimized yet. At the same time, the flexibility is improved tremendously, making it possible to obtain an extremely flexible supercapacitor. Additionally, in the current work, the material system of nickel iron oxide has been studied systematically. The effect of nickel iron ratio on the electrochemical performance has been examined. The resulting extremely bendable or flexible current collector with the active material grown on it showed a high volumetric capacitance of 11.6 F cm−3 and energy and power densities of 4.12 mW h cm−3 and 236.25 mW cm−3, respectively. It should be noted that these volumetric values are truly high because they are calculated based on the whole device volume of the two current collectors, rather than that of the active material itself only. Such a strategy might be readily extended to other systems with nano-structured materials grown on metal foams.
1. Introduction
In the evaluation of the specific capacitance (energy density) of a supercapacitor, there are mainly 3 different units used in the literature: F g−1 (kW h Kg−1),1–4 F cm−2 (kW h cm−2)5–7 and F cm−3 (kW h cm−3).8–10 Nowadays, the electrode performances of supercapacitors are usually reported on a gravimetric basis, i.e. using the units of F g−1 and kW h kg−1. Sole comparisons made based on gravimetric capacitance would be inappropriate due to the overestimated values at significantly low masses (less than 0.5 mg), as well as the difficulty in comparing the overall device performances when the samples have different loadings of electrodes. It needs to be mentioned here that achieving a very high loading when directly growing electrode materials on a current collector is challenging. Areal capacitance is a crucial parameter for miniaturized energy storage devices in modern gadget applications. However, by using areal specific capacitance (energy density) F cm−2 (kW h cm−2), it is difficult to compare the performances especially for devices utilizing different planar and foam current collectors. Volumetric specific capacitance (energy density) with a unit of F cm−3 (kW h cm−3) can be used for easy comparison of the performances of whole devices but is mostly ignored regrettably. Gogotsi and Simon11 recommended that the volumetric performance should be a more reliable parameter than the gravimetric one to evaluate the real potential of electrode materials for supercapacitors, especially for compact and portable energy storage devices. Recently a growing number of research groups12–14 have started to use capacitance per volume (F cm−3) in reporting the performance of a supercapacitor especially when it is used in practical applications. In our opinion, a combination of reporting gravimetric, areal and volumetric specific capacitance and the corresponding energy/power density for a device can better describe the performance of both the electrode material and the supercapacitor device.The realization of high volumetric specific capacitance and energy density is challenging, especially for the advanced approach of directly growing electrode materials on current collectors (substrates). The advantage of direct growth is the maximization of the electrode/collector conductivity and carrier transportation. To date, foam-based collectors have been adopted in many actual device applications, due to their high surface areas and flexible nature. However, the relatively big pores in conventional commercial metal foams lead to much unused space when employing the direct growth approach of electrode materials, thereby foams are very poorly utilized and the supercapacitor volumetric performance is poor. Commercial foams with big pores are abundant and cheap possibly because industries use them in the mass electrode fabrication process, especially in fabricating slurry-derived supercapacitor electrodes. Nevertheless, it is hard to find commercial foams with suitable small pore sizes to satisfy the needs. In this paper, we will demonstrate that the volumetric performance can be greatly improved by an easy and economical treatment of a piece of conventional commercial foam. The aim of this process is to minimize the unused or wasted pore space of a piece of foam and optimize the realizable specific capacitance per volume, while guaranteeing sufficient volume for the accumulation of the electrolyte at the same time. We also hope that this success can trigger the enthusiasm of foam manufacturers to explore technologies in producing cheap metal foams with small pores suitable for advanced supercapacitors.
In addition to the exploration to achieve high volumetric performance, this paper will also investigate the potential of nickel iron oxide as an effective supercapacitor electrode material. Nickel oxide has emerged as a cheap and ecologically benign electrode material in recent years.15,16 However, the results obtained to date are still not satisfactory. Complete or partial substitution of Ni with low-cost and benign elements to form binary oxides/hydroxides is a very interesting topic, and high supercapacitor performance is achieved for some compounds like NiCo2O4.17–19 In the periodic table, iron (Fe) is in the same row of Ru and adjacent to Mn and Co. These latter three elements (when form oxides or hydroxides) have been demonstrated to be good pseudocapacitance electrode materials. Moreover, iron oxide, which is renowned for its high theoretical charge storage capability (1005 mA h g−1 in LIB (lithium ion battery) anodes and 1342 F g−1 in EC (electrochemical capacitors)) and natural abundance, has been developed as an attractive electrode material.20–22 The mixed valence states of iron provide readily accessible redox couples. Besides, enlightened by the previous reports that the specific capacitance of the binary Mn–Fe oxide is enhanced by 21% with appropriate Fe addition23 and the superior performance of the nickel hydroxide@iron oxide core–shell structure,24 it is anticipated that nickel iron oxide may be a candidate. However, only a limited number of papers on nickel iron oxide supercapacitors have been published.25–27 These papers synthesized NiFe2O4 with limited fixed discrete compositions of Ni–Fe atomic ratios. It is unclear about the effect of Ni–Fe ratio of the nickel iron oxide system on the supercapacitor performance. Therefore, it is necessary to evaluate this material system with different nickel–iron ratios for understanding its potential as an effective supercapacitor material.
In this paper, two issues are addressed and problems are solved for achieving high performance supercapacitors. One issue is how to achieve high volumetric specific capacitance and energy density of a supercapacitor through a proper treatment of cheap conventional commercial metal foam current collectors (substrates) before the growth of the material. Another is to understand the potential of the nickel iron oxide electrode material system in supercapacitor applications through a systematic study of the effect of nickel–iron ratio.
2. Experimental
2.1. Direct synthesis of the nickel iron hybrid material
The typical reaction process for the synthesis of Ni–Fe–O/r-NF was as follows. First, a piece of nickel foam (20 cm × 20 cm × 0.15 cm) was rolled by the rolling machine. Then, they were cut into 2 cm × 2 cm and cleaned in an ultrasonic bath with absolute ethanol and deionized water to remove adsorbed dust and surface contamination. Then, an aqueous solution was prepared by mixing NiSO4 and FeSO4 with urea together. After a few minutes of stirring, the treated rolled nickel foam was suspended in the prepared solution.After the solution was stirred for 30 min, the final solution was transferred into a Teflon-lined stainless steel autoclave. Hydrothermal synthesis was run at a temperature of 120 °C for 6 h and then cooled down to room temperature. The rolled nickel foam with the grown material was filtered, washed with distilled water several times, and then dried at 80 °C in an oven for 24 h.
For the synthesis of the material on commercial conventional nickel foam, all the experimental parameters remained to be the same except for the substrate being changed to the unrolled conventional one.
2.2. Fabrication of asymmetric supercapacitors (ASCs)
ASCs were assembled by using Ni–Fe–O/r-NF (Ni–Fe–O/NF) as the positive electrode (after optimization, here 3 mg) and reduced graphene oxide (after optimization, here 11 mg) as the negative electrode. The dimensions of the positive/negative electrode are 2 cm × 1 cm × 0.05 cm. The original 2 cm × 2 cm positive electrode with the active material on it has been cut into 2 cm × 1 cm when assembling the full cell device. The detailed calculation can be found in the ESI.†2.3. Characterization
The morphology of the prepared samples was examined by using a scanning electron microscope with an X-ray energy dispersive spectrometer (SEM, Zeiss SUPRA40). The crystal structure was identified using X-ray diffraction. Transmission electron microscopy and electron diffraction (TEM, JEOL 2000FX) were employed to obtain high resolution images and structural information. The surface functional groups were investigated by X-ray photoelectron spectroscopy (XPS) (AXIS Ultra). N2 adsorption at a temperature of 77 K with relative pressure (P/P0) ranging from 0.02 to 1 (Micromeritics ASAP2020) was employed to measure the specific surface area. Cyclic voltammetry (CV), galvanostatic charge–discharge and electrochemical impedance spectroscopy (EIS) were performed using an electrochemical analyzer (Solartron S1 1287) under ambient conditions. Electrochemical measurements were performed using 1 M KOH as the electrolyte. Electrochemical impedance spectroscopy (EIS) measurements were conducted by applying an AC voltage with 5 mV amplitude in the frequency range of 0.01 Hz to 100 kHz at the open circuit potential. Full cell galvanostatic cycling with potential limitation was duplicated over a potential window from 0 to 1.6 V for charge experiments.3. Results and discussion
3.1. Significant increase in volumetric specific capacitance
Metal foam has a larger surface area than that of metal foil, making it a preferred type of current collector for supercapacitors. Direct growth of materials on current collectors is especially important for metal oxide electrode materials. There are several advantages of direct growth of electrode materials on current collectors (substrates), including the elimination of the use of binders that can cover some electrode active area, and the avoidance of an additional binder layer that is harmful to the carrier transport between the active material and the current collector.28 However, it is very difficult or impossible to grow a porous and thick enough electrode material layer that can fill the whole pore space of a foam collector. Conventional commercial metal foams (100–110 PPI) have pores with sizes in the range of 50–500 μm (Fig. 1a). After the growth of the optimum amount of electrode materials, most space is still empty (Fig. 1a′), leading to undesirable small volumetric specific capacitance and energy density of supercapacitors. To solve this problem, there are two possible approaches. One is to use special foams with very small pores, and the other is to minimize the unused space of the conventional foams. For the former, either it is difficult or impossible to find suitable commercial metal foams with very small pores or such foams (if existing) are very expensive. For the latter, no report has been found and we would like to try it to increase volumetric performance based on the conventional commercial foams.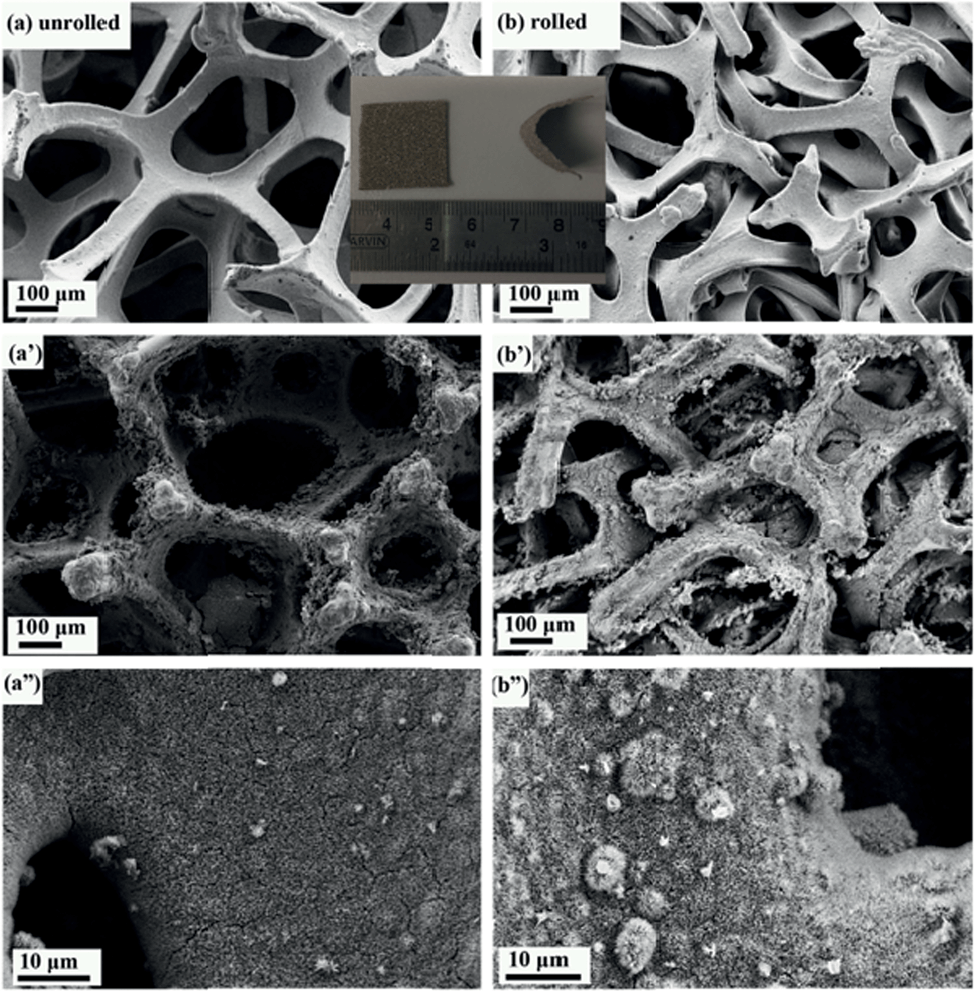 | ||
| Fig. 1 Comparison of SEM images of unrolled and rolled nickel foam before and after the growth of the material. | ||
The method we employed here was to roll the nickel foam with a compression, strong enough for the nickel foam to surpass elastic distortion but without breaking. We compressed the nickel foam from 1.5 mm to 0.5 mm in thickness and the resulting compressed nickel foam is shown in Fig. 1b. The unused pore space after the growth of electrode materials significantly decreased as seen in Fig. 1b′ when compared with Fig. 1a′. It can be seen that the pores of the rolled nickel foam became considerably smaller along the compressing direction (Fig. 1a′ and b′), and the coverage of the electrode material was similar for the rolled and unrolled samples (Fig. 1a′′ and b′′). As we all know, pseudo-capacitance is mainly produced by a fast faradaic reaction occurring near the solid electrode surface. As we hoped, the material grown on the rolled nickel foam does not agglomerate (pile up) together and the pores of the foam are much better used than the unrolled counterpart. The arrays grown on the substrates did not suffer from the problem of collapsing towards the neighbors which leads to the decrease in the functional surface and limited accessibility to the electrolyte. Herein, the volumetric capacitance of the active material directly grown on nickel foam is increased. Another advantage of the rolled nickel foam is that it is very flexible and can be bent sharply as shown in Fig. 2i and 2ii. To evaluate the flexibility, the original and rolled nickel foams were wound tightly and removed from cylinders of different radii as shown in Fig. 2i. It can be seen that the foams were intact after winding on cylinders of radii 14 mm (Fig. 2i: a1′ and b1′) and 9 mm (a2′ and b2′). However, the original nickel foam was damaged and lost elasticity (Fig. 2i: a3′) after winding on a cylinder of 2 mm radius. Interestingly, the rolled foam was still intact (Fig. 2i: b3′). The rolled foam was then further tightly wounded on wires with smaller radii (Fig. 2ii). After winding on a wire of 1 mm radius (b4) and then a wire of 0.5 mm radius (b5), the removed rolled foam still had very good elasticity (b4′ and b5′). After straightening the curved foam, the rolled foam did not show damage traces (Fig. 2ii: b5′-s). The elasticity has been maintained after releasing the foam free (Fig. 2ii: b5′–f). The great improvement in flexibility after rolling the foam can be explained as follows. Bending generates forces on the material through the thickness of the sheet in the bend region. The materials towards outside and inside in the bend region are under opposite tension and compression forces, respectively. There is subsequently a neutral layer or axis, which is roughly near the sheet center, with zero force on it during bending. The magnitude of the bend force increases with the distance from the neutral layer. Since the rolled foam is much thinner than the original foam, the maximum compression and extension on the outer layers are much smaller. Therefore, the rolled foam possesses a much greater bendability. The superior flexible properties of the rolled foam have great application significance and potential as there is a huge demand for bendable or flexible energy storage devices.
 | ||
| Fig. 2 (i) Images of the (a) original and (b) rolled nickel foams after tightly winding on and removal (a′ and b′) from cylinders of radii (1) 14 mm, (2) 9 mm, and (3) 2 mm. (ii) Images of the rolled nickel foam after further tightly winding on and removal from wires of radii 1 mm (b4) and 0.5 mm (b5). (b5′-s) and (b5′-f) show the images of the firstly straightened and then released foam (the foam in b5′). | ||
The evaluation of supercapacitor performance for the unrolled and rolled nickel foams with the same electrode material directly grown on them is carried out. All these tested foams have areas of 2 cm × 2 cm. The loading of NiO for both samples was measured to be almost the same: 5.0 mg. Fig. 3a shows the CV curves (at a scan rate of 5 mA) and Fig. 3b shows the discharge curves (at a constant current density of 2 mA cm−2) of NiO directly grown on unrolled and rolled nickel foams. From Fig. 3a, the capacitance of the material on rolled nickel foam is 1146 F g−1 while that of the unrolled one is 1390 F g−1 based on the CV profiles. The calculated capacitance from Fig. 3b for the rolled nickel foam and unrolled one are 1430 F g−1 and 1400 F g−1, respectively. Therefore, the volumetric energy density for the rolled sample is 2.47 times that of the unrolled sample. While from the charging–discharging (CD) results (Fig. 3b), the capacitances were 1430 F g−1 and 1400 F g−1 at a constant current of 8 mA, respectively. Thus the improvement (based on the CV curve results) is 2.47 times, and 3.06 times based on the discharging results. The difference in these two types of results can be because the surface is not so easily accessible for ions in the electrolyte at a high current or scan rate for the rolled foam. It is known that the specific capacitance value depends on the constant current density imposed for the charging–discharging (CD) plot just as the specific capacitance value varies with the scan rate used for the CV loops. However, the dependence of different samples on each technique (CV and CD) is not comparable, many factors will affect the calculated values, in our case, such as the porosity of the current collector (one is rolled, the other is unrolled), and the degree of irreversibility (from CV curves, the separation of the peak for our two samples are different). These can be the reasons why the ratios from CV and CD measurements are different. It should be mentioned that a comparison of various nickel iron oxides (with different Ni/Fe ratios) grown on the unrolled and rolled nickel foams also reveals that the rolled nickel foam does not affect the total capacitances much. The thickness of the rolled nickel foam is 1/3 of that of the unrolled one, and the volumetric specific capacitance of the rolled sample is almost 3 times higher than that of the unrolled one. More quantitative data to demonstrate the benefit of this rolling approach can be found in Section 3.2.2.
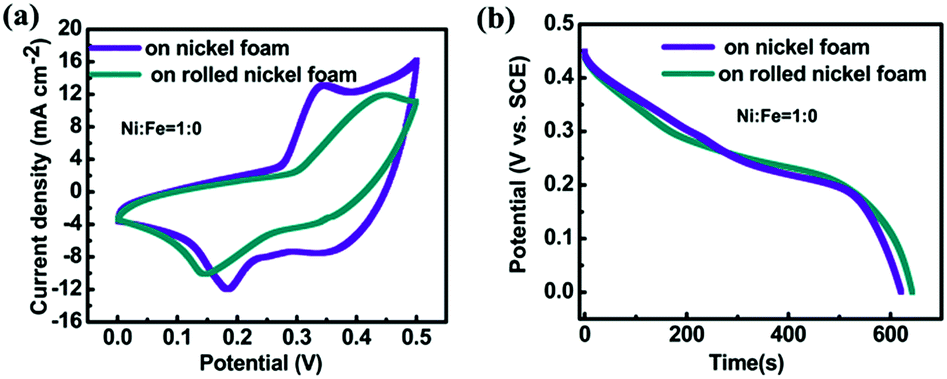 | ||
Fig. 3 Comparison of (a) CV curves and (b) discharge curves of NiO (Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe = 1:0) directly grown on unrolled and rolled nickel foams. :Fe = 1:0) directly grown on unrolled and rolled nickel foams. | ||
3.2. On the nickel iron oxide system as a potential supercapacitor electrode
:Fe ratios then appears necessary. Nickel iron oxides with different Ni:Fe ratios are grown on nickel foams, under similar experimental conditions except for using different amounts of Ni and Fe containing precursors.
Fig. 4 displays the scanning electron microscopy (SEM) images of the nickel iron compound grown on nickel foams. Without iron (Ni:Fe = 1:0, i.e. only nickel oxide), the electrode layer is porous and the nanobelts are thin (Fig. 4a). With a Ni:Fe ratio of 0.8:0.2, a similar morphology is maintained but there are some big particles on the surface (Fig. 4b). With the addition of more iron such that the Ni:Fe ratio is 0.6:0.4, thin sheets are formed and pores of the network become bigger (Fig. 4c). When the Ni:Fe ratio decreases to 0.4:0.6, thick sheets together with even bigger pores are formed (Fig. 4d). When the Ni:Fe ratio decreases to 0.2:0.8, well-defined nanowires or nanowire bundles are formed (Fig. 4e). Without Ni (Ni:Fe = 0:1, i.e. iron oxide), big particles with step terrace surface morphology are obtained (Fig. 4f). It is noted that when the Ni:Fe ratio is equal to or smaller than 0.6:0.4, SEM images become blurred. The blurred SEM images are due to the well-known charging effect caused by the poor electrical conductance of a sample. An impedance measurement reveals that Z′ increases or the conductance decreases with the Fe component in the nickel iron oxide system as shown in Fig. S6.†
 | ||
| Fig. 4 Typical SEM images of the as-obtained nanosheets synthesized at different Ni:Fe feed molar ratios: (a) 1:0, (b) 0.8:0.2, (c) 0.6:0.4, (d) 0.4:0.6, (e) 0.2:0.8, and (f) 0:1. | ||
Transmission electron microscopy (TEM) images of the hybrid nanosheets display the nanostructures explicitly. Except for the Ni–Fe–O sample with Ni:Fe = 0:1 (Fig. 5f), other Ni–Fe–O hybrid samples show overlapping silk-like or flake-like morphologies. Fig. 5a and b reveal thin ribbons with a width of less than 30 nm. Fig. 5c suggests the wafery flakes while Fig. 5d exhibits the bulky flakes. It can be observed from Fig. 5e that the dense grass-like film is composed of delicate groups of needles emanating from a point. The morphology variation trend corresponds well with the SEM observations.
 | ||
| Fig. 5 Typical TEM images of the as-obtained nanosheets obtained at different Ni:Fe feed molar ratios: (a) 1:0, (b) 0.8:0.2, (c) 0.6:0.4, (d) 0.4:0.6, (e) 0.2:0.8, and (f) 0:1. | ||
To determine the phase of the as-prepared nickel iron oxides, X-ray diffraction (XRD) measurements are conducted. For samples with Ni:Fe ratios of 1:0 and 0:1, XRD peaks can be readily indexed to NiO (JCPDS Card no. 47-1049) and Fe2O3 (JCPDS Card no. 33-0664) (ESI Fig. S2†). However, it is unfortunate that, for the other nickel iron oxide materials grown, no XRD peaks appear except the Ni peaks from Ni foam substrates. The reasons can be that the materials are poorly crystallized or the amount of loading is insufficient. Then we perform selected area electron diffraction (SAED) (Fig. 6) to acquire crystal structure information. When the precursors do not contain iron, the electron diffraction rings (Fig. 6a) can match those of NiO (JCPDS Card no. 47-1049) very well as expected. When the precursors do not contain nickel, the electron diffraction pattern (Fig. 6f) reveals the material to be Fe2O3 (JCPDS Card no. 33-0664). These results are in agreement with XRD observations. For samples containing both nickel and iron, electron diffraction ring patterns similar to those of NiO are obtained. Such results are out of our expectation, especially for Ni–Fe–O samples which contain a lot of Fe. For a smaller amount of Fe in Ni–Fe–O, it is easy to understand why the NiO crystal structure is maintained. For very high Fe content in the sample of Ni–Fe–O, the maintenance of the NiO structure, rather than the Fe2O3 structure, is difficult to understand. One possible reason for not having the Fe2O3 structure is that the ionic charge of Ni is 2+, while that of Fe in Fe2O3 is 3+ and it is difficult for Ni2+ to take the Fe3+ position in Fe2O3.
 | ||
| Fig. 6 SAED patterns of the hybrid nanosheets prepared with different Ni:Fe feed molar ratios of (a) 1:0, (b) 0.8:0.2, (c) 0.6:0.4, (d) 0.4:0.6, (e) 0.2:0.8, and (f) 0:1. | ||
A survey of various iron oxides reveals that iron(II) oxide (FeO) has the same crystal structure as that of Fm–3m (225) cubic NiO (JCPDS Card no. 47-1049). The lattice constant of NiO is 0.4177 nm. The reported lattice constant of FeO ranges from 4.32 (ref. 29) to 4.35.30 Lattice constants for Fe deficient Fe1−xO are smaller than these values. It is then not surprising that various Ni–Fe–O samples have similar crystal structures to that of NiO due to 3 reasons: the same crystal structures, similar lattice constants and the same valences of the metal ions in NiO and FeO. A closer inspection reveals that the diameters of the rings decreased slightly with the incorporation of Fe. The decrease of ring diameter corresponds to the increase of lattice spacing. At the first sight, this lattice expansion appears strange as Fe (atomic number of 26) should have a smaller atomic radius than that of Ni (atomic number of 28). It is known that the crystal ionic radius is different from the atomic radius generally. Lang et al.31 listed ionic radii for various elements, and showed that crystal ionic radii of Fe and Ni (both with ionic charge 2+ and high spin) are 92 and 83 pm, respectively. Therefore, Fe2+ taking the position of Ni2+ in the NiO crystal structure can lead to lattice expansion. Due to the incorporation of the foreign element into the NiO structure, the crystallinity of the crystals is deteriorated, which may explain why there are no peaks appearing in XRD patterns of Ni–Fe–O samples. It is known that FeO is prone to be iron deficient with compositions ranging from Fe0.84O to FeO;32 it is then possible that a more stable phase Fe2O3, rather than FeO, is formed for pure iron oxide (Fig. 6f).
The surface chemical compositions and the valence states of the samples are characterized by using X-ray photoelectron spectroscopy (XPS). From the XPS results, both Fe and Ni peaks appear for all the nickel iron oxides except for the pure ones. The XPS spectra for these samples are similar. In the Ni 2p spectrum (Fig. S3†), two obvious shakeup satellites close to two spin–orbit doublets at 872 and 854 eV can be identified as Ni 2p1/2 and Ni 2p3/2 signals of Ni2+.33 In the Fe 2p spectrum (Fig. S4†), two major peaks at 711 and 724 eV corresponding to the 2p3/2 and 2p1/2 core levels are obtained. It is noteworthy that no satellites appear in the Fe 2p spectrum, except for the sample of Ni:Fe = 0.2:0.8 that contains a lot of Fe. It is known that for Fe3+ in Fe2O3, there is a satellite at about 719 eV.34,35 However, with the existence of both Fe2+ and Fe3+ in Fe3O4, no satellites appear in Fe 2p spectra.34,35 The absence of satellites in our Ni–Fe–O samples may indicate the existence of both Fe2+ and Fe3+, supporting the NiO structure of Ni–Fe–O samples. The appearance of a satellite at about 719 eV for the sample of Ni:Fe = 0.2:0.8 can be associated with the dominance of the Fe amount in this material, because Fe2O3 can be formed on the pure FeO surface.36 From the SEM images (Fig. 4 inset), some flakes are observed, which may prove that a little bit Fe2O3 can be formed.
The mesoporous nature of the products (Ni–Fe–O/r-NF) is characterized using N2-adsorption/desorption measurements. As measured by the BET (Brunauer–Emmet–Teller) method, the surface area of the Ni–Fe–O/rolled nickel foam is listed in Table S1 (ESI).† The BET surface area of the material grown on the current collector is not high when compared with the literature. However, it should be noted that most of the BET values reported in the literature are based on the active material in the powder phase without the current collector; therefore the literature values and our values cannot be compared. The addition of Fe does not provide any beneficial effects.
:Fe feed molar ratios at different current densities based on different criteria such as gravimetric, areal and volumetric capacitances. The results are similar to those grown on the unrolled nickel foam. It further corroborates that the treatment of nickel foam by rolling does not significantly deteriorate the performance for various electrode materials. The morphologies also remain almost the same (Fig. S1†). The highest capacitance we obtained is about 45 F cm−3 (three electrode configuration) for the pure nickel oxide grown on rolled foam. It should be noted that this paper reports the novel idea and effect of rolling the foams for the improvement of volumetric capacitance mainly and there is still a huge space for further significant improvement of the volumetric supercapacitor performance because the space is still not well utilized as seen in Fig. 1. Some papers report volumetric capacitance up to 70 F cm−3 based on the carbon itself,37–40 however, such results should not be compared with our data because the volume for our data is the total volume of the electrode material and current collector.
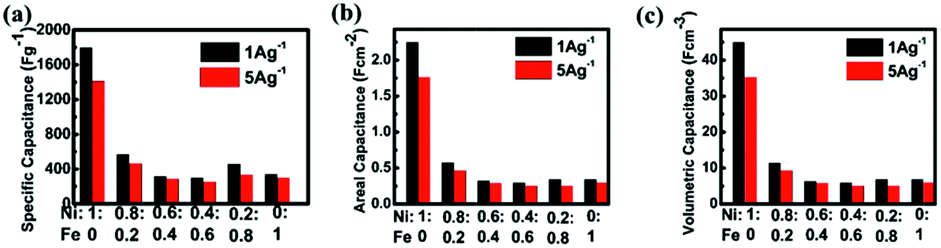 | ||
| Fig. 7 Relationships between specific (a) gravimetric capacitances, (b) areal capacitance, and (c) volumetric capacitance and current densities of different hybrid electrodes prepared with different Ni:Fe feed molar ratios. | ||
Specific capacitances of samples with various Fe/Ni ratios measured at 1 A g−1 and 5 A g−1 are shown in Fig. 7. It is observed that the pure nickel oxide exhibits the highest performance, achieving almost 1800 F g−1 at 1 A g−1. It is necessary to investigate this system and we are the first one to report. We also hoped to differentiate the effects of Ni–Fe ratio and surface morphology, but found it is almost impossible. In the literature, several papers indicated that it is very difficult or impossible to differentiate. Even for the simple case of carbon, it has been indicated that “in some cases no clear linear relationship between the specific capacitance and total surface area was observed”.41a,b The situation for pseudocapacitance is far more complicated, e.g. Roberts and Slade41c concluded that “no correlation was found between the SBET of MnO2 powders and specific capacitances”. Actually, we have measured the BET surface areas of the different samples and listed the data in Table S1 of the ESI.† We have tried but are also unable, similar to the above authors, to find a reasonable correlation between the surface morphology and supercapacitor performance. Since the electrochemical behaviors of iron oxide are complicated, the reasons for this undesirable phenomenon could be explained just based on the literature and our own experimental results. One is that the published results for iron oxide thus far as the supercapacitor electrode do not show a high capacitance.41–43 The second reason is that some of the iron oxides are used as negative electrodes.44 The potential window is in the negative range of −0.1 V to −0.6 V.45 In the potential window of our test, iron oxide does not contribute to the pseudocapacitance of the binary oxide material. The third reason is that the surface area is not high. Finally, some of the tests of iron oxide are in other kinds of electrolytes,46,47 and the highest capacitance of 510 F g−1 is achieved for Fe3O4 using sodium sulphite as the electrolyte.48 The intrinsic problem for iron oxide and nickel oxide to be used as electrodes of an electrochemical capacitor is to obtain high enough electrical conductivity even though we have already adopted the direct growth of the material.49 In our case, nickel oxide can only demonstrate pseudo-capacitance in the positive voltage range (vs. SCE) to generate redox reactions in the alkaline electrolyte. However, in this range, iron oxide could not afford pseudo-capacitance. Therefore, the incorporation of iron into nickel oxide does not produce positive results.
In order to understand the electrochemical behavior of different Ni–Fe–O/r-NF compounds, resistance components associated with the supercapacitors are analyzed by electrochemical impedance spectroscopic (EIS) studies after stabilization and oxidation upon cycling (50 cycles). The spectra display quasi-straight lines for all the samples. Counterintuitively, well shaped CV peaks arising from redox reactions should be observed with a usual convex downward semi-circle, but they are absent here. The first intersection point on the real axis of the Nyquist spectrum in the high frequency region represents the total resistance due to ionic resistance from the electrolyte (Rs), intrinsic resistance of electrodes (Re) and the contact resistance (Rc) between the electrode and current collector.51–53 This phenomenon means that the conductivity difference between the solid electrode (electronic conductivity) and electrolyte phase (ionic conductivity) is small. The charge transfer of the electrons on the electrode is quick and the charge transfer resistance is small.50 The Nyquist plots in Fig. S6† indicate that this value is almost the same for all the impedance spectra. It can be pointed out that the resistance increases with the addition of Fe. It further specifies the reason why capacitance decreases in Fig. 7.
To evaluate the volumetric (here the total volume of the positive electrode and negative electrode is considered) energy density, an asymmetric supercapacitor device is assembled. In the two electrode configuration of our asymmetric supercapacitor, the positive electrode is nickel oxide on 2 cm2 r-NF, and the negative electrode is commercialized reduced graphene oxide (RGO from Graphene Supermarket) on another 2 cm2 NF. Weights of the positive electrode and the negative electrode are 3 and 11 mg, respectively. CV curves at different scan rates shown in Fig. 8ia indicate that the asymmetrical supercapacitor can be reversibly cycled within a voltage window of 0–1.6 V, possibly due to the high over-potential for dihydrogen evolution. Galvanic charging–discharging (GC) curves are shown in Fig. 8ib. It should be mentioned that the capacitance does not change much when the current densities are changed. The discharge time has almost a linear correlation with the current density. This further demonstrates the good rate performance of the asymmetric full cell. The power density and energy density are critical parameters for the investigation of the electrochemical performance of electrochemical cells. They have been used to evaluate the performance of supercapacitor devices. The Ragone plot, which is used to relate the power density to the energy density, is calculated from charging–discharging curves. The calculated specific capacitance of the hybrid nanostructured ASCs is about 165 F g−1 at a current density of 2 mA cm−2 (the current is 8 mA), while the maximum energy density and maximum power density are improved to 58.8 W h kg−1 and 3.37 kW kg−1, respectively. The volumetric energy density and power density are calculated based on the total volume of both of the current collectors for positive and negative electrodes. The highest energy density is about 4.12 mW h cm−3. In the literature, most values are based on the active material only, thus it is hard to compare. One group fabricated a solid-state device and the volumetric energy density is about 0.234 mW h cm−3, and the electrolyte is included.54 Another group used Ultrathin-Graphite Foam (UGF) as the current collector, and they calculated the volumetric energy density based on the total weight of electrodes including the UGF current collector, just the same as ours, which was equal to 8.7 mW h cm−3.8 Our nickel foam supercapacitor could reach half of the result compared with graphite foam, even though it is far from optimized. Energy densities do not decrease much with the increase of power density, which can mostly be attributed to the relatively good rate capability of the electrode. Mostly in the literature, the performance of a device is merely attributed to the active material. This is not enough, because at the application level, electrode active materials must be incorporated with current collectors. To investigate the effect of Fe, we also fabricated the Ni–Fe asymmetric device (with the feed ratio of 0.8:0.2); the result can be found in Fig. 8ii. The capacitance of the full cell is about 78 F g−1, and the maximum energy density and power density are 27.8 W h kg−1 and 3.6 kW kg−1. The corresponding volumetric energy density is 1.94 mW h cm−3.
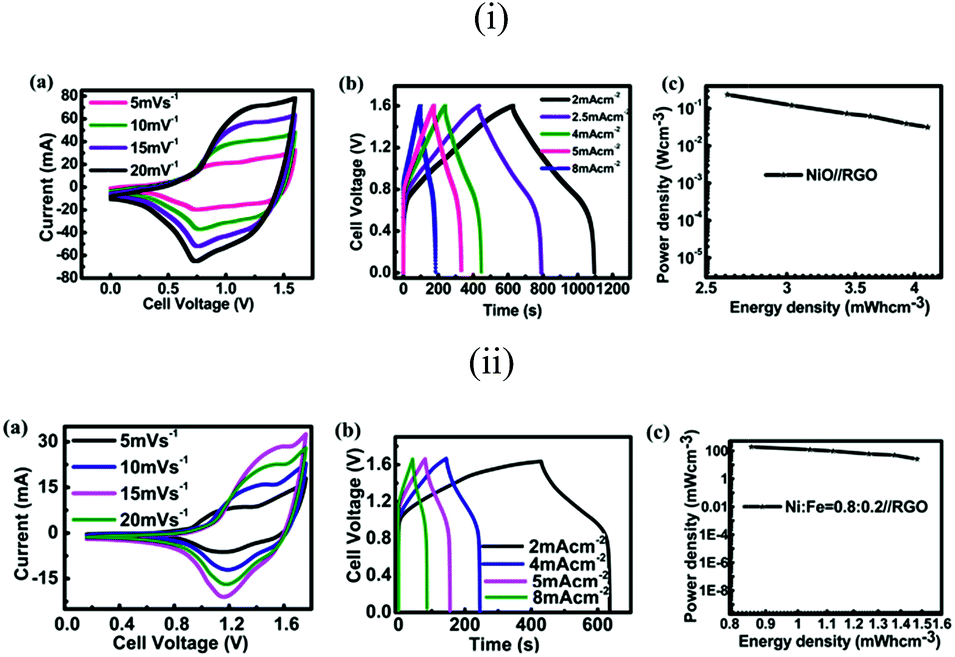 | ||
| Fig. 8 (i) The performance of the nickel oxide (Ni:Fe = 1:0)//RGO asymmetric supercapacitor: (a) CV curves at different scan rates, (b) galvanostatic charge–discharge curves at different current densities, and (c) power density as a function of energy density on a volumetric basis. (ii) The performance of the nickel iron oxide (Ni:Fe = 0.8:0.2)//RGO asymmetric supercapacitor: CV curves at different scan rates, (b) galvanostatic charge–discharge curves at different current densities, and (c) power density as a function of energy density on a volumetric basis. | ||
4. Conclusions
In summary, we have reported a simple way to increase the volumetric performance of a nickel foam based direct growth material by rolling the nickel foam before the direct growth of materials. The flexibility is also increased tremendously at the same time. The pretreatment of nickel foam does not result in significant morphological and electrochemical changes of the material. On the contrary, the volumetric performance is remarkably enhanced by almost three times. Besides that, the potential of nickel iron oxide as the supercapacitor electrode has been evaluated by further studying the effect of nickel iron ratio systematically for the first time. Moreover, the versatility of this protocol provides a platform to fabricate nanomaterials grown on nickel foam with a high volumetric performance and extremely high flexibility in a more benign and easy way. The highest energy density we achieved even when considering the volume of current collectors is 4.12 mW h cm−3 and the highest capacitance of the full cell device is 11.6 F cm−3.Acknowledgements
The support of Singapore MOE Tier 2 grant R-284-000-125-112 is appreciated.Notes and references
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- Z. Q. Niu, P. S. Luan, Q. Shao, H. B. Dong, J. Z. Li, J. Chen, D. Zhao, L. Cai, W. Y. Zhou, X. D. Chen and S. S. Xie, Energy Environ. Sci., 2012, 5, 8726–8733 CAS.
- L. Y. Yuan, X. H. Lu, X. Xiao, T. Zhai, J. J. Dai, F. C. Zhang, B. Hu, X. Wang, L. Gong, J. Chen, C. G. Hu, Y. X. Tong, J. Zhou and Z. L. Wang, ACS Nano, 2012, 6, 656–661 CrossRef CAS PubMed.
- G. D. Moon, J. B. Joo, M. Dahl, H. Jung and Y. Yin, Adv. Funct. Mater., 2014, 24, 848–856 CrossRef CAS.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS PubMed.
- H. Wang, X. Sun, Z. Liu and Z. Lei, Nanoscale, 2014, 6, 6577–6584 RSC.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- B. Anothumakkool, A. A. T. Torris, S. N. Bhange, S. M. Unni, M. V. Badiger and S. Kurungot, ACS Appl. Mater. Interfaces, 2013, 5, 13397–13404 CAS.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- J. Yan, Q. Wang, T. Wei, L. Jiang, M. Zhang, X. Jing and Z. Fan, ACS Nano, 2014, 8, 4720–4729 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- S. Murali, N. Quarles, L. L. Zhang, J. R. Potts, Z. Tan, Y. Lu, Y. Zhu and R. S. Ruoff, Nano Energy, 2013, 2, 764–768 CrossRef CAS PubMed.
- L. Yang, L. Qian, X. Tian, J. Li, J. Dai, Y. Guo and D. Xiao, Chem.–Asian J., 2014, 9, 1579–1585 CrossRef CAS PubMed.
- J. Y. Son, Y. H. Shin, H. Kim and H. M. Jang, ACS Nano, 2010, 4, 2655–2658 CrossRef CAS PubMed.
- W. K. Hu and D. Noreus, Chem. Mater., 2003, 15, 974–978 CrossRef CAS.
- H. C. Chien, W. Y. Cheng, Y. H. Wang and S. Y. Lu, Adv. Funct. Mater., 2012, 22, 5038–5043 CrossRef CAS.
- C. Guan, X. Li, Z. Wang, X. Cao, C. Soci, H. Zhang and H. J. Fan, Adv. Mater., 2012, 24, 4186–4190 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- L. Wang, H. Ji, S. Wang, L. Kong, X. Jiang and G. Yang, Nanoscale, 2013, 5, 3793–3799 RSC.
- J. X. Zhu, Z. Y. Yin, D. Yang, T. Sun, H. Yu, H. E. Hoster, H. H. Hng, H. Zhang and Q. Y. Yan, Energy Environ. Sci., 2013, 6, 987–993 CAS.
- M. T. Lee, J. K. Chang, Y. T. Hsieh and W. T. Tsai, J. Power Sources, 2008, 185, 1550–1556 CrossRef CAS PubMed.
- W. Tian, X. Wang, C. Zhi, T. Zhai, D. Liu, C. Zhang, D. Golberg and Y. Bando, Nano Energy, 2013, 2, 754–763 CrossRef CAS PubMed.
- Z. Wang, X. Zhang, Y. Li, Z. Liu and Z. Hao, J. Mater. Chem. A, 2013, 1, 6393–6399 CAS.
- Z. Y. Yu, L. F. Chen and S. H. Yu, J. Mater. Chem. A, 2014, 2, 10889–10894 CAS.
- S. Anwar, K. S. Muthu, V. Ganesh and N. Lakshminarasimhan, J. Electrochem. Soc., 2011, 158, A976–A981 CrossRef CAS PubMed.
- M. Heon, S. Lofland, J. Applegate, R. Nolte, E. Cortes, J. D. Hettinger, P.-L. Taberna, P. Simon, P. Huang, M. Brunet and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 135–138 CAS.
- T. Katsura, B. Iwasaki, S. Kimura and S. Akimoto, J. Chem. Phys., 1967, 47, 4559–4560 CrossRef CAS PubMed.
- W. L. Roth, Acta Crystallogr., 1960, 13, 140–149 CrossRef CAS.
- P. F. Lang and B. C. Smith, Dalton Trans., 2010, 39, 7786–7791 RSC.
- N. N. Greenwood and A. Earnshaw, Chemistry of Elements, Butterworth-Heinemann, 2nd edn, 1997, ISBN 0080379419 Search PubMed.
- L. Zhang, C. Tang, X. Yin and H. Gong, J. Mater. Chem. A, 2014, 2, 4660–4666 CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS PubMed.
- S. Palchoudhury, W. An, Y. Xu, Y. Qin, Z. Zhang, N. Chopra, R. A. Holler, C. H. Turner and Y. Bao, Nano Lett., 2011, 11, 1141–1146 CrossRef CAS PubMed.
- A. N. Mansour and R. A. Brizzolara, Surf. Sci. Spectra, 1996, 4, 345–350 CrossRef CAS.
- J. A. Lee, M. K. Shin, S. H. Kim, S. J. Kim, G. M. Spinks, G. G. Wallace, R. Ovalle-Robles, M. D. Lima, M. E. Kozlov and R. H. Baughman, ACS Nano, 2012, 6, 327–334 CrossRef CAS PubMed.
- E. Raymundo-Pinero, F. Leroux and F. Beguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef CAS.
- G. Lota, K. Lota and E. Frackowiak, Electrochem. Commun., 2007, 9, 1828–1832 CrossRef CAS PubMed.
- X. Yu, J.-G. Wang, Z.-H. Huang, W. Shen and F. Kang, Electrochem. Commun., 2013, 36, 66–70 CrossRef CAS PubMed.
- (a) H. Shi, Electrochim. Acta, 1996, 41, 1633–1639 CrossRef CAS; (b) G. Gryglewicz, J. Machnikowski, E. L-Grabowska, G. Lota and E. Frackowiak, Electrochim. Acta, 2005, 50, 1197–1206 CrossRef CAS PubMed; (c) A. J. Roberts and R. C. T. Slade, Electrochim. Acta, 2010, 55, 7460–7469 CrossRef CAS PubMed; (d) N. Nagarajan and I. Zhitomirsky, J. Appl. Electrochem., 2006, 36, 1399–1405 CrossRef CAS.
- P. M. Hallam, M. G'omez-Mingot, D. K. Kampouris and C. E. Banks, RSC Adv., 2012, 2, 6672–6679 RSC.
- K. Y. Xie, J. Li, Y. Q. Lai, W. Lu, Z. A. Zhang, Y. X. Liu, L. M. Zhou and H. T. Huang, Electrochem. Commun., 2011, 13, 657–660 CrossRef CAS PubMed.
- D. Liu, X. Wang, X. Wang, W. Tian, J. Liu, C. Zhi, D. He, Y. Bando and D. Golberg, J. Mater. Chem. A, 2013, 1, 1952–1955 CAS.
- B. Sethuraman, K. K. Purushothaman and G. Muralidharan, RSC Adv., 2014, 4, 4631–4637 RSC.
- J. Mu, B. Chen, Z. Guo, M. Zhang, Z. Zhang, P. Zhang, C. Shao and Y. Liu, Nanoscale, 2011, 3, 5034–5040 RSC.
- S. Chaudhari, D. Bhattacharjya and J.-S. Yu, RSC Adv., 2013, 3, 25120–25128 RSC.
- N. L. Wu, S. Y. Wang, C. Y. Han, D. S. Wu and L. R. Shiue, J. Power Sources, 2003, 113, 173–178 CrossRef CAS.
- W. Shi, J. Zhu, D. H. Sim, Y. Y. Tay, Z. Lu, X. Zhang, Y. Sharma, M. Srinivasan, H. Zhang, H. H. Hng and Q. Yan, J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- S. H. Aboutalebi, A. T. Chidembo, M. Salari, K. Konstantinov, D. Wexler, H. K. Liu and S. X. Dou, Energy Environ. Sci., 2011, 4, 1855–1865 CAS.
- V. Ganesh, S. Pitchumani and V. Lakshminarayanan, J. Power Sources, 2006, 158, 1523–1532 CrossRef CAS PubMed.
- C. Portet, P. L. Taberna, P. Simon, E. Flahaut and C. Laberty Robert, Electrochim. Acta, 2005, 50, 4174–4181 CrossRef CAS PubMed.
- M. Hughes, G. Z. Chen, M. S. P. Shaffer, D. J. Fray and A. H. Windle, Chem. Mater., 2002, 14, 1610–1613 CrossRef CAS.
- Z. Wang, Z. Zhu, J. Qiu and S. Yang, J. Mater. Chem. C, 2014, 2, 1331–1336 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of capacitance, energy density and power density. The BET surface area of the nickel iron hybrid together with the nickel foam. XRD profiles of pure nickel oxide and iron oxide. XPS data, CV data and EIS data of nickel iron oxide with different ratios. See DOI: 10.1039/c4ta06649a |
| This journal is © The Royal Society of Chemistry 2015 |