
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrathin oxide shell coating of metal nanoparticles using ionic liquid/metal sputtering†
Tsukasa
Torimoto
*a,
Yasuhiro
Ohta
a,
Kazuki
Enokida
a,
Daisuke
Sugioka
a,
Tatsuya
Kameyama
a,
Takahisa
Yamamoto
a,
Tamaki
Shibayama
b,
Kazuki
Yoshii
c,
Tetsuya
Tsuda
c and
Susumu
Kuwabata
*c
aGraduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: torimoto@apchem.nagoya-u.ac.jp
bCenter for Advanced Research of Energy Conversion Materials, Hokkaido University, Sapporo 060-8628, Japan
cGraduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
First published on 11th February 2015
Abstract
The surface coating of metal nanoparticles resulting into core–shell structures is expected to improve the physicochemical properties of the nanoparticle cores without changing their size and shape. Here, we developed a novel strategy to coat Au, AuPd or Pt catalyst cores having average sizes smaller than 2.5 nm, which were pre-synthesized in ionic liquids by corresponding metal sputtering, with an extremely thin In2O3 layer (ca. <1.5 nm) by sputter deposition of indium in a room-temperature ionic liquid. The metal cores of Au or AuPd in core–shell particles exhibited superior stability against heat treatments or during electrocatalytic reactions compared to the corresponding bare metal particles. The In2O3 shell coating considerably enhanced the durability of electrocatalytically active Pt particles (1.2 nm). This sequential metal sputter deposition of different metals in ionic liquids will considerably contribute to the exploitation of key nanostructured components for next-generation energy-conversion systems.
Introduction
Metal nanoparticles exhibit tunable catalytic properties depending on the particle size, shape, and their chemical composition.1–4 Many strategies have been reported to improve the catalytic activities of metal nanoparticles and their tolerance to deactivation for fabrication of high-performance devices, such as fuel cells5,6 and automotive exhaust gas purification systems.7,8 The formation of core–shell structures by particle coating with shell layers has proven advantageous for modifying the catalytic properties of nanoparticles without affecting their dimensions.9–15 So far, many strategies have been developed to prepare core–shell-structured particles. Sol–gel methods have been intensively investigated for coating noble metal catalyst particle surfaces with metal oxides such as SiO2 and TiO2.11,14,16,17 The layer-by-layer approach has enabled the deposition of polyelectrolytes, such as ionic polymers or inorganic nanosheets as shell materials on surface-charged particles.18–22 The partial oxidation of metal particle surfaces has produced metal oxide shell layers on metal cores.23,24 However, coating a metal particle with an extremely thin layer remains difficult by conventional techniques, especially for particles smaller than ca. 3 nm exhibiting high catalytic activity. This is related to inter-particle coalescence and/or thick shell formation on the surface, which sometimes induce considerable deterioration of catalytic activity of core particles.Room-temperature ionic liquids (RTILs) exhibited unique features compared with those of water or conventional organic solutions and they have attracted much attention as media for preparation of inorganic nanoparticles.25–33 Metal sputter deposition in RTILs under reduced pressure31,32,34–42 has recently produced noble metal nanoparticles, such as Au,34 Ag,35 Pd,36 and Pt,39 without additional stabilizing agents. This RTIL/metal sputtering technique has also enabled the preparation of alloy nanoparticles, such as AuAg,35 AuPd,41 and AuPt,40 by simultaneous sputtering of two different metal plates. The resulting nanoparticles displaying particle size of several nanometers in diameter did not present any stabilizing agent adsorption on their surface and were expected to show high catalytic activities. However, the presence of the bare surface on the nanoparticles suggested that the particles exhibited a strong tendency to coalesce into larger structures, which decreases catalytic activity. Therefore, a nanoparticle stabilization technique is required for their long-term catalyst life.
In this study, we prepared novel core–shell particles via RTIL/metal sputtering, in which noble metals, such as Au, AuPd, and Pt, and a base metal of In were sequentially deposited in RTILs. The obtained particles consisted of a small noble metal core (ca. <2.5 nm) surrounded by an ultrathin indium oxide shell (ca. <1.5 nm) and then the thermal stability of noble metal cores in core–shell particles was much more enhanced than that of bare particles. The In2O3-coated AuPd nanoparticles acted as electrocatalysts for ethanol oxidation and oxygen reduction. Furthermore the durability of electrocatalytically active Pt particles was improved by coating with the In2O3 shell.
Experimental
Preparation of core–shell-structured Au@In2O3 and AuPd@In2O3 particles
Fig. 1 shows the schematic illustration of preparation of noble metal (M) core–In2O3 shell nanoparticles (M@In2O3). First, noble metals, such as an Au or an AuPd alloy, were sputter-deposited on RTILs under reduced Ar pressure using the corresponding metal targets to prepare uniform metal nanoparticle dispersions in RTILs. Next, the noble metal target plate was replaced with an In target plate and this second metal was sputter-deposited onto the thus-obtained metal-particle-dispersed RTILs.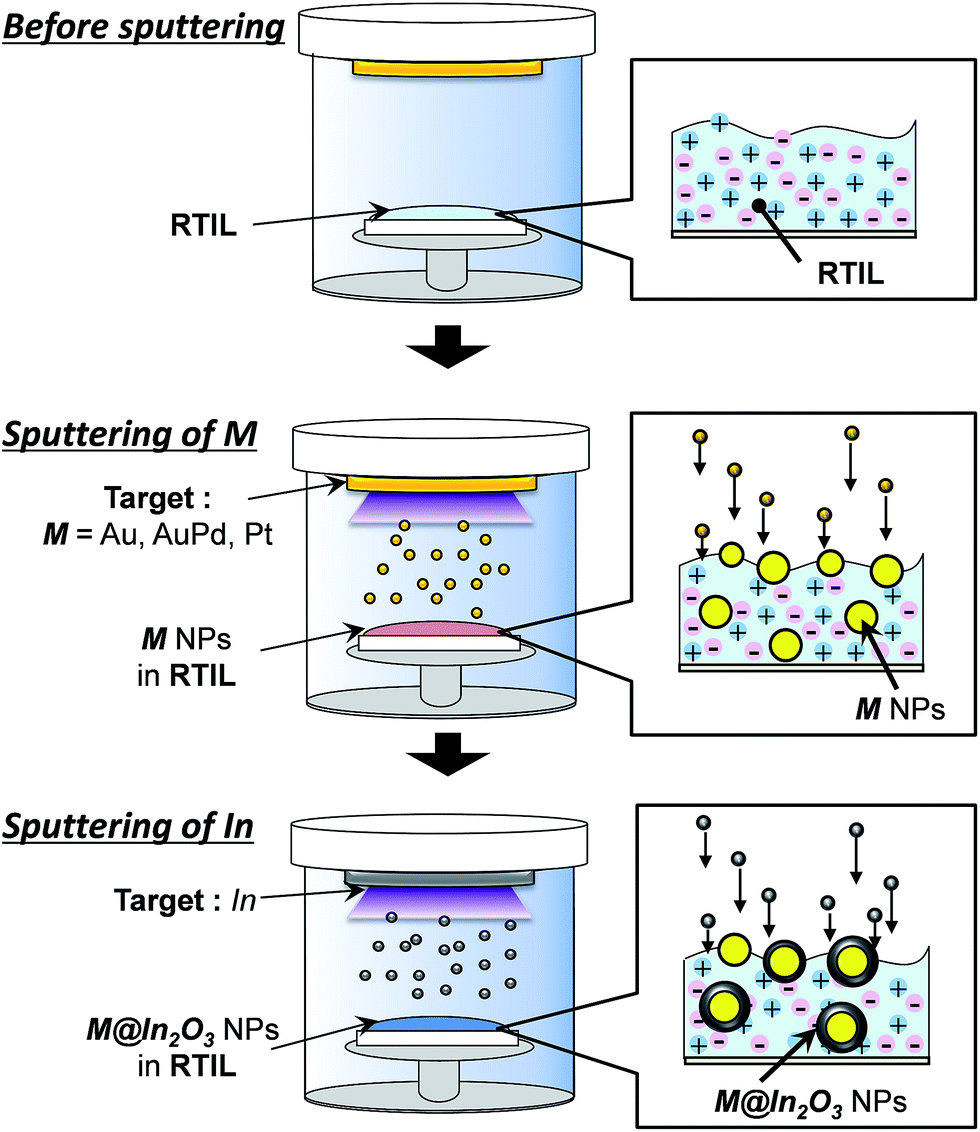 | ||
| Fig. 1 Schematic for the preparation procedure of noble metal (M) core–In2O3 shell nanoparticles (M@In2O3, M = Au, AuPd, or Pt). | ||
Gold nanoparticles were sputter-deposited in 1-ethyl-3-methylimidazolium tetrafluoroborate (EMI-BF4) used as an RTIL by a previously-reported method34,35,41 with a slight modification. A portion of EMI-BF4 (0.60 cm3), that was vacuum-dried for 3 h at 150 °C before use, was spread on a glass plate (10 cm2) horizontally set in a sputter coater (Sanyu Electron Co. Ltd., SC-701HMCII). The plate was located 25 mm from an Au target (diameter: 49 mm). The sputter deposition of Au in EMI-BF4 was conducted for 10 min at 10 mA under an argon pressure of ca. 2.0 Pa at room temperature. The thus-obtained Au nanoparticles had the average size of 2.2 nm, in which the concentration of particles amounted to 19 μmol (particle) dm−3, measured using X-ray florescence spectroscopy. On the other hand, for the preparation of AuPd alloy nanoparticles,34,35,41 the binary Au–Pd metal target was prepared by radially and alternately arranging Au (99.99% in purity) and Pd plates (99.99%), which each had a fan-like shape with a central angle of 30°. This target was used for the simultaneous sputter deposition instead of a pure Au target. The area fraction of Au plates in targets, fAu (= AAu/(AAu + APd)), equalled 0.50, where AAu and APd were the total surface areas of Au and Pd plates, respectively. After the simultaneous sputter deposition of Au and Pd under the same sputtering condition, the resulting EMI-BF4 contained AuPd particles (average size: 2.0 nm) at a particle concentration of 26 μmol (particle) dm−3.
The subsequent In sputter deposition on thus-obtained EMI-BF4 solutions containing noble metal particles was carried out by replacing a noble metal target plate by an In metal one. The In sputter deposition on EMI-BF4 containing Au particles lasted for 30 min at 10 mA under an argon pressure of ca. 2.0 Pa at room temperature to obtain In2O3-coated Au nanoparticles (Au@In2O3). For the preparation of AuPd@In2O3, the In sputtering time for AuPd nanoparticle EMI-BF4 dispersions was varied from 10 to 30 min under the same sputtering condition. Thus-obtained EMI-BF4 solutions containing Au@In2O3 or AuPd@In2O3 were heat-treated at various temperatures for 30 min in air with vigorous stirring if necessary.
The shape and size distribution of the obtained particles was examined using a Hitachi H7650 transmission electron microscope (TEM) with an acceleration voltage at 100 kV. Samples for TEM measurements were prepared by dropping a particle-containing EMI-BF4 solution onto a carbon-coated copper grid, which was washed with acetonitrile and then dried under reduced pressure. High resolution images of high-angle-annular-dark-field scanning transmission microscopy (HAADF-STEM) were obtained by Cs-corrected HR-STEM (ARM-200F, JEOL Co. Ltd.) with an acceleration voltage at 200 kV. The energy-dispersive X-ray spectroscopy (EDX) analysis was simultaneously carried out during TEM measurements using an electron probe of ca. 1 nm in diameter.
Electrochemical measurements of the electrocatalytic activity of AuPd@In2O3-modified HOPG electrodes
Synthesized nanoparticles were immobilized by heat treatment as reported previously.41 An aliquot (20 mm3) of EMI-BF4 solution containing bare AuPd nanoparticles or AuPd@In2O3 (the concentration of AuPd particles: 26 μmol (particle) dm−3) was spread on highly-oriented pyrolytic graphite (HOPG) (1.4 cm2) before heat treatment at 100 or 200 °C for 30 min under vacuum. These modified HOPG electrodes were washed several times with acetonitrile to remove unattached nanoparticles and dried at room temperature. The surface morphology of the nanoparticle films on HOPG was examined in air by an atomic force microscope (AFM) (Veeco Instruments Inc., NanoScope IIIa).The electrocatalytic activities of nanoparticle-modified HOPG electrodes were assessed using an electrochemical analyser (BAS Instruments Inc., ALS model 701C). Electrochemical measurements were conducted using a three-electrode electrochemical cell, in which the AuPd-immobilized HOPG electrode acted as the working electrode, a Pt wire as the counter electrode, and a reversible hydrogen electrode (RHE) as the reference electrode. The electrolyte used for oxygen reduction reaction (ORR) was an O2-saturated 0.50 mol dm−3 KOH aqueous solution, while the one for ethanol oxidation consisted of a degassed aqueous solution containing 0.50 mol dm−3 ethanol and 0.50 mol dm−3 KOH. Cyclic voltammograms were obtained at a sweep rate of 100 mV s−1. The apparent area of the working electrode was set to 0.16 cm2 using an O-ring. The current density was calculated by dividing the obtained current by the electrochemically active surface area (ECSA) of AuPd particles obtained by the literature procedure.43
Preparation of Pt@In2O3-loaded carbon particles and their durability for polymer electrolyte fuel cells
Platinum nanoparticles measuring 1.2 nm in diameter were sputter-deposited in 1-butyl-3-methylimidazolium tetrafluoroborate (BMI-BF4) (0.40 cm3), spread on a glass plate (6.3 cm2), for 15 min at 40 mA under an argon pressure of ca. 9.5 Pa. The In2O3 surface coating was carried out by the subsequent In metal sputtering for 2 min at 10 mA under an argon pressure of ca. 2.0 Pa. Pt@In2O3 particles were immobilized on carbon black particles (VULCAN XC-72) (Pt@In2O3/C) for a Pt content of ca. 5 wt% by a previously-reported method with heat treatment at 150 °C.39 For comparison, bare Pt particles (size: 1.2 nm) were similarly immobilized on carbon black particles (Pt/C, ca. 5 wt%-Pt). After mixing the resulting powders (1.0 mg) with 0.05 wt% Nafion® solution (0.50 cm3), catalyst powders (Pt@In2O3/C or Pt/C, 10 μg) were immobilized on a glassy carbon rotating disk electrode (0.20 cm2). The durability test of Pt catalysts was performed according to the test protocol recommended by the Fuel Cell Commercialization Conference of Japan (FCCJ).44,45 The step-like potential cycling between 0.6 and 1.0 V vs. RHE was carried out in 0.1 mol dm−3 HClO4 aqueous solution degassed by N2 bubbling at a room temperature, in which the holding period was 3 s at each potential application. The ECSA of Pt particles was measured in 0.10 mol dm−3 HClO4 aqueous solution saturated with N2 after at least every 500 cycles of the potential step. The electrocatalytic activity for ORR was evaluated by measuring hydrodynamic voltammograms for ORR in O2-saturated 0.10 mol dm−3 HClO4 aqueous solution at a rotational speed of 1600 rpm with a potential scan rate of 10 mV s−1 after every 1000 cycles of the potential step.Results and discussion
Surface coating of noble metal nanoparticles with thin indium oxide shells
Particles of the noble metal (M) core–In2O3 shell nanostructure (M@In2O3) were prepared by the procedure shown in Fig. 1. Fig. 2a–c show representative TEM images of nanoparticles formed in an EMI-BF4 solution at individual steps. The simple sputter deposition of Au produced spherical 2.2 nm-diameter Au nanoparticles in the solution without particle aggregation as shown in Fig. 2a. Subsequent In sputtering covered individual Au nanoparticle surfaces with thin layers without changing the core size (ca. 2.2 nm, Fig. 2b). Whole core–shell particles displayed an average diameter of ca. 4.4 nm by TEM. The shell thickness was assumed to be equal to half the difference between the Au core (ca. 2.2 nm) and Au@In2O3 particle diameters, which amounted to ca. 1.1 nm. X-ray fluorescence spectroscopy showed that the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In molar ratio amounted to 1:3.9 in the thus-obtained EMI-BF4 dispersion, consistent with that of Au@In2O3 particles comprising a 2.2 nm-diameter Au-core surrounded by a ca. 1.1 nm-thick In2O3 shell.
:In molar ratio amounted to 1:3.9 in the thus-obtained EMI-BF4 dispersion, consistent with that of Au@In2O3 particles comprising a 2.2 nm-diameter Au-core surrounded by a ca. 1.1 nm-thick In2O3 shell.
 | ||
| Fig. 2 Representative TEM images of sputter-deposited Au particles (a) and Au@In2O3 (b and c) in EMI-BF4 and those of heat-treated particles of bare Au (d) and Au@In2O3 (e and f). The heat treatment of the solutions was conducted at 250 °C for 1 h. Panels c and f show high-magnification HAADF-STEM images of particles in panels b and e, respectively. | ||
A high-angle-annular-dark-field scanning transmission microscopy (HAADF-STEM) image of the core–shell particles at high magnification is shown in Fig. 2c. Cores exhibited clear lattice fringes with a 0.24 nm interplanar spacing assignable to Au (111) lattice planes. The shell layers displaying a thickness ranging between 1 and 1.5 nm exhibited an amorphous structure and completely covered the individual cores. The energy-dispersive X-ray spectroscopy (EDX) analysis simultaneously obtained by TEM measurements (electron probe diameter, ca. 1 nm) revealed that the shell layer predominantly contained In as a metallic element, while the core consisted of Au. It has been previously reported46 that the sputter deposition of the In metal in EMI-BF4 produced In metal core–In2O3 shell particles (In@In2O3), in which the 4.6 nm-diameter In metal core was covered by an amorphous 1.7 nm-thick indium oxide shell. Thus the valence states of In in the Au@In2O3 particles were analysed using X-ray photoelectron spectroscopy (XPS) (Fig. S1†). The In 3d5/2 and 3d3/2 signals presented two sets of peaks. One set consisted of two small signals at binding energies of 444.1 and 451.7 eV, which agreed with In 3d5/2 and 3d3/2 signals for the In(0) metal observed at 443.49 and 451.18 eV,47 respectively. The second set comprised two large signals at 446.0 and 453.6 eV for In 3d5/2 and 3d3/2, respectively and was assigned to In(III) in In2O3, though the observed signals exhibited energies higher than those reported for In2O3 (444.5 and 452.1 eV, respectively).48 This may stem from the strong adsorption of EMI-BF4 and/or the electronic interaction between the Au core and the In2O3 shell. Therefore, it was concluded that the sequential sputter deposition of Au and In produced Au core–In2O3 shell particles, in which the shell was composed of an ultrathin amorphous In2O3 layer. It is worth noting that the signal intensity ratio of In(III):In(0) in the XPS spectrum was ca. 1:0.12, suggesting that In(III) species predominantly contributed to the thermal stability of Au particle cores below-mentioned.
The order of this sequential deposition was critical to prepare noble metal core–metal oxide shell particles. The In@In2O3 particles obtained by simple sputter deposition of the In metal onto pure EMI-BF4 displayed much larger diameters (ca. 14 nm) than Au@In2O3 (4.4 nm, Fig. 2b). The following Au sputtering onto the EMI-BF4 solution containing In@In2O3 produced a simple mixture of In@In2O3 and bare 2.2 nm-diameter Au particles (Fig. S2†) instead of Au-coated In@In2O3. A mechanism may therefore be proposed for the Au@In2O3 formation (Fig. 3). Sputtered In species are injected into the Au particle-dispersed RTIL. Small Au particles act as nuclei for sputter-deposited In species in the RTIL, and then a thin In metal layer is predominantly deposited on the Au nanoparticle surface via heterogeneous nucleation in place of homogeneous nucleation to produce large In metal particles. Subsequently, the In layer rapidly oxidized into a thin In2O3 shell by reactions with H2O or O2 molecules present in RTIL as dissolved impurities or incorporated upon air exposure.
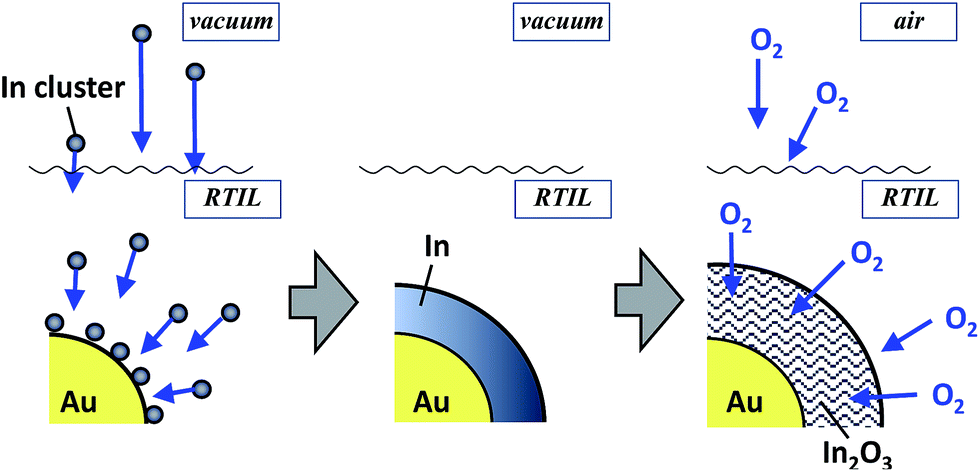 | ||
| Fig. 3 Schematic for the formation of Au@In2O3 particles via oxidation of In metal sputter-deposited on Au nanoparticle cores. | ||
The noble metal cores exhibited remarkably improved thermal stability despite their extremely thin In2O3 shells. The bare 2.2 nm-diameter Au nanoparticles readily coalesced into larger particles (ca. >200 nm) upon heat treatment at 250 °C (Fig. 2d). In contrast, Au@In2O3 maintained its core–shell structure even when heated in EMI-BF4 at 250 °C for 1 h in air (Fig. 2e). TEM images provided Au core diameters of 2.2 and 2.5 nm and In2O3 shell thicknesses of 1.1 and 1.6 nm before and after heat treatment, respectively, indicating that the Au@In2O3 nanostructure remained almost unchanged. The high-resolution HAADF-STEM image (Fig. 2f) shows that, in heat-treated Au@In2O3, the Au core, exhibiting a lattice fringe corresponding to the Au (111) plane, remained completely covered with the amorphous In2O3 shell.
This sequential metal sputter deposition in ionic liquids enabled us to prepare core–shell particles incorporating different kinds of core metals. For example, the In2O3 shell coating was successfully carried out for Au, AuPd, AuPt, or Pt particles as a noble metal core. Furthermore, the shell thickness could be varied by controlling the In sputtering time. Fig. 4a–c show HAADF-STEM images of AuPd@In2O3 particles formed after different In sputtering times. The original spherical AuPd nanoparticle cores (Fig. 4a) were covered by a thin In2O3 layer after 10 (Fig. 4b) and 30 min of In sputter deposition (Fig. 4c). Average AuPd core sizes, In2O3 shell thicknesses, and overall AuPd@In2O3 particle sizes were estimated from TEM measurements and plotted in Fig. 4d as a function of In sputtering time. The overall particle size increased from 2.0 to 4.0 nm when the sputtering time rose from 0 to 30 min. The In2O3 shell thickness, which was assumed equal to half the difference between the AuPd core and overall particle average diameters, increased slightly from 0.79 to 0.95 nm with increasing In sputtering time. EDX analyses at the same time as the TEM observations gave an Au:Pd molar ratio of 0.71:0.29 for the AuPd core in AuPd@In2O3 obtained after In sputtering for 30 min, which was in good agreement with that of original bare AuPd particles (0.70:0.30). In addition, the shell predominantly contained In2O3.
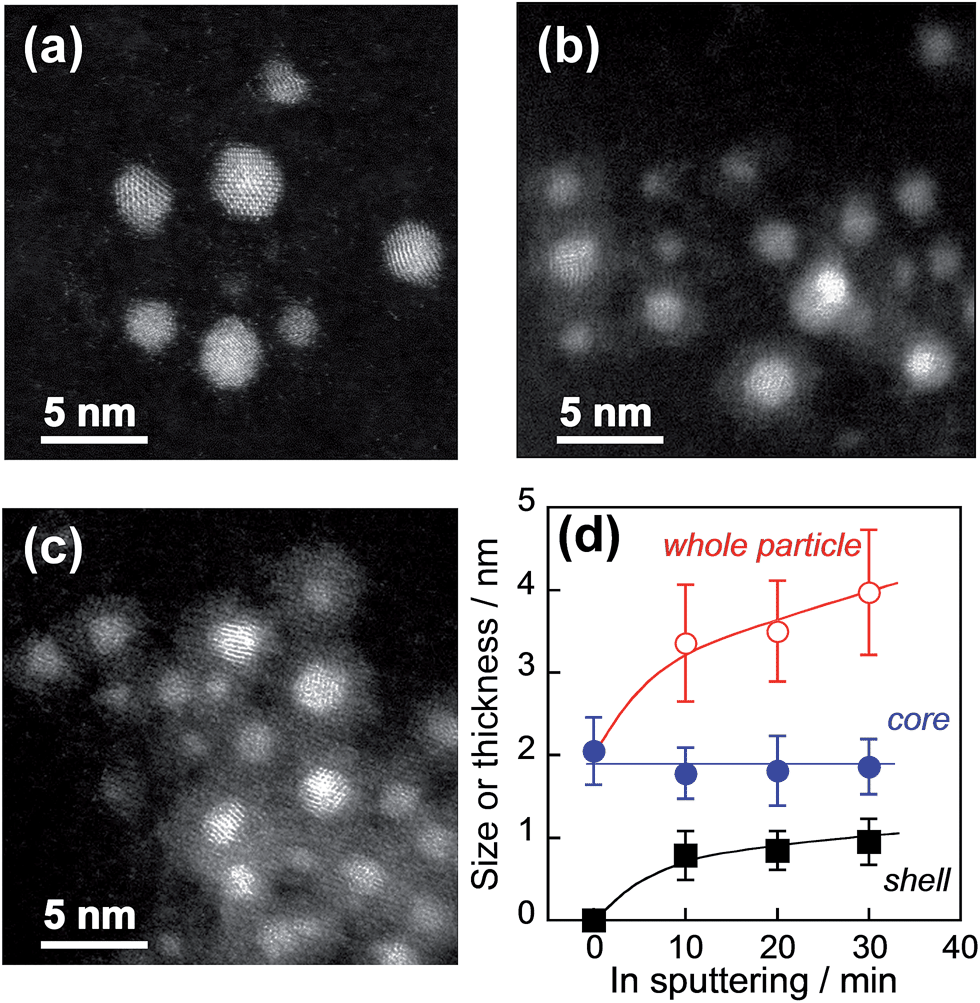 | ||
| Fig. 4 Representative HAADF-STEM images of original AuPd particles (a) and AuPd@In2O3 particles formed after different In sputtering times (b and c). The In sputtering time amounted to 10 (b) and 30 min (c). (d) Overall AuPd@In2O3 size, AuPd core sizes, and In2O3 shell thicknesses as a function of In sputtering time. | ||
The thermal stability of AuPd nanoparticles was also greatly improved by the surface coating with a thin In2O3 shell as expected. Fig. 5 shows the XRD patterns of original AuPd particles and AuPd@In2O3 particles before and after heat treatment at various temperatures. As-prepared bare AuPd particles exhibited broad diffraction peaks due to their small crystallite size, and the diffraction angle of each peak was consistent with that of the bulk AuPd (1:1) alloy with an fcc crystal structure, being similar to AuPd alloy particles sputter-deposited in an IL of 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide (BMI-TFSA) reported in our previous paper.41 The heat treatment of bare AuPd particles caused the narrowing of each diffraction peak due to the enlargement of the crystallite size with coalescence between nanoparticles, the degree being enhanced with an increase in the heating temperature. In contrast, the XRD patterns of AuPd@In2O3 particles did not show any remarkable changes after heat treatment at 100 or 200 °C, indicating that the In2O3 shell layer on AuPd cores prevented the coalescence of core particles. The HAADF-STEM measurements revealed that the amorphous In2O3 shell still covered the AuPd core even after the heat treatment of AuPd@In2O3 with 250 °C (Fig. S3†). It is noteworthy that no diffraction peaks assigned to the In2O3 crystal were observed in Fig. 5b, being in good agreement with the formation of an amorphous shell layer on the surface of AuPd core particles as observed in HAADF-STEM images (Fig. 4 and S3†).
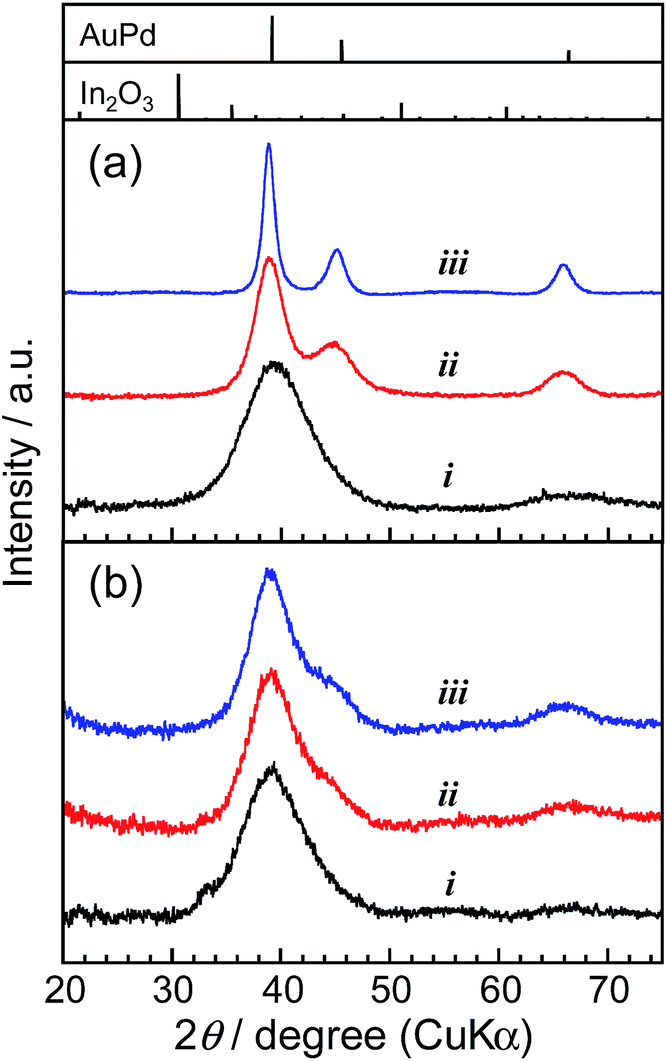 | ||
| Fig. 5 XRD patterns of (a) bare AuPd particles and (b) AuPd@In2O3 particles covered with a 0.79 nm-thick In2O3 shell. The samples were as-prepared (i) and heat-treated at 100 (ii) and 200 °C for 30 min (iii). Standard diffraction patterns of fcc AuPd (1:1) alloy and cubic In2O3 (PDF card# 01-072-537 and 00-006-0416, respectively) are also shown. | ||
Electrocatalytic activities of AuPd@In2O3
The electrocatalytic activity of AuPd nanoparticles has attracted intensive research interest for the development of highly efficient fuel cells.41,49 When AuPd@In2O3 core–shell particles are used as electrocatalysts, it is expected that the presence of a metal oxide shell layer and its thickness influence the activity of AuPd cores. In fact, it has been reported for core–shell-structured SiO2-coated CdS particles (CdS@SiO2)50 that the increase in the SiO2 shell thickness significantly decreased the reaction rate of CdS cores for photocatalytic hydrogen evolution. Furthermore, the In2O3 shell can also be expected to enable an electron transfer between AuPd catalyst cores and a collecting electrode due to its semiconductor behaviour.Nanoparticles dispersed in RTILs were immobilized on HOPG electrodes by heat treatment at 100 or 200 °C. The AFM measurements (Fig. 6) revealed that bare AuPd and AuPd@In2O3 particles prepared by 10 min-In sputtering (shell thickness: 0.79 nm) were successfully immobilized on HOPG surfaces without large aggregate formation at 100 °C. A similar uniform immobilization was observed for AuPd@In2O3 (Fig. 6d) but not for bare AuPd particles (Fig. 6b) upon heat treatment at 200 °C, the latter of which formed large aggregates.
 | ||
| Fig. 6 AFM images of the HOPG surface modified with bare AuPd particles (a and b) or AuPd@In2O3 covered with a 0.79 nm-thick In2O3 shell (c and d). The immobilization temperature equalled 100 (a and c) or 200 °C (b and d). | ||
Fig. 7a shows the cyclic voltammograms for ORR involving AuPd@In2O3 particles presenting different shell thicknesses and immobilized at 100 °C. Bare AuPd particles showed an oxygen reduction peak at around 0.81 V vs. RHE in the negative potential scan but no anodic current peak appeared in the positive potential scan, indicating that oxygen mainly underwent reduction to water on the AuPd surface (4e− reduction). At the AuPd@In2O3 electrode, the peak potential slightly shifted to ca. 0.77 V vs. RHE and the peak current decreased with an increasing In2O3 shell thickness on the AuPd core. When the shell thickness was as thick as 0.95 nm, the peak current amounted to ca. half of that obtained for bare AuPd particles. Interestingly, In2O3 hollow shell particles prepared by In metal sputter deposition exhibited negligible electrocatalytic activity for ORR (not shown). These results suggested that the amorphous In2O3 shell was probably porous enough to enable the diffusion of oxygen molecules from the bulk solution to the AuPd core surface acting as an ORR site inside the shell, and that the increase in In2O3 shell thickness lowered the diffusion rate of oxygen molecules.
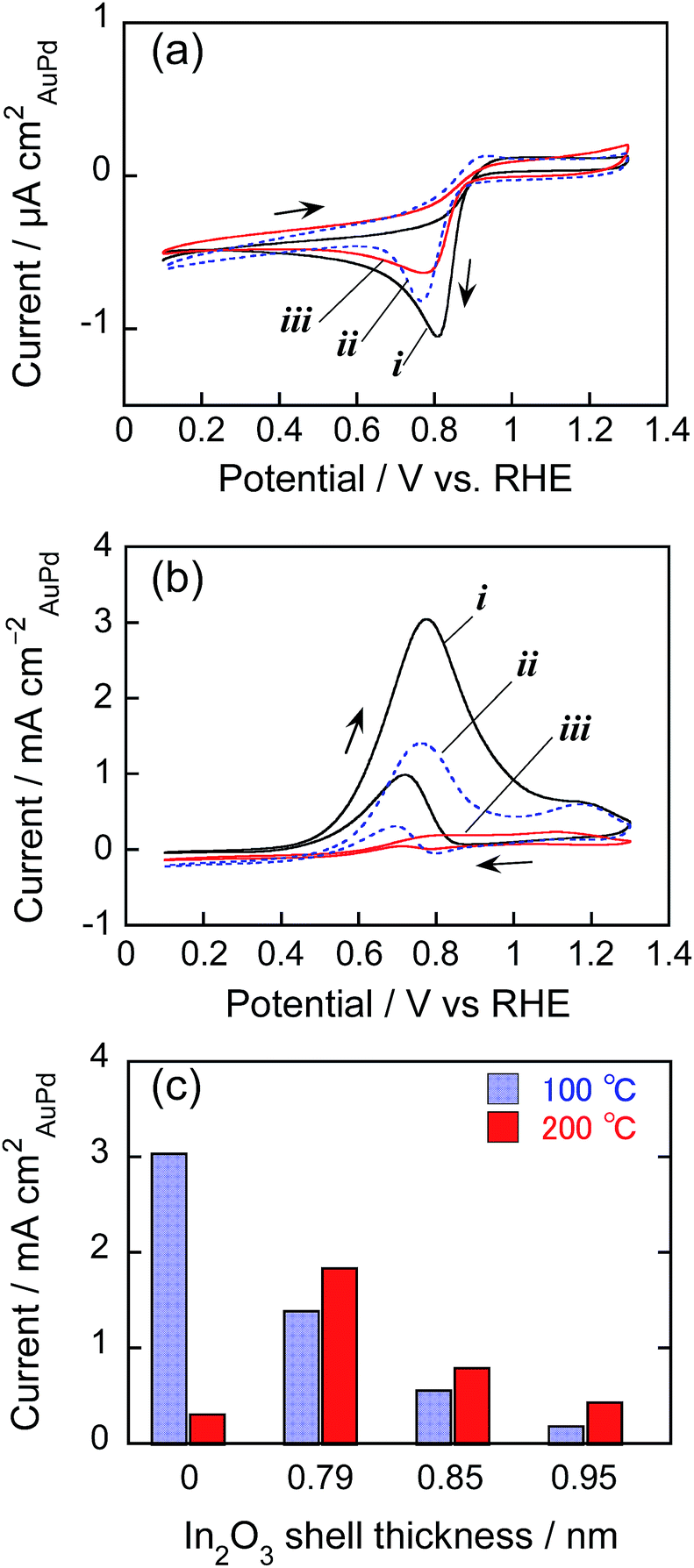 | ||
| Fig. 7 (a and b) Cyclic voltammograms for (a) ORR in O2-saturated 0.50 mol dm−3 KOH aqueous solution and (b) ethanol oxidation in 0.50 mol dm−3 KOH aqueous solution containing 0.50 mol dm−3 ethanol using HOPG electrodes modified with bare AuPd particles (i) and AuPd@In2O3 (ii and iii) at 100 °C. The In2O3 shell thickness equalled 0.79 (ii) or 0.95 nm (iii). Potential sweep rate: 100 mV s−1. (c) Relationship between the peak current density of AuPd@In2O3-modifed HOPG electrodes for ethanol oxidation and In2O3 shell thickness. The immobilization temperature is shown in the figure. | ||
Fig. 7b shows the cyclic voltammograms of AuPd particles for ethanol oxidation. Both bare AuPd and AuPd@In2O3 particles exhibited an oxidation peak at around 0.8 V vs., RHE, being in good agreement with previous reports,41 but the oxidation current decreased with increasing In2O3 shell thickness. Fig. 7c shows the change in peak current for ethanol oxidation in the positive potential scan as a function of In2O3 shell thickness. For AuPd@In2O3 immobilized at 100 °C, the peak current declined notably due to the presence of thicker In2O3 shells. The AuPd@In2O3 covered with a 0.95 nm shell produced an oxidation peak current that equalled ca. one-tenth of that for bare AuPd particles. This difference in electrocatalytic activity between bare AuPd particles and AuPd@In2O3 became more remarkable than that observed for ORR, where the decrease in electrocatalytic activity by ca. half was caused by covering AuPd cores with a 0.95 nm shell (Fig. 7a). This was probably due to the difference in the diffusion rate of target molecules through the In2O3 shell layer: increasing In2O3 shell thickness more remarkably hindered the diffusion of ethanol molecules from the bulk solution to the reactive AuPd core surface rather than that of O2 ones. It is worth mentioning that Au@In2O3 particles could be similarly immobilized on HOPG electrodes at 100 °C without the formation of large aggregates and then exhibited a peak for ethanol oxidation in the cyclic voltammogram (Fig. S4a†) at the potential of ca. 1.15 V vs. RHE, being similar to bare Au particles immobilized on HOPG at 100 °C. However the peak current obtained with Au@In2O3 was much smaller than that of corresponding AuPd@In2O3 with a 0.79 nm shell (Fig. 7b), because of lower electrocatalytic activity of Au particles than AuPd ones for ethanol oxidation.41
XPS measurements were carried out for AuPd-immobilized HOPG electrodes before and after cyclic voltammogram measurements in Fig. 7b with 100 cycles. As shown in Fig. S5,† as-prepared HOPG electrodes immobilized with bare AuPd or AuPd@In2O3 particles exhibited the signals of Au 4f5/2 and 4f7/2 at binding energies of 86.8 eV and 83.1 eV, respectively, which were in good agreement with those of corresponding signals for bare AuPd alloy particles reported in our previous paper.41 The positions of Au 4f signals were unchanged even after the electrochemical measurement, regardless of the presence of the In2O3 shell layer. Since the binding energy of Au 4f7/2 has been reported to decrease with a decrease in the fraction of Au in the AuPd alloy,51,52 it was indicated that dealloying of the AuPd alloy or segregation of Au in alloy particles did not occur when the particles of bare AuPd or AuPd@In2O3 were used as electrocatalysts for ethanol oxidation.
The temperature adopted for the nanoparticle immobilization also significantly affected the electrocatalytic activity for ethanol oxidation as shown in Fig. 7c. With an increase in the immobilization temperature from 100 to 200 °C, the peak current of ethanol oxidation for bare AuPd alloy particle electrodes remarkably decreased, accompanied with the positive shift of the peak potential from 0.8 to ca. 1.1 V vs. RHE as shown in Fig. S4b.† Since bare AuPd alloy particles immobilized at 200 °C gave the oxidation peak at a potential similar to that of Au particle electrodes, ca. 1.15 V vs. RHE (Fig. S4a†), it was suggested that the coalescence of alloy particles by the heat treatment at higher temperature (Fig. 6b) could produce a Au-rich surface on bare AuPd alloy particles due to the partial segregation of Au, resulting in the deterioration of ethanol oxidation activity of the AuPd alloy surface. In contrast, the electrocatalytic activity of AuPd@In2O3 in this reaction rose slightly when the immobilization temperature increased from 100 to 200 °C, indicating that the heat resistance of AuPd@In2O3 (Fig. 6 and S3†) resulted in maintaining their electrocatalytic activity after heat treatments at different temperatures.
Durability of Pt@In2O3-immobilized carbon catalysts for polymer electrolyte fuel cells
Platinum is one of the metal catalysts exhibiting the highest electrocatalytic activity in fuel cells. Especially Pt-loaded carbon particles are widely utilized in polymer electrolyte fuel cells. The improvement of the durability of those catalysts is earnestly investigated as an important issue to achieve large-scale commercialization. The In2O3 surface coating of these particles into core–shell nanostructures (Pt@In2O3), the structure of which prevents direct contact between Pt and carbon, is expected to increase the durability of Pt-loaded carbon materials under electrochemically oxidizing conditions.TEM images of bare Pt particles and Pt@In2O3 particles used for electrocatalysts are shown in Fig. 8. Spherical particles of 1.2 nm in average size were observed for bare Pt particles. The high resolution TEM images in the inset of Fig. 8a revealed that these Pt particles exhibited clear lattice fringes with a 0.23 nm interplanar spacing assignable to the Pt (111) lattice plane. On the other hand, Fig. 8b shows a TEM image of Pt@In2O3 particles which had a core–shell structure composed of a 1.2 nm-diameter Pt particle core and a thin In2O3 shell layer exhibiting lighter contrast. The high-resolution HAADF-STEM image of these particles in Fig. 8c clearly indicates that Pt cores exhibiting a lattice fringe were covered with the amorphous In2O3 shell with a thickness less than ca. 0.8 nm. The thickness of the In2O3 shell layer in Pt@In2O3 was less uniform than those observed for Au@In2O3 or AuPd@In2O3 which showed dense In2O3 layers on metal cores (Fig. 2c and 4c), probably due to the short sputtering time of In, 2 min, used for the Pt@In2O3 preparation. The elemental analysis of Pt@In2O3 revealed that the particles contained Pt and In with a Pt:In molar ratio of 1:0.28, being much smaller than that of Au@In2O3 prepared with In sputtering for 30 min, Au:In = 1:3.9. The thus-obtained Pt@In2O3 particles were loaded in the carbon black powder so as to obtain the Pt content of 5 wt%. The resulting catalyst powder was immobilized on a glassy carbon rotating-disk electrode using the ionomer Nafion®. The durability of the Pt@In2O3/C catalyst was examined according to the FCCJ protocol44,45 for the accelerated durability test of fuel cells.
 | ||
| Fig. 8 (a) TEM images of as-prepared bare Pt particles sputter-deposited in BMI-BF4. The high resolution TEM image is shown in the inset. (b and c) Images of Pt@In2O3 prepared in BMI-BF4 obtained by conventional TEM (b) and HAADF-STEM (c). | ||
Fig. 9a and b show the hydrodynamic voltammograms for the ORR of Pt/C and Pt@In2O3/C catalysts, respectively, under the rotation of 1600 rpm at a room temperature. In the case of the Pt/C catalyst, the cathodic currents due to ORR were gradually deteriorated during the durability test: with an increase in the number of potential step cycles, the onset potential of ORR became more negatively shifted. On the other hand, interestingly, the Pt@In2O3/C catalyst exhibited improvement of the catalytic activities at the first 4000 cycles, followed by some deterioration till 6000 cycles. To quantitatively evaluate the change in the ORR activity, the half-wave potential (E1/2), at which the cathodic current is one-half of the limiting current, was estimated from the voltammograms in Fig. 9a and b. Fig. 9c shows the relationship between E1/2 and the number of potential step cycles. The E1/2 obtained for Pt/C catalysts was rapidly negative-shifted by increasing the potential step cycle up to 5000, whereas the Pt@In2O3/C catalyst gave roughly constant E1/2 within 3000 cycles, except for the positive shift of E1/2 at the initial stage of the durability test. The negative shift of E1/2 was also observed for the Pt@In2O3/C electrode when the potential cycles exceeded 3000. It is likely that the deterioration of the catalytic activities observed for the Pt/C electrode was partially due to decrease in the surface of the Pt catalyst because the diffusion-limited current steadily decreased with potential cycling.
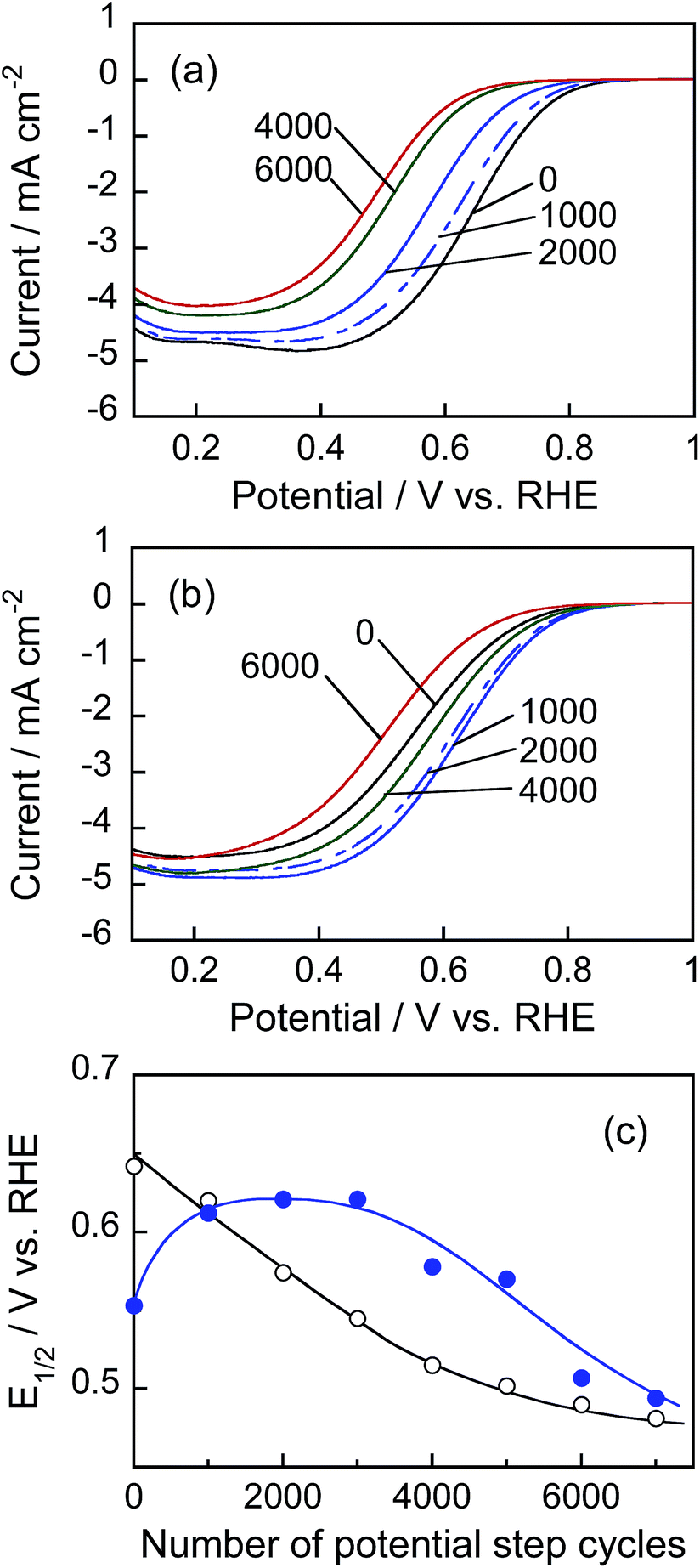 | ||
| Fig. 9 (a and b) Hydrodynamic voltammograms for ORR in O2-saturated 0.1 mol dm−3 HClO4 aqueous solution using rotating disc electrodes immobilized with Pt/C (a) or Pt@In2O3/C catalysts (b). Potential sweep rate: 10 mV s−1. The number of potential step cycles is indicated in the figure. (c) Plots of half-wave potential (E1/2) for ORR obtained with Pt/C (open circles) and Pt@In2O3/C catalysts (solid circles) as a function of the number of potential step cycles in the accelerated durability test. | ||
In order to find out information on Pt surface changes, the ECSA of Pt was determined using the hydrogen adsorption peak in the cyclic voltammograms (Fig. S6†) and the charge required for hydrogen adsorption on unit platinum area (210 μC cm−2).45Fig. 10 shows the changes in ECSA during potential cycle tests for Pt/C and Pt@In2O3/C. For the Pt/C catalyst, the ECSA initially increased with the rising cycle number up to 500 probably because of the removal of strongly absorbed ionic liquid species on the Pt surface. Meanwhile, the further potential cycling gradually deteriorated the ECSA. A similar behaviour has been widely reported previously,45,53–55 in which the decrease in ECSA was usually interpreted as the coalescence of loaded Pt nanoparticles into larger ones and/or the Pt particle detachment caused by the carbon support damage. In contrast, the In2O3 coating of Pt nanoparticles contributed to higher durability. The very small initial ECSA in Pt@In2O3 may stem from the strong adsorption of ionic liquid species on Pt@In2O3. However, it rapidly increased when the number of cycles rose up to ca. 2000 before becoming constant without any detectable deterioration up to 7000 cycles. These results strongly suggest that the ultrathin In2O3 shells covering the Pt cores effectively inhibited the deterioration of the Pt@In2O3/C.
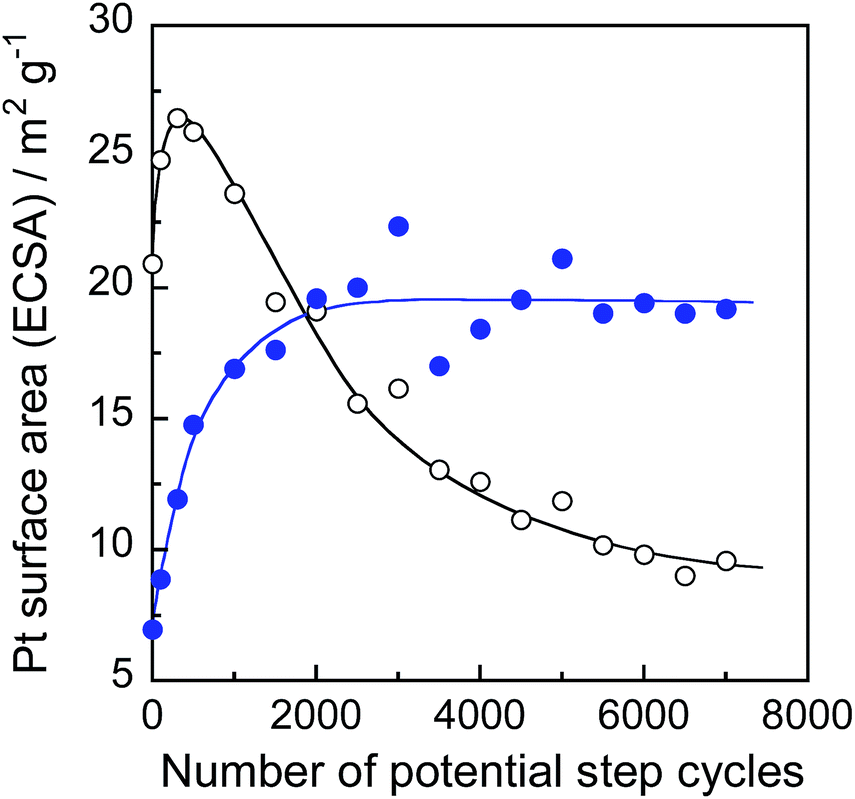 | ||
| Fig. 10 ECSA changes of Pt/C (open circles) and Pt@In2O3/C composites (solid circles) during the accelerated durability test of fuel cells. | ||
Conclusions
The coating of noble metal nanoparticles smaller than ca. 2.5 nm using an ultrathin metal oxide shell layer (thickness < ca. 1.5 nm), which was difficult to be prepared by conventional chemical strategies such as sol–gel methods, was achieved by the RTIL/sequential metal sputter deposition. In this technique, the shell thickness was readily controlled by changing the sputtering time. Coating with a thin In2O3 shell increased the stability of noble metal cores without losing electronic conduction between noble metal cores and collecting electrodes. Metal core particles could work as electrocatalysts, probably because thin In2O3 shell layers might be porous for small target molecules enough to penetrate through the shell from bulk solution toward metal core surfaces on which the electrocatalytic reaction occurred. These features are attractive for the development of highly active electrocatalysts. Therefore though only preparation of noble metal particles surface-coated with a thin In2O3 shell is described in this paper, the sequential sputter deposition of different metals in RTILs is expected to contribute significantly to the exploitation of novel nanostructured materials that will be key components for next-generation energy-conversion devices, such as fuel cells, solar cells, or photocatalysts.Acknowledgements
This work was partially supported by a Grant-in-Aid for a Funding Program for Next Generation World-Leading Researchers (NEXT Program) and Grant-in-Aid for Challenging Exploratory Research (no. 26620191) from the Japan Society for the Promotion of Science.Notes and references
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343 CrossRef CAS.
- C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS PubMed.
- M. Crespo-Quesada, A. Yarulin, M. S. Jin, Y. N. Xia and L. Kiwi-Minsker, J. Am. Chem. Soc., 2011, 133, 12787 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- J. Kaspar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419 CrossRef CAS.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433 CrossRef CAS.
- T. Torimoto, J. P. Reyes, K. Iwasaki, B. Pal, T. Shibayama, K. Sugawara, H. Takahashi and B. Ohtani, J. Am. Chem. Soc., 2003, 125, 316 CrossRef CAS PubMed.
- S. Ikeda, S. Ishino, T. Harada, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., Int. Ed., 2006, 45, 7063 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. D. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126 CrossRef CAS PubMed.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750 CrossRef CAS PubMed.
- N. Zhang, S. Q. Liu and Y. J. Xu, Nanoscale, 2012, 4, 2227 RSC.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. D. Yin, Acc. Chem. Res., 2013, 46, 1816 CrossRef CAS PubMed.
- S. Q. Liu, N. Zhang and Y. J. Xu, Part. Part. Syst. Charact., 2014, 31, 540 CrossRef CAS.
- S. H. Liu and M. Y. Han, Chem.–Asian J., 2010, 5, 36 CAS.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746 RSC.
- F. Caruso, Adv. Mater., 2001, 13, 11 CrossRef CAS.
- L. Wang, T. Sasaki, Y. Ebina, K. Kurashima and M. Watanabe, Chem. Mater., 2002, 14, 4827 CrossRef CAS.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831 CrossRef CAS PubMed.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- H. T. Sun, X. Sun, T. Hu, M. P. Yu, F. Y. Lu and J. Lian, J. Phys. Chem. C, 2014, 118, 2263 CAS.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711 CrossRef CAS PubMed.
- K. Y. Niu, J. Park, H. M. Zheng and A. P. Aivisatos, Nano Lett., 2013, 13, 5715 CrossRef CAS PubMed.
- K. Richter, A. Birkner and A. V. Mudring, Angew. Chem., Int. Ed., 2010, 49, 2431 CrossRef CAS PubMed.
- K. Ueno, A. Inaba, Y. Sano, M. Kondoh and M. Watanabe, Chem. Commun., 2009, 3603 RSC.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- C. Vollmer, E. Redel, K. Abu-Shandi, R. Thomann, H. Manyar, C. Hardacre and C. Janiak, Chem.–Eur. J., 2010, 16, 3849 CrossRef CAS PubMed.
- M. Brettholle, O. Hofft, L. Klarhofer, S. Mathes, W. Maus-Friedrichs, S. Z. El Abedin, S. Krischok, J. Janek and F. Endres, Phys. Chem. Chem. Phys., 2010, 12, 1750 RSC.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780 RSC.
- S. Kuwabata, T. Tsuda and T. Torimoto, J. Phys. Chem. Lett., 2010, 1, 3177 CrossRef CAS.
- T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196 CrossRef CAS PubMed.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184 CrossRef CAS.
- T. Torimoto, K. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka and S. Kuwabata, Appl. Phys. Lett., 2006, 89, 243117 CrossRef PubMed.
- K. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka, S. Kuwabata and T. Torimoto, Chem. Commun., 2008, 691 RSC.
- Y. Oda, K. Hirano, K. Yoshii, S. Kuwabata, T. Torimoto and M. Miura, Chem. Lett., 2010, 39, 1069 CrossRef CAS.
- Y. Hatakeyama, S. Takahashi and K. Nishikawa, J. Phys. Chem. C, 2010, 114, 11098 CAS.
- H. Wender, R. V. Goncalves, A. F. Feil, P. Migowski, F. S. Poletto, A. R. Pohlmann, J. Dupont and S. R. Teixeira, J. Phys. Chem. C, 2011, 115, 16362 CAS.
- K. Yoshii, T. Tsuda, T. Arimura, A. Imanishi, T. Torimoto and S. Kuwabata, RSC Adv., 2012, 2, 8262 RSC.
- S. Suzuki, T. Suzuki, Y. Tomita, M. Hirano, K. Okazaki, S. Kuwabata and T. Torimoto, CrystEngComm, 2012, 14, 4922 RSC.
- M. Hirano, K. Enokida, K. Okazaki, S. Kuwabata, H. Yoshida and T. Torimoto, Phys. Chem. Chem. Phys., 2013, 15, 7286 RSC.
- H. Wender, P. Migowski, A. F. Feil, S. R. Teixeira and J. Dupont, Coord. Chem. Rev., 2013, 257, 2468 CrossRef CAS PubMed.
- Y. W. Lee, M. Kim, Y. Kim, S. W. Kang, J. H. Lee and S. W. Han, J. Phys. Chem. C, 2010, 114, 7689 CAS.
- Fuel Cell Commercialization Conference of Japan (FCCJ), Proposals of the development targets, research and development challenges and evaluations methods concerning PEFCs, http://fccj.jp/pdf/23_01_kt.pdf.
- H. Yano, T. Akiyama, P. Bele, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 3806 RSC.
- T. Suzuki, K. Okazaki, S. Suzuki, T. Shibayama, S. Kuwabata and T. Torimoto, Chem. Mater., 2010, 22, 5209 CrossRef CAS.
- B. Balamurugan, F. E. Kruis, S. M. Shivaprasad, O. Dmitrieva and H. Zahres, Appl. Phys. Lett., 2005, 86, 083102 CrossRef PubMed.
- A. Monopoli, A. Afzal, C. di Franco, N. Ditaranto, N. Cioffi, A. Nacci, P. Cotugno and L. Torsi, J. Mol. Catal. A: Chem., 2014, 386, 101 CrossRef CAS PubMed.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS PubMed.
- B. Pal, T. Torimoto, K. Okazaki and B. Ohtani, Chem. Commun., 2007, 483 RSC.
- P. A. P. Nascente, S. G. C. Decastro, R. Landers and G. G. Kleiman, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 4659 CrossRef CAS.
- S. Deki, K. Akamatsu, Y. Hatakenaka, M. Mizuhata and A. Kajinami, Nanostruct. Mater., 1999, 11, 59 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285 CrossRef CAS PubMed.
- K. C. Neyerlin, R. Srivastava, C. F. Yu and P. Strasser, J. Power Sources, 2009, 186, 261 CrossRef CAS PubMed.
- F. Hasche, M. Oezaslan and P. Strasser, Phys. Chem. Chem. Phys., 2010, 12, 15251 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, XPS spectra, and cyclic voltammograms of metal@In2O3 particles. See DOI: 10.1039/c4ta06643j |
| This journal is © The Royal Society of Chemistry 2015 |