
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and the structure of freeze-dried MgO nanoparticle foams and their electrical behaviour in polyethylene†
L. K. H.
Pallon
a,
R. T.
Olsson
*a,
D.
Liu
a,
A. M.
Pourrahimi
a,
M. S.
Hedenqvist
a,
A. T.
Hoang
b,
S.
Gubanski
b and
U. W.
Gedde
a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Fibre and Polymer Technology, SE-100 44 Stockholm, Sweden. E-mail: rols@kth.se
bChalmers University of Technology, Department of Materials and Manufacturing Technology, High Voltage Engineering, SE-412 96 Göteborg, Sweden
First published on 25th February 2015
Abstract
Electrically insulating low-density polyethylene (LDPE) nanocomposites based on dispersed MgO nanoparticle foams are reported. The foams were obtained via freeze-drying aqueous suspensions of precipitated ca. 40 nm wide and 10 nm thick Mg(OH)2 nanoparticles and dewatering (calcining) at 400 °C, resulting in a 25 times more voluminous powder compared to conventionally dried nanoparticles. This powder handling prior to extrusion melt-processing greatly facilitated the nanocomposite preparation since no particle grinding was necessary. Large quantities of particles were prepared (>5 g), and the nanoparticle foams showed improved dispersion in the LDPE matrix with 70% smaller aggregate sizes compared to the conventionally dried and ground nanopowders. The nature of the nanoparticle foams was evaluated in terms of their dispersion on Si-wafers using ultrasonication as a dispersing aid, which showed to be detrimental for the nanoparticle separation into solitary particles and induced severe aggregation of the calcined nanoparticles. The grind-free MgO nanoparticles/LDPE-composite was evaluated by electrical measurement. The prepared composite showed an initial ca. 1.5 orders of magnitude lower charging current at 102 s, and a 4.2 times lower charging current after 16 hours compared to unfilled LDPE. The results open a way for improved insulation to be implemented in the future high-voltage cable system and present a new promising nanoparticle powder handling technique that can be used on a large scale.
1. Introduction
The need for clean and accessible electrical energy is a huge challenge for the modern world. A major problem in developing a modern energy grid system is the need to increase long-distance transmission capacity (>2000 km) with a limited amount of energy loss (<10%).1 For densely populated areas it is suggested that underground megavolt HVDC-cables are the best technical solution.2 The high voltage requires the development of better cable insulation that avoids excessive heat generation, which result in electrical breakdown. Heat generation due to leakage currents is a mechanism that limits a rise from the present 320 kV transmission voltages. The relative temperature increase (ΔT) in the insulation can be approximated as ΔT ∝ σU2, where σ is the conductivity of the leakage current and U the voltage. The applied voltage can thus only be increased if the material conductivity is decreased. To increase the voltage to 1 MV, the conductivity of today's cable low density polyethylene (LDPE) needs to be reduced by a factor of ten.3 This may be achieved with further development of magnesium oxide based nanocomposite cable insulations, which have been reported to show lower electrical conductivity and higher electrical breakdown strength due to a suppressed space charge accumulation as compared to the analogous pristine LDPE.4–6Magnesium oxide (MgO) nanoparticles are industrially prepared by thermal decomposition of magnesia-rich minerals such as magnesium carbonate, or salt deposits containing Mg(OH)2.7,8 The Mg(OH)2 precursor may also be produced in the laboratory by sol–gel reactions,9 hydration of MgO,10 hydrothermal synthesis,11 spinning disc synthesis12 or wet precipitation.13 The wet precipitation is normally carried out from a mixture of a magnesium salt solution and an alkaline medium,13–16 resulting in hexagonal platelets of Mg(OH)2 (rods and other morphologies with additives17). It is a versatile and inexpensive method that allows the use of low temperature conditions for various precursor stoichiometries, yielding large quantities (>20 g). However, one of the challenges in producing uniform individual nanoparticles of MgO is the necessary post-synthesis calcination, where the hexagonal unit cell of brucite Mg(OH)2 is transformed into the cubic unit cell of periclase MgO via dewatering and crystal lattice rearrangement.7 This dewatering at conventional drying of the large hydroxide functional surfaces (>70 m2 g−1)12 leads to the formation of aggregates, via either hydrogen bridging or covalent bonds due to the condensation of hydroxide groups upon the release of water.18,19 The formed solid bridges between particles yield hard unbreakable clusters that may show an even more aggravated aggregation after the calcination process. Such aggregates and clusters make electrical characterization of any MgO nanoparticle composite inconceivable because of the unpredictable internal particle organization.20 It is thus important to develop methods that avoid the formation of hard unbreakable aggregates during the synthesis and calcination of the nanoparticles. To be of industrial relevance the methods need to be applicable on a large-scale, and preferably do not involve extensive and energy consuming grinding procedures that introduce particle contamination21–23 or extensive water adsorption to the pristine high surface area nanoparticles.24
It is herein described how freeze-drying can be used as a method for powder handling, i.e. before preparation of insulating LDPE nanocomposites with ultra-low electrical conductivity. The sub-50 nm Mg(OH)2 nanoparticles were prepared by an aqueous synthesis in quantities sufficient for preparation of the nanocomposites. Dewatering of the Mg(OH)2 platelets was carried out by (a) freeze-drying and (b) conventional drying, followed by calcination at 400 °C into solitary sub-70 nm MgO-nanoparticles. The freeze-drying gave nanoparticle foams after drying, whereas the second approach resulted in a hard and compact cake that needed grinding to yield a powder of unevenly sized aggregates. The nanoparticles obtained were compounded with LDPE and the distribution of the nanoparticles in the composite was assessed. It is demonstrated that an extrusion of pre-calcination freeze-dried MgO can more easily be dispersed in a polymer matrix, resulting in smaller aggregates. A reduction in electrical conductivity with a factor ca. 4 compared to unfilled cable LDPE was obtained.
2. Experimental
2.1. Synthesis of precursor Mg(OH)2 hexagonal platelets
Aqueous precipitated Mg(OH)2 was synthesized by adding 1 L dissolved magnesium chloride (0.75 M; MgCl2·6H2O, ACS Reagent, Sigma-Aldrich) to a 1 L sodium hydroxide solution (1.5 M; NaOH, ≥98%, Sigma-Aldrich) in stoichiometric balance under rapid stirring (400 rpm). The reaction was allowed to proceed for 30 min at 23 °C. The precipitate was washed with milliQ-water in three cycles in order to remove Na+, Cl− and other residual reagents by centrifugation, using a Rotina 420 centrifuge (Hettich) and ultrasonic bath to break clusters (DTH 2510, Branson).25 All the particles studied were produced in a single batch synthesis.2.2. Drying of precursor Mg(OH)2 platelets
The washed precipitate of Mg(OH)2 platelets was divided into two parts; one part was dried in air at 90 °C and then ground into a fine powder using a pestle and mortar (sample Mg(OH)2-CD), and the second part was freeze-dried (sample Mg(OH)2-FD). The freezing was carried out in liquid nitrogen as 5 mL batches for 10 min, and was then dried for 12 h in a CoolSafe freeze drier (Scanvac) operating at 0.060 hPa and −96 °C. The small volume was used in order to minimize diffusion problems.2.3. Calcination of the precursor hexagonal platelets
The calcination of the Mg(OH)2 platelets was performed for 1 h at 400 °C in a muffle furnace (ML Furnaces) and the time and temperature were chosen in order to reduce the risk of sintering. The MgO powder obtained was stored under vacuum in a desiccator to prevent the uptake of CO2 and H2O.262.4. Wide-angle X-ray diffraction
The crystal phase of the dry powders was determined by X-ray powder diffractomety (XRD) with a PANalytical X'pert Pro using monochromotized Cu Kα radiation (λ = 0.154021 nm) with a step size of 2θ = 0.017° every 51 s (45 kV, 35 mA). Line broadening was removed using Si as an internal standard. Due to the anisotropic nature of the Mg(OH)2 platelets the crystallite size of Mg(OH)2 was determined by the Warren-modified Scherrer equation:27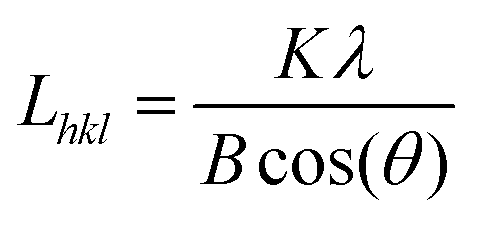 | (1) |
where B is the peak width in radians at half the peak height (FWHM) and Lhkl is the crystallite size along the normal of the (hkl) plane. The shape factor (K) is dependent on the analysed plane (hkl), giving K = 1.84 for (hk0) and 0.94 for (00l) peaks for Mg(OH)2.27,28 For MgO, anisotropy need not be considered and a shape factor of 0.94 was used.
2.5. Scanning electron microscopy
A field emission scanning electron microscope (FE-SEM), Hitachi S-4800, was used to assess the size of nanoparticles deposited as 10 μL individual droplets of 2-propanol (C3H8O, ASC Reagent, VWR) onto Si-wafers (Polished Wafer, MEMC Electronic Material). 2-Propanol was used since MgO is not stable in water, and a volume of 10 μL was sufficient to cover the Si-wafer without overflow. The droplets were allowed to evaporate to ensure that the material was statistically representative of all types of particle associations. This method made it possible to differentiate between individual particles, flocks formed in the 2-propanol and remaining aggregates, while avoiding clusters being flushed away from the sample, as reported by Rodriguez-Nawarro et al.28 The area of the Si-wafer for one droplet varied between 42 and 64 mm2. To achieve representative data for the individual samples; 20 micrographs with 500× magnification were analysed, covering a total area of 0.9 (mm)2, corresponding to the area covered by ca. 8000 micrographs at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification, constituting up to 1/50 of the Si-wafer. The size of the solitary particles of MgO was determined using the same procedure as for the aggregates, where the atomic flat surface of the Si-wafer made it possible to assess the particle size distribution including more than 1000 measured primary particles. The particle diameter was determined as the greatest distance across the primary platelet particles. The melt-processed nanocomposites were characterized by FE-SEM. The rod shaped samples were immersed in liquid nitrogen for 20 min to enable cryo-fractured surfaces.
000× magnification, constituting up to 1/50 of the Si-wafer. The size of the solitary particles of MgO was determined using the same procedure as for the aggregates, where the atomic flat surface of the Si-wafer made it possible to assess the particle size distribution including more than 1000 measured primary particles. The particle diameter was determined as the greatest distance across the primary platelet particles. The melt-processed nanocomposites were characterized by FE-SEM. The rod shaped samples were immersed in liquid nitrogen for 20 min to enable cryo-fractured surfaces.
2.6. Transmission electron microscopy
The size distributions of the Mg(OH)2 platelets were determined from never-dried samples, using a Hitachi HT7700 transmission electron microscope operated at 100 kV. The MgO samples were dispersed by depositing 1 droplet of particles suspended in 2-propanol onto a carbon-coated 400-mesh ultrathin holey carbon film grid (Ted Pella) before drying under vacuum. The Mg(OH)2 was deposited on a carbon-coated Formvar 200 μm Cu-grid (Agar Scientific) dried under vacuum.2.7. Surface area determination by nitrogen adsorption measurements
The specific surface area (SSA) was determined by liquid nitrogen adsorption–desorption isotherms using a Micromeritics ASAP 2000 at 77 K. Before measurement, the samples were degassed at 200 °C until the pressure reached 0.003 hPa.292.8. Thermal analysis
A Mettler-Toledo TGA/DSC 1 thermogravimetric analyser (TGA and DSC) was used to assess the mass loss, the onset temperature of the reaction, and the change in enthalpy of the thermal decomposition of Mg(OH)2 to MgO. Samples were run at a rate of 10 °C min−1 from 30 to 600 °C in oxygen using 70 μL aluminium oxide crucibles. Prior to the thermogravimetry, all the samples were dried under reduced pressure at 100 °C for 24 h to remove crystallite water. The mass of each sample was 5 ± 0.2 mg.2.9. Dispersion of MgO
The calcined samples (0.1 g) were dispersed in 2 mL of 2-propanol with a concentration of 50 gL−1, using an ultrasonication bath (DTH 2510, Branson) for 30, 60 and 120 min. The ultrasonic bath treatment was standardized with regard to energy input, vial position, volume of water, temperature, time and sample taking for further dilution. After ultrasonication all the samples were diluted 5000 times to reduce the risk of particle flocculation.2.10. Melt processing and preparation of films for conductivity measurements
Ground low-density polyethylene (Borealis) powder was mixed with antioxidant Irganox 1076 dissolved in n-heptane (>99%, VWR) and shaken for 60 min using a Vortex Genie 2 shaker (G560E, Scientific Industries). The concentration of Irganox 1076 was 200 ppm (mass) in the LDPE to limit its influence in subsequent conductivity measurements, and heptane and thorough mixing helped to facilitate good dispersion of the antioxidant. Each MgO sample powder was added to the LDPE and shaken for another 30 min before it was dried overnight at normal pressure at 80 °C, and then shaken for a further 30 min before extrusion. The blend was extruded at 115 °C and 150 °C for 6 min at 100 rpm in a Micro 5cc Twin Screw Compounder (DSM Xplore) forming a composite with 3 wt% MgO. The weight percentage MgO was chosen based on previous reports that it is sufficient as a filler to influence the electrical behaviour.30 The extrusion parameters were set to evaluate the dispersion of the MgO at 115 °C or at 150 °C. Compression moulding was performed in an ambient atmosphere on a LabPro 400 (Fontijne Grotnes) at 130 °C for 10 min contact pressure, 10 min at 200 kN, and with the pressure maintained until cooling to 30 °C. The films were compression moulded in a 75 μm thick stainless steal mould. A reference sample of unfilled LDPE for conductivity measurement, with the same concentration of the antioxidant, was extruded at 150 °C and hot pressed with the same settings as the nanocomposites.2.11. Conductivity measurements
The volume electrical conductivity measurements were performed following a standard procedure31 by applying direct current (DC) voltage from a power supply (Glassman FJ60R2) over the film sample and measuring the charging current by means of an electrometer (Keithley 6517A), as sketched in Fig. 1. The detected current signal was recorded by LabVIEW software incorporated in a personal computer and stored for further analyses. An oven was used to control temperature, whereas an overvoltage protection secured the electrometer from damaging due to possible overshoots and a low-pass filter removed high frequency disturbance. A three-stainless steel electrode system was used, in which the high voltage electrode was a cylinder with a diameter of 45 mm; the current measuring electrode was 30 mm in diameter, whereas the guard ring allowed for eliminating surface currents. The good contact of the high voltage electrode and the film sample was obtained by placing an Elastosil R570/70 (Wacker) layer between them. The experiments were conducted on the LDPE/MgO-FD composite and unfilled LDPE as a reference sample at 60 °C for ∼6 × 104 s (16.7 h). The applied voltage was 2.6 kV corresponding to the electric field of 32.5 kV (mm)−1, giving conditions (40–90 °C) of temperature and electric field resembling the stress conditions in the insulation of a real HVDC cable.32 The test was repeated three times for each material and good repeatability was achieved.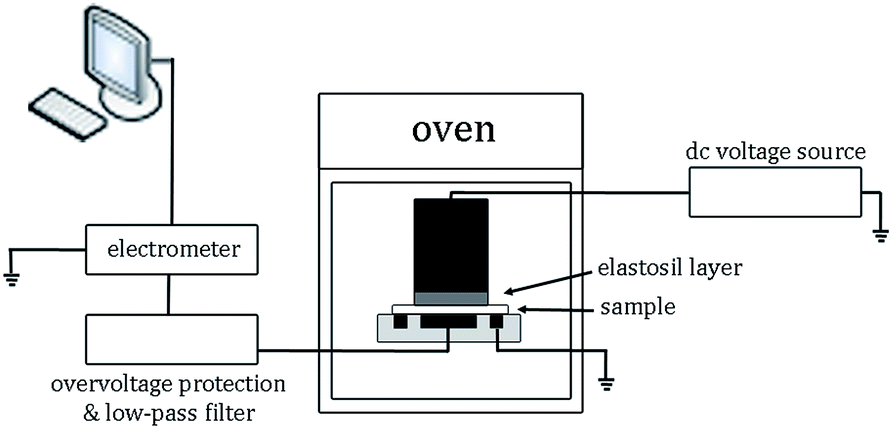 | ||
| Fig. 1 Schematic view of the test setup for measurements of volume electrical conductivity. | ||
3. Results and discussion
3.1. Mg(OH)2 platelet foams and cakes after drying
The Mg(OH)2 nanoparticles precipitate consisted of hexagonal platelets with sizes ranging from ca. 15 to 80 nm, and a relatively narrow size distribution with an average size of 43 nm (Fig. 2a and 4a). This platelet size range was the same as that of platelets precipitated from aqueous solutions by Giorgi et al.13 Occasional platelets with a round shape were observed, in agreement with Henrist et al.14 The thickness of the Mg(OH)2 platelets was 10–20 nm along the [001] direction, as determined from the platelets vertically positioned on their edges. | ||
| Fig. 2 The difference in structures between freeze-dried and conventionally dried Mg(OH)2. (a) Transmission electron micrograph of as precipitated Mg(OH)2 with an average size of 43 nm (inset), determined from >600 particles. The freeze-dried Mg(OH)2 powder (b) had a volume 25 times larger than the conventional dried Mg(OH)2 powder (c). Scanning electron micrograph at high and low magnification of (d and e) the conventionally dried Mg(OH)2 and (f and g) the freeze-dried foam. In (d) and (f), both drying procedures lead to Mg(OH)2-particles arranged in an epitaxial manner, and in (d) the conventionally dried cake has pores, as revealed by the black holes. | ||
 | ||
| Fig. 3 (a and b) Scanning electron micrographs at low and high magnification of freeze-dried Mg(OH)2 intensely shaken for 10 s. The thin flakes are easily separated from the original foam. The >100 μm large holes seen in the surface in (a) belong to carbon tape used for sample preparation. | ||
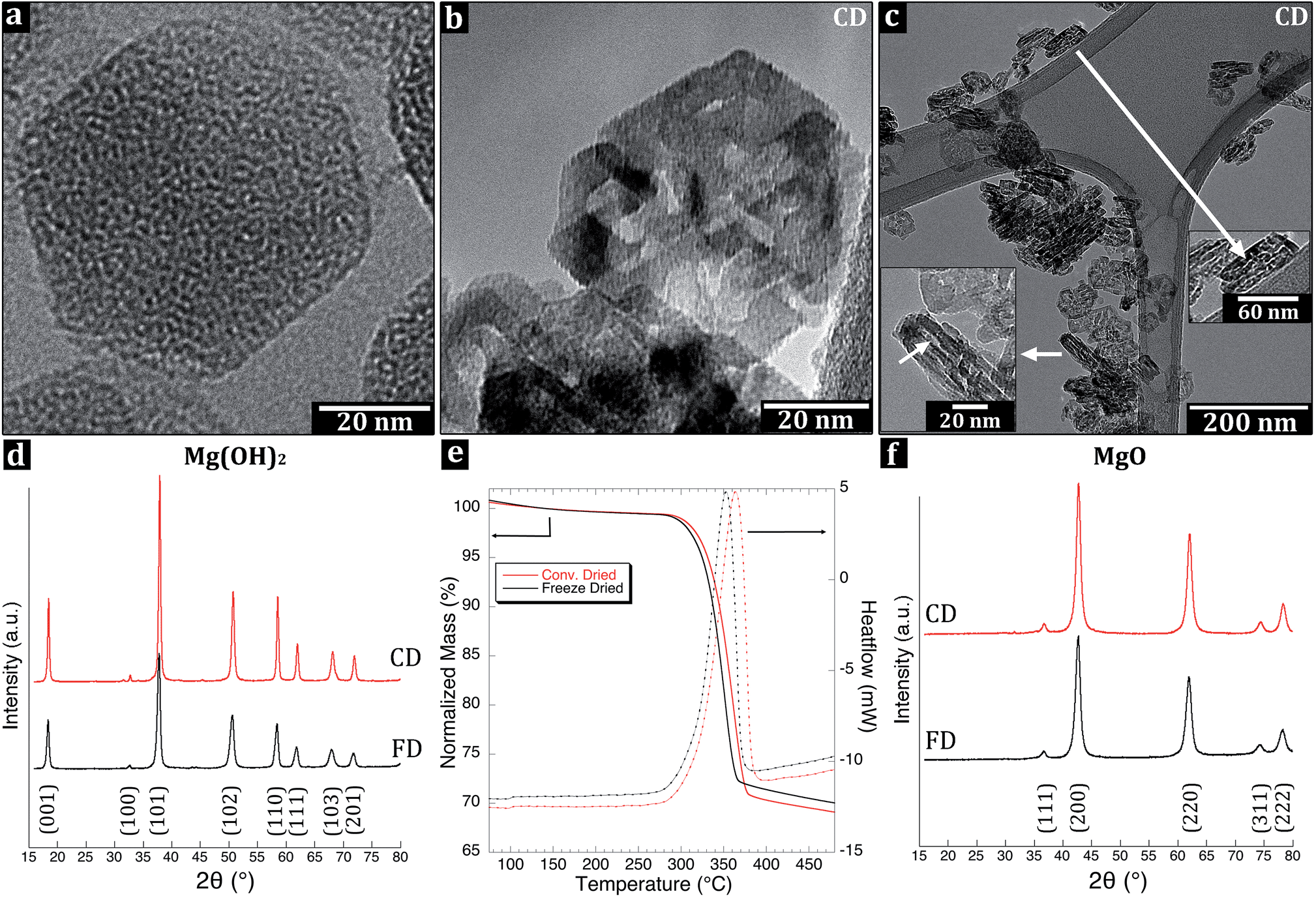 | ||
| Fig. 4 Transmission electron micrographs of the uniform (a) Mg(OH)2 and of the (b and c) polycrystalline conventionally dried MgO at high and low magnification. Arrows in (c) mark porous parts of the MgO formed during the thermal decomposition. X-ray diffractograms for (d) pure Mg(OH)2. The TGA/DSC-response (e) for the thermal decomposition normalized at 140 °C of Mg(OH)2 to MgO, revealing a delayed conversion of the conventionally dried (red) compared to the freeze-dried (black) Mg(OH)2 and (f) XRD for MgO, confirming the phase without displaying any residues. | ||
Fig. 2b and c show the powders of the Mg(OH)2 platelets and their structure immediately after freeze drying (FD), and conventional drying (CD) at 90 °C in air. The method for removal of the aqueous phase had a major impact on the porosity of the nanoparticle powder. The volume of the freeze dried Mg(OH)2 sample was ca. 25 times larger than that of the conventionally dried Mg(OH)2 sample. Microscopy revealed that the platelets in the freeze dried sample had formed a porous and fragile foam structure (Fig. 2g), whereas the conventional dried platelets had formed a hard cake, which cracked in a brittle manner when it was ground with a pestle and mortar (Fig. 2e). The foam consisted of a network of inter-bridging ca. 1 to 100 μm flakes that consisted of the associated platelets (Fig. 2g). The overall structure showed 20 μm wide channels emanating from the sublimed ice crystals, with anisotropy depending on the water crystal growth direction. Fig. 2f shows a highlighted area of a flake of associated platelets. The platelet configuration was determined by the displacement of the platelets into segmented regions as the ice crystals were formed during the liquid nitrogen quenching prior to the drying. For comparison, the compact cake showed inter-condensed platelets with occasional 50–100 nm pores that penetrated the macrostructure (Fig. 2d and e). For both samples, the particles were predominantly assembled in an epitaxial manner due to the platelet morphology. This epitaxial arrangement is energetically favourable due to the overlapping of the same crystal planes (in this case the basal plane (001)) of adjacent platelets.28
The formation of the nanoparticle foam relied not only on the utilization of freeze-drying as a method for removal of the water phase, but also on the selection of the precursor salt used for the wet-precipitation of the platelets. When the Mg(OH)2 was prepared from MgSO4 as a precursor for the synthesis, a porous network was not supported and the structure collapsed into a loosely aggregated powder (ESI, S1†). Accordingly, the “card-house” structure only resulted when the Mg(OH)2 platelets were made from the MgCl2 inorganic salt, followed by the lyophilisation. Small amounts of surface located counter ions from the original salts were therefore interpreted as a cause for the sustained stability of the porous foam after drying. Surface located counter ions from the original salts are known to remain from the synthesis even after extensive ultrasonic cleaning procedures.25Fig. 3 shows how the fragile open assembly of the particles could be fragmented into separated flakes merely by intense shaking for 10 s, with a subsequent 10% additional increase in the total volume of the nanopowder (Fig. 3a and b).
3.2. Phase characterization of the Mg(OH)2
Fig. 4a shows the brucite Mg(OH)2 with a uniform layered in-plane internal structure of hydroxyl (OH−) groups in hexagonal close packing extending over the entire platelets.The X-ray diffraction confirmed that the platelets were brucite-structured Mg(OH)2 (ICSD reference code 00-007-0239) independent of the drying method, Fig. 4d. The freeze-dried (FD) sample exhibited broader Bragg peaks at 18.43°, 32.69°, 37.93°, 50.75°, 58.51°, 61.94°, 68.08° and 72.06° with small shifts in the peak positions (0.102°) to lower 2θ angles – for the otherwise identical pattern with the conventionally dried (CD) sample. The broadening was accompanied by a small expansion of the unit cell ((001) – 0.55%; (100) – 0.30%; (110) – 0.16%) with d values of d100 – 2.739 Å, d001 – 4.819 Å for FD and d100 – 2.731 Å, d001 – 4.793 Å for CD. The calculated lattice constants were equal to: a = b = 3.163 Å and c = 4.819 Å for FD and corresponding values for CD were 3.153 Å and 4.793 Å. The shift in the peak position could be interpreted as the result of strain in the foam structure due to the bridging of Mg(OH)2 platelets, i.e. related to the absence of internal stress relaxation in the particles upon the rapid quenching of the system prior to drying.33 The platelets size was 63 nm in the CD compared to 39 nm-sized platelets in the FD sample (Table 1), according to the Warren-modified Scherrer equation (eqn (1)). The larger platelet size in the CD sample was likely due to the rearrangements and partial inter-particle condensation of some platelets during the drying at 90 °C, which was not allowed to occur in the FD sample. The freeze-dried platelets only deviated by 4 nm in lateral dimensions from their determined average size of 43 nm according to TEM (Fig. 2a).
| Sample | Shape | Surface areaa (m2 g−1) | Primary particle sizeb,c (nm) | Plane (hkl) | Platelet/crystallite sized (nm) |
|---|---|---|---|---|---|
| a Particle surface area from nitrogen-adsorption measurement using the BET-method. b Platelet size by TEM as measured over the maximum cross-section. c Platelet size by SEM as the suspension deposit on a Si-wafer measured over the maximum cross-section. d Platelet/crystallite size from the Scherrer equation. | |||||
| Mg(OH)2 FD | Plate | 52 | 43b | (001) | 19 |
| (100) | 39 | ||||
| Mg(OH)2 CD | Plate | 44 | 43b | (001) | 26 |
| (100) | 63 | ||||
| MgO FD | Plate | 118 | 59c | (200) | 8.1 |
| MgO CD | Plate | 166 | 66c | (200) | 9.2 |
3.3. Calcination of precursor Mg(OH)2 to MgO nanoparticles
Fig. 4b shows a platelet after the Mg(OH)2 high-temperature transformation into periclase MgO via hydroxylation (OH− condensation), dissipation of water, and rearrangement of the lattice into its cubic close packed structured crystallites inside the platelets.The thermogravimetric analysis (TGA) of the transformation revealed an initial mass loss of 1.4 wt% (FD) and 1.1 wt% (CD) due to the evaporation of loosely bound water from the platelet surfaces (75–140 °C), and the TG-plot was therefore normalized to the value of 140 °C,34Fig. 4e. Between 140 and 270 °C, the loss of an additional 0.6 wt% (FD) and 0.5 wt% (CD) was assigned to the release of more strongly associated water partially formed from condensation reactions. The total mass loss prior to the actual decomposition/reorganization of the lattice structure from brucite Mg(OH)2 into periclase MgO was 2.0 wt% (FD) and 1.6 wt% (CD), acquired from the normalized values. The mass loss related to the transformation of Mg(OH)2 into MgO occurred between 270 °C and 380 or 395 °C (for the FD and CD samples, respectively) according to the DSC and TGA data presented in Fig. 4e. In this region, the freeze-dried platelets lost 27.9 wt%, whereas the conventionally dried platelets lost 29.0 wt% due to the release of water molecules formed by hydroxylation. An additional mass loss of 1.4 wt% (FD) and 1.3 wt% (CD) was recorded between 395 °C and 480 °C, resulting in a total mass loss of 30.0 wt% for the FD sample and 30.9 wt% for the CD sample, with only the latter in agreement with the theoretical decomposition value of 30.9 wt%.
The difference between the samples was confirmed by repeated differential scanning calorimeter (DSC) scans. The scans consistently generated a transformation enthalpy of 920 kJ (kg)−1 for the FD sample and 1055 kJ (kg)−1 for the CD sample (Fig. 4e). Both values were smaller than 1304 kJ (kg)−1 at 600 K required for the transformation, i.e. based on the enthalpy of decomposition of MgO into Mg(OH)2.35 Considering the mass loss values at 395 °C, the reached transformations should have shown 90.3 and 94.3 wt% for FD and CD samples, respectively, but only showed 70.6 and 80.9 wt% according to the integrated endotherms. The reason for the discrepancies was unclear, in particular since the X-ray diffraction results showed no residual Mg(OH)2 phase post calcination, see Fig. 4f and section 3.5. It was, however, apparent that the last 1.4 wt% (FD) and 1.3 wt% (CD) mass loss (above 395 °C) did not show any signs of an endothermic reaction, only slight inclination in the curves in Fig. 4e. Based on the fact that the TG data for the conventionally dried (CD) powder indicated a complete stoichiometric transformation, the enthalpy associated with such full conversion was calculated but reached only a value of 1115 kJ (kg)−1. It was therefore concluded that the small amount of Mg(OH)2 phase transformation must have occurred also at lower temperatures for the freeze dried Mg(OH)2. The results are consistent with TG mass loss values presented by Huyen et al.36 who showed by coupled mass spectroscopy that a small amount of H2O is released already at 150–200 °C from the Mg(OH)2. The specific surface area created during the calcination must also have affected transformation enthalpy since it increased from 44 m2 g−1 (before) to 166 m2 g−1 (after) during the high temperature treatment (Table 1). Inevitably, the formed interface related to the crystallites inside the hexagonal platelets cannot condense into perfectly cubic structured crystalline lattices at this temperature. Variations in the magnitude of the decomposition endotherms and the decomposition temperature have previously been reported to also depend on the sample-crucible contact due to the porous nature of powders.35,37–39
In the case of the FD sample, neither the mass loss data nor the enthalpy for the full conversion into MgO showed complete transformation. Due to the discrepancies related to MgO formation enthalpy, only the mass loss data were considered. For the FD sample 0.9 wt% water loss was never showed in the TG data. Some Mg(OH)2 transformation into dehydrated MgO must therefore have occurred in the FD sample already during the extensive drying under vacuum, i.e. below the starting temperature for the TG measurement. The endothermic peak at 352 °C for the FD sample (compared to 364 °C for the CD sample) also suggested more extensive evaporation of water in the more open and dry freeze-dried material (1–2 min earlier at a heating rate of 10 °C min−1). Green et al. explained that the delay in the endothermic peak for conventionally dried MgO is related to the diffusion of a more substantial water phase from the interior of the MgO powder.40 In conclusion, the heating to 480 °C completely transformed Mg(OH)2 phase into MgO platelets for both samples (Fig. 4f), whereas a small release of water molecules at temperatures significantly below the major transformation occurred. Given the fact that the platelets originated from the same synthesized batch and that the CD sample showed the correct mass loss value, any remaining hydroxylation reactions with the formation of water above 480 °C must have been insignificant.29 This small amount of water loss was to some extent released already during the sublimation of the ice in the lyophilisation process, which may have had an influence on the formation of the slightly deformed/expanded unit cells (0.6%; Section 3.2) in the c-direction from 4.79 to 4.82 Å.
To further investigate the effect of the freeze-drying on the prepared platelets, Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption isotherms together with the corresponding Barret–Joyner–Halenda (BJH) pore size distributions were obtained for the FD and the CD nanoparticle samples before and after calcination. Prior to calcination, the samples were characterized by pores with a size between 10 and 100 nm and peak values of 23 and 32 nm for the Mg(OH)2-FD and Mg(OH)2-CD samples, respectively (ESI, S2†). These pores were attributed to the inter-distances between aggregates of the plate-like particles, and are commonly referred to as slit-shaped pores, as suggested by the type III isotherm with H3 hysteresis loops.41 The smaller inter-aggregate distances for the Mg(OH)2-FD sample were ascribed to a better packing of the flakes during the nitrogen adsorption measurement, whereas the cracked and ground cake (Mg(OH)2-CD) had remains of larger aggregates in the powder that were more difficult to pack. The measurements also showed very small 3 nm pores related to the internal structure of the platelets. These pores were twice as many in the Mg(OH)2-FD sample as in the Mg(OH)2-CD sample, which may have contributed to the broadening and the shift of the peaks in the XRD for Mg(OH)2-FD. Upon calcination, the pores increased in relative frequency (normalized with respect to their presence in the Mg(OH)2 samples), by factors of 27 and 233 for the MgO-FD and MgO-CD samples, respectively. The arrows in Fig. 4c indicate the pores developed between the crystallites inside the hexagonal platelets. Similar pores were reported by Horlock et al.42 and are formed upon contraction in the [001] direction in the arrangement of cubic crystallites from the hexagonal hydroxide lattice. The arrangement of the crystallites and their specific formation was not described by Horlock et al.42 but it was clear that the difference in the drying prior to the calcination of the Mg(OH)2 had an effect on their formation and frequency in the final MgO material. It is suggested that the more extensive and early release of the H2O in the freeze-dried nanoparticle foam allowed for partial initiation of some of the porosity associated with the crystallite borders, which led to a smaller degree of novel pore formation during the calcination. To what extent the higher reaction enthalpy for the CD sample could be related to the pore expansion during the high-temperature treatment, or the dissociation of water from the compact cake structure (e.g. vapour pressure build-up), remained an open question. It has been reported that water catalyses the thermal decomposition of Mg(OH)2 to MgO, but due to the correlation between DSC and the TG measurements for both the CD and FD samples, it should only have had a minor influence on the reaction enthalpy.8,43
3.4. MgO – phase characterization and morphology
Fig. 4f shows the X-ray diffraction pattern of the calcined periclase MgO (ICSD code 009863, ref. 01-071-1176) nanoparticles prepared by freeze-drying (FD) and conventional drying (CD). None of the samples showed any Mg(OH)2 residues. The calculated lattice constants were equal to: a = b = c: 4.234 Å and 4.228 Å for FD and CD, respectively. After calcination, the platelets showed more intensified rhombic lateral habits with angles of 80° and 100° and the subdomains of cubic close packed crystallites which formed at 350–400 °C (Fig. 4b). The size across the largest distance of the platelet increased with the high temperature treatment from an average size of 43 nm to an average size of ca. 60 nm for the MgO-FD and MgO-CD nanoparticles, see Table 1. The size distribution of the primary particles of MgO after calcination was only slightly broader than that of the Mg(OH)2-particles before the calcination, Fig. 2a, 5c and f, insets. The thicknesses' of the platelets were between 10 and 20 nm for both samples and showed no apparent contraction, which previously has been reported in the [001] direction.42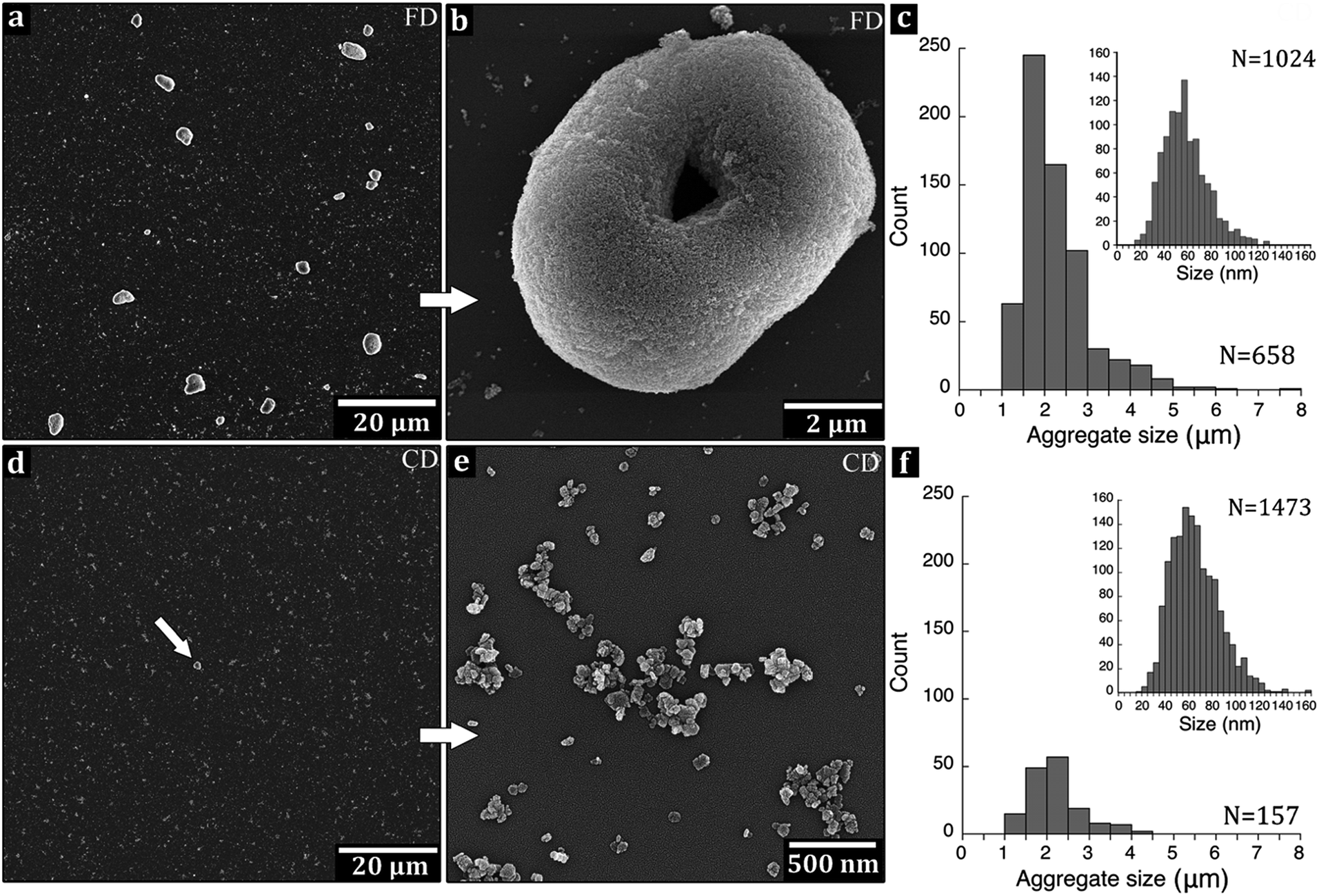 | ||
| Fig. 5 Scanning electron micrographs of MgO dispersed by ultrasonication and deposited on Si-wafer for (a and b) the freeze-dried and (d and e) the conventionally dried sample. The donut-shaped aggregate (b), more frequently found in the freeze-dried sample was formed during the ultrasonication. The arrow in (d) indicates a single aggregate with the remainder of the MgO dispersed as solitary particles, seen as a “night sky” at low magnification (d) or as flocculation's in (e). The number and size distribution of remaining aggregates after 120 min ultrasonication of freeze-dried (c) and conventionally dried (f) samples are displayed in the histograms. The numbers of aggregates were normalized with respect to the area of the Si-wafers. The size distributions of the primary particles are seen in the insets. | ||
The small cubic crystallites formed inside the hexagonal platelets were 8.1 and 9.2 nm for MgO-FD and MgO-CD, respectively, based on the Scherrer equation (for the most intense peaks: 2θ = 42.70° and 2θ = 42.76°, hkl 200) (Table 1). Fig. 4c shows the ca. 3–5 nm pores visible as bright lines inside the sintered vertically positioned primary particles, which were displayed by the BJH. It was unclear whether this internal porosity was entirely related to formation of the crystallites or if some pores also originated from the inter-particle sintering during the calcination at 400 °C. If the latter would have been the case, this would partially explain the significantly larger increase in porosity associated with the MgO-CD sample. The cumulative pore volumes were 0.84 cm3 g−1 for the MgO-CD samples, whereas the MgO-FD sample showed only 0.78 cm3 g−1. It has previously been reported that precursor strain in the transformation leads to smaller crystallites.44
Assuming non-porous cubic crystallites with a density (ρ) of 3580 kg m−3 within the hexagonal platelets, and the specific surface area (SSA) obtained by nitrogen absorption experiments' (Table 1), the crystallite diameters (D) were calculated to be 14.2 and 10.0 nm for MgO-FD and MgO-CD nanoparticles, according to eqn (2), which is valid for both spheres and cubes.:45
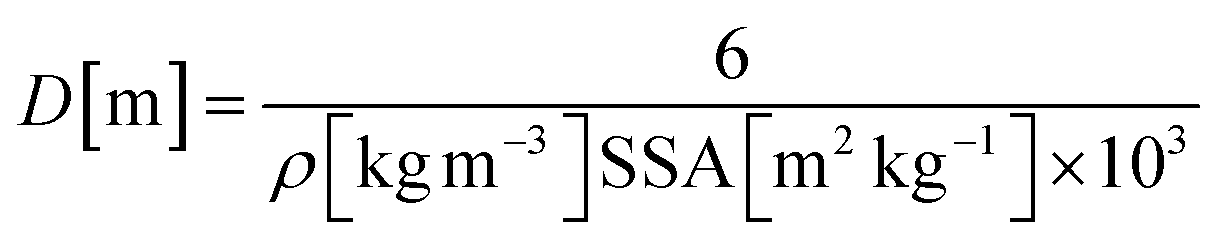 | (2) |
Whereas the MgO-FD value of 14.2 nm deviated for the value obtained by the Scherrer equation, the value for the MgO-CD crystallites was in relatively good agreement with the cubic crystallite size determined from the X-ray diffraction pattern and micrographs (Fig. 4b). The cause for the deviation in the FD sample may stem from the different porosities of the samples (8%, see above), which suggested that the sizes obtained by X-ray diffraction were more reliable. It has also been reported that some pores may not show up in the BET measurements due to their extremely small character as possibly closed.8 For comparison, the platelet sizes of the Mg(OH)2 prior to calcination are listed, which also deviated from their true size (determined by TEM) due to their platelet morphology. These values refer to the entire hexagonal platelets since no crystallites were present in the Mg(OH)2 platelets before calcination.
3.5. Evaluation of powder aggregation on silicon wafer
The ability to break up the calcined dried platelets into individual and separated nanoparticles was evaluated by placing 10 μL ultrasonicated MgO/2-propanol suspensions on the surface of a Si-wafer (Fig. 5). High temperature calcination often leads to sintering of nanoparticles into unbreakable hard aggregates,46 and to verify whether solitary MgO nanoparticles could be released from the aggregates of the different particle powders a sonication study was performed. The 2-propanol is polar and commonly used as a large surface area nanoparticle carrier in catalyst reactions with MgO47 and was therefore used as solvent. Fig. 5a and b show that a considerable amount of large aggregates were present in the freeze-dried sample after intense ultrasonic dispersion in the 2-propanol suspension. The numbers (from 20 micrographs of each sample) of the aggregates after 120 min sonication are shown in Fig. 5c, where an aggregate was defined as a cluster of MgO nanoparticles with a size larger than 1 μm. For comparison, the conventionally dried (CD) and ground sample showed better dispersion (Fig. 5d and e), which was surprising since sonication of nanoparticle suspensions usually lead to fragmentation as a mixture of fracture and erosion.48 This was expected to occur to a larger extent for the fragile MgO-FD foam compared to MgO-CD powder. The donut shaped aggregate structures (Fig. 5b) were not present in any of the original foam structures of the freeze-dried (FD) particles. It is therefore suggested that sonication of the FD sample opened for additional large interactive surface area to be exposed (when the foams broke apart) to the 2-propanol, which as a non-perfect solvent allowed for re-aggregation into donut shapes. Accordingly, the flaky structure of the FD material showed higher ability to reform into new aggregate structures under the ultrasound, i.e. than the conventionally dried (CD) ground powders. Note: the freeze-dried nanoparticle foam could not be ground due to its soft texture that made it stick to the pistil. In conclusion, it was evident that the large interactive surface of the nanoparticles in their foam conformation allowed for disintegration under the combination of 2-propanol with ultrasonication (commonly used dispersion tool), with the drawback that the nanoparticles also re-aggregated very easily. Statistics on the frequencies of aggregates with sonication times is available in the ESI, S3.† Ultrasound was therefore not considered further in any of the pre-melt processing steps before extrusion.3.6. The LDPE/MgO composite
Fig. 6 shows the non-ground flakes of MgO-FD platelets (a and b) and the ground MgO-CD platelets (c and d) dispersed by 150 °C extrusion as 3 wt% in the LDPE. While some nanoparticles and smaller clusters were evenly distributed, most of the MgO for both the freeze-dried (FD) and conventionally dried (CD) materials was still tied up in 1–3 μm aggregates after 6 min of internal re-circulation in the extruder. No obvious difference in dispersion quality could be observed as related to the differently dried nanoparticles. The temperature was then lowered to 115 °C. After 6 min, the liquid nitrogen cracked surfaces of the extruded bar revealed aggregates up to 30 μm for the freeze-dried MgO, whereas the conventionally dried MgO sample showed 100 μm aggregates (ESI, S4†). It could therefore be concluded that the freeze-drying into nanoparticle foams was not only beneficial from powder handling perspective (avoiding extensive grinding procedures) but also facilitated the dispersion of the platelets in the LDPE at 115 °C. It was, however, clear from a processing point of view that the LDPE incompatibility with the hydrophilic platelet surfaces caused major aggregation, regardless of the drying method. This incompatibility likely resulted in platelet aggregate slippage in the LDPE matrix, which may explain why the lower extrusion temperature and more viscous nature of the LDPE was less efficient in releasing solitary particles than the 150 °C extrusion.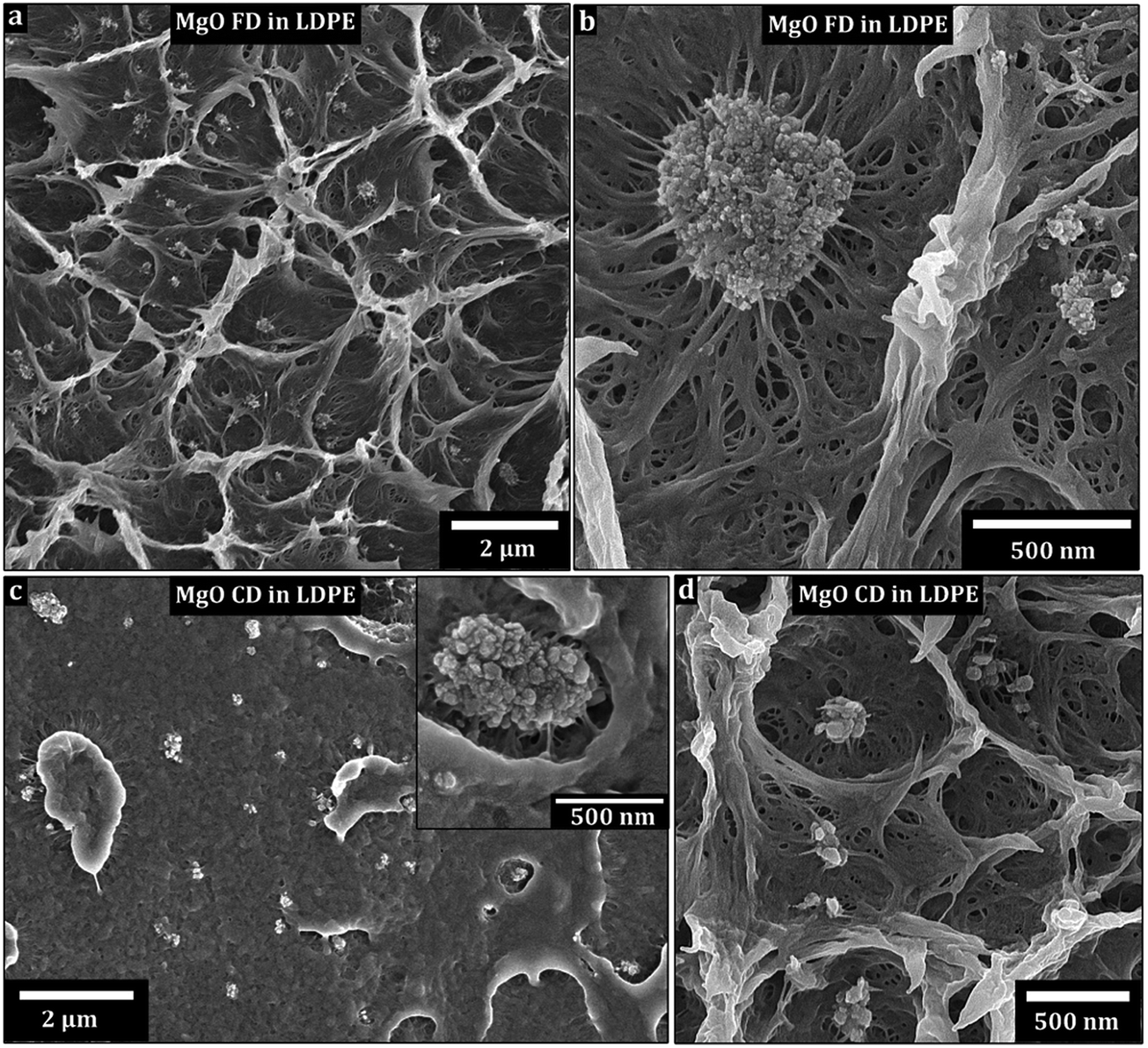 | ||
| Fig. 6 Scanning electron micrographs of freeze-fractured LDPE composites with 3 wt% of MgO for (a and b) freeze-dried and (c and d) conventionally dried samples. The fibrous surface (a, b and d) is due to plastic deformation during cracking. | ||
Fig. 6b and c inset shows the apparent low wettability of the platelet aggregates to the LDPE (regardless of initial nanopowder structure), with a significant amount of void formation surrounding the aggregates. A complementary explanation to the better dispersion achieved in the less viscous LDPE at 150 °C (even if the shear force is lower) could be a more ready penetration of the porous aggregate at the lower LDPE viscosity in the melt, which in turn facilitated more efficient break up of the aggregated structure into smaller clusters and individual particles. In Fig. 6b and c the LDPE shows the aggregates forming arms to the surrounding. It is suggested that the LDPE is soaked into the aggregates, which further enhanced the rupture of aggregates into solitary particles. No donut formed aggregates are seen in the composites.
3.7. Results of electrical conductivity measurements
The electrical insulation capacity of the LDPE composite based on the freeze-dried MgO (MgO-FD/LDPE) was compared to unfilled LDPE. The obtained data of the measured charging currents are displayed in Fig. 7. The charging current was always lower for the nanocomposite during the entire measurement, as compared with the unfilled LDPE.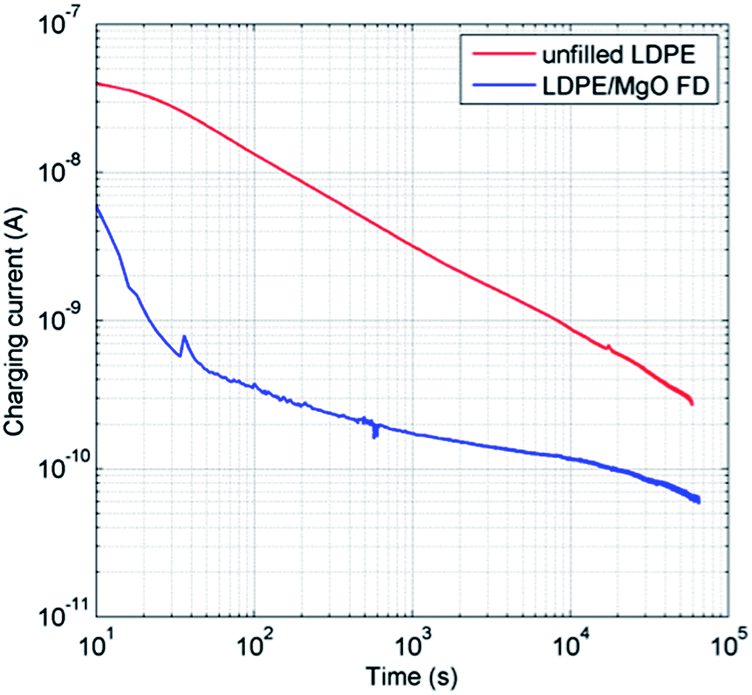 | ||
| Fig. 7 Charging currents of LDPE films with (blue) and without (red) freeze-dried MgO nanoparticles obtained at 2.6 kV (E = 32.5 kV mm−1) at 60 °C. | ||
The values of charging currents recorded at 6 × 104 s (16.7 h) were utilized for deriving the material bulk conductivity (Table 2). At this time, a reduction with a factor of 4.2 in the volume conductivity (σ) was determined as related to incorporated freeze-dried MgO nanoparticles. The charging currents were still declining for the samples after the extended measurement duration of 6 × 104 s (16.7 h), at which the measurements were terminated for practical reasons. The test duration was motivated by the fact that steady state conduction current of unfilled LDPE under a DC electrical stress of 30 kV mm−1 may not be achieved even after 17 days.49 In addition, results obtained in the present work for the unfilled LDPE are in line with those published earlier.5 Some experiments were also run for much longer time (2 × 105 s) and a continuously decaying character of the charging current was still observed with an electrical conductivity ratio in the expected range of 3.3–3.6 (σLDPE/σcomposite).
| Sample | Charging current i (at 6 × 104 s), A | Conductivity σ, S m−1 |
|---|---|---|
| LDPE/MgO FD | 6.4 × 10−11 | 2.79 × 10−15 |
| Unfilled LDPE | 2.7 × 10−10 | 1.18 × 10−14 |
The mechanism of conductivity reduction and the rapid drop in charging current of the nanocomposite at the beginning of the measurement has been suggested to originate from a nanoparticle interphase region with lower mobility of charge carriers.50 This lower mobility has been reported to be causing the suppressed charge accumulation, with improved internal electrical field distribution due to scattering of charge carriers.51,52 The decrease in conduction current has also been attributed to the formation of deeper traps for charge carriers in the particle/matrix interface.53 The influence of surface area, morphology and porosity of the nanoparticles and their ability to affect the charge accumulation from the polymer matrix is currently under investigation.
4. Conclusions
Highly insulating LDPE/MgO nanocomposite materials for HVDC cables are presented in detail, showing 1.5 orders of magnitude lower charging current at 102 s, and 4.2 times lower charging current after 16 hours compared to virgin low density polyethylene (LDPE). A key to avoiding MgO nanoplatelets contamination induced by the grinding procedure was to apply freeze-drying as a powder handling technique for the precipitated Mg(OH)2 nanoplatelets, prior to high-temperature calcination of the platelets into the highly crystalline and pure MgO nanoparticles. The application of freeze-drying generated a previously never reported highly porous nanofoam consisting of wide and 40–1000 nm thin flakes of Mg(OH)2-nanoparticles arranged in an epitaxial manner. The volume of the freeze-dried foam was 25 times greater than the volume of conventionally dried Mg(OH)2. The foam could more easily be disrupted into solitary particles, facilitating the extrusion of the nanocomposites, leading to fewer and smaller particle aggregates. Fracture analysis was performed to study the interfaces within the cryo-fractured nanocomposites, revealing 70% smaller aggregates for the particles dispersed as nanoplatelet foams of MgO. An extensive characterization of the synthesised Mg(OH)2 nanoplatelets and the formed MgO nanofoam is presented, with additional information on the limitation of the sensitive foam structure in its capacity to disintegrate into solitary particles under ultrasound dispersion.Acknowledgements
The Swedish Foundation for Strategic Research (EM11-0022) is acknowledged for providing financial support for this study.References
- O. Saksvik, in 9th IET International Conference on Advances in Power System Control, Operation and Management, Hong Kong, China, 2012 Search PubMed.
- Frost & Sullivan, European Smart Grid Market – Advanced Components M 521-14, 2009.
- T. Hjertberg, V. Englund, P.-O. Hagstrand, W. Loyens, U. Nilsson and A. Smedberg, in Jicable-HVDC'13, Perpignan, France, 2013 Search PubMed.
- Y. Murakami, S. Okuzumi, M. Nagao, M. Fukuma, Y. Sekiguchi, M. Goshowaki and Y. Murata, in Proceedings of 2008 International Symposium on Electrical Insulating Materials, 2008, pp. 159–162 Search PubMed.
- Y. Murakami, M. Nemoto, S. Okuzumi, S. Masuda, M. Nagao, N. Hozumi, Y. Sekiguchi and Y. Murata, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 33–39 CrossRef CAS PubMed.
- W. Zhang, Y. Dai, H. Zhao and L. Zhong, J. Nanomater., 2014, 146260 Search PubMed , in press.
- W. R. Eubank, J. Am. Ceram. Soc., 1951, 34, 225–229 CrossRef CAS PubMed.
- M. G. Kim, U. Dahmen and A. W. Searcy, J. Am. Ceram. Soc., 1987, 70, 146–154 CrossRef CAS PubMed.
- T. Baird, P. S. Braterman, H. D. Cochrane and G. Spoors, J. Cryst. Growth, 1988, 91, 610–616 CrossRef CAS.
- M. Laska, J. Valtyni and P. Fellner, Cryst. Res. Technol., 1993, 28, 931 CrossRef CAS.
- Z. Q. Wei, Q. Hua, P. H. Ma and J. Q. Bao, Inorg. Chem. Commun., 2002, 5, 147–149 CrossRef CAS.
- C. Y. Tai, C. T. Tai, M. H. Chang and H. S. Liu, Ind. Eng. Chem. Res., 2007, 46, 5536–5541 CrossRef CAS.
- R. Giorgi, C. Bozzi, L. Dei, C. Gabbiani, B. W. Ninham and P. Baglioni, Langmuir, 2005, 21, 8495–8501 CrossRef CAS PubMed.
- C. Henrist, J. P. Mathieu, C. Vogels, A. Rulmont and R. Cloots, J. Cryst. Growth, 2003, 249, 321–330 CrossRef CAS.
- W. Jiang, X. Hua, Q. Han, X. Yang, L. Lu and X. Wang, Powder Technol., 2009, 191, 227–230 CrossRef CAS PubMed.
- J. W. Mullin, J. D. Murphy, O. Söhnel and G. Spoors, Ind. Eng. Chem. Res., 1989, 28, 1725–1730 CrossRef CAS.
- D. Chen, L. Zhu, H. Zhang, K. Xu and M. Chen, Mater. Chem. Phys., 2008, 109, 224–229 CrossRef CAS PubMed.
- M. J. Ready, R. R. Lee, J. W. Halloran and A. H. Heur, J. Am. Ceram. Soc., 1990, 73, 1499–1503 CrossRef PubMed.
- L. S. Jones and C. J. Norman, J. Am. Ceram. Soc., 1988, 71, C190–C191 CrossRef PubMed.
- J. I. Hong, L. S. Schadler, R. W. Siegel and E. Mårtensson, J. Mater. Sci., 2006, 41, 5810–5814 CrossRef CAS PubMed.
- A. Alves, C. P. Bergmann and F. A. Berutti, Novel Synthesis and Characterization of Nanostructured Materials, Springer-Verlag, Berlin Heidelberg, 2013, ch. 7, p. 81 Search PubMed.
- C. L. De Castro and B. S. Mitchell, Synthesis, Functionalization and Surface Treatment of Nanoparticles, American Scientific Publishers, 2002, ch. 1, pp. 1–15 Search PubMed.
- T. P. Yadav, R. M. Yadav and D. P. Singh, Nanosci. Nanotechnol., 2012, 2, 22–48 CrossRef CAS PubMed.
- D. Fabiani and G. C. Montanari, IEEE Trans. Dielectr. Electr. Insul., 2010, 17, 221–230 CrossRef CAS.
- A. M. Pourrahimi, D. Liu, L. K. H. Pallon, R. L. Andersson, A. Martinez Abad, J.-M. Lagaron, M. S. Hedenqvist, V. Ström, U. W. Gedde and R. T. Olsson, RSC Adv., 2014, 4, 35568–35577 RSC.
- D. Scarano, S. Bertarione, F. Cesano, G. Spoto and A. Zecchina, Surf. Sci., 2004, 570, 150–166 CrossRef PubMed.
- H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures: for Polycrystalline and Amorphous Materials, John Wiley & Sons, New York, 2nd edn, 1974, ch. 9 Search PubMed.
- C. Rodriguez-Navarro, E. Ruiz-Agudo, M. Ortega-Huertas and E. Hansen, Langmuir, 2005, 21, 10948–10957 CrossRef CAS PubMed.
- J. C. Yu, A. Xu, L. Zhang, R. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64–70 CrossRef CAS.
- M. Nagao, N. Takamura, M. Kurimoto, Y. Murakami, Y. Inoue and Y. Murata, in Annual Report Conference on Electrical Insulation and Dielectric Phenomena (CEIDP), 2012, pp. 311–314 Search PubMed.
- IEC, in Methods of Test for Volume Resistivity and Surface Resistivity of Solid Electrical Insulating Materials, Standard 60093, 1980 Search PubMed.
- C. C. Reddy and T. S. Ramu, IEEE Trans. Dielectr. Electr. Insul., 2006, 13, 1236–1244 CrossRef.
- D. Balzar, J. Res. Natl. Inst. Stand. Technol., 1993, 98, 321–353 CrossRef CAS.
- R. Mueller, H. K. Kammler, K. Wegner and S. E. Pratsinis, Langmuir, 2003, 19, 160–165 CrossRef CAS.
- P. R. Hornsby and R. N. Rothon, Fire Retardancy of Polymers: New Applications of Mineral Filler, Royal Society of Chemistry, London, 2006, ch. 2 Search PubMed.
- P. T. Huyen, E. Callone, R. Campostrini, G. Carturan, T. T. Hong, H. N. Nhat and H. D. Chini, Eur. Phys. J.: Appl. Phys., 2013, 64, 10405 CrossRef.
- R. N. Rothon, Particulate Filled Polymer Composites, Longman, New York, 1995, ch. 6 Search PubMed.
- P. R. Hornsby and C. L. Watson, in IOP Short Meetings Series No 4, Institute of Physics, London, April 1997, p. 17 Search PubMed.
- P. R. Hornsby and A. Mthupha, Proceedings from Society of plastics Engineering, Annual Technical Conference, ANTEC '93, New Orleans, May 9–13 1993, pp. 1954–1956 Search PubMed.
- J. Green, J. Mater. Sci., 1983, 18, 637–651 CrossRef CAS.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, UK, 1982 Search PubMed.
- R. F. Horlock, P. L. Morgan and P. J. Anderson, Trans. Faraday Soc., 1963, 59, 721–728 RSC.
- P. J. Anderson and R. F. Horlock, Trans. Faraday Soc., 1964, 60, 930–937 RSC.
- Z. Librant and R. Pampuch, J. Am. Ceram. Soc., 1968, 51, 109–110 CrossRef CAS PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids, Academic Press, London, 1999, ch. 1 Search PubMed.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175–181 CrossRef CAS.
- M. A. Aramendía, V. Borau, C. Jiménez, J. M. Marinas, J. R. Ruiz and F. J. Urbano, Appl. Catal., A, 2003, 244, 207–215 CrossRef.
- K. A. Kusters, S. E. Pratsinis, S. G. Thoma and D. M. Smith, Powder Technol., 1994, 80, 253–263 CrossRef.
- J. Viertel, L. Petersson, A. Friberg, G. Dominguez, and C. Törnkvist, in IEEE International Conference on Solid Dielectrics (ICSD), 2013, pp. 1048–1051 Search PubMed.
- M. Roy, J. K. Nelson, R. K. MacCrone and L. S. Schadler, J. Mater. Sci., 2007, 42, 3789–3799 CrossRef CAS PubMed.
- T. J. Lewis, IEEE Trans. Dielectr. Electr. Insul., 2004, 11, 739 CrossRef CAS.
- F. Fujita, M. Ruike and M. Baba, in Conference record of the IEEE Intl Symp Elec Ins, San Fransico, 1996, vol. 2, p. 738 Search PubMed.
- Y. Cao and P. C. Irwin, in IEEE Conference on Electrical Insulation and Dielectric Phenomena (CEIDP), 2003, pp. 116–119 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta06362g |
| This journal is © The Royal Society of Chemistry 2015 |