
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 capture by dry alkanolamines and an efficient microwave regeneration process†
J.
Yang
a,
H. Y.
Tan
a,
Q. X.
Low
a,
B. P.
Binks
*b and
J. M.
Chin
*ab
aInstitute of Materials Research and Engineering, Agency for Science Technology and Research, 3 Research Link, Singapore 117602, Singapore. E-mail: j.chin@hull.ac.uk
bSurfactant & Colloid Group, Department of Chemistry, University of Hull, HU6 7RX, UK. E-mail: b.p.binks@hull.ac.uk
First published on 17th February 2015
Abstract
Removal of acidic gases such as H2S and CO2 is performed during the purification of raw natural gas, most commonly using amine gas treatment. However, this industrially entrenched method is limited by significant shortcomings including low operational capture efficiency, amine pipeline corrosion and a large energy penalty due to the sorbent regeneration process. To address these shortcomings, we have studied the use of perfluorinated silica-stabilized dry alkanolamines (DAf) for CO2 capture. Due to their micronized liquid domains, DAf display high operational CO2 capture efficiency. Further, to minimize energy requirements for sorbent regeneration, microwave-assisted regeneration of the spent DAf sorbent was also studied and shown to decrease the energy requirements by about ten times. In contrast to very recent work, our results show that the use of DAf exhibits extraordinary recyclability, with a negligible decrease in absorption capacity over at least ten absorption–regeneration cycles, indicating the potential of this material for gas treatment applications.
Introduction
Carbon dioxide capture is crucial not only for mitigating greenhouse gas emissions but also for natural gas processing.1–4 The recent United States shale gas boom has precipitated an increase in the production of natural gas, making the need for effective CO2 capture technologies more pressing than ever.5 Examples of solid sorbents for CO2 capture include metal–organic frameworks,6–8 porous polymers9–12 and zeolites.13 Such sorbents have the advantage of a high surface area to accelerate CO2 uptake.14 Nevertheless, liquid sorbents, which often require agitation to achieve high gas–liquid interfacial areas, are still predominant in industrial processes. In fact, amongst various CO2 capture technologies, the most established is amine gas treatment, which relies on the use of alkanolamines to separate acidic gases such as H2S and CO2 from raw natural gas.15Despite amine-based gas sweetening being an already well-established industrial process, it faces multiple shortcomings including low operational CO2 capture efficiency, amine corrosivity and a significant energy penalty due to the sorbent regeneration process.1,16 Although primary and secondary alkanolamines react with CO2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio to form their corresponding carbamates with a theoretical CO2 absorption capacity of 50 mol% (Scheme 1),17–19 industrial systems utilizing alkanolamines typically only operate at 15 to 30 mol% capture efficiency.20 In addition, the high viscosity and corrosivity of alkanolamines necessitate their dilution with water to approximately 30 wt% of amines.6 This leads to an associated energy penalty during sorbent regeneration, whereby CO2-loaded alkanolamine sorbents are heated to regenerate the amines, since the additional water content increases the overall heat capacity of the sorbent. To address this problem, researchers have turned to utilizing ionic liquid–alkanolamine blends21 or to amine-functionalized porous solids for CO2 capture.22–24
:1 mole ratio to form their corresponding carbamates with a theoretical CO2 absorption capacity of 50 mol% (Scheme 1),17–19 industrial systems utilizing alkanolamines typically only operate at 15 to 30 mol% capture efficiency.20 In addition, the high viscosity and corrosivity of alkanolamines necessitate their dilution with water to approximately 30 wt% of amines.6 This leads to an associated energy penalty during sorbent regeneration, whereby CO2-loaded alkanolamine sorbents are heated to regenerate the amines, since the additional water content increases the overall heat capacity of the sorbent. To address this problem, researchers have turned to utilizing ionic liquid–alkanolamine blends21 or to amine-functionalized porous solids for CO2 capture.22–24
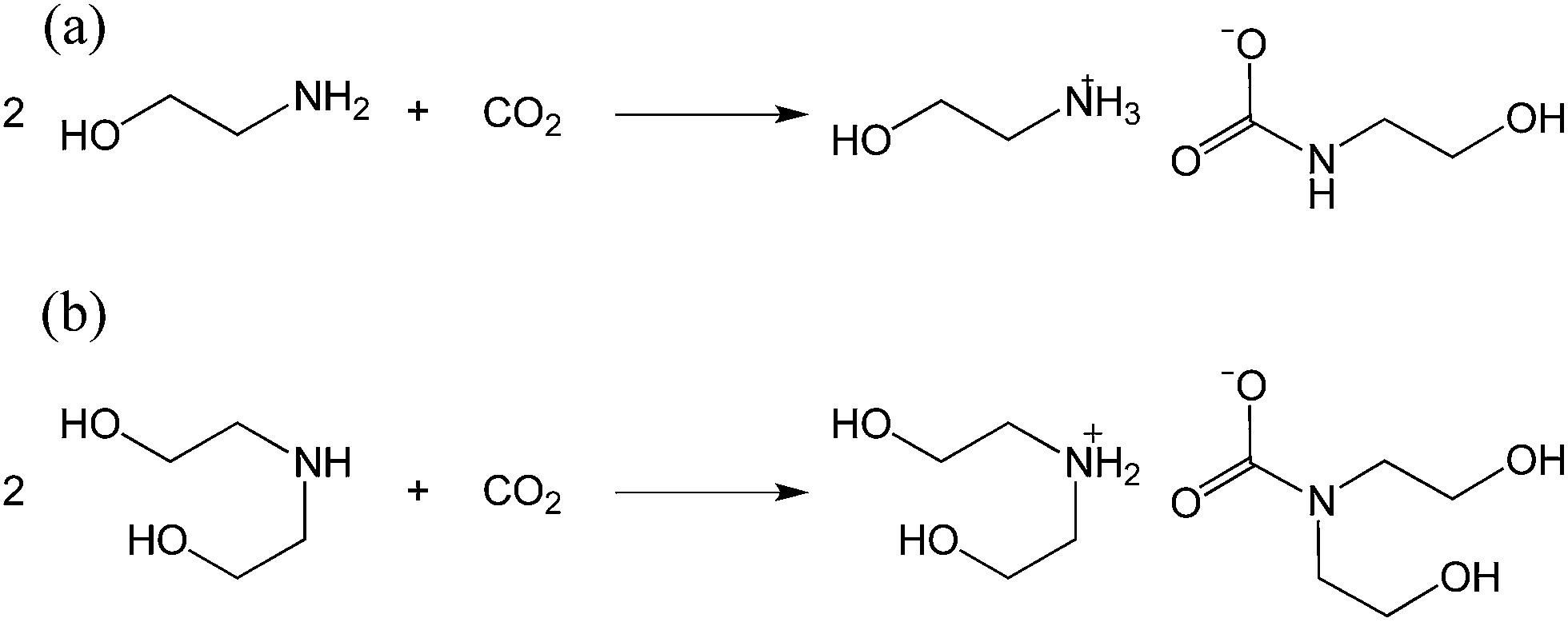 | ||
| Scheme 1 Reaction of (a) monoethanolamine (MEA) and (b) diethanolamine (DEA) with CO2 to form the corresponding carbamate salts. | ||
Dawson et al. recently reported an alternative approach whereby they used hydrophobic silica-stabilized dry alkanolamines (DAh) for CO2 capture.25 However, the DAh exhibited low recyclability and substantial sorbent loss during regeneration, as well as significantly decreased CO2 uptake after the first cycle. Nevertheless, this approach circumvents the need for dilution of the sorbent with water, thereby decreasing the energy penalty incurred during sorbent regeneration. Amine-related corrosion problems are also potentially reduced because the amine is isolated by the silica particles from pipelines.
We herein report the use of dry, undiluted alkanolamines for CO2 capture whereby instead of hydrocarbon-functionalized, hydrophobic fumed silica, perfluoroalkyl-functionalized oleophobic fumed silica was utilized for dry alkanolamine, DA, formation. These findings were previously outlined in our patent filed in May 2013,26 prior to the work being reported by Dawson et al. We refer to this DA as DAf. In contrast to the findings by Dawson et al.,25 we found that DAf showed excellent recyclability and stability. Studies were conducted on monoethanolamine (MEA) and diethanolamine (DEA) due to their widespread industrial usage. Furthermore, a novel method using microwave-assisted heating for DAf regeneration was also investigated as an energy-efficient alternative to the conventional (convective and conductive heating) regeneration method.
DAs are powdered materials and can be thought of as inverted foams, i.e. liquid droplets dispersed in air, whereby microscopic liquid droplets stabilized by nanoparticles form the dispersed phase and air is the continuous matrix (Fig. 1). They appear as a free-flowing powder and are analogous in form to dry water (DW) which has received significant attention in recent years.27–31
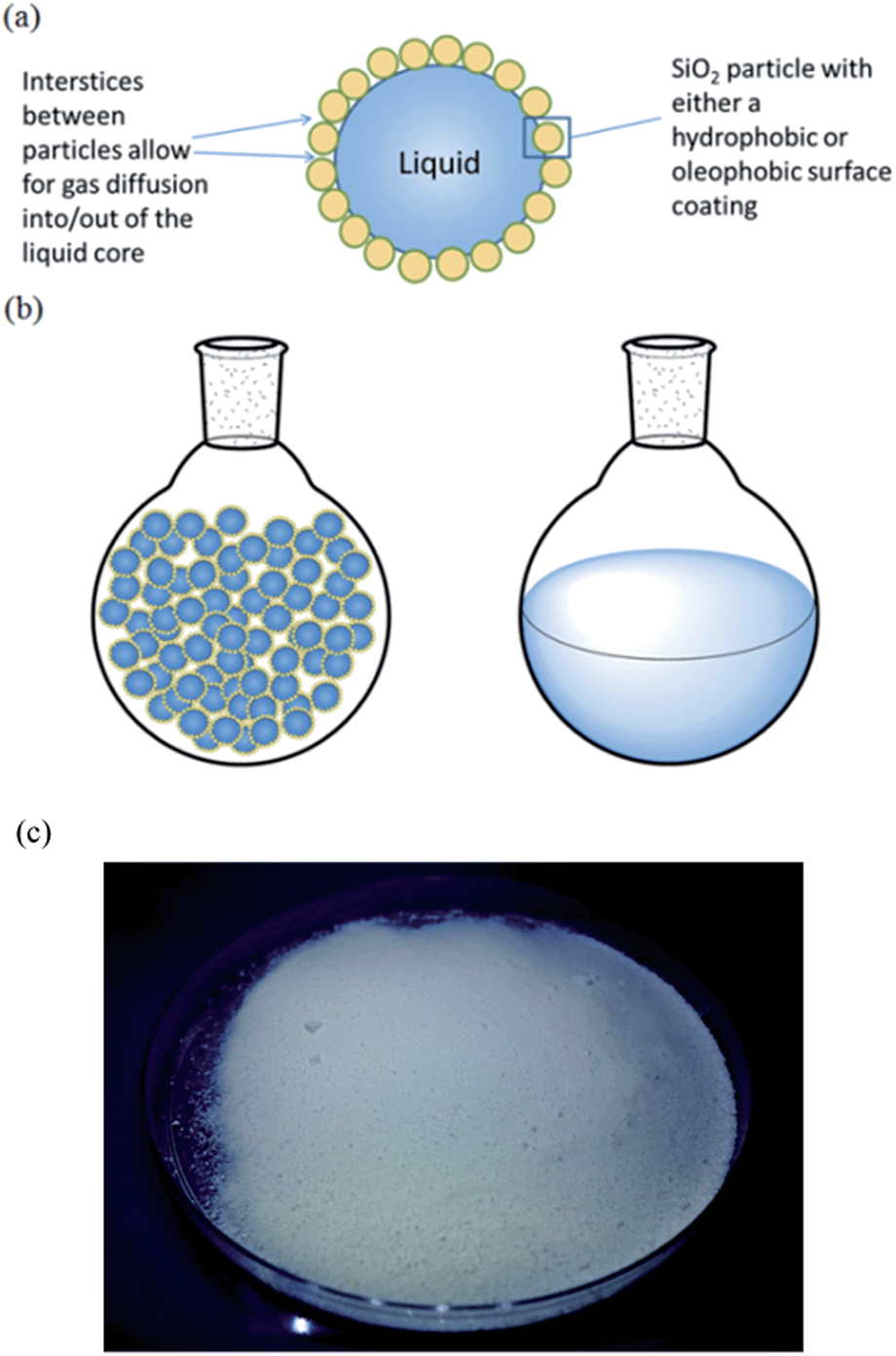 | ||
| Fig. 1 (a) Sketch of a liquid droplet in air encapsulated by particles of low surface energy. Since the particles form a porous shell on the surface of the liquid, there is rapid diffusion of gases into and out of the liquid through the particle interstices. (b) Sketch of a dry liquid (left) and bulk liquid (right) in round-bottomed flasks. (c) Photograph of the DAf powder containing DEA. | ||
For dry liquid formation to occur, the stabilizing particles must be hydrophobic towards the encapsulated liquid, otherwise a foam or paste may ensue.27 Generally, for a flat surface to be hydrophobic towards a liquid (contact angle > 90°), the solid surface tension γS must be γS ≪ γL/4, where γL is the surface tension of the liquid.32 It has recently been demonstrated that the formation of dry oils with liquids of decreasing surface tension requires the use of nanoparticles with increasing degrees of fluorination on their surfaces.33 Based on the surface tensions of MEA and DEA (Table S1†), it can be deduced that oleophobic perfluoroalkyl-functionalized nanoparticles are preferable over oleophilic hydrocarbon-functionalized nanoparticles for stabilization of DAs (Fig. S1†).
Experimental
Materials
All materials were purchased from commercial sources and used as received. 1H,1H,2H,2H-Perfluorooctyltriethoxysilane (purity 97%) was purchased from Fluorochem. Ethanolamine (purity 98%), diethanolamine (purity 98%) and hydrophilic fumed silica powder with an average particle size (aggregate) of 0.2–0.3 μm (product number S5505) were purchased from Sigma Aldrich. Ethanol (purity 99%) was purchased from International Scientific. Aqueous ammonia (28%) was purchased from Malayan Acid Works. Compressed CO2 (purity 99.8%) was purchased from Singapore Oxygen Air Liquide.Methods
:1 alkanolamine to CO2.
Results and discussion
Brunauer–Emmett–Teller (BET) analysis performed on perfluorinated SiO2 particles showed that CO2 adsorption on these particles at room temperature was negligible (Fig. 2), because 1 g of perfluorinated SiO2 nanoparticles absorbed approximately 6 mg of CO2 at atmospheric pressure while 1 g of DAf (containing 0.48 g of perfluorinated silica) absorbed approximately 100 mg of CO2. Of this 100 mg, about 3% is attributed to adsorption by silica. Hence, the mass increase is largely due to CO2 uptake by the alkanolamine.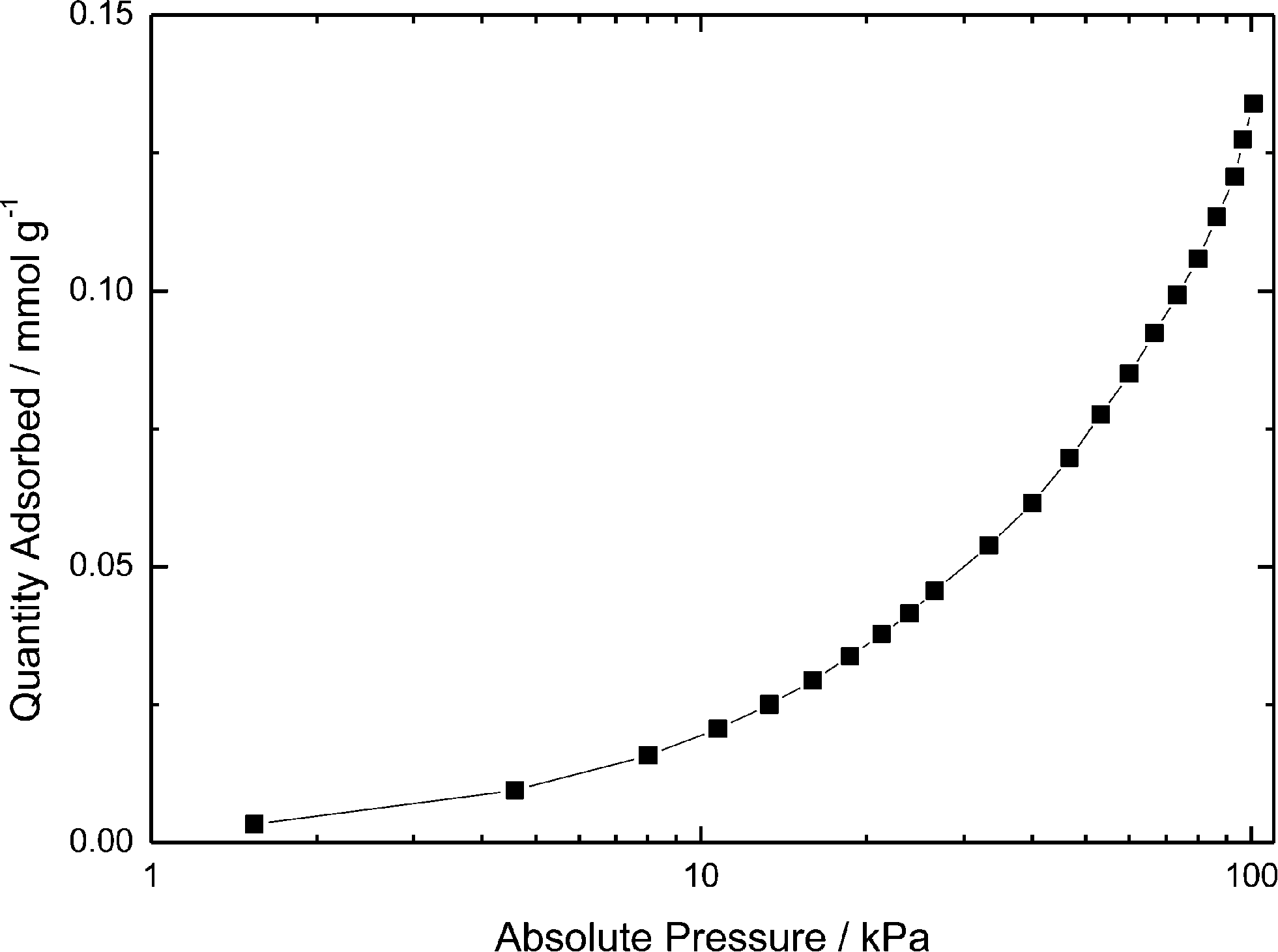 | ||
| Fig. 2 Isothermal CO2 adsorption at 22 °C by perfluorinated SiO2 nanoparticles showing negligible CO2 uptake (0.135 mmol g−1 at 100 kPa). | ||
Carbon dioxide absorption results for both DAf and bulk liquid are shown in Fig. 3. The theoretical maximum alkanolamine CO2 uptake (in the absence of water) was calculated by assuming a reaction mole ratio of 2:1 alkanolamine to CO2. Hence, 1 g MEA theoretically absorbs about 0.36 g CO2 at the maximum, and 1 g DEA theoretically absorbs 0.21 g CO2. As shown in Fig. 3, CO2 absorption by dry and bulk MEA in the first 10 min of CO2 exposure reached 98 and 60 wt% of the theoretical maximum absorption, respectively. Similarly, dry and bulk DEA achieved about 90 and 35 wt% of the theoretical maximum within 20 min. Mass changes due to the replacement of air in the reaction vessel headspace with CO2 were taken into account in the calculations.
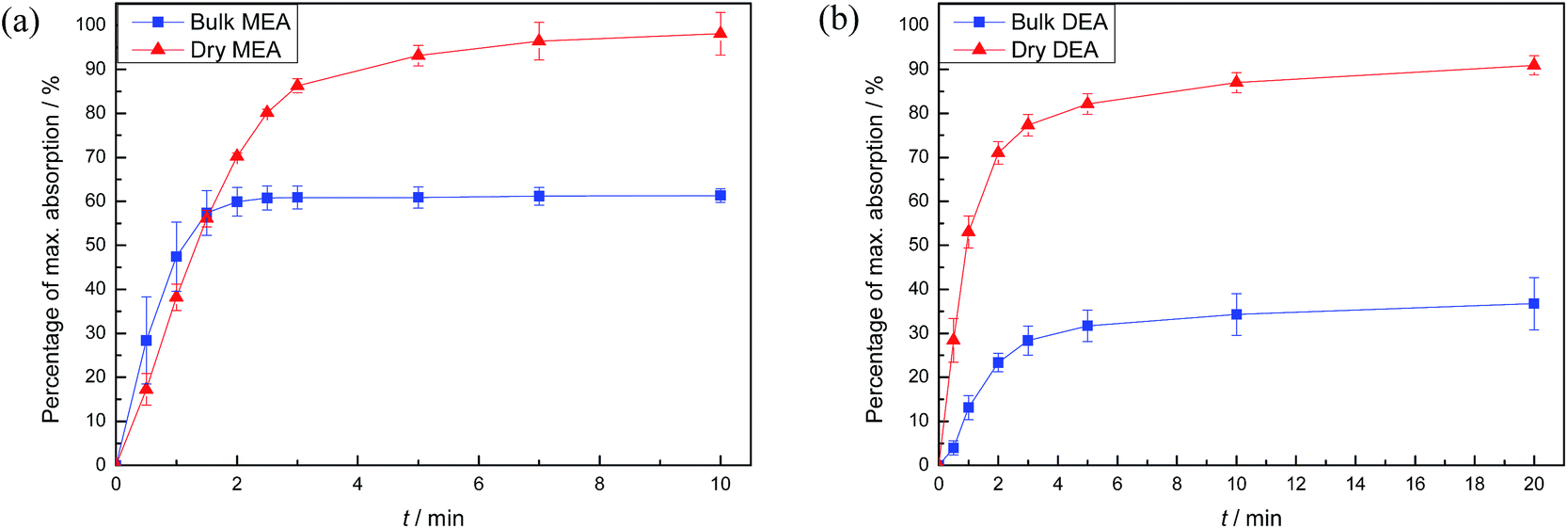 | ||
| Fig. 3 Results of the absorption profile of CO2 into bulk alkanolamine or DAf for (a) MEA and (b) DEA at room temperature and pressure. | ||
For both amines, the DAf showed significantly higher CO2 absorption than their corresponding bulk liquid counterparts (Fig. 3). The lower absolute absorption of the bulk amines is due to mass transport effects arising from the large increase in the liquid viscosity caused by carbamate salt formation, which was verified by measuring the viscosities of the alkanolamine liquids before and after CO2 absorption (Table S3†). The effect of viscosity of a liquid on the mass transfer kinetics is described by the Stokes–Einstein equation:34
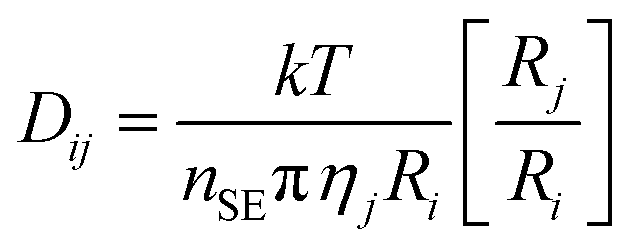 | (1) |
In this equation, Dij is the diffusion coefficient of solute i in pure solvent j, k is the Boltzmann constant, T is the temperature, nSE is the Stokes–Einstein number, ηj is the solvent viscosity, Ri is the radius of the solute molecule and Rj is the radius of the solvent molecule. According to eqn (1), the diffusion coefficient of CO2 gas in the amine sorbent decreases with increasing solvent viscosity. Further, given that the system studied is stationary and unagitated, the increased viscosity also significantly affects the replenishing of the unreacted amine near the gas–liquid interface, which is dependent upon the movement of alkanolamine molecules towards the interface via convection currents or diffusion.
However, due to the small domain sizes within DAf (approximate diameter of a microscopic liquid marble is 80 ± 30 μm, as measured by cryo-SEM), the required distance of CO2 diffusion within the droplets to contact the unreacted alkanolamine is significantly less than for the bulk liquid. This helps to explain why the DAf achieve a much larger gas absorption relative to the theoretical capacity within the timeframe of the experiment than do the bulk amines. In addition, the reason for the slightly higher absorption capacity of dry MEA compared to dry DEA is due to the inherently lower viscosity of the former compared to the latter.
Based on the work of Wang et al.,31 who postulated that the higher methane gas hydrate formation rate in dry versus bulk water was due to the higher surface area to volume ratio in dry water, and taking the view that the rate of gas uptake is dependent upon the gas–liquid interfacial area, we expect that the CO2 absorption rate by DAf would follow a similar trend (estimated surface area to volume ratio in DAf is 75000 m−1 based on the volume of alkanolamine present). Nevertheless, the gas–liquid interfacial area of the microscopic liquid droplets is reduced by the presence of the silica shell around each droplet; CO2 must diffuse across this porous shell to react with the encapsulated alkanolamines.
Our CO2 uptake studies showed that for MEA, the absorption rate by the bulk liquid in the first minute was faster than that by dry MEA (Fig. S4†). This is presumably because the lower liquid viscosity of MEA compared with DEA means that CO2 mass transfer effects do not outweigh the effect of the silica barrier initially. However, as carbamate formation proceeds, the viscosity of the liquid mixture increases (Table S3†), and mass transfer effects become more important. The rate of CO2 uptake therefore becomes faster for dry over bulk MEA. For DEA, dry DEA exhibits faster CO2 absorption than bulk DEA throughout the entire absorption cycle (Fig. S4†), because the mass transfer rate in the more viscous DEA liquid was much slower and hence the effect of higher surface area to volume ratio of dry DEA was more prominent.
Since the carbamate salts decompose in the temperature range of 100–150 °C,35,36 the CO2-loaded bulk alkanolamines were heated to 120 °C to release the absorbed CO2 gas and regenerate the alkanolamines. The decrease in mass was attributed to both CO2 and H2O release, which was verified by thermogravimetric mass spectroscopy (TG-MS) analysis (see Fig. 4).
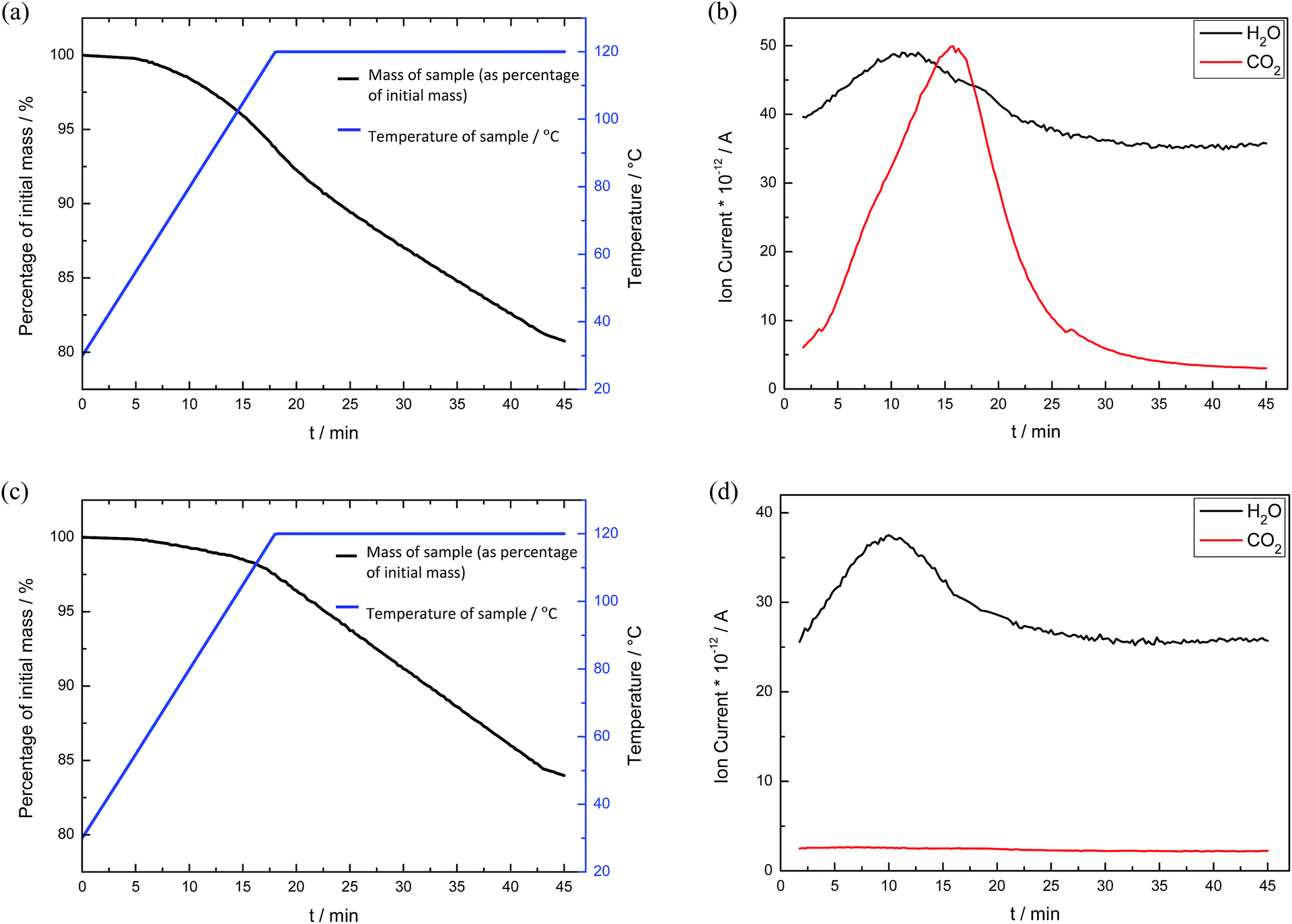 | ||
| Fig. 4 (a and b) TG-MS graphs of CO2-loaded bulk DEA, and (c and d) TG-MS graphs of bulk DEA without CO2 loading. (a) The black line shows a decrease of 18.92% of the initial sample mass for CO2-loaded bulk DEA with a concomitant increase in the sample temperature from 30 to 120 °C as depicted by the blue line, (b) graph corresponding to (a), depicting ion current detection of both CO2 and H2O. (c) The black line shows a decrease of 16.03% of the initial sample mass for bulk DEA, (d) graph corresponding to (c) depicting detection of only H2O but not CO2. | ||
For each sample of bulk alkanolamine and DAf, three cycles of absorption and desorption of CO2 were performed (Fig. 5). As shown in Fig. 5, DEA appears to be more recyclable than MEA, with a negligible change in CO2 uptake capacity over three cycles. This is because secondary amines like DEA form weaker bonds with CO2 than primary amines like MEA, allowing for easier regeneration of the free amines.37 This is supported by the findings of McCann et al., where the enthalpy of carbamate formation for MEA and DEA was found to be −29.7 kJ mol−1 and −23.7 kJ mol−1, respectively.38 Moreover, MEA has a lower boiling point than DEA (170 °C vs. 271 °C) and a much higher vapour pressure, leading to a greater degree of sorbent loss per cycle.
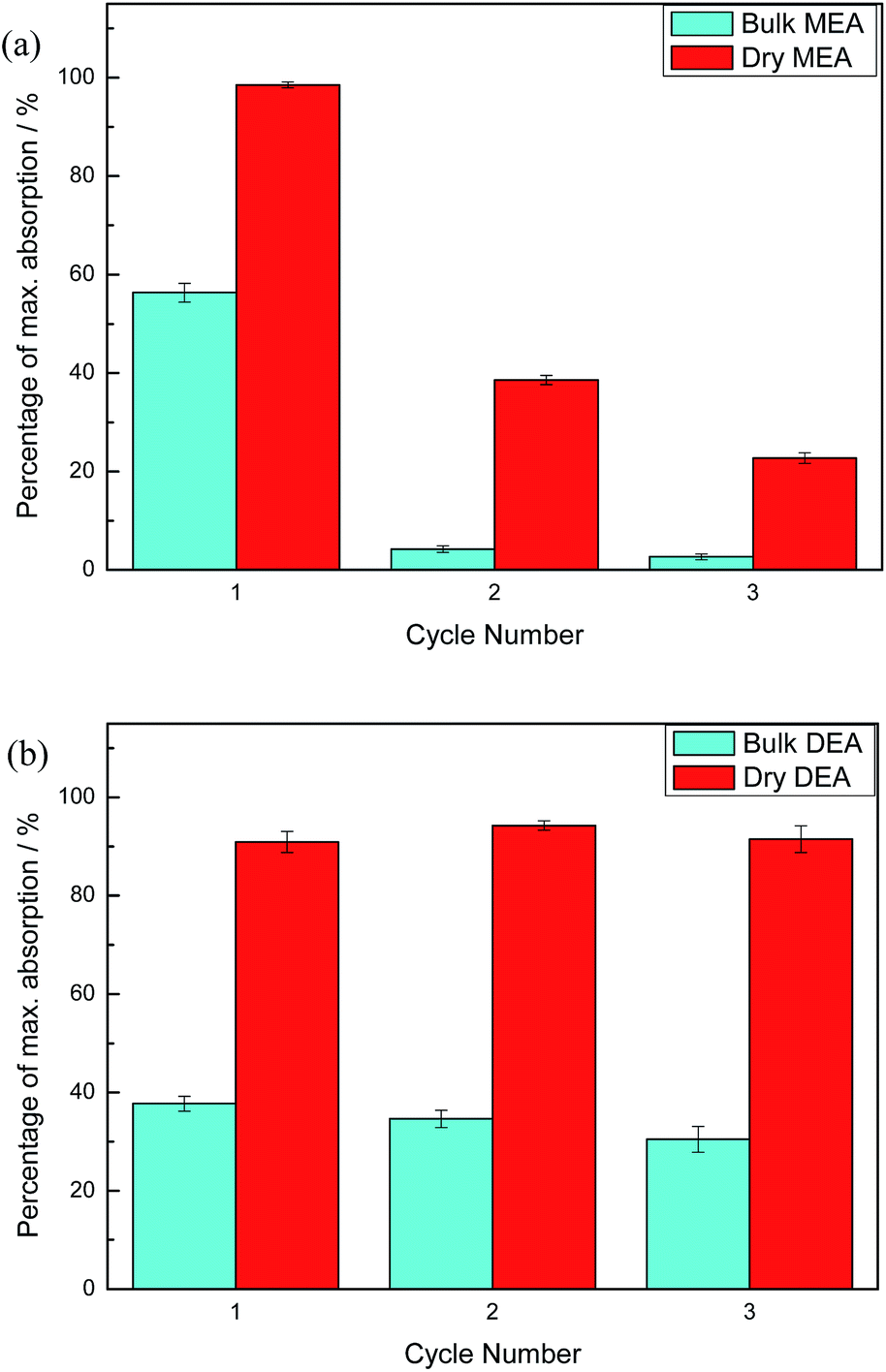 | ||
| Fig. 5 Comparison of solvent recyclability of (a) bulk MEA versus dry MEA, whereby heating lasts for 2 h for each cycle of regeneration and (b) bulk DEA versus dry DEA, whereby heating lasts for 1 h for each cycle of regeneration. | ||
As heating is essential for the sorbent regeneration process, it is imperative to maximize the heat transfer efficiency within the amine sorbents. This is dependent on the three main mechanisms of heat transfer, namely conduction, convection and radiation. In the case of DAf, these three mechanisms happen to be hindered by the silica shell encasing the micronized alkanolamine droplets and the air pockets throughout the dry liquids. For heat transfer via conduction, fumed silica particles exhibit low thermal conductivity due to the nanoscale pores among them in their fractal arrangement,39,40 causing the silica shell to block effective heat conduction from the heat source to the droplets. Importantly, many air pockets between the microscopic particle-coated liquid marbles in DAf also limit heat conductivity. With regard to convection, the liquid in DAf exists as dispersed microscopic droplets, thereby limiting convective heat transfer. As for radiative heat transfer, Taylan and Berberoglu have shown that the silica shell in dry water greatly attenuates infrared radiative heat transfer due to its high single-scattering albedo, i.e. a significantly larger proportion of heat radiation is scattered compared to that absorbed by the silica shell,41 thus preventing infrared radiative heat transfer from the heat source to the droplets.
Since the three major forms of heat transfer are significantly inhibited for DAf, we turned to microwave heating as an alternative method to improve the efficiency of heat transfer during the regeneration process. This method is dependent on the ability of the irradiated material, e.g. a solvent, to absorb microwave energy and convert it to heat, which is quantified by a dielectric parameter called the loss factor tanδ. This loss factor is expressed as:42
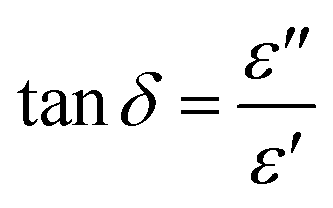 | (2) |
δ value of more than 0.5 is required for efficient microwave absorption. DEA, with a high tanδ value of ∼0.6 at 2.45 GHz at 278 K can be rapidly heated to temperatures >100 °C within a minute when irradiated under microwave conditions.43
Owing to the higher recyclability of DEA over MEA, we chose to study DEA for microwave heating-based regeneration. Samples of approximately 10 g of dry DEA containing 5 mL of liquid DEA were loaded with CO2 until the absorption plateaued, and the CO2-loaded samples were subsequently heated in a programmable microwave synthesizer at 120 °C for approximately 1 h until no further mass loss occurred. To test the recyclability of the sorbent and confirm that the observed mass loss was correctly attributed to the CO2 loss, 10 repeated cycles of CO2 absorption and removal were carried out, as shown in Fig. 6. The level of CO2 uptake by dry DEA was found to be stable over at least 10 cycles, showing an excellent recyclability of dry DEA. Similar regeneration results were obtained using a simple household microwave instrument, although the heating periods were limited to 10–20 s in order to avoid overheating.
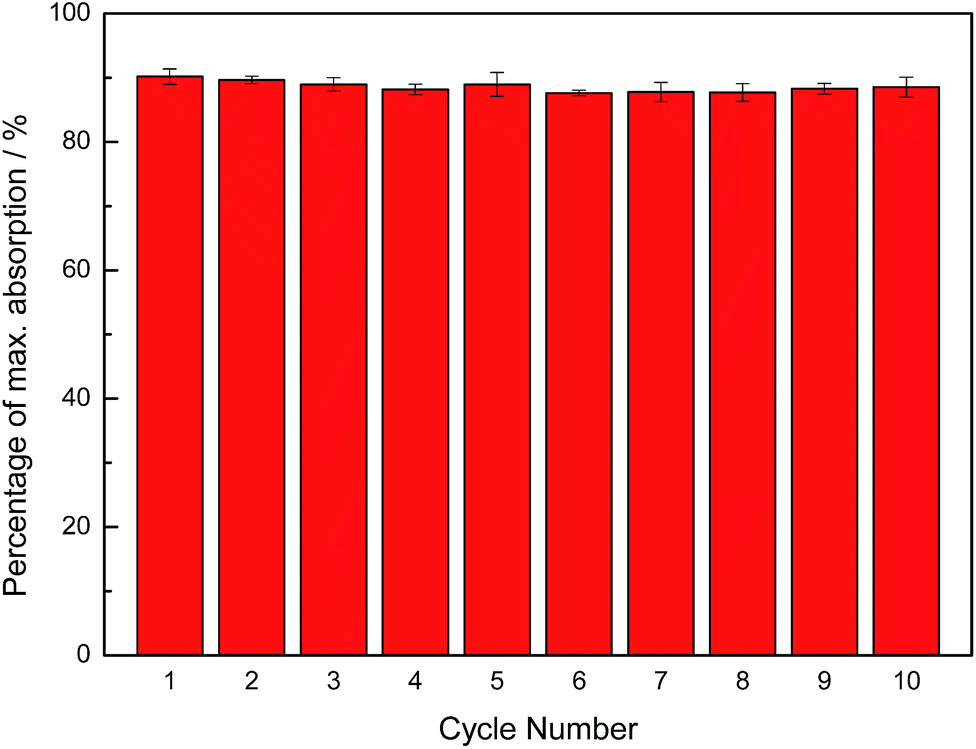 | ||
| Fig. 6 Sorbent recyclability of dry DEA using dielectric heating (1 h per cycle) for regeneration, showing a negligible drop in absorption capacity over 10 cycles. | ||
Regeneration of DAfvia microwave heating was found to be more efficient than conventional hotplate heating under the same conditions (whereby no purge gas was used). Conventional hotplate heating took more than 3 h and consumed about 0.38 kW h per cycle on average. Microwave heating on the other hand took about 1 h and consumed about 0.034 kW h per cycle on average, which is less than 10% of the energy consumption by conventional hotplate heating.
Finally, the recyclability of the perfluorinated silica particles was investigated. Alkanolamines typically undergo degradation over multiple absorption and regeneration cycles after which they must be replaced.44 Nevertheless, the perfluorinated silica particles could be separated from the amines by dispersion of DAf in ethanol and then centrifuged, and re-used in at least three subsequent batches of DAf. This leads to significant cost-savings since only the alkanolamines need to be replaced.
Conclusions
In conclusion, the dry alkanolamines described herein demonstrate significantly enhanced performance with regard to CO2 absorption capacity and sorbent recyclability. Furthermore, the novel dielectric heating technique for sorbent regeneration proposed here has proven to possess the benefits of reduced energy consumption and minimal use of purge gas. The perfluorinated silica particles can also be recycled and re-used to prepare DAf with a negligible change in their wettability and overall quality for at least three cycles. Moreover, as suggested by the recent work of Dawson et al.,25 the amine component of the DAf is also isolated by the silica particles from pipelines and other corrosion sensitive materials, leading to further reduction in maintenance costs associated with the addition of corrosion inhibitors and repair of damage to pipelines. Last but not least, this novel method of using DAf is not restricted to MEA and DEA; in principle any dry liquid can be formed provided that nanoparticles of appropriate surface energy are selected.33 The implications are that this idea can be extended to other liquid CO2 sorbents, possibly leading to a similar improvement in the absorption rate and capacity. However, there still exist several challenges for the application of DAf industrially. Firstly, the powdery nature of DAf will likely require significant changes in the current process technology and facilities specialized in handling liquid amine sorbents. Secondly, due to the nanoscale primary particle size of fumed silica, careful confinement of the powder needs to be put in place to prevent contamination in the outlet gas stream. Thirdly, stripper columns need to be modified to incorporate dielectric heating capabilities. Nevertheless, given the multiple advantages of utilizing DAf for CO2 absorption and desorption, we believe that they should still be considered promising candidates for practical applications in CO2 capture.Acknowledgements
JMC would like to acknowledge the Institute of Materials Research and Engineering, Agency for Science Technology and Research (A*STAR) for funding this work. The authors thank Mr Anthony Sinclair for the cryo-SEM analysis, Ms Sophie A. Darragh, Dr Michael R. Reithofer, Dr Tommy S. Horozov, Dr Robert A. Lewis and Dr R. McDonald for helpful discussions.Notes and references
- S. Anderson and R. Newell, Annu. Rev. Environ. Resour., 2004, 29, 109–142 CrossRef.
- C. Yu, C. Huang and C. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 CAS.
- H. D. Jacoby, F. M. O'Sullivan and S. Paltsev, MIT Joint Program on the Science and Policy of Global Change, 2011.
- Z. Zhang, Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 CAS.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- H. Hea, W. Lia, M. Zhong, D. Konkolewicza, D. Wub, K. Yaccato, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, Energy Environ. Sci., 2013, 6, 488–493 Search PubMed.
- H. He, M. Zhong, D. Konkolewicz, K. Yacatto, T. Rappold, G. Sugar, N. E. David, J. Gelb, N. Kotwal, A. Merkle and K. Matyjaszewski, Adv. Funct. Mater., 2013, 23, 4720–4728 CrossRef CAS.
- F. Sua and C. Lu, Energy Environ. Sci., 2012, 5, 9021–9027 Search PubMed.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- Y. E. Kim, J. A. Lim, S. K. Jeong, Y. Yoon, S. T. Bae and S. C. Nam, Bull. Korean Chem. Soc., 2013, 34, 783–787 CrossRef CAS.
- S. S. Laddha and P. V. Danckwerts, Chem. Eng. Sci., 1981, 36, 479–482 CrossRef CAS.
- P. V. Danckwerts, Chem. Eng. Sci., 1979, 34, 443–446 CrossRef CAS.
- E. Sada, H. Kumazawa, Z. Q. Han and H. Matsuyama, AIChE J., 1985, 31, 1297–1303 CrossRef CAS.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS.
- M. Hasib-ur-Rahman and F. Larachi, Environ. Sci. Technol., 2012, 46, 11443–11450 CrossRef CAS PubMed.
- C. Chen, J. Kim and W. Ahn, Korean J. Chem. Eng., 2014, 3, 1919–1934 CrossRef PubMed.
- M. Pera-Titus, Chem. Rev., 2014, 114, 1413–1492 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- R. Dawson, L. A. Stevens, O. S. A. Williams, W. Wang, B. O. Carter, S. Sutton, T. C. Drage, F. Blanc, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2014, 7, 1786–1791 CAS.
- Dry Liquids for Gas Absorption and Purification, J. M. Chin, S. W. Tay, J. W. Xu and A. Y. X. Tan, Assigned to Agency for Science, Technology and Research, Singapore, SG. Pat. 2013040316, 23 May 2013.
- B. P. Binks and R. Murakami, Nat. Mater., 2006, 5, 865–869 CrossRef CAS PubMed.
- B. O. Carter, D. J. Adams and A. I. Cooper, Green Chem., 2010, 12, 783–785 RSC.
- L. Forny, K. Saleh, R. Denoyel and I. Pezron, Langmuir, 2009, 26, 2333–2338 CrossRef PubMed.
- G. Hu, Y. Ye, C. Liu, Q. Meng, J. Zhang and S. Diao, Fuel Process. Technol., 2011, 92, 1617–1622 CrossRef CAS PubMed.
- W. Wang, C. L. Bray, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 11608–11609 CrossRef CAS PubMed.
- K. Tsujii, T. Yamamoto, T. Onda and S. Shibuichi, J. Colloid Interface Sci., 1998, 208, 1011–1012 Search PubMed.
- B. P. Binks, T. Sekine and A. T. Tyowua, Soft Matter, 2014, 10, 578–589 RSC.
- M. J. W. Frank, J. A. M. Kuipers and W. P. M. van Swaaij, J. Chem. Eng. Data, 1996, 41, 297–302 CrossRef CAS.
- P. Zhang, Y. Shi, J. Wei, W. Zhao and Q. Ye, J. Environ. Sci., 2008, 20, 39–44 CrossRef CAS.
- D. Bonenfant, M. Mimeault and R. Hausler, Ind. Eng. Chem. Res., 2003, 42, 3179–3184 CrossRef CAS.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- N. McCann, M. Maeder and H. Hasse, J. Chem. Thermodyn., 2011, 43, 664–669 CrossRef CAS PubMed.
- H. Abe, I. Abe, K. Sato and M. Naito, J. Am. Ceram. Soc., 2005, 88, 1359–1361 CrossRef CAS PubMed.
- T. Kashiwagi, J. W. Gilman, K. M. Butler, R. H. Harris and J. R. Shields, Fire Mater., 2001, 24, 277–289 CrossRef.
- O. Taylan and H. Berberoglu, J. Quant. Spectrosc. Radiat. Transfer, 2013, 120, 104–113 CrossRef CAS PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- A. V. Patil and V. P. Pawar, J. Mol. Liq., 2013, 188, 1–4 CrossRef CAS PubMed.
- G. S. Goff and G. T. Rochelle, Ind. Eng. Chem. Res., 2006, 45, 2513–2521 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, ESI Fig. S1–S4 and ESI Tables S1–S3. See DOI: 10.1039/c4ta06273f |
| This journal is © The Royal Society of Chemistry 2015 |