
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of shaped Pt nanoparticles using common anions or small molecules as shape-directing agents: observation of a strong halide or pseudo-halide effect†
Jason A.
Michel‡
,
William H.
Morris III§
and
Charles M.
Lukehart
*
Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: chuck.lukehart@vanderbilt.edu; Fax: +1 615 3431234; Tel: +1 615 3222935
First published on 2nd December 2014
Abstract
Chemical reduction of Pt(II) and Pt(IV) salts by H2, hydrazine, or borohydride ion, in the presence of common anions or small molecules is surveyed to discover shape-directing agents suitable for selective formation of Pt metal nanocubes or nanotetrahedra. Surfactants tested include a variety of amino acids, HSCH2CO2H, Ph2PCH2CO2H, polycarboxylate species, such as glycolate or tartrate ion, and several common anions (including CF3SO3−, ClO4−, NO3−, CO32−, PO43−, SO42−, Cl−, Br−, or I−). Although carboxylate, chloride, bromide, carbonate, nitrate, perchlorate, phosphate, sulfate, or triflate ions show poor shape-directing influence, the presence of hydroxide and iodide ions gives an unexpected halide (or pseudo-halide) effect. Hydrogen reduction of [Pt(OH)6]2− ion at basic pH gives high yields of cubic Pt nanoparticles, while hydrogen reduction of [PtI6]2− ion at basic pH gives high yields of tetrahedral Pt nanoparticles.
Introduction
Utilizing platinum nanoparticle shape selectivity in chemical catalysis and electrochemical catalysis gives more efficient chemical processing at lower energy cost and is the subject of two recent reviews.1,2 To achieve such benefits, convenient methods of preparing catalytically active Pt metal nanocrystals of desired shape are needed.A vast literature is available on the synthesis and catalytic reactivity of Pt metal catalysts, and synthesis strategies for preparing shape- and size-controlled Pt metal colloids have been reviewed.3 More recent methods of preparing shaped Pt metal nanoparticles include: Pt-ion reduction by H2 in the presence of carboxylic acid4–6 or amine7 capping agents; Pt-ion reduction by citrate ion4 or formic acid;8 polyol reduction of Pt-ion in the presence of polyvinylpyrrolidone (PVP),9 PVP/AgNO3,10 PVP/NaNO3,11 or oleylamine;12 thermal decomposition of organometallic Pt precursors;13–15 Pt(acetylacetonate)2 reduction by oleylamine,16 alkenes,17 or metal carbonyls in the presence of oleylamine/oleic acid;18 photo-induced reduction of Pt(IV) ion within micelles;19 reduction of Pt-ion with wood nanofibers;20 and supported Pt nanoparticles formed by borohydride reduction,21 electrochemical reduction,8 or electron-beam deposition.22 Control of Pt nanoparticle shape is usually achieved by the choice of surfactant or shape-directing agent used and by careful regulation of experimental conditions, such as surfactant or shape-directing agent concentration, pH, temperature, ambient light intensity, and reaction time. A wide variety of Pt particle shapes have been prepared through experimental control of metal particle nucleation and growth events.
Recent reports of chemical23–28 or electrochemical29–37 catalysis employing shaped Pt metal nanoparticles commonly utilize Pt nanoparticles prepared by colloidal synthesis strategies using polymeric surfactants, such as PVP or polyacrylic acid (PAA), as shape-directing agents. While these synthesis methods are convenient to perform and provide shaped Pt metal nanoparticles at sufficient mass scale, complete removal of surfactant material remains a challenge.38 While extensive solvent washing39,40 or electrochemical stripping41 procedures reduce surfactant contamination, UV irradiation treatments40,42 lead to Pt nanoparticle aggregation or surface damage.
In this study, common anions or small molecules are surveyed as possible shaped-directing agents with the goal of finding polymer-free methods for convenient preparation of shaped Pt nanoparticles. Given the known catalytic reactivity differences of Pt(100) and Pt(111) surfaces, formation of Pt metal nanocubes or nanotetrahedra is of primary interest. The outcome of this study was unexpected. Carboxylate, polycarboxylate, hydroxy-, amino-, phosphino-, or mercaptocarboxylate anions all showed poor shape-directing influence (even though such functional groups are present in shape-directing polymeric surfactants) as did chloride, bromide, carbonate, nitrate, perchlorate, phosphate, sulfate, or triflate ions. However, hydroxide and iodide anions serve as endogenous shape-directing agents in the H2 reduction of Pt(IV) complexes to form, respectively, Pt nanocubes and nanotetrahedra. This strong “halide (or pseudo-halide) effect” is surprising, though not unprecedented. A recent review of the complex role that halide ions play in controlling anisotropic noble metal nanocrystal growth emphasizes the need for additional investigation of this phenomenon.43
Experimental
Reagents and general methods
All reagents were used as purchased without further purification. Potassium tetrachloroplatinate(II), 99.9%, was purchased from Strem Chemicals. Potassium hexaiodoplatinate(IV), 99.9%, potassium iodide, 99%, β-alanine, 98%, sodium glycolate, 97%, glycolic acid, 99%, and stearic acid, 95%, were purchased from Sigma Aldrich. Dihydrogen hexahydroxyplatinate(IV), 99.9%, and potassium tartrate, 99%, were purchased from Alfa Aesar. Glycine, 97%, and perchloric acid, 70%, were purchased from Fisher. Sarcosine, 98%, N,N-dimethylglycine, 97%, and betaine (inner salt), 98%, and tetrabutylammonium hydroxide solution, 40% w/w, were purchased from Acros. Potassium hydroxide, 85%, and hydrochloric acid, 38%, were purchased from EMD. Vulcan XC-72R and Black Pearls 2000 were purchased from Cabot. Water was singly distilled in air over 5% NaOH and 2.5% KMnO4 by weight.All routine transmission electron microscopy (TEM) was carried out on a Philips CM-20 TEM equipped with a LaB6 thermionic electron source and an energy dispersive spectrometer (EDS) and operating at 200 kV accelerating voltage. High-resolution transmission electron microscopy (HR-TEM) was performed on a Philips CM-30 TEM equipped with a LaB6 thermionic emitter and operating at 300 kV. Samples for TEM were dropped onto carbon-coated grids and allowed to dry in air. All powder X-ray diffraction (XRD) was carried out on a Scintag X-1 XRD in the θ–θ configuration and equipped with a Peltier-cooled solid-state detector. Samples for XRD were deposited onto a Si(510) plate and scanned in air. Ultraviolet-visible spectroscopy (UV-Vis) was carried out using a Hewlett-Packard 8452A photodiode array spectrophotometer.
Pt colloid syntheses
Formation of Pt metal colloids were surveyed using: Pt2(C2H4)2Cl4, K2[PtCl4], PtI2(glycinato)2, K2[PtI6], or H2[Pt(OH)6] as sources of Pt ion; aqueous or non-aqueous solvent; H2, hydrazine (N2H4) hydrate, or NaBH4 as reducing agents; and a variety of carboxylates, amino acids, Ph2PCH2CO2H, HSCH2CO2H, or common anions (CF3SO3−, ClO4−, NO3−, CO32−, PO43−, SO42−, Cl−, Br−, or I−) as shape-directing agents. A listing of syntheses performed along with nanoparticle shape outcomes is provided in Table 1. Detailed synthesis procedures are available as ESI.† In a typical synthesis, Pt reagent is dissolved in water at a desired temperature and pH in the presence of a surface-directing agent under subdued lighting. Reducing agent is introduced, and the reaction is monitored until a golden yellow or brown color develops. Various control syntheses were conducted, as discussed below.| Pt Source | Shaping agent | Reducing agent | React. conditions | Particle shape outcome |
|---|---|---|---|---|
| a THF = tetrahydrofuran; RT = room temperature; DPPA = diphenylphosphinoacetic acid; Td = tetrahedral; Oh = octahedral; “trun” = truncated. | ||||
| Pt2(C2H4)2Cl4 | [Bu4N]glycolate | None | 60 °C; THF | Fig. S1 heterogeneous shapes |
| K2[PtCl4] | [Na]glycolate | None | 75 °C | Fig. S2 heterogeneous shapes |
| K2[PtCl4] | [K]tartrate | None | 75 °C | Fig. S2 heterogeneous shapes |
| K2[PtCl4] | [Na]glycolate | H2 | RT | Fig. S3 heterogeneous shapes; some cubes & Td |
| K2[PtCl4] | Stearic acid | N2H4 | RT; emulsion | Fig. S4 spheroidal |
| PtI2(glycinato)2 | Glycine | H2 | 70 °C/KI/pH 10.5 | Fig. S5 heterogeneous shapes |
| PtI2(glycinato)2 | Glycine | H2 | 70 °C/KI/pH 6 | Fig. S6 very small; some Td |
| PtI2(glycinato)2 | Glycine | H2 | 70 °C/KI/pH 3.5 | Fig. S7 multi-Td; stars; multipods |
| K2[PtCl4] | DPPA | H2 | RT/KI | Fig. S8 trun. Td & cubo-Oh |
| K2[PtCl4] | HSCH2CO2H | NaBH4 | RT/pH 9 | Fig. S8 too small & heterogeneous |
| K2[PtCl4] | L-Serine | H2 | RT | Fig. S9 spheroidal & oblong |
| K2[PtCl4] | L-Proline | H2 | RT | Fig. S9 spheroidal & oblong |
| K2[PtCl4] | Phenylalanine | H2 | RT | Fig. S9 spheroidal & oblong |
| K2[PtCl4] | Dimethylglycine | H2 | 70 °C/KI/pH 10.5 | Fig. S10 mostly Td and some cubes |
| K2[PtI6] | Glycine | H2 | RT/pH 10 | Fig. S11A Td |
| K2[PtI6] | Sarcosine | H2 | RT/pH 10 | Fig. S11B Td |
| K2[PtI6] | Dimethylglycine | H2 | RT/pH 10 | Fig. S11C Td |
| K2[PtI6] | Betaine | H2 | RT/pH 10 | Fig. S11D Td |
| K2[PtI6] | β-Alanine | H2 | RT/pH 10 | Fig. S11E Td |
| K2[PtI6] | None | H2 | RT/pH 10 | Fig. S11F Td |
| H2[Pt(OH)6] | Glycine | H2 | RT/pH 10 | Fig. S12A cubes & cubo-Oh |
| H2[Pt(OH)6] | Sarcosine | H2 | RT/pH 10 | Fig. S12B cubes & cubo-Oh |
| H2[Pt(OH)6] | Dimethylglycine | H2 | RT/pH 10 | Fig. S12C cubes & cubo-Oh |
| H2[Pt(OH)6] | Betaine | H2 | RT/pH 10 | Fig. S12D cubes |
| H2[Pt(OH)6] | β-Alanine | H2 | RT/pH 10 | Fig. S12E cubes |
| H2[Pt(OH)6] | None | H2 | RT/pH 10 | Fig. S12F 90+% cubes |
| H2[Pt(OH)6] | K[CF3SO3] | H2 | RT/pH 10 | Fig. S13A cubo-Oh |
| H2[Pt(OH)6] | KClO4 | H2 | RT/pH 10 | Fig. S13B cubo-Oh |
| H2[Pt(OH)6] | KNO3 | H2 | RT/pH 10 | Fig. S13C cubo-Oh |
| H2[Pt(OH)6] | K2CO3 | H2 | RT/pH 10 | Fig. S13D cubo-Oh |
| H2[Pt(OH)6] | Dimethylglycine | H2/N2H4 | RT/pH 10 | Fig. S14 mostly cubes |
| H2[Pt(OH)6] | KCl | H2/N2H4 | RT/pH 9.9 | Fig. S15A trun cubes; cubo-Oh, wires |
| H2[Pt(OH)6] | KBr | H2/N2H4 | RT/pH 9.8 | Fig. S15B more wires mostly cubo-Oh |
| H2[Pt(OH)6] | K2CO3 | H2/N2H4 | RT/pH 10.7 | Fig. S15C even more wires |
| H2[Pt(OH)6] | K3PO4 | H2/N2H4 | RT/pH 11.0 | Fig. S15D see dendrites |
| H2[Pt(OH)6] | K2SO4 | H2/N2H4 | RT/pH 9.8 | Fig. S15E wider wires |
| H2[Pt(OH)6] | K[CF3SO3] | H2/N2H4 | RT/pH 10.0 | Fig. S15F most wires of all |
| H2[Pt(OH)6] | None | H2/NaBH4 | RT/pH 10 | Fig. S16 cubo-Oh, Oh, decahedra |
Results and discussion
Pt colloid syntheses
Formation of shaped Pt nanoparticles by El-Sayed, et al.,44 and shaped Ni and Pd nanoparticles by Reetz, et al.,45 using polycarboxylate or α-hydroxycarboxylate shape-directing agents prompted a survey of Pt-ion reductions in the presence of simple bi-functional or poly-functional carboxylate ions. Reaction of Pt2(C2H4)2Cl4 and [Bu4N]glycolate in THF solution, using glycolate ion as both reducing agent and shape-directing agent, produces Pt nanoparticles of heterogeneous shapes (see ESI, Fig. S1†), suggesting that the Reetz method does not apply to Pt-ion reduction. Reduction of [PtCl4]2− in water by either glycolate or tartrate ion requires elevated temperatures and gives poorly faceted, highly agglomerated Pt nanoparticles (see ESI, Fig. S2†). Reduction of aqueous [PtCl4]2− solutions at room temperature using H2 as reducing agent and either glycolate or tartrate ion as shape-directing agent gives less agglomerated Pt nanoparticles exhibiting heterogeneous shapes but includes some identifiable cubic and tetrahedral particles (see ESI, Fig. S3†). Hydrazine reduction of [PtCl4]2− within a stearic acid/water-in-oil emulsion at room temperature forms relatively large (ave. dia. ≈ 20 nm), spheroidal Pt nanoparticles that apparently form within a poly-carboxylate, spherical micelle (see ESI, Fig. S4†).Amino acid conjugate bases represent another class of potentially bifunctional carboxylate shape-directing agents. Basic pH is required to ensure a high degree of deprotonation (glycine; pKa = 9.60). When using glycine as a possible shape-directing agent, Pt colloid formation is accelerated by in situ displacement of chloro ligands of K2[PtCl4] by iodide ion (KI) giving to form (glycinato)2PtI2. H2 reduction of (glycinato)2PtI2 at pH 10.5 yields highly faceted Pt nanoparticles exhibiting a variety of shapes with sizes ranging from 5–20 nm (see ESI, Fig. S5†). Unfortunately, this method is not very shape selective. Control reactions indicate that while varying free glycine concentration has little effect on Pt nanoparticle shape, variation in reaction pH has a dramatic effect. Pt particles formed at pH 6 are sufficiently small (1–3 nm) that particle shape is not easily discerned (see ESI, Fig. S6†). Pt particles formed at pH 3.5 exhibit a variety of shapes, though the most common are multi-tetrahedral, star-like shapes and Pt multi-pods (see ESI, Fig. S7†).46,47 While such complex nanoparticle shapes are interesting, they possess a mixture of surface facets and are not desirable for shape-selective catalysis.
Diphenylphosphinoacetic acid (DPPA) and mercaptoacetic acid proved to have only limited usefulness as shape-directing agents. DPPA is soluble only at very high pH, which destabilizes Pt colloids, leading to particle precipitation. Mercaptoacetic acid apparently binds sufficiently strongly to Pt ion that NaBH4 is required as reducing agent. The presence of DPPA produces faceted Pt nanoparticles as a mixture of truncated tetrahedral and cubo-octahedral shapes, while mercaptoacetic acid gives very small Pt particles (1–2 nm) of irregular shape (see ESI, Fig. S8†).
Proline, serine, and phenylalanine are also poor shape-directing agents. Each of these amino acids produces 2–5 nm Pt particles having a variety of spheroidal and oblong shapes (see ESI, Fig. S9†).
N-substituted amino acids [sarcosine (N-methylglycine), N,N-dimethylglycine, betaine (inner salt, 2-trimethylammonio-acetate), and β-alanine (3-aminopropanoic acid)] represent a series of shape-directing agents containing α-amino groups having different Lewis and Brønsted basicities. H2 reduction of K2[PtCl4] solutions in the presence of sarcosine, betaine, or β-alanine gives Pt colloids of primarily spheroidal or oblong shape or particles too small for easy shape identification. However, H2 reduction of [PtCl4]2− in the presence of N,N-dimethylglycine is accelerated by addition of eight equivalents of KI affording mainly tetrahedral Pt nanoparticles along with far fewer nanocubes than that observed when using glycine. This observation suggests a possible special influence by iodide ion. The presence of N,N-dimethylglycine gives far fewer irregular shapes and a nearly complete absence of very small nanoparticles (see ESI, Fig. S10†). H2 reduction of basic solutions of K2[PtI6] in the presence of glycine, sarcosine, N,N-dimethylglycine, betaine, or β-alanine forms tetrahedral Pt nanoparticles as primary product, and in some cases, almost entirely Pt nanotetrahedra (see ESI, Fig. S11†).
Quite unexpectedly, however, control experiments reveal that Pt nanotetrahedra are formed by H2 reduction of basic [PtI6]2− solutions even in the absence of any intentional surface-directing agent (see ESI, Fig. S11F†)! Likewise, H2 reduction of basic solutions of K2[Pt(OH)6] in the absence of iodide ion and any other intentional shape-directing agent gives predominantly Pt nanocubes (see ESI, Fig. S12 & S13†). Fig. 1 & 2 show TEM and HR-TEM images of Pt nanocubes and nanotetrahedra prepared by H2 reduction of [Pt(OH)6]2− and [PtI6]2−, respectively. HR-TEM images reveal lattice fringe spacings of 0.20 nm for cubic Pt nanoparticles and of 0.23 nm for tetrahedral Pt nanoparticles consistent with {100} and {111} d-spacings reported by others for similarly shaped Pt nanoparticles.48 The expected {111} cross fringe pattern is also observed for tetrahedral Pt nanoparticles. Given that hydroxo and iodo ligands serve as highly selective shape-directing agents in H2 reduction of Pt(IV) ion, knowing the kinetic stability of [Pt(OH)6]2− and [PtI6]2− precursor complexes in basic aqueous solutions and in the presence of H2 becomes important.
 | ||
| Fig. 1 As-prepared Pt nanoparticles deposited from aqueous colloids. (Left) Cubic Pt nanoparticles prepared by H2 reduction of [Pt(OH)6]2− at pH 9.7. Note the presence of a minority of tetrahedra and cubo-octahedra. (Right) Tetrahedral Pt nanoparticles prepared by H2 reduction of [PtI6]2− at pH 10. Many truncated tetrahedra are present, but cubes are rare. | ||
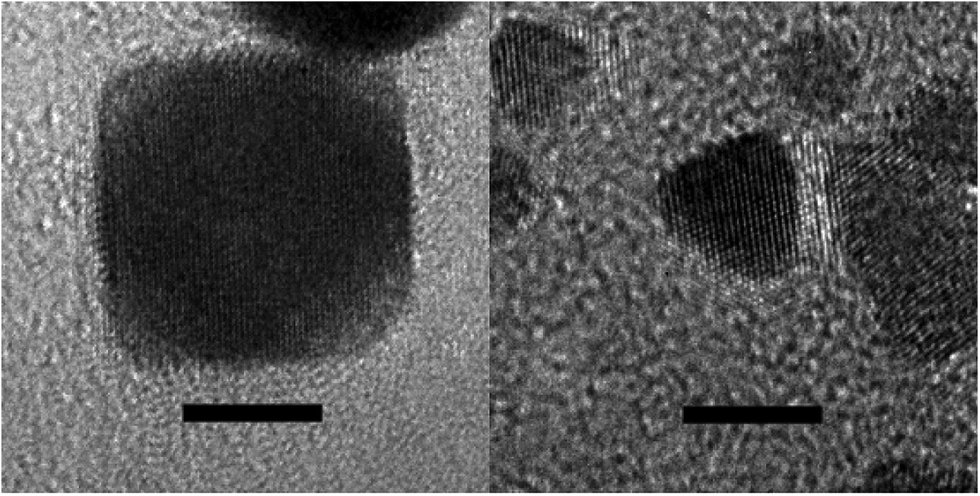 | ||
| Fig. 2 High-resolution TEM images of a typical cubic Pt nanoparticle (left image) and of a typical tetrahedral Pt nanoparticle (right image). Scale bars = 5 nm. | ||
Kinetic stability and H2 reduction of [Pt(OH)6]2− solutions
As shown in Fig. 3, the UV-Vis spectrum of [Pt(OH)6]2− is dominated by a single absorption band centered near 200 nm, characteristic of an intense charge-transfer band. A similar band appears at 262 nm in [PtCl6]2− ion.49 [Pt(OH)6]2− solutions appear visibly to be unaffected by the presence of iodide ion. UV-Vis spectra of mixtures of solutions containing [Pt(OH)6]2− and iodide ion show no new bands above 260 nm over two days. Intense absorption centered near 225 nm by iodide ion obscures UV-Vis band detection from ca. 190–260 nm.50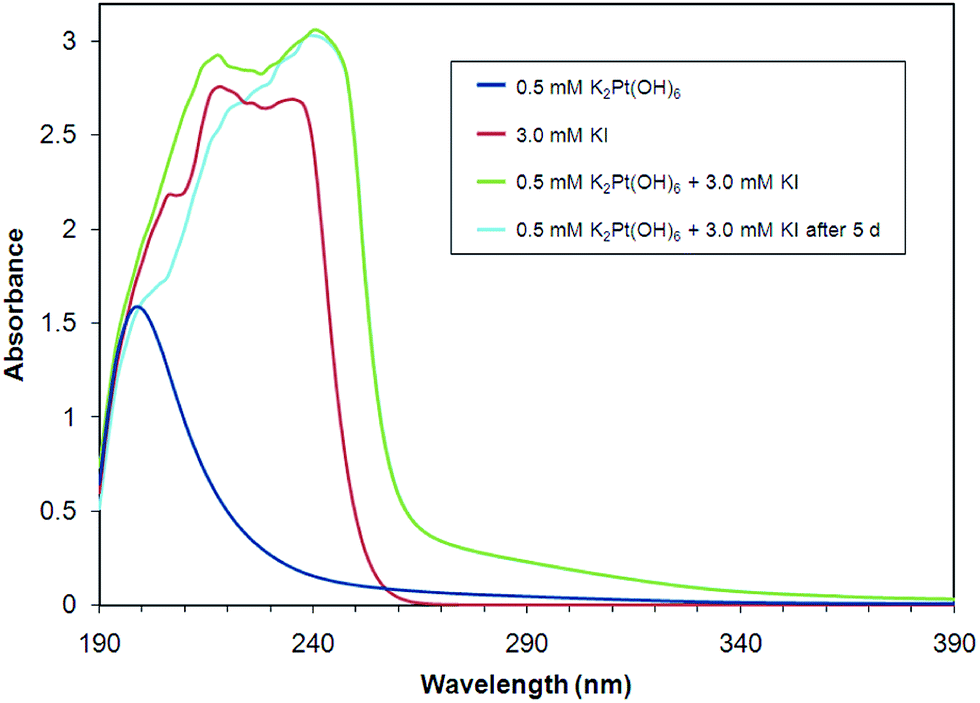 | ||
Fig. 3 UV-Vis spectra of K2[Pt(OH)6] and KI aqueous solutions and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar mixture of K2[Pt(OH)6] and KI over a five-day period. :6 molar mixture of K2[Pt(OH)6] and KI over a five-day period. | ||
H2 reduction of aqueous [Pt(OH)6]2− occurs with lengthy induction periods that are strongly pH dependent (ca. 7 days at pH 9.7; ca. 20 days at pH 10.2). Pt nanoparticle growth is visibly observed as the emergence and gradual darkening of a deep golden color associated with formation of well-dispersed Pt nanoparticles. Complete reaction is achieved after 36 h. Even in the presence of six equivalents of added iodide ion, H2 reduction of [Pt(OH)6]2− consistently forms predominantly Pt nanocubes.
The apparent rate of H2 reduction of aqueous [Pt(OH)6]2− is significantly increased when amino acids or other ionic species are present but substantial loss of shape selectivity occurs. In the presence of weakly coordinating anions, such as triflate, nitrate, perchlorate, and carbonate, cubo-octahedral Pt nanoparticles are primarily formed (see ESI, Fig. S13†). Hydroxide ion appears to inhibit Pt particle nucleation giving relatively large particles and favors formation of (100) facets – an effect diminished somewhat by the presence of other ions (vide infra).
Kinetic stability and H2 reduction of [PtI6]2− solutions at basic pH
[PtI6]2− solutions in neutral or acidic water readily decompose to form metallic Pt and are highly light sensitive. At basic pH, rapid reaction is evident from UV-Vis spectral changes, as shown in Fig. 4. Initial spectra reveal three absorption bands near 340 nm, 440 nm, and 490 nm characteristic of [PtI6]2− aqueous solutions.50 These bands disappear with a first-order rate constant of ca. 15 × 10−3 s−1 (τ1/2 = ca. 46 s; determined from the rate of disappearance of the 490 nm band) with concomitant appearance of a single band centered at ca. 390 nm and a recognizable isosbestic point at ca. 375 nm. A previous kinetics study of [PtI6]2− ion hydrolysis over a pH range of 6–10.5 identified a first-stage iodide/water ligand exchange reaction forming [PtI5(H2O)]− showing nearly identical spectral changes and a similar first-order rate constant of 16.5 × 10−3 s−1 at pH 10 and 21.0 °C.50 Given that [PtI5(H2O)]− has a first-ionization pKa value of 8.55 at 25.5 °C,50 this complex would be 96% ionized at pH 10 giving [PtI5(OH)]2− ion as the solution species assigned to the product absorption band centered at ca. 390 nm.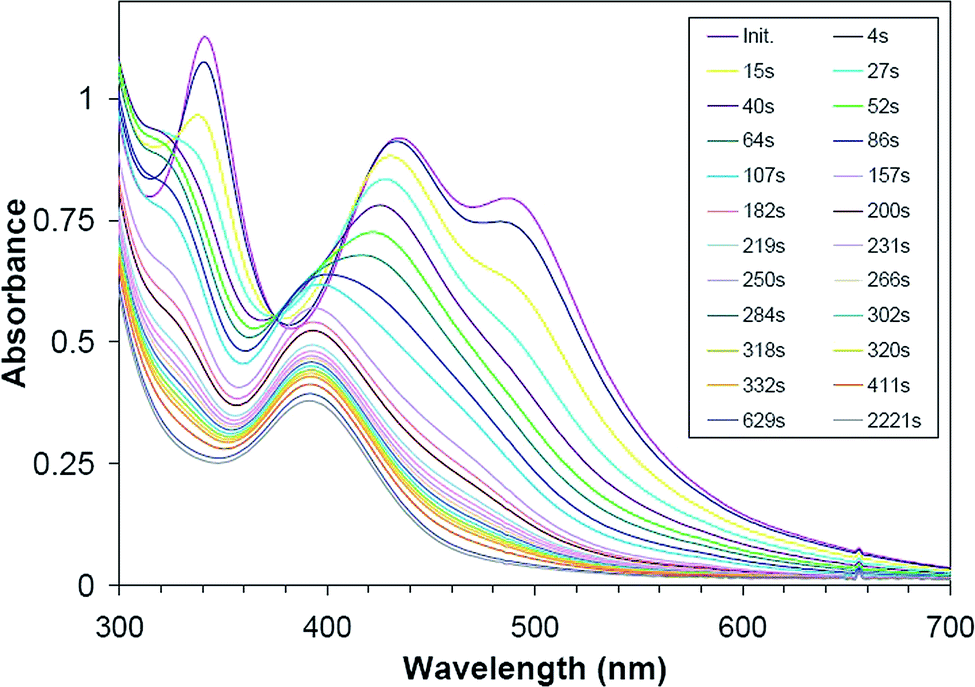 | ||
| Fig. 4 UV-Vis spectra collected over various reaction time intervals showing overall kinetic displacement of one iodo ligand of [PtI6]2− by hydroxide ion at pH 10 and 22 °C giving [PtI5(OH)]2− as product. | ||
The relatively high lability of iodo ligands in Pt(IV) complexes leads to very rapid reduction by H2. Upon addition to basic water, the deep violet color of [PtI6]2− begins to change to orange as OH− begins to displace I−. Typically, the orange color of the [PtI6−xOHx]2− precursor solutions begins to change within minutes of H2 introduction, and Pt nanoparticle formation is complete within 2 h. To achieve high yields of tetrahedral Pt nanoparticles at pH 10, H2 must be introduced within a few minutes of the addition of K2PtI6 salt to basic water. The shape selectivity of Pt nanotetrahedra appears to be highly dependent on the composition of solution species during nanoparticle nucleation. If the reaction solution is allowed to reach near equilibrium before exposure to H2, a dramatic decrease in the number of tetrahedra is observed along with an increase in the number of cubo-octahedra and cubic shapes. Detailed kinetics study of [PtI6]2− hydrolysis at pH 6 reveals a complex three-stage reaction mechanism involving hydrolysis and redox reactions generating as many as eight different Pt solution species.50
Reduction of [Pt(OH)6]2− or [PtI6]2− solutions using hydrazine or borohydride
Attempts to accelerate Pt nanoparticle formation by using stronger reducing agents, such as hydrazine and borohydride ion, leads to diminished shape selectivity with the following outcomes: (1) hydrazine or borohydride accelerate Pt nanoparticle formation relative to H2 reduction but without a reduction in Pt average nanoparticle size; (2) reduction of [Pt(OH)6]2− at increasing concentrations of hydrazine gives higher number fractions of Pt nanotetrahedra, cubo-octahedra, and nanorods (see ESI, Fig. S14†); (3) hydrazine reductions of [Pt(OH)6]2− in the presence of chloride, bromide, carbonate, phosphate, sulfate, or triflate ions give similar mixtures of Pt nanoparticle shapes, including worm-like nanowires (see ESI, Fig. S15†); and, (4) borohydride reduction of [Pt(OH)6]2− gives primarily Pt nanocubo-octahedra along with small number fractions of octahedral and decahedral (pentagonal bipyramidal) nanoparticles (see ESI, Fig. S16†). Images of a particularly large Pt nanodecahedron are shown in Fig. 5. | ||
| Fig. 5 Three views, at different angles of TEM stage tilt, of a particularly large decahedron formed by reduction of [Pt(OH)6]2− with borohydride ion. | ||
Roles of hydroxide and iodide ions in influencing Pt nanoparticle shape
Given that the adsorption enthalpy of hydroxide ion on a Pt(100) surface (ca. −280 kJ mol−1),51 is greater than that on a Pt(111) surface (ca. −255 kJ mol−1),52 formation of cubic Pt nanoparticles might be expected from H2 reduction of [Pt(OH)6]2− at basic pH. While hydroxide adsorption enthalpy on Pt(111) surfaces decreases with increasing surface coverage, this effect is not observed on Pt(100) surfaces, further supporting formation of Pt nanocubes at basic pH.51 This surface coverage effect is attributed to differences in hydrogen bonding networks established among OH groups on these two surfaces and to the fact that a OH–H2O hydrogen bond is much stronger than a OH–OH hydrogen bond. Species that disrupt hydrogen bonding networks at Pt(100) surfaces could inhibit selective growth of that surface, possibly explaining the formation of Pt cubo-octahedral and tetrahedral nanoparticle shapes when Pt-ion reduction occurs in the presence of simple anions or amino acid additives.Iodide ion binds more strongly to top and bridge sites of Pt(111) surfaces (bonding enthalpies of ca. −245 kJ mol−1 and −286 kJ mol−1, respectively)53 than to similar sites of Pt(100) surfaces (bonding enthalpies of ca. −225 kJ mol−1 and −265 kJ mol−1, respectively).52 During H2 reduction of [PtI6]2−, iodide ions would presumably establish monolayer coverage on the surface of a growing nanoparticle, thus preventing adsorption of other species and enforcing retention of (111) surfaces to give Pt nanotetrahedra.
Conclusions
Efficient methods of preparing cubic or tetrahedral Pt nanoparticles via solution reduction of common Pt(II) or Pt(IV) salts in the absence of polymeric surfactants have been found. Hydrogen reduction of [Pt(OH)6]2− ion at basic pH gives high yields of cubic Pt nanoparticles, while hydrogen reduction of [PtI6]2− ion at basic pH gives high yields of tetrahedral Pt nanoparticles. Although minor shape effects are observed when Pt salts are reduced in the presence of simple carboxylate species, amino acids, or common inorganic ions, the exceptional shape effect of hydroxide ion and iodide ion is noteworthy. The presence of surface-bound hydroxide or iodide ions on Pt nanoparticle surface sites can be mitigated to promote chemical or electrochemical catalysis. Hydroxylated Pt metal surfaces are catalytically active toward methanol oxidation,54 and CO displacement of chemisorbed iodine on pristine Pt metal surfaces is used in protocols for cleaning Pt metal surfaces.55,56 Although atomic-level understanding of how hydroxide and iodide direct Pt nanoparticle growth toward cubic or tetrahedral shapes, respectively, at basic pH has not been determined, it is hoped that shaped Pt metal nanoparticles prepared by this method prove to be effective catalysts under convenient operating conditions.Acknowledgements
Financial support from the DuPont Science & Engineering University Grant Program and from the Honda Initiation Grant Award Program (C.M.L.) is gratefully acknowledged. We thank Dr James Bentley for access to HR-TEM instrumentation at the Oak Ridge National Laboratory.References
- I. Lee and F. Zaera, Top. Catal., 2013, 56, 1284–1298 CrossRef CAS PubMed.
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS PubMed.
- C. Coutanceau, P. Urchaga, S. Brimaud and S. Baranton, Electrocatalysis, 2012, 3, 75–87 CrossRef CAS PubMed.
- G. A. Attard, J.-Y. Ye, P. Jenkins, F. J. Vidal-Iglesias, E. Herrero and S.-G. Sun, J. Electroanal. Chem., 2013, 688, 249–256 CrossRef CAS PubMed.
- J. Zeng, J. Mater. Chem., 2012, 22, 3170–3176 RSC.
- M. A. El-Sayed and J. W. Yoo, ChemCatChem, 2010, 2, 268–271 CrossRef CAS.
- L.-M. Lacroix, C. Gatel, R. Arenal, C. Garcia, S. Lachaize, T. Blon, B. Warot-Fonrose, E. Snoeck, B. Chaudret and G. Viau, Angew. Chem., Int. Ed., 2012, 51, 4690–4694 CrossRef CAS PubMed.
- S. Sattayasamitsathit, Y. Gu, K. Kaufmann, W. Jia, X. Xiao, M. Rodriguez, S. Minteer, J. Cha, D. B. Burckel and C. Wang, et. al. , J. Mater. Chem. A, 2013, 1, 1639–1645 CAS.
- S. Alayoglu, C. Aliaga, C. Sprung and G. A. Somorjai, Catal. Lett., 2011, 141, 914–924 CrossRef CAS.
- N. V. Long, C. M. Thi, M. Nogami and M. Ohtaki, New J. Chem., 2012, 36, 1320–1334 RSC.
- Y. Yang, Z. Huang, D. Li and M. Nogami, Proc. SPIE, 2010, 7658, 76580H CrossRef PubMed.
- N. Shukla, M. M. Nigra, M. A. Bartel, T. Nuhfer, C. Phatak and J. A. Gellman, Catal. Lett., 2010, 140, 85–89 CrossRef CAS.
- K. Pelzer, M. Haevecker, M. Boualleg, J.-P. Candy and J.-M. Basset, Angew. Chem., Int. Ed., 2011, 50, 5170–5173 CrossRef CAS PubMed.
- K. Mase, H. Kondo, S. Kondo, M. Hori, M. Hiramatsu and H. Kano, Appl. Phys. Lett., 2011, 98, 193108 CrossRef PubMed.
- M. R. Axet, K. Philippot, B. Chaudret, M. Cabie, S. Giorgio and C. R. Henry, Small, 2011, 7, 235–241 CrossRef CAS PubMed.
- X. Gong, Y. Yang, L. Zhang, C. Zou, P. Cai, G. Chen and S. Huang, J. Colloid Interface Sci., 2010, 352, 379–385 CrossRef CAS PubMed.
- C. Kim, S. S. Kim, S. Yang, J. W. Han and H. Lee, Chem. Commun., 2012, 48, 6396–6398 RSC.
- Y. Kang, J. B. Pyo, X. Ye, R. E. Diaz, T. R. Gordon, E. A. Stach and C. B. Murray, ACS Nano, 2013, 7, 645–653 CrossRef CAS PubMed.
- S. Kundu and H. Liang, Langmuir, 2010, 26, 6720–6727 CrossRef CAS PubMed.
- X. Lin, M. Wu, D. Wu, S. Kuga, T. Endo and Y. Huang, Green Chem., 2011, 13, 283–287 RSC.
- Y. He and H. Cui, Chem.–Eur. J., 2012, 18, 4823–4826 CrossRef CAS PubMed.
- V. Komanicky, H. Iddir, K.-C. Chang, A. Menzel, G. Karapetrov, D. C. Hennessy, P. Zapol and H. You, Electrochim. Acta, 2010, 55, 7934–7938 CrossRef CAS PubMed.
- V. V. Pushkarev, N. Musselwhite, K. An, S. Alayoglu and G. A. Somorjai, Nano Lett., 2012, 12, 5196–5201 CrossRef CAS PubMed.
- S. Takeda, Appl. Phys. Express, 2011, 4, 065001 CrossRef.
- F. Vines and A. Goerling, Angew. Chem., Int. Ed., 2011, 50, 4611–4614 CrossRef CAS PubMed.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed.
- I. Lee and F. Zaera, J. Catal., 2010, 269, 359–366 CrossRef CAS PubMed.
- A. M. T. Silva, B. F. Machado, H. T. Helder, J. L. Figueiredo, G. Drazic and F. Goran, J. Nanopart. Res., 2010, 12, 121–133 CrossRef CAS.
- P. Urchaga, S. Baranton, C. Coutanceau and G. Jerkiewicz, Langmuir, 2012, 28, 13094–13104 CrossRef CAS PubMed.
- F. J. Vidal-Iglesias, R. M. Aran-Ais, J. Solla-Gullon, E. Herrero and J. M. Feliu, ACS Catal., 2012, 2, 901–910 CrossRef CAS.
- O. Guillen-Villafuerte, G. Garcia, A. G. Orive, B. Anula, A. H. Creus and E. Pastor, Electrocatalysis, 2011, 2, 231–241 CrossRef CAS PubMed.
- M.-H. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
- I. J. Hsu, D. V. Esposito, E. G. Mahoney, A. Black and J. G. Chen, J. Power Sources, 2011, 196, 8307–8312 CrossRef CAS PubMed.
- C. Susut and Y. Y. J. Tong, Electrocatalysis, 2011, 2, 75–81 CrossRef CAS PubMed.
- K. R. Beyerlein, J. Solla-Gullon, E. Herrero, E. Garnier, F. Pailloux, M. Leoni, P. Scardi, R. L. Snyder, A. Aldaz and J. M. Feliu, Mater. Sci. Eng., A, 2010, 528, 83–90 CrossRef PubMed.
- V. Grozovski, J. Solla-Gullon, V. Climent, E. Herrero and J. M. Feliu, J. Phys. Chem. C, 2010, 114, 13802–13812 CAS.
- C. M. Sanchez-Sanchez, J. Solla-Gullon, F. J. Vidal-Iglesias, A. Aldaz, V. Montiel and E. Herrero, J. Am. Chem. Soc., 2010, 132, 5622–5624 CrossRef CAS PubMed.
- E. Schmidt, W. Kleist, F. Krumeich, T. Mallat and A. Baiker, Chem.–Eur. J., 2010, 16, 2181–2192 CrossRef CAS PubMed.
- S. O. Blavo, E. Qayyum, L. M. Baldyga, V. A. Castillo, M. D. Sanchez, K. Warrington, M. A. Barakat and J. N. Kuhn, Top. Catal., 2013, 56, 1835–1842 CrossRef CAS PubMed.
- J. M. Krier, W. D. Michalak, L. R. Baker, K. An, K. Komvopoulos and G. A. Somorjai, J. Phys. Chem. C, 2012, 116, 17540–17546 CAS.
- P. S. Fernandez, D. S. Ferreira, C. A. Martins, H. E. Troiani, G. A. Camara and M. E. Martins, Electrochim. Acta, 2013, 98, 25–31 CrossRef CAS PubMed.
- F. J. Vidal-Iglesias, J. Solla-Gullon, E. Herrero, V. Montiel, A. Aldaz and J. M. Feliu, Electrochem. Commun., 2011, 13, 502–505 CrossRef CAS PubMed.
- S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzan and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 222, 1924–1926 Search PubMed.
- J. S. Bradley, B. Tesche, W. Busser, M. Maase and M. T. Reetz, J. Am. Chem. Soc., 2000, 122, 4631–4636 CrossRef CAS.
- T. Herricks, J. Chen and Y. Xia, Nano Lett., 2004, 4, 2367–2371 CrossRef CAS.
- X. Teng and H. Yang, Nano Lett., 2004, 4, 2367–2371 CrossRef.
- B. Zhang, D. Wang, Y. Hou, S. Yang, X. H. Yang, J. H. Zhong, J. Liu, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Sci. Rep., 2013, 3, 1836 Search PubMed.
- C. K. JØrgensen, Acta Chem. Scand., 1956, 10, 518–534 CrossRef PubMed.
- B. Corain and A. J. Poe, J. Chem. Soc. A, 1967, 329, 1633–1641 RSC.
- V. Climent, R. Gomez, J. M. Orts and J. M. Feliu, J. Phys. Chem. B, 2006, 110, 11344–11351 CrossRef CAS PubMed.
- A. B. Anton and D. C. Cadogan, Surf. Sci., 1990, 239, L548–L560 CrossRef CAS.
- S.-B. Zhu, Surf. Sci., 1995, 329, 276–284 CrossRef CAS.
- A. Kuzume, Y. Mochiduki, T. Tsuchida and M. Ito, Phys. Chem. Chem. Phys., 2008, 10, 2175–2179 RSC.
- M. L. Lynch and R. M. Cave, J. Phys. Chem., 1990, 94, 4382–4385 CrossRef CAS.
- D. Zurawski, L. Rice, M. Hourani and A. Wieckowski, J. Electroanal. Chem., 1987, 230, 221–231 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: See experimental procedures and TEM images. See DOI: 10.1039/c4ta05952b |
| ‡ Present address: Department of Chemistry, George Washington University, Washington, DC, 20052, USA. |
| § Present address: Kemira, Atlanta, GA, 30313, USA. |
| This journal is © The Royal Society of Chemistry 2015 |