
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diselenogermole as a novel donor monomer for low band gap polymers†
Zhuping
Fei
a,
Raja Shahid
Ashraf
a,
Yang
Han
ac,
Sarah
Wang
b,
Chin Pang
Yau
a,
Pabitra S.
Tuladhar
a,
Thomas D.
Anthopoulos
c,
Michael L.
Chabinyc
b and
Martin
Heeney
*a
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London, SW7 2AZ, UK. E-mail: m.heeney@imperial.ac.uk
bMaterials Department, University of California, Santa Barbara, CA 93106-5050, USA
cDepartment Physics and Centre for Plastic Electronics, Imperial College London, SW7 2AZ, UK
First published on 8th December 2014
Abstract
We report the synthesis of a new diseleno[3,2-b:2′,3′-d]germole (DSG) monomer containing branched 2-ethylhexyl sidechains on the bridging germanium group. The utility of this new electron rich monomer is explored by co-polymerisation of a distannylated DSG monomer with N-octylthienopyrrolodione to afford a soluble low band gap polymer. The DSG containing polymer shows a broader and more red-shifted absorption spectrum compared its thiophene analogue. Polymer solar cells are fabricated from blends with PC71BM exhibiting initial power conversion efficiencies of 5.2% in inverted bulk heterojunction solar cells.
Introduction
There has been significant interest in the development of fused aromatic units for incorporation into conjugated polymer backbones.1 In particular the development of building blocks in which aromatic units such as benzene or thiophene are rigidly held in a co-planar arrangement by the use of bridging heteroatoms has been a successful approach to the development of polymers for field effect transistor (FET) or photovoltaic (OPV) applications.2 The use of fully co-planar building blocks prevents any torsional twisting between the adjacent aromatic units, which potentially leads to a reduction in conjugation length and an increase in reorganizational energy. More rigid co-monomers can also enhance polymer persistence length, which has been correlated with better transport.3 In addition, the bridging atom fulfils an important role as the point of attachment for solubilising alkyl sidechains and as a defining factor in the geometry of the backbone by modifying the bond angles in the monomer.4Many of the best performing classes of these ladder-like monomers contain thiophene as the aromatic unit, and a variety of bridging heteroatoms. For example bridged 2,2′-bithiophenes have been well investigated, with polymers of cyclopentadithiophene2d (which contain a C bridge) exhibiting very promising FET5 and OPV6 performance. The ability of the polymers to crystallise, as well as the ionisation and reduction potentials can be influenced by changing the bridging heteroatom.7 For example, changing from C to Si (dithienosilole, DTS) or Ge (dithienogermole, DTG) whilst maintaining identical sidechains has been shown to enhance polymer crystallinity and solar cell performance, whilst simultaneously lowering both the HOMO and LUMO energy levels.8 The increased crystallinity has been rationalised by the longer C–Si/C–Ge bond compared to the C–C bond, which alters the geometry of the monomer building block, enhancing intramolecular interactions.8 These interesting observations have resulted in the co-polymerisation of DTS and DTG with a wide range of monomers in an effort to further fine tune to polymer optoelectronic properties and solid state organisation.9
Another approach to control the properties of semiconducting polymers has been to change the nature of the heterocycle, for example changing from thiophene to selenophene. The replacement of S for Se, which is larger and more polarisable has been proposed to improve intermolecular contacts in polymer thin films,10 resulting in improved charge carrier mobility in some cases. The reduced aromaticity of selenophene in comparison to thiophene also improves delocalisation of the conjugated system into the polymer backbone, often resulting in reduction in the optical band gap.11 These desirable properties have prompted much interest in selenophene containing polymers for both solar cell and transistor applications.12 Despite this interest, to the best of our knowledge there have been no reports of the incorporation of bridged diselenophene monomers into conjugated polymers. There has been notable work on the development of diselenopheno[3,2-b:2,3-d]pyrrole13 (N bridge) as well as cyclopenta[2,1-b:3,4-b′]diselenophene-4-one14 ((C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bridge) by Marder and co-workers, but so far their properties have only been reported as monomeric materials.
O) bridge) by Marder and co-workers, but so far their properties have only been reported as monomeric materials.
Here we report the first synthesis of diseleno[3,2-b:2′,3′-d]germole, (DSG), the selenophene analogue to DTG, and its co-polymerisation with N-octylthienopyrrolodione (TPD) to afford a new low band gap polymer (PDSGTPD). Recently we, and others, have been interested in the potential of DTG based donor–acceptor materials,9a–d,9g–j,15 because the incorporation of the bridging dialkylgermanium improves the chemical stability of the monomer in comparison to the silicon bridged material leading to greater tolerance of synthetic conditions.9b We have further developed this stable unit by synthesizing the selenophene analogue and using it as a co-monomer in a model donor–acceptor polymer. TPD was chosen as a suitable co-monomer because of the presence of the additional solubilising octyl group, which was expected to counter the reduction in solubility often encountered upon the inclusion of the heavier Se heteroatom. The reduction in solubility is related to the enhanced quinoidal character of selenophene containing polymers,11,16 which planarises the polymer backbone, as well as the reduction in the percentage of solubilising alkyl content due to the heavier mass of Se. TPD also has a relatively strong electron withdrawing effect due to the presence of the imide group and this was expected to lead to polymers with relatively high ionisation potentials, which are desirable in both OPV and FET applications.17 We report the optoelectronic properties of this new polymer, and compare them to the previously reported thiophene analogue, as well as report the properties of the polymer in FET and OPV devices.
Experimental section
General
Reagents and chemicals were purchased from Aldrich and Acros unless otherwise noted. 3,3′-Dibromo-5,5′-bis(trimethylsilyl)-2,2′-biselenophene13b (1) dibromo-di-(2-ethylhexyl) germane9b and 1,3-dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione17a,18 were synthesized by the reported method. The thiophene polymer PDTGTPD was identical to that reported in ref. 21.All reactions were carried out under Ar using solvents and reagents as commercially supplied, unless otherwise stated. 1H and 13C NMR spectra were recorded on a Bruker AV-400 (400 MHz), using the residual solvent resonance of CDCl3 or d-1,1,2,2-tetrachloroethane and are given in ppm. Number-average (Mn) and weight-average (Mw) were determined by Agilent Technologies 1200 series GPC running in chlorobenzene at 80 °C, using two PL mixed B columns in series, and calibrated against narrow polydispersity polystyrene standards. Electrospray mass spectrometry was performed with a Thermo Electron Corporation DSQII mass spectrometer. UV-vis spectra were recorded on a UV-1601 Shimadzu UV-vis spectrometer. Flash chromatography was performed on silica gel (Merck Kieselgel 60 F254 230–400 mesh). Photo Electron Spectroscopy in Air (PESA) measurements were recorded with a Riken Keiki AC-2 PESA spectrometer with a power setting of 5 nW and a power number of 0.5. Samples for PESA were prepared on glass substrates by spin-coating. Differential Scanning Calorimetry (DSC) measurements: ∼4 mg material was used for the DSC experiments, which was conducted under nitrogen at scan rate of 10 °C min−1 with a TA DSC Q20 instrument. Cyclic voltammetry (CV) were carried out with standard three-electrode cell in a 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) solution in acetonitrile at room temperature with a scanning rate of 0.05 V s−1. A Pt wire counter electrode, a glass carbon working electrode, and an Ag/AgCl reference electrode were used. The oxidation potentials were calibrated with a standard ferrocene/ferrocenium (FOC) redox system as the internal standard (assuming the energy level of FOC is 4.8 eV below vacuum) for estimating the HOMO energy level of polymers. The polymer film was drop cast onto the working electrode from a 5 mg mL−1 chlorobenzene solution, and dried at room temperature.
OFET (organic field effect transistors) devices fabrication
Bottom gate/top contact devices were fabricated on heavily doped n+-Si (100) wafers with a 400 nm-thick thermally grown SiO2 gate dielectric. The Si/SiO2 substrates were treated with OTS to form a self-assembled monolayer before deposition of the polymer. PDSGTPD was dissolved in dichlorobenzene (5 mg mL−1) and spin cast at 2000 rpm for 60 s in nitrogen atmosphere. No annealing procedure was carried out. Au (30 nm) source and drain electrodes were deposited under vacuum through shadow masks. The channel width and length of the transistors are 1000 μm and 30 μm, respectively. Mobility was extracted from the slope of ID1/2vs. VG.OPV fabrication and characterization
Organic photovoltaic devices were fabricated using PDSGTPD and [6,6]-phenyl C71 butyric acid methyl ester (PC71BM, purchased from Nano C Inc.) as the donor and acceptor materials. Devices were fabricated onto the indium tin oxide (ITO) coated substrates with the device structures of ITO/PEDOT:PSS/PDSGTPD:PC71BM/LiF/Al and ITO/ZnO/PDSGTPD:PC71BM/MoO3/Ag. For the conventional ITO/PEDOT:PSS/PDSGTPD:PC71BM/LiF/Al device, after sequential cleaning of the ITO with the detergent (Mucasol), acetone and isopropyl alcohol, a 30 nm layer of PEDOT:PSS (AI4083) was spin-coated onto the plasma-treated ITO substrate and annealed at 150 °C for 30 min. For inverted ITO/ZnO/PDSGTPD:PC71BM/MoO3/Ag, after sequential cleaning of the ITO with the detergent (Mucasol), acetone and isopropyl alcohol, ZnO was deposited by spin-coating a zinc acetate dihydrate precursor solution (2 mL 2-methoxyethanol with 60 μL monoethanolamine) followed by annealing at 150 °C for 10–15 min. PDSGTPD:PC71BM solution in dichlorobenzene (ODCB) at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:4 were stirred overnight at 80 °C. 1,8-diiodooctane (DIO, 3% v/v) was used as an additive and added prior to spin coating. The blend solution (12 mg mL−1 for PDSGTPD) was spin coated onto the PEDOT:PSS or ZnO coated ITO substrate (2000 rpm, 1 min). For the power conversion efficiency (PCE) measurement of OPV devices, we used the thermally deposited LiF (1 nm)/Al (100 nm) or MoO3 (10 nm)/Ag (100 nm) cathode. Electrical characteristics were measured by Keithley 236 source/measure units under AM 1.5 solar illumination (Oriel 300 W solar simulator) at an intensity of 100 mW cm−2 with a device area of 0.045 cm2. All electrical measurements of OPVs were executed in the inert nitrogen purged devices chamber.
:1 to 1:4 were stirred overnight at 80 °C. 1,8-diiodooctane (DIO, 3% v/v) was used as an additive and added prior to spin coating. The blend solution (12 mg mL−1 for PDSGTPD) was spin coated onto the PEDOT:PSS or ZnO coated ITO substrate (2000 rpm, 1 min). For the power conversion efficiency (PCE) measurement of OPV devices, we used the thermally deposited LiF (1 nm)/Al (100 nm) or MoO3 (10 nm)/Ag (100 nm) cathode. Electrical characteristics were measured by Keithley 236 source/measure units under AM 1.5 solar illumination (Oriel 300 W solar simulator) at an intensity of 100 mW cm−2 with a device area of 0.045 cm2. All electrical measurements of OPVs were executed in the inert nitrogen purged devices chamber.
Synthesis of monomer and polymer
To the resulting oil in THF (100 mL) was added N-bromosuccinimide (NBS) (1.42 g, 8.0 mmol) in one portion at 0 °C and the mixture was stirred for 1 h in the absence of light. An aqueous saturated solution of Na2SO3 (50 mL) was added to quench the reaction. The mixture was extracted with hexane (3 × 50 mL). The combined organics were dried (MgSO4), filtered and the solvent removed solvent under reduced pressure. The residue was purified by silica gel chromatography (eluent: hexane) to afford 2 as a pale yellow oil (2.14 g, yield: 52%). 1H NMR (CDCl3, 400 MHz), δ (ppm): 7.14 (s, 2H), 1.47–1.41 (m, 2H), 1.27–1.12 (m, 20H), 0.87–0.77 (m, 12H). 13C NMR (CDCl3, 100 MHz), δ (ppm): 160.6, 152.6, 144.3, 135.1, 114.9, 36.9, 35.4, 28.9, 28.8, 23.0, 20.9, 14.1, 10.9. HRMS (EI)+ calculated for C24H36Br2GeSe2: 715.8726. Found: 715.8743.
ε) 661 (4.66), 722 (4.76) nm. Anal. calcd: (C38H53GeNO2SSe2)n: C, 55.76; H, 6.53; N, 1.71. Found: C, 55.89; H, 6.50; N, 1.60.
Results and discussion
The synthesis of diselenogermole (DSG), and its subsequent polymerisation to afford PDSGTPD is shown in Scheme 1. To enhance the polymer solubility, branched 2-ethylhexyl side chains were employed as the bridging groups. The starting material 3,3′-dibromo-5,5′-bis(trimethylsilyl)-2,2′-biselenophene 1 was prepared as reported by the base catalysed rearrangement and subsequent oxidative coupling of 5-bromo-2-trimethylsilylselenophene.13b Our initial attempts to synthesise DSG repeated the conditions used to prepare the DTG, namely the treatment of 1 with 2.2 equivalents of n-BuLi in THF at −78 °C, followed by the addition of the dibromo-di-(2-ethylhexyl)germane. However only very low yields of DSG were isolated under these conditions, with the majority of the mass balance being decomposition products, presumably formed by the ring opening of the lithiated selenophene.19 Cooling the reaction to −100 °C improved the stability of the 3-lithiated selenophene, and the reaction proceeded smoothly under these conditions. Purification of the crude product was complicated by the proto-desilylation of the trimethylsilyl groups during column chromatography over silica. Therefore, the crude mixture of partially desilylated diselenogermoles resulting from the reaction was brominated directly with excess NBS. The intermediate dibromodiselenogermole (2) could be readily purified by column chromatography, and was isolated in a yield of 52% from 1.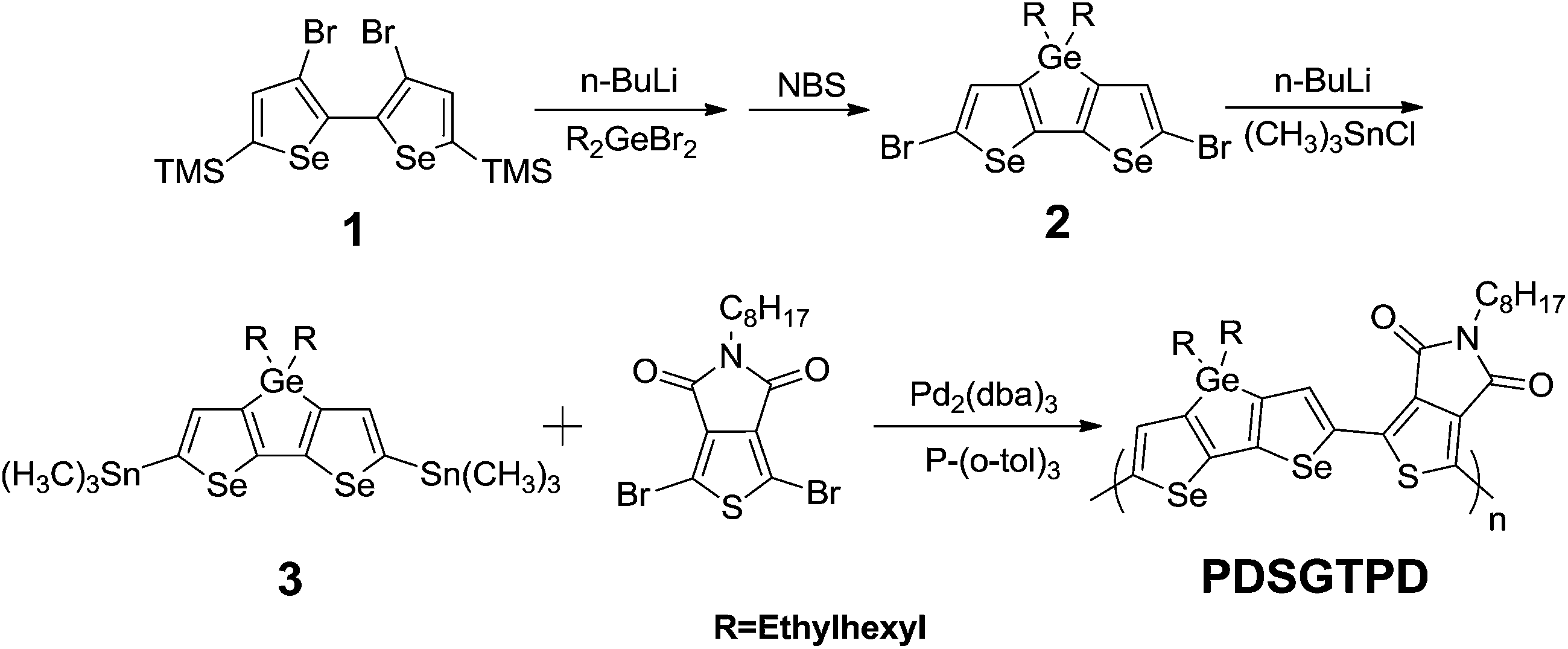 | ||
| Scheme 1 Synthetic route to PDSGTPD. | ||
The dibromodiselenogermole (2) was stannylated by treatment with 2.4 equivalents of n-BuLi again at −100 °C, followed by the addition of trimethyltin chloride. Similar to the thiophene analogue, the crude product destannylated on normal phase silica, so we purified by preparative gel permeation chromatography on cross-linked polystyrene to afford the final monomer 3 in a yield of 84%. Polymerisation of 3 and 1,3-dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione was performed by Stille coupling under microwave-assisted condition.20 After precipitation and solvent extraction to remove lower weight oligomers and catalyst residues, the polymer was purified by preparative GPC (chlorobenzene) and reprecipitation. The resulting polymer exhibited reasonable solubility in chlorinated solvents like chlorobenzene upon gentle warming. PDSGTPD had a number average molecular weight (Mn) of 41 KDa with a PDI of 1.5 as determined by GPC against polystyrene standards. Differential scanning calorimetry (DSC) (Fig. S1†) showed no obvious thermal transitions for the polymer upon cycling between 0 and 300 °C.
The UV-vis absorption spectra of PDSGTPD in dilute chlorobenzene (CB) and as a spun-cast thin film are shown in Fig. 1. The polymer exhibits an absorption maximum at 722 nm with a pronounced shoulder at 661 nm in solution, and the thin film exhibits a very similar spectral shape, with a modest red shift in λmax to 728 nm, with the shoulder at 662 nm. The separation of peaks of approximately 0.15 eV is consistent with a vibronic progression, as observed in many ordered semiconducting polymers. The similarity between the solution and thin film spectra suggest polymer aggregation in solution. The UV spectra of the analogous thiophene polymer PDTGTPD9a,21 are included for comparison in Fig. 1. It can be seen that PDSGTPD exhibits a near identical spectral shape but both absorption peaks are red-shifted by approximately 50 nm. The extinction coefficient of the polymer in solution (ε 58000 cm−1 at λmax) is approximately equal to that reported for the thiophene analogue (ε 51900 cm−1 at λmax) of similar molecular weight in chlorobenzene under equivalent dilution.21 The absorption onset of PDSGTPD in the solid state is 775 nm, corresponding to an optical band gap of 1.60 eV. This is 0.1 eV less than the optical band gap of the thiophene analogue. The red shift in the absorption peaks and reduction in optical band gap, are consistent with previous experimental and theoretical studies on selenophene substitution.10–12
 | ||
| Fig. 1 UV-vis spectra of PDTGTPD and PDSGTPD in dilute chlorobenzene and as spun cast thin films. | ||
The ionization potential of a thin film of this polymer was measured by photo electron spectroscopy in air (PESA) to be 5.12 (±0.05) eV. This is equivalent, within experimental error, to the value measured by the same technique for films of the thiophene analogue (I.P. = 5.1 ± 0.05 eV).21 This suggests that the most significant effect of replacing thiophene for selenophene is to stabilize the polymer LUMO, leading to the observed reduction in band gap. This observation is further supported by cyclic voltammetry (CV) measurements of the films on glassy carbon electrodes in anhydrous, degassed solutions of acetonitrile with tetrabutylammonium hexafluorophosphate (0.1 M) electrolyte. The cyclic voltammogram of the polymer is shown in Fig. S2† (potentials were measured against an Ag/Ag + reference electrode). The HOMO and LUMO levels were estimated to −5.29 and −3.40 eV, respectively (by the onset of the first oxidation and reduction potential, assuming the ferrocene/ferrocenium reference redox system is 4.8 eV below the vacuum level). This compares to values of 5.28 and 3.18 eV measured for the analogous thiophene polymer by CV.9a
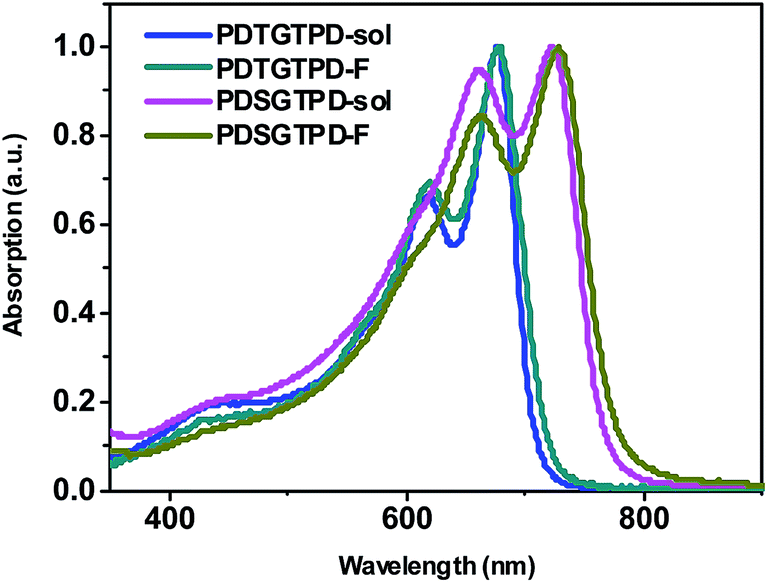
In order to further probe the influence of the heteroatom on the polymer structure and energy levels, quantum mechanical calculations at the B3LYP/6-31G* level of theory were performed on model trimers using Gaussian 09. The side chains were modified to methyl groups in order to simplify the calculations. Frequency calculations were performed on the lowest energy conformations to ensure the geometry was not the result of a local minimum. For comparative purposes, the thiophene analogue DTGTPD was also calculated, and the structure is shown in the ESI.† The minimum energy conformation of the DSGTPD trimer (Fig. 2a) reveals the planar structure of this molecule. The thiophene analogue is similarly fully coplanar (Fig. S3†) The HOMO and LUMO of DSGTPD are predicted to be delocalized over the entire conjugated backbones (Fig. 2b and c), although due to the asymmetry of the oligomer, a clear edge effect is observed with the HOMO localizing towards the electron rich DSG end and the LUMO towards the electron deficient TPD end. The HOMO is predominantly aromatic and the LUMO has quinoidal character. Comparing the selenophene to the thiophene polymer, the HOMO is predicted to rise in energy slightly (−4.87 eV for DSGversus −4.91 eV for DTG) upon selenophene inclusion, and the LUMO lower (−3.03 eV for DSGversus −2.98 eV for DTG), resulting in a net reduction of band gap of 0.09 eV, which is close to the shift in the optical gap observed experimentally. The small difference in HOMO levels is within the experimental error of our PESA experiments, although as discussed later, the selenophene polymer does have a slightly lower open circuit voltage in solar cells, consistent with a slightly higher lying HOMO level.
 | ||
| Fig. 2 Energy-minimized conformation (B3LYP/6-31G*) (a), HOMO (b) and LUMO (c) distributions of a methyl substituted DSGTPD trimer. | ||
Interestingly the DFT calculations also predict subtle differences in the molecular geometries of the two polymers. For comparative purposes we base our discussions here on the central DSG or DTG unit of the modeled trimers, since this is most representative of a section of the polymer chain and minimizes the edge effects mentioned above. As expected the inclusion of the larger Se atom, leads to a lengthening of the C–X (S or Se) bond in the fused unit, from 1.77 Å in DTG to 1.90 Å in DSG. The longer C–Se bond also has the effective of making the DSG unit more linear in comparison to DTG, thus the angle between the 5,5′ positions is approximately 28° for DSG and 36° for DTG (see Fig. S4†). The lengthening of C–Se bond is also predicted to place the Se in slightly closer proximity (2.94 Å) to the oxygen of the carbonyl of the TPD, than for DTG (2.99 Å) despite the larger size of Se. In both cases the predicted interaction is less than the sum of the van der Waal's radii, and although we note caution should be applied to the interpretation of DFT results, they do suggest that the inclusion of the Se does not cause significant disruption to the planarity of the backbone.
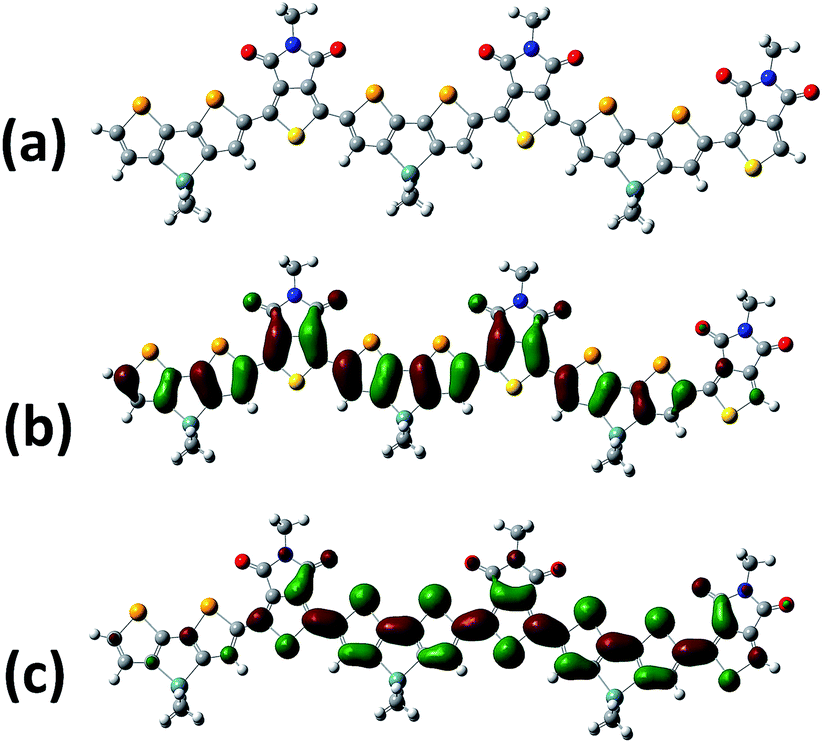
The charge carrier transport behavior of PDSGTPD was investigated via electrical field-effect measurements using bottom-gate, top-contact transistor architectures made on doped heavily doped Si++ wafers as the gate electrode and a 400 nm-thick thermally oxidized silicon dioxide as the gate dielectric layer. The dielectric was treated with HMDS or OTS prior to deposition of the semiconductor, with the best results obtained for OTS treated SiO2 based transistors. The devices exhibit typical p-type behavior, with typical transfer and output characteristics shown in Fig. 3. The polymers exhibited average as cast saturated and linear mobilities of 8.4 × 10−3 cm2 V s−1 and 2.6 × 10−3 cm2 V s−1, respectively, with a peak saturated hole mobility of ∼1 × 10−2 cm2 V s−1. The bottom-gate, top-contact performance of the analogous thiophene polymer has not been reported, but the transistor performance of the thiophene polymer with a silicon bridge rather than a germanium bridge has been reported in this device configuration. In that case, an average saturated mobility of 6 × 10−4 cm2 V s−1 was observed.22 The hole mobility observed for PDSGTPD in TFTs suggested that the polymer would have reasonable transport characteristics as a donor in BHJ solar cells.
 | ||
| Fig. 3 Transfer (left) and output (right) characteristics of bottom gate, top contact organic field-effect transistor with channel length = 30 μm and channel width = 1 mm under N2. | ||
The photovoltaic performance of PDSGTPD was investigated in devices using both conventional (ITO/PEDOT:PSS/PDSGTPD:PC71BM/LiF/Al) and inverted (ITO/ZnO/PDSGTPD:PC71BM/MoO3/Ag) device structures and their power conversion efficiency was measured under 100 mW cm2 AM 1.5 illumination. Polymer:fullerene blends were solution processed from o-dichlorobenzene (ODCB), and due to the low band gap absorption of the polymer, PC71BM was used to complement and capture the low wavelength of the visible spectrum.23 The polymer was initially tested in a 1:1, 1:2, 1:3 and 1:4 weight ratio against PC71BM in the conventional device structure. Initial performance was rather poor, with the 1:2 blend giving the best efficiency around 1.5%. The addition of 4 vol% of 1,8-diiodooctane (DIO) as a processing additive24 resulted in a significant improvement in performance, with device efficiency improving to 4.6% (Fig. S5†).
In an effort to further improve performance, inverted devices were also fabricated using the optimal 1:2 ratio. Fig. 4 shows the external quantum efficiency (EQE) corrected current density vs. voltage (J–V) curves along with the actual EQE spectra, and the data is summarized in Table 1. Inverted devices showed improved efficiencies over the conventional architecture, both for films fabricated with and without DIO (Table 1). Slightly lower volumes of DIO gave the best performance in the inverted devices. The highest performing devices had an efficiency of 5.23%, with reasonable Jsc (12.2 mA cm−2) and voltage (Voc) but a moderate fill factor of 0.58.
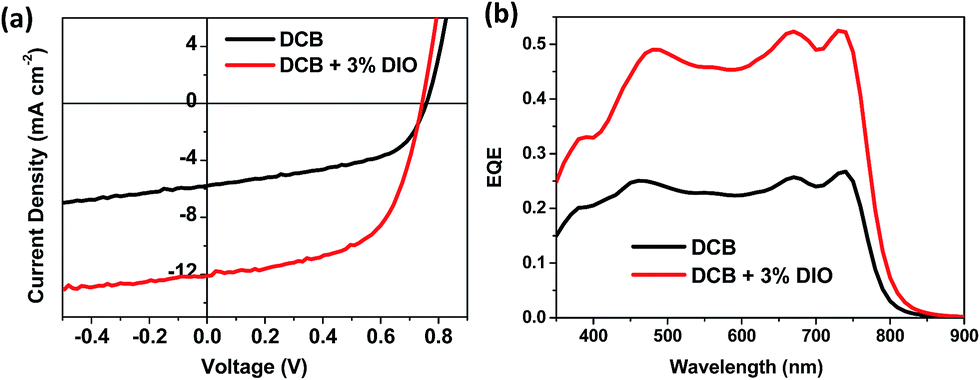 | ||
| Fig. 4 (a) EQE corrected J–V curve and (b) EQE of inverted polymer solar cells based on PDSGTPD:PC71BM blends (1:2 wt%) with or without solvent additives. | ||
:2 ratio (PDSGTPD:PC71BM) with or without additive. The average values of 6 devices values are shown, with the best performing device shown in parenthesis
|
PDSGTPD:PC71BM (1:2) |
J sc mA cm−2 | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|
| a EQE corrected. | ||||
| DCB | 5.8 ± 0.1 (5.9) | 0.76 ± 0.01 (0.76) | 0.50 ± 0.02 (0.51) | 2.2 ± 0.1 (2.3) |
| DCB + 3% DIO | 12.1 ± 0.15 (12.2) | 0.74 ± 0.01 (0.74) | 0.58 ± 0.02 (0.58) | 5.2 ± 0.1 (5.2) |
The external quantum efficiency (EQE) curves for the best devices are depicted in Fig. 4. Both devices show a broad spectral response in the 450–800 nm region, with EQE values doubled for devices with 3% DIO compared to the one without any additive. The EQE response in the 650–800 nm region closely resembles the absorption of the polymer spectrum, suggesting that significant photocurrent is generated from the polymer.
It is instructive to compare the performance of the PDSGTPD devices to that reported for the thiophene analogue (PDTGTPD) in blends with PC71BM. Reynolds and So found that PDTGTPD exhibited the best performance in a 1:1.5 ratio with PC71BM, in this instance for films cast from chlorobenzene (CB) with 5% vol of DIO.9a,25 They found a similar photocurrent (12.6 mA cm−2), but a significantly higher voltage (Voc 0.85 V) and FF (0.68) leading to high efficiencies of 7.3%. Two studies of PDTGTPD:PC71BM 1:2 wt% blends processed from CB/DIO in conventional device structure have also been reported.9j,26 They both found slightly reduced performance, with photocurrents of 9.7 or 10.4 mA cm−2, voltages of 0.80 or 0.81 V and fill factors of 0.53 or 0.59 leading to overall efficiency of 4.1 or 4.9%. It is difficult to fully determine the origin of the variation of the performance due to differences in processing conditions, film thicknesses and polymer molecular weights between the studies. Nevertheless in all three cases in it clear that PDTGTPD exhibits higher open circuit voltages than the selenophene polymer, in agreement with the DFT calculations, which suggest a slightly higher lying HOMO for the selenophene polymer. Similar reductions in voltage upon selenophene substitution have been previously reported.12n,12y The photocurrent of PDSGTPD is also already close to the best reported for the thiophene polymer under highly optimised processing conditions, suggesting that the red shift of the absorption upon selenophene substitution can help improve photocurrent. Further improvements in the performance of PDSGTPD may be obtainable by further device optimisation.
In order to probe the role of the DIO, atomic force microscopy (AFM) was used to investigate the surface morphology of the blends of PDSGTPD:PC71BM (1:2) with and without solvent additives, and their topography images are shown in Fig. 5. The AFM images show clear differences in the surface topography, with the DIO processed film exhibiting a much finer and more homogeneous distribution of feature sizes than the film without the additive. The surface roughness of the films with and without DIO are 2.69 and 4.55 nm, respectively. The rather large domains and increased surface roughness in the absence of DIO are suggestive of poor intermixing between the fullerene and the polymer, which is generally considered non-optimal for good device performance.
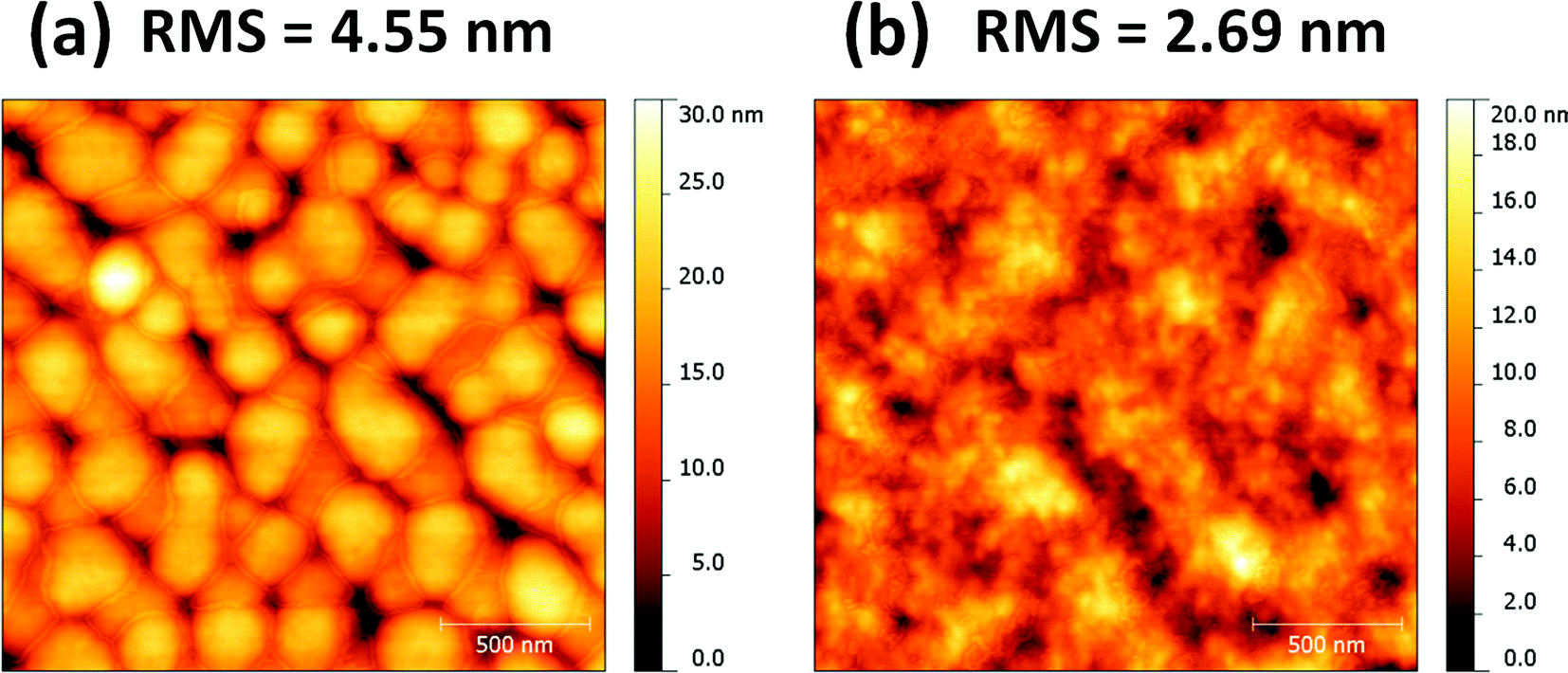 | ||
| Fig. 5 AFM topography images (2 × 2 μm) based on PDSGTPD:PC71BM blends (1:2 wt%) without (a) and with (b) solvent additives. | ||
In conclusion, we report the preparation of a novel diselenogermole (DSG) monomer and its polymerisation with 1,3-dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione by Stille polycondensation to afford a soluble low bad gap polymer. The polymer shows a broader and more red-shifted absorption spectrum compared to its thiophene analogue. The resulting polymer exhibits initial power conversion efficiencies of 5.2% in inverted bulk heterojunction solar cells, demonstrating that the DSG monomer is a promising building block for the design of new high performance conjugated polymers.
Acknowledgements
This work was supported by EPSRC grants EP/G060738/1 and EP/I002936/1, and NSF ICC Award CHE1026664.References
- (a) Y. He, W. Hong and Y. Li, J. Mater. Chem. C, 2014, 2, 8651 RSC; (b) K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2014, 26, 587 CrossRef CAS.
- (a) I. McCulloch, R. S. Ashraf, L. Biniek, H. Bronstein, C. Combe, J. E. Donaghey, D. I. James, C. B. Nielsen, B. C. Schroeder and W. M. Zhang, Acc. Chem. Res., 2012, 45, 714 CrossRef CAS PubMed; (b) C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326 CrossRef CAS PubMed; (c) X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS PubMed; (d) P. Coppo and M. L. Turner, J. Mater. Chem., 2005, 15, 1123 RSC; (e) J. S. Wu, S. W. Cheng, Y. J. Cheng and C. S. Hsu, Chem. Soc. Rev., 2014 10.1039/c4cs00250d.
- P. Carbone and A. Troisi, J. Phys. Chem. Lett., 2014, 5, 2637 CrossRef CAS.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605 CrossRef CAS PubMed.
- D. Muehlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Guadiana and C. Brabec, Adv. Mater., 2006, 18, 2884 CrossRef CAS.
- G. L. Gibson and D. S. Seferos, Macromol. Chem. Phys., 2014, 215, 811 CrossRef CAS.
- (a) M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367 CrossRef CAS PubMed; (b) H. Y. Chen, J. H. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371 CrossRef CAS PubMed; (c) J. S. Kim, Z. P. Fei, D. T. James, M. Heeney and J. S. Kim, J. Mater. Chem., 2012, 22, 9975 RSC; (d) J. S. Kim, Z. Fei, S. Wood, D. T. James, M. Sim, K. Cho, M. J. Heeney and J. S. Kim, Adv. Energy Mater., 2014, 1400116 Search PubMed.
- (a) C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062 CrossRef CAS PubMed; (b) Z. Fei, J. S. Kim, J. Smith, E. B. Domingo, T. D. Anthopoulos, N. Stingelin, S. E. Watkins, J.-S. Kim and M. Heeney, J. Mater. Chem., 2011, 21, 16257 RSC; (c) Y.-M. Hwang, J. Ohshita, Y. Harima, T. Mizumo, Y. Ooyama, Y. Morihara, T. Izawa, T. Sugioka and A. Fujita, Polymer, 2011, 52, 3912 CrossRef CAS PubMed; (d) X. Guo, N. Zhou, S. J. Lou, J. W. Hennek, R. Ponce Ortiz, M. R. Butler, P.-L. T. Boudreault, J. Strzalka, P.-O. Morin, M. Leclerc, J. T. Lopez Navarrete, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 18427 CrossRef CAS PubMed; (e) Z. P. Fei, Y. Kim, J. Smith, E. B. Domingo, N. Stingelin, M. A. McLachlan, K. Song, T. D. Anthopoulos and M. Heeney, Macromolecules, 2012, 45, 735 CrossRef CAS; (f) Z. P. Fei, M. Shahid, N. Yaacobi-Gross, S. Rossbauer, H. L. Zhong, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Commun., 2012, 48, 11130 RSC; (g) Q. Wang, S. Zhang, L. Ye, Y. Cui, H. Fan and J. Hou, Macromolecules, 2014, 47, 5558 CrossRef CAS; (h) C. P. Yau, Z. Fei, R. S. Ashraf, M. Shahid, S. E. Watkins, P. Pattanasattayavong, T. D. Anthopoulos, V. G. Gregoriou, C. L. Chochos and M. Heeney, Adv. Funct. Mater., 2014, 24, 678 CrossRef CAS; (i) F.-B. Zhang, J. Ohshita, M. Miyazaki, D. Tanaka and Y. Morihara, Polym. J., 2014, 46, 628 CrossRef CAS; (j) D. Gendron, P.-O. Morin, P. Berrouard, N. Allard, B. R. Aich, C. N. Garon, Y. Tao and M. Leclerc, Macromolecules, 2011, 44, 7188 CrossRef CAS; (k) J. Ohshita, M. Nodono, T. Watanabe, Y. Ueno, A. Kunai, Y. Harima, K. Yamashita and M. Ishikawa, J. Organomet. Chem., 1998, 553, 487 CrossRef CAS; (l) H. Usta, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 9034 CrossRef CAS PubMed; (m) J. W. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS; (n) L. Liao, L. Dai, A. Smith, M. Durstock, J. Lu, J. Ding and Y. Tao, Macromolecules, 2007, 40, 9406 CrossRef CAS; (o) P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Muellen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514 CrossRef CAS PubMed; (p) J. Ding, N. Song and Z. Li, Chem. Commun., 2010, 46, 8668 RSC; (q) C. Cui, X. Fan, M. Zhang, J. Zhang, J. Min and Y. Li, Chem. Commun., 2011, 47, 11345 RSC; (r) P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J.-L. Bredas, F. So, K. Muellen and J. R. Reynods, J. Am. Chem. Soc., 2012, 134, 8944 CrossRef CAS PubMed; (s) T.-Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345 CrossRef CAS; (t) X. Guo, N. Zhou, S. J. Lou, J. W. Hennek, R. Ponce Ortiz, M. R. Butler, P.-L. T. Boudreault, J. Strzalka, P.-O. Morin, M. Leclerc, J. T. López Navarrete, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 18427 CrossRef CAS PubMed; (u) J. Min, Z.-G. Zhang, M. Zhang and Y. Li, Polym. Chem., 2013, 4, 1467 RSC.
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422 RSC.
- S. S. Zade, N. Zamoshchik and M. Bendikov, Chem.–Eur. J., 2009, 15, 8613 CrossRef CAS PubMed.
- (a) M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061 RSC; (b) M. Al-Hashimi, M. A. Baklar, F. Colleaux, S. E. Watkins, T. D. Anthopoulos, N. Stingelin and M. Heeney, Macromolecules, 2011, 44, 5194 CrossRef CAS; (c) J. Hollinger, D. Gao and D. S. Seferos, Isr. J. Chem., 2014, 54, 440 CrossRef CAS; (d) A. Patra, R. Kumar and S. Chand, Isr. J. Chem., 2014, 54, 621 CrossRef CAS; (e) A. A. B. Alghamdi, D. C. Watter, H. Yi, S. Al-Faifi, M. S. Almeataq, D. Coles, J. Kingsley, D. G. Lidzey and A. Iraqi, J. Mater. Chem. A, 2013, 1, 5165 RSC; (f) D. Gao, J. Hollinger, A. A. Jahnke and D. S. Seferos, J. Mater. Chem. A, 2014, 2, 6058 RSC; (g) H. Yan, J. Hollinger, C. R. Bridges, G. R. McKeown, T. Al-Faouri and D. S. Seferos, Chem. Mater., 2014, 26, 4605 CrossRef CAS; (h) A. Bedi, S. P. Senanayak, K. S. Narayan and S. S. Zade, Macromolecules, 2013, 46, 5943 CrossRef CAS; (i) H. Kong, Y. K. Jung, N. S. Cho, I.-N. Kang, J.-H. Park, S. Cho and H.-K. Shim, Chem. Mater., 2009, 21, 2650 CrossRef CAS; (j) Z. Y. Chen, H. Lemke, S. Albert-Seifried, M. Caironi, M. M. Nielsen, M. Heeney, W. M. Zhang, I. McCulloch and H. Sirringhaus, Adv. Mater., 2010, 22, 2371 CrossRef CAS PubMed; (k) J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364 CrossRef CAS PubMed; (l) W.-H. Lee, S. K. Son, K. Kim, S. K. Lee, W. S. Shin, S.-J. Moon and I.-N. Kang, Macromolecules, 2012, 45, 1303 CrossRef CAS; (m) H. A. Saadeh, L. Lu, F. He, J. E. Bullock, W. Wang, B. Carsten and L. Yu, ACS Macro Lett., 2012, 1, 361 CrossRef CAS; (n) M. Shahid, R. S. Ashraf, Z. Huang, A. J. Kronemeijer, T. McCarthy-Ward, I. McCulloch, J. R. Durrant, H. Sirringhaus and M. Heeney, J. Mater. Chem., 2012, 22, 12817 RSC; (o) C.-C. Chang, C.-P. Chen, H.-H. Chou, C.-Y. Liao, S.-H. Chan and C.-H. Cheng, J. Polym. Sci., Part A-1: Polym. Chem., 2013, 51, 4550 CAS; (p) T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960 CrossRef CAS PubMed; (q) I. Kang, T. K. An, J.-a. Hong, H.-J. Yun, R. Kim, D. S. Chung, C. E. Park, Y.-H. Kim and S.-K. Kwon, Adv. Mater., 2013, 25, 524 CrossRef CAS PubMed; (r) A. E. Rudenko, S. Noh and B. C. Thompson, Nanotechnology, 2013, 24, 484002 CrossRef PubMed; (s) J. Cao, S. Chen, Z. Qi, Z. Xiao, J. Wang and L. Ding, RSC Adv., 2014, 4, 5085 RSC; (t) J.-H. Kim, J. B. Park, S. A. Shin, M. H. Hyun and D.-H. Hwang, Polymer, 2014, 55, 3605 CrossRef CAS PubMed; (u) R. L. Uy, L. Yan, W. Li and W. You, Macromolecules, 2014, 47, 2289 CrossRef CAS; (v) A. M. Ballantyne, L. C. Chen, J. Nelson, D. D. C. Bradley, Y. Astuti, A. Maurano, C. G. Shuttle, J. R. Durrant, M. Heeney, W. Duffy and I. McCulloch, Adv. Mater., 2007, 19, 4544 CrossRef CAS; (w) M. Shahid, T. McCarthy-Ward, J. Labram, S. Rossbauer, E. B. Domingo, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2012, 3, 181 RSC; (x) J. H. Bannock, M. Al-Hashimi, S. H. Krishnadasan, J. J. M. Halls, M. Heeney and J. C. de Mello, Mater. Horiz., 2014, 1, 214 RSC; (y) K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130 CrossRef CAS PubMed; (z) H.-Y. Chen, S.-C. Yeh, C.-T. Chen and C.-T. Chen, J. Mater. Chem., 2012, 22, 21549 RSC; (a a) K. A. Mazzio, M. Yuan, K. Okamoto and C. K. Luscombe, ACS Appl. Mater. Interfaces, 2011, 3, 271 CrossRef CAS PubMed; (a b) Z. Zeng, Y. Li, J. Deng, Q. Huang and Q. Peng, J. Mater. Chem. A, 2014, 2, 653 RSC; (a c) L. Dou, W.-H. Chang, J. Gao, C.-C. Chen, J. You and Y. Yang, Adv. Mater., 2013, 25, 825 CrossRef CAS PubMed; (a d) J. Warnan, A. El Labban, C. Cabanetos, E. T. Hoke, P. K. Shukla, C. Risko, J.-L. Brédas, M. D. McGehee and P. M. Beaujuge, Chem. Mater., 2014, 26, 2299 CrossRef CAS.
- (a) Y. A. Getmanenko, T. A. Purcell, D. K. Hwang, B. Kippelen and S. R. Marder, J. Org. Chem., 2012, 77, 10931 CrossRef CAS PubMed; (b) Y. A. Getmanenko, P. Tongwa, T. V. Timofeeva and S. R. Marder, Org. Lett., 2010, 12, 2136 CrossRef CAS PubMed.
- (a) Y. A. Getmanenko, C. Risko, P. Tongwa, E.-G. Kim, H. Li, B. Sandhu, T. Timofeeva, J.-L. Brédas and S. R. Marder, J. Org. Chem., 2011, 76, 2660 CrossRef CAS PubMed; (b) S. Tarutani and K. Takahashi, Bull. Chem. Soc. Jpn., 2004, 77, 463 CrossRef CAS.
- S. V. Kesava, Z. Fei, A. D. Rimshaw, C. Wang, A. Hexemer, J. B. Asbury, M. Heeney and E. D. Gomez, Adv. Energy Mater., 2014, 4, 1400116 Search PubMed.
- H. S. Chandak and S. S. Zade, Org. Electron., 2014, 15, 2184 CrossRef CAS PubMed.
- (a) C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595 CrossRef CAS PubMed; (b) J. Warnan, C. Cabanetos, A. E. Labban, M. R. Hansen, C. Tassone, M. F. Toney and P. M. Beaujuge, Adv. Mater., 2014, 26, 4357 CrossRef CAS PubMed; (c) H. L. Zhong, Z. Li, F. Deledalle, E. C. Fregoso, M. Shahid, Z. P. Fei, C. B. Nielsen, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, J. R. Durrant and M. Heeney, J. Am. Chem. Soc., 2013, 135, 2040 CrossRef CAS PubMed; (d) D. H. Wang, A. Pron, M. Leclerc and A. J. Heeger, Adv. Funct. Mater., 2013, 23, 1297 CrossRef CAS.
- C. B. Nielsen and T. Bjørnholm, Org. Lett., 2004, 6, 3381 CrossRef CAS PubMed.
- S. Gronowitz and T. Frejd, Acta Chem. Scand., 1970, 24, 2656 CrossRef CAS PubMed.
- S. Tierney, M. Heeney and I. McCulloch, Synth. Met., 2005, 148, 195 CrossRef CAS PubMed.
- C. P. Yau, S. Wang, N. D. Treat, B. J. Tremolet de Villers, M. L. Chabinyc and M. Heeney, Adv. Energy Mater., 2014, 1400116 Search PubMed.
- Y. Zhang, J. Zou, H.-L. Yip, Y. Sun, J. A. Davies, K.-S. Chen, O. Acton and A. K. Y. Jen, J. Mater. Chem., 2011, 21, 3895 RSC.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371 CrossRef CAS PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115 CrossRef CAS.
- Y. Zhang, J. Zou, H.-L. Yip, Y. Sun, J. A. Davies, K.-S. Chen, O. Acton and A. K. Y. Jen, J. Mater. Chem., 2011, 21, 3895 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta05703a |
| This journal is © The Royal Society of Chemistry 2015 |