
Nanocomposites based on Hofmann-type structure NiII(pz)[NiII(CN)4] (pz = pyrazine) nanoparticles for reversible iodine capture†
G.
Massasso
a,
M.
Rodriguez-Castillo
a,
J.
Long
a,
A.
Grandjean
b,
B.
Onida
c,
Y.
Guari
*a,
Ch.
Guerin
a and
J.
Larionova
a
aInstitut Charles Gerhardt Montpellier, ICGM, UMR 5253 CNRS-UM2-ENSCM-UM1, Chimie Moléculaire et Organisation du Solide, Université Montpellier II, Place E. Bataillon, 34095, Montpellier cedex 5, France. E-mail: yannick.guari@um2.fr
bCEA/DEN/DTCD/SPDE, Laboratoire des Procédés Supercritiques et de Décontamination, BP 17171, 30207 Bagnols sur Cèze, France
cInstitute of Chemistry, Department of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
First published on 22nd October 2014
Abstract
Hybrid nanocomposites based on nanoparticles with the Hofmann-type structure NiII(pz)[NiII(CN)4] (where pz = pyrazine) confined into mesoporous silica or porous glass pearls were synthesised by sequential coordination of the molecular precursors into the pores of the functionalized matrices. Infrared (IR) and UV/Visible (UV-Vis) spectroscopy, powder X-ray diffraction (PXRD), and transmission electron microscopy (TEM) reveal the presence of uniformly-sized spherical NiII(pz)[NiII(CN)4] nanoparticles of 3–6 nm, which are homogeneously dispersed into the matrices. These nanocomposites are able to efficiently capture iodine from cyclohexane solutions with a maximum sorption capacity of 1.75 mmol per g of material. A particular emphasis is given on the mechanism of iodine sorption as well as on the sorption cycling ability of the materials.
Introduction
Radioactive iodine isotopes produced during the fission of 235U and in particular the γ-ray emitter 129I, which have a long half-life (1.57 × 107 years), are dangerous for health due to their volatility, toxicity and persistence in the environment.1 Nuclear industry attaches a great deal of attention to their efficient and selective capture during fuel reprocessing or in case of off-normal working conditions in order to minimize their environmental impact. Several processes for iodine treatment in its elemental and diatomic (I2) form are currently used in reprocessing plants. These consist of the following: (i) scrubbing with an alkaline solution and the precipitation of stable and insoluble compounds, like NaI and NaIO3; (ii) the “Mercurex” process, which uses a mercuric nitrate–nitric acid solution to precipitate HgI2; or (iii) the “Iodox” process employing an hyperazeotropic nitric acid scrub solution to solubilize and oxidize iodine to (IO3)− species with their further precipitation and encapsulation as Ba(IO3).2,3 These methods present various unattractive features such as multiple-step technology, indirect iodine treatment in the liquid phase, corrosiveness, potential formation of nitrated organics, and a significant volume of radioactive wastes, which should be then retreated. Recently, direct gas phase iodine trapping using silver-impregnated zeolites with formation of insoluble and stable AgI was also applied in industry. But this process is relatively expensive and presents a limited weight capacity of 33%.4,5 For these reasons, more efficient, stable and less costly materials for iodine capture in the gas phase are required. In this line of thought, numerous research efforts were devoted to the design of new thermally stable nanoporous materials, such as aluminium-6,7 or alkali-8 zeolites, zeolite-related structures (“zeoballs”)9 or metal–organic-frameworks (MOFs)10–14 able to selectively and efficiently capture I2 in the gas and liquid phases. Recently, we also demonstrated that the thermally stable and porous cyano-bridged coordination polymer called Hofmann-type clathrate with the formula NiII(pz)[NiII(CN)4] (where pz = pyrazine) presents a high affinity to iodine capture due to a selective insertion of I2 into the cavities of the crystalline network through the formation of weak interactions with pyrazine and the cyanometallate. This compound is able to reversibly capture iodine in both the liquid and gas phase, with a maximum capacity of 1 mole of I2 per mole of Hofmann-type structure, which is higher or comparable to other efficient nanoporous materials.15However, the direct use of bulk coordination polymers as a microcrystalline powder in decontamination processes presents several drawbacks due to their instability under irradiation, and the difficulties arising from the management and storage of the ultimate radioactive wastes. In order to avoid these problems, the synthesis of nanocomposites based on coordination polymer nanoparticles inserted into an appropriate porous matrix seems to be a promising approach. In this line of thought, nanocomposite materials containing prussian blue type nanoparticles of 3–6 nm that were grown directly within mesoporous silica or porous glass pearls were recently proposed for the selective and effective Cs+ absorption in a column process.16–19 In this system, good control over the nanoparticle composition, nanoparticle size and the quantity of nanoparticles inserted into the silica matrix was obtained. The nanocomposites not only absorb caesium ions more efficiently in comparison with their bulk materials (per gram of the prussian blue), but also permit the implementation of decontamination in a column process.
The present manuscript describes the synthesis and characterization of mesoporous silica- and porous glass pearl- based nanocomposites containing covalently linked nanoparticles with a Hofmann-type structure NiII(pz)[NiII(CN)4] inside the pores and their use as an efficient sorbent of molecular iodine. The choice of these matrices is explained by the high mesoporosity of silica, as well as by the possibility of easily anchoring various functional groups to the silica walls. For these reasons, mesostructured silica can be used as a model matrix for the nanocomposites prepared in this study. On the other hand, even if porous glass pearls have a lower surface area than silica, they present better thermal and chemical stability, higher mechanical hardness and greater resistance to damage caused by irradiation.20 In addition, they are compatible with current radioactive waste management.21 The Hofmann-type structure on a nanoscale was chosen due to its high affinity to molecular iodine and its high efficiency for I2 retention (1 molecule of I2 per unit cell), its relatively high thermal stability (400 °C), the possibility of reversible iodine capture and the simplicity of synthesis at room temperature. It should be noted that the synthesis of nanoparticles with analogous structures to FeII(pz)[PtII(CN)4] was previously reported inside chitosan beads22 or by reverse micelles methods,23 but so far no one has attempted the preparation of mesoporous silica- and porous glass pearl- based nanocomposites. In this work, the NiII(pz)[NiII(CN)4] nanoparticles are covalently anchored to the pores walls of the silica-based supports through grafted diamino functionality avoiding the risk of their leaching during the iodine sorption process. Iodine uptake by these nanocomposites is carried out in a liquid cyclohexane phase to investigate the sorption kinetics and isotherms as well as to understand the sorption mechanism. The iodine sorption capacity of the nanocomposites was also compared with that of the corresponding bulk Hofmann-type structure and the parent functionalized silica matrices. A study on the potential reversibility and cyclability of the iodine capture was also performed.
Experimental section
A. Synthesis
All materials were purchased from commercially available sources, and used without further purification. Ni(BF4)2·6H2O, (C4H9)4NBr, K2[Ni(CN)4], pyrazine, HCl, TEOS, mesitylene, Pluronic 123, and 2-aminoethyl 3-aminopropyl triethoxysilane were purchased from Alfa Aesar. Iodine, cyclohexane, methanol and toluene were purchased from Sigma-Aldrich. All the reactions were carried out under aerobic conditions using analytical grade solvents. Glass pearls were purchased from Vitrabio®. The synthesis of (TBA)2[Ni(CN)4] (TBA = tetrabutylammonium) has been carried out using a metathesis reaction as previously reported.24The grafting of mesoporous silica and glass pearls was carried out under inert conditions in order to avoid oxidation of the grafting agent.29 2.00 g of the support and 2.00 g of the grafting agent (2-aminoethyl-3-aminopropyl trimethoxysilane) were mixed in a large amount (250 mL) of distilled toluene under an Ar atmosphere. Reflux conditions were set at 110 °C, for 48 hours. After filtration, the materials were washed with toluene and dried at 90 °C.
Elemental analysis. Grafted silica (Sil): Si, 38.18%; C, 10.50%; N, 4.11%; H, 2.37% (grafting of 17 wt% organic groups).
Grafted glass pearls (Glass): Si, 41.84%; C, 5.45%; N, 2.16%; H, 1.42% (grafting of 9 wt% organic groups).
Elemental analysis. Sil@NP: Si, 29.35%; Ni, 14.58%; C, 14.79%; N, 10.76%; H, 2.18%.
Glass@NP: Si, 29.47%; Ni, 7.00%; C, 9.44%; N, 5.18%; H, 1.23%.
In order to determine the sorption isotherms, the materials were kept in dynamic contact with different concentrations of iodine i.e. 2.6 × 10−3, 8 × 10−3, 1.5 × 10−2, 4 × 10−2, and 7 × 10−2 M in cyclohexane (the iodine solubility limit in cyclohexane is around 8 × 10−2 M, so that it was not possible to use higher concentrations), at an equilibrium time determined in kinetic experiments (24 h and 48 h for nanocomposites and functionalized matrices, respectively). Experiments were performed with a powder-solution ratio of 1 mg mL−1. The amount of iodine absorbed in materials was determined by measuring the residual concentration of iodine in solution by UV-Vis spectroscopy at the end of the experiment. After sorption, the materials were placed in a cartridge in a Soxhlet apparatus and thoroughly washed with cyclohexane for 24 h in order to remove the surface absorbed iodine.
B. Characterization methods
Thermal analyses were performed using a STA 409 Luxx Instrument under nitrogen flow with a heating rate of 5 °C min−1. Infrared spectra were recorded on a Perkin Elmer 1600 spectrometer with a 4 cm−1 resolution in KBr disks. UV-Vis spectra were recorded on an Analityk Jena Specord 210 spectrometer. Elemental analyses were performed by the Service Central d'Analyse (CNRS, Villeurbanne, France). Nitrogen and carbon content in the samples was determined using a LECO instrument. The samples were heated at 3000 °C under oxygen. Nitrogen and carbon were transformed into NOx and CO, respectively and detected using an IR detector. Iodine content was determined by measuring the residual concentration of iodine in solution using UV-Visible spectroscopy. Powder X-ray diffraction patterns were measured on a Bruker® D8 advanced diffractometer in Bragg-Bentano geometry with Ni-filtered Cu-Kα radiation. The measurement parameters are: step size 0.02008; counting time 15 s. Samples for transmission electron microscopy (TEM) measurements were prepared using extractive replicas or ultramicrotomy techniques. The extractive replica technique consists of depositing a suspension of the nanocomposite in ethanol onto a freshly cleaved mica plate. After evaporation of ethanol, a carbon film is deposited onto the mica plate. Then, the latter is immersed in a dilute HF solution. The carbon film was then detached from the mica plate and floats on the surface of the solution, which allows dissolving the silica part of the nanocomposite whilst keeping the Hofmann-type clathrate nanoparticles on the carbon film. After washing twice, the carbon film was deposited onto copper grids for TEM observations. Thus, this technique allows visualization of the Hofmann-type structure nanoparticles after the removal of silica. The ultramicrotomy technique consists of suspending the material in a resin, which is polymerized at low temperature (i.e. 70 °C), then slices of ca. 60 to 100 nm are cut with an ultramicrotome apparatus equipped with a diamond knife. This technique allows the visualization of the silica matrix especially because of the slow difference of the electronic density between the matrix and nanoparticles. TEM measurements were carried out at 100 kV with a JEOL 1200 EXII microscope. The nanoparticle size distribution histograms were determined using enlarged TEM micrographs taken at a magnification of ×50k. A large number of nanoparticles were counted in order to obtain a size distribution with good statistics. Scanning electron microscopy measurements were performed using a Philips Quant 200 operating at 15 kV equipped with a Bruker detector. System software for EDX analysis was developed by Bruker. The surface area was obtained using nitrogen adsorption isotherms on an ASAP2020 analyser from Micromeritics. Samples were outgassed under vacuum at 60 °C overnight prior to analysis. The surface area was determined using the Brunauer–Emmet–Teller (BET) method.Results and discussion
A. Synthesis and characterisation of the silica-based nanocomposites containing Hofmann-type structure NiII(pz)[NiII(CN)4] nanoparticles
The synthesis of the hybrid nanocomposites supporting NiII(pz)[NiII(CN)4] Hofmann-type structure nanoparticles confined into both mesoporous silica or porous glass pearls was performed using a multistep approach consisting of the successive coordination of the molecular precursors to the functionalized silica matrix (porous silica or glass pearls) (Fig. 1 and S1, ESI†).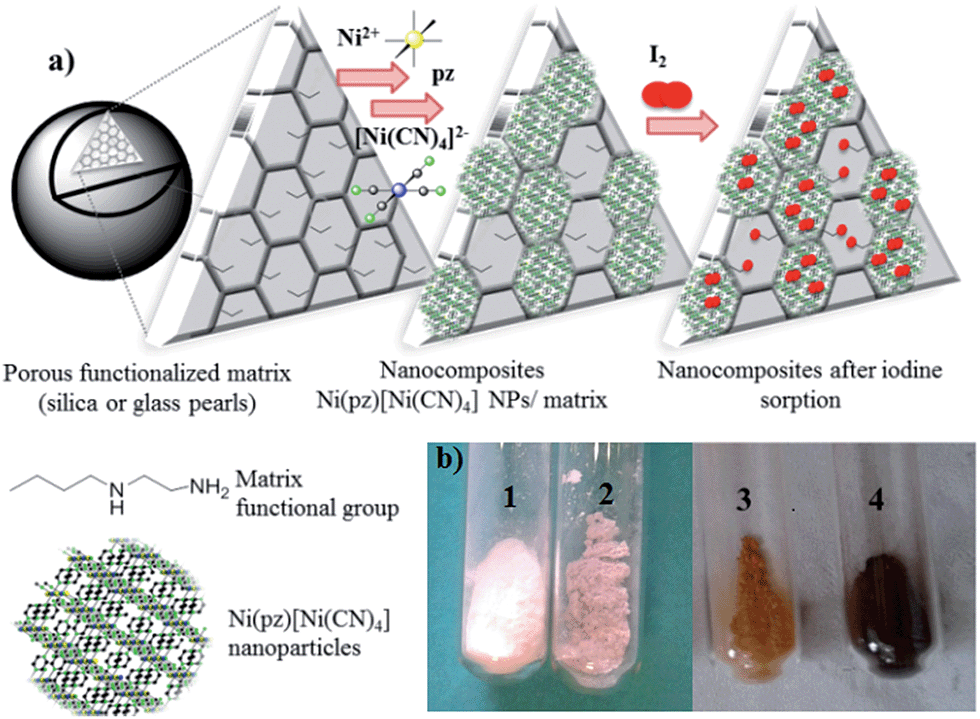 | ||
| Fig. 1 (a) A schematic representation of the approach used for the synthesis of silica-based nanocomposites containing Hofmann-type structure nanoparticles and their employment for iodine capture; (b) photographs of: (1) functionalised silica (Sil), (2) the silica-based nanocomposite Sil@NP, (3) functionalised silica (Sil) after iodine capture, (4) the silica-based nanocomposite Sil@NP after iodine capture. | ||
In the first step, the mesoporous silica and glass pearls were functionalized with a diamine functionality (matrix functional group in Fig. 1).30 The diamine-grafted mesoporous silica (Sil) was prepared as described in the Experimental section. Its preparation differs from a typical SBA-15 (ref. 27 and 28) synthesis due to the use of mesitylene that allows an open and disordered porous network with larger pores to be achieved, as can be seen by the transmission electronic microscopy (TEM) image shown in Fig. 2a. Thermogravimetric analysis (TGA) indicates the grafting of 16 wt% of (2-aminoethyl)-3-aminopropyl functionality to the silica walls (Fig. S2-a, ESI†). The closed amount of grafted organic groups (17%) was also confirmed by elemental analyses (see Experimental section). The N2 adsorption isotherms performed at 77 K were analyzed using the BET (Brunauer–Emmett–Teller) method to find the specific surface area and with the BJH (Barrett–Joyner–Halenda) method to determine the pore size distribution. The isotherms and pore size distribution of the silica before and after grafting are shown in Fig. 3. Porous silica initially shows a specific surface of 646 cm2 g−1 with an average pore size of 9 nm. As expected, a decrease of the specific surface to 270 cm2 g−1 with an average pore size of 7 nm was observed after anchoring of the organic functionality (Fig. 3).
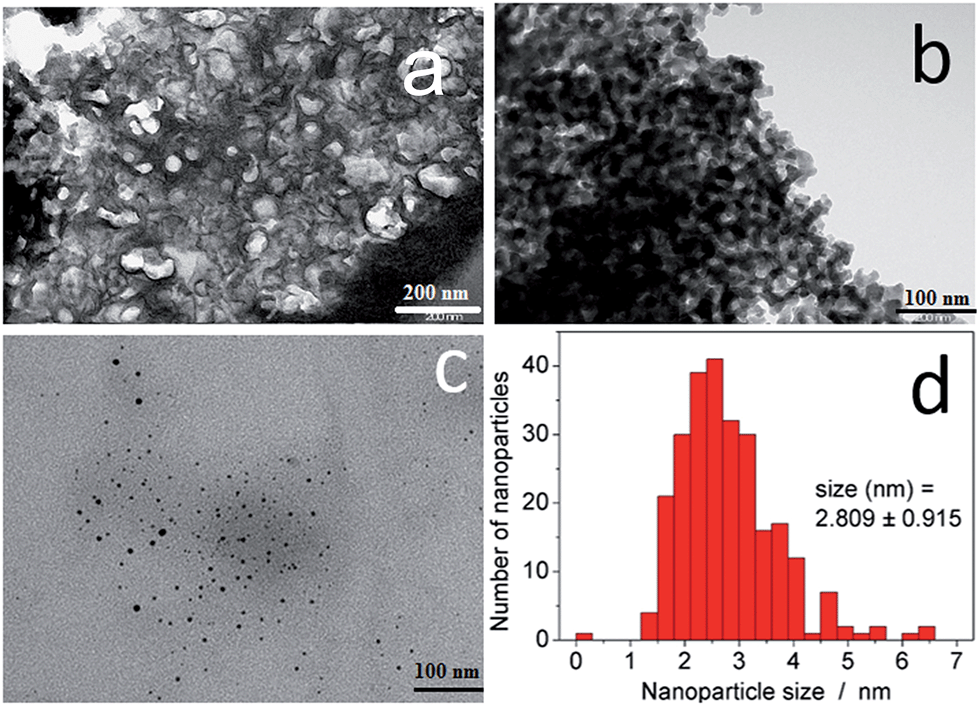 | ||
| Fig. 2 TEM images of (a) functionalized mesoporous silica (Sil); (b) functionalized glass pearls (Glass); (c) Hofmann-type NiII(pz)[NiII(CN)4] nanoparticles after extractive replicas on Sil@NP; and (d) the size distribution for NiII(pz)[NiII(CN)4] nanoparticles in Sil@NP. | ||
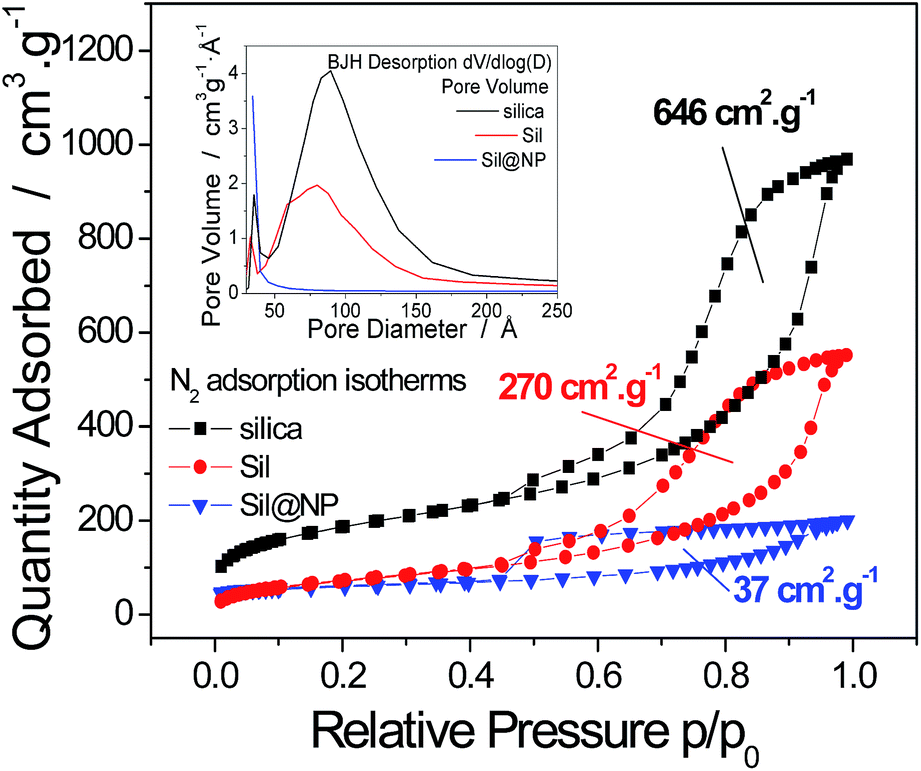 | ||
| Fig. 3 Nitrogen adsorption isotherms at 77 K for the mesoporous silica before and after diamine groups grafting (Sil) and the silica-based Sil@NP nanocomposite. Inset: pore size distribution for the mesoporous silica before and after diamine groups grafting (Sil) and the Sil@NP nanocomposite. | ||
Porous glass pearls purchased from Vitrabio® with a pore size of approximately 40 nm (Fig. 2b), a specific area of 130 cm2 g−1 (Fig. S3, ESI†) and grain sizes from 200 to 500 μm were also considered as a matrix to prepare the nanocomposites. The preparation of the diamine-grafted glass beads (Glass) by the (2-aminoethyl)-3-aminopropyl functionality was performed using the same procedure to that used in the case of the silica matrix and described in the Experimental section. TGA and elemental analyses performed for the Glass sample indicate an anchoring of 9 wt% of organic moieties, which means that expressed as grams per unit surface, a grafting of 6.9 × 10−4 g m−2 was carried out on Glass. This value is higher than the one of 2.4 × 10−4 g m−2 obtained for Sil. The Glass sample exhibits a decrease in the specific surface to 83 cm2 g−1 and the pores size to 30 nm.
The second step of the strategy used (Fig. 1) consists of the intrapore growth of NiII(pz)[NiII(CN)4] nanoparticles by adapting a previously described step-by-step procedure for the prussian blue analogues.16–18,31,32 For this, the functionalized matrices (Sil and Glass) were shaken at first in a 4 × 10−2 M methanolic solution of Ni(BF4)2·6H2O and pyrazine for 2 h. Then, the solid was filtered, thoroughly washed with methanol and shaken with a 4 × 10−2 M methanolic solution of (TBA)2[Ni(CN)4] (TBA = tetrabutylammonium) for 2 h. This procedure was repeated 4 more times and the color of the matrices turn to a pink color due to the growth of the Hofmann-type nanoparticles inside the pores (Fig. 1 and S1†). The elemental analysis of the final nanocomposite Sil@NP indicates a weight ratio Ni/Si = 0.55 (molar ratio Ni/Si = 0.28) meaning a loading of 39 wt% of nanoparticles into the silica pores. For Glass@NP, a weight ratio of Ni/Si = 0.24 (molar ratio Ni/Si = 0.11) corresponds to the presence of 18 wt% of Hofmann-type nanoparticles, that is in accordance to the lower quantity of grafted organic functionalities in glass. Indeed, even though the amount of organic functionality grafted per unit surface on the glass matrix is higher than the silica one, the significantly higher specific surface area of the latter leads to a higher final content of organic moieties per gram of material and thus to a higher content of the Hofmann-type structure nanoparticles. It is estimated that around 1.1 wt% and 0.3 wt% of the grafted amines are not coordinated with nanoparticles on Sil@NP and Glass@NP, respectively (Table 1 and Experimental section).
| Samples | SSA, cm2 g−1 | Free diamine functionality, wt% | NPs loading, wt% | t eq, h | Q max, mmol g−1 | Q max, wt% |
|---|---|---|---|---|---|---|
| a Calculated from iodine desorption experiments. | ||||||
| Ni(pz)[Ni(CN)4] | — | — | 2 | 3.28 | ||
| Sil | 270 | 17 | — | 48 | 4.01 (on free diamines) | 99 (on free diamines) |
| Sil@NP | 37 | 1.1 (residual) | 39 | 14 | 1.75 (1.49 on NPs and 0.25 on residual diaminesa) | 46 (39.1 on NPs and 6.9 on residual diamines) |
| Glass | 83 | 9 | — | 48 | 2.10 (on free diamines) | 53 (on free diamines) |
| Glass@NP | 66 | 0.3 (residual) | 18 | 14 | 1.10 (1.01 on NPs and 0.07 on residual diaminesa) | 28 (25.7 on NPs and 2.3 on residual diamines) |
For both nanocomposites, the TGA curves show a two-step decomposition profile with the organic functionality decomposition at 250 °C in the first step followed by the Hofmann-type structure nanoparticles decomposition at 300 °C in the second step. The two-step decomposition is better distinguished for the glass-based nanocomposite (Fig. S2-a and S2-b, ESI†). Note that a lower decomposition temperature was observed for the nanoparticles with respect to that found with the NiII(pz)[NiII(CN)4] bulk materials (400 °C), which can be associated with the smaller grain size and higher surface energy.33 The specific surface area of the Sil@NP and Glass@NP nanocomposites was determined using the BET method on the nitrogen isotherms at 77 K. As expected, a decrease of the specific surface to 37 cm2 g−1 for Sil@NP and to 66 cm2 g−1 for Glass@NP was observed in comparison to the corresponding nanoparticles-free functionalized matrices (Sil and Glass, respectively) and was assigned to the filling of the pores with nanoparticles (Table 2, Fig. 3 & S3, ESI†).
| Samples after iodine capture | Raman, cm−1 | UV-Vis, nm |
|---|---|---|
| Ni(pz)[Ni(CN)4] | 103 (I3− on defects) 165 (confined I2) | 550 (clathrate) |
| Sil | 110, 150 (I3−) | 360 (I3−) |
| Sil@NP | 110, 150 (I3−), 165 (confined I2) | 360 (I3−), 500 (confined I2) 550 (clathrate) |
| Glass | 110, 150 (I3−), 160 (I2 in I5− complex) | 360 (I3−), 450 (I2 in I5− complex) |
| Glass@NP | 110, 150 (I3−), 160 (I2 in I5− complex), 165 (clathrate confined I2) | 360 (I3−), 450 (I2 in I5− complex), 520 (clathrate confined I2) 550 (clathrate) |
The presence of the nanoparticles in the matrix pores of the Sil@NP was confirmed by X-ray powder diffraction (XRPD), where the diffraction pattern of NiII(pz)[NiII(CN)4] (tetragonal P4/m space group)34 is clearly recognized by comparison with the XRPD pattern of the bulk material (Fig. 4). Solid state UV-Vis spectra in reflectance mode also indicated an absorption band at 550 nm for Sil@NP and Glass@NP, respectively, matching with the d–d transitions in the spectrum of the bulk Hofmann-type structure (Table 1, inset of Fig. 5 and S4, ESI†). In addition, infrared spectra of both samples show the cyanide vibration ν (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) at 2172 cm−1, characteristic of the bridging coordinated cyanides in the parent bulk Hofmann-type structure (Table 2, Fig. S5, ESI†).15,34 Direct observation of the nanoparticles by TEM was possible only for Sil@NP using the HF etching procedure devoted to dissolving the silica matrix (see Experimental section). The representative images show the presence of spherically shaped and non-aggregated nanoparticles with a mean size distribution of 2.8 ± 0.9 nm (Fig. 2c–d). All these results are in accordance with the successful growth of small Hofmann-type nanoparticles into the pores of both matrices.
N) at 2172 cm−1, characteristic of the bridging coordinated cyanides in the parent bulk Hofmann-type structure (Table 2, Fig. S5, ESI†).15,34 Direct observation of the nanoparticles by TEM was possible only for Sil@NP using the HF etching procedure devoted to dissolving the silica matrix (see Experimental section). The representative images show the presence of spherically shaped and non-aggregated nanoparticles with a mean size distribution of 2.8 ± 0.9 nm (Fig. 2c–d). All these results are in accordance with the successful growth of small Hofmann-type nanoparticles into the pores of both matrices.
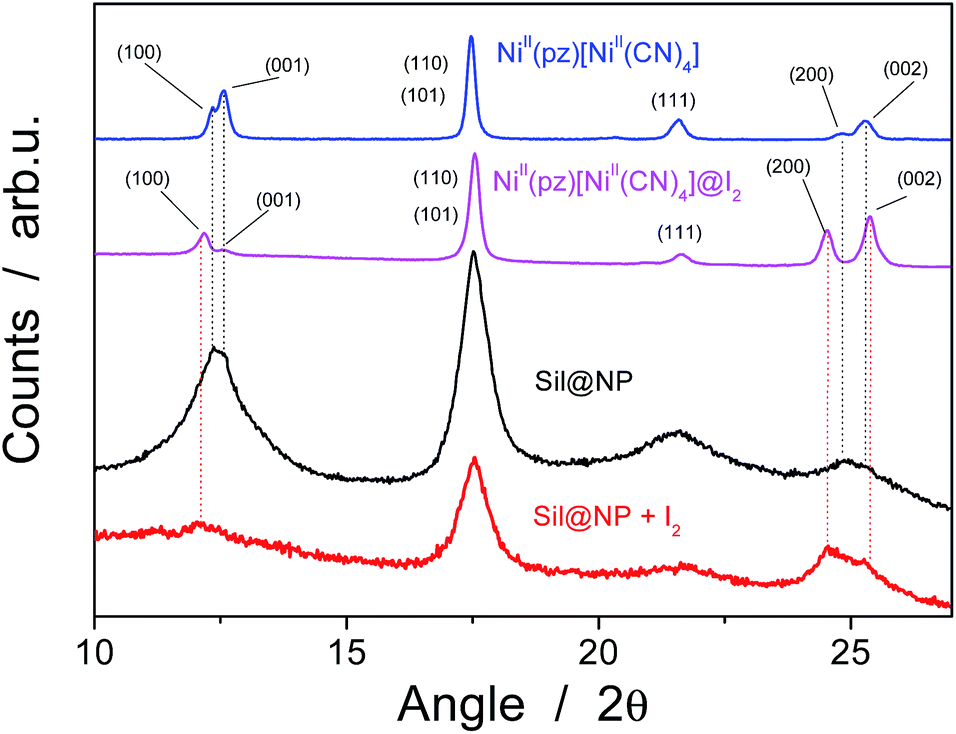 | ||
| Fig. 4 X-ray powder diffraction (XRPD) patterns for the Sil@NP nanocomposite and the bulk NiII(pz)[NiII(CN)4] before and after iodine uptake. | ||
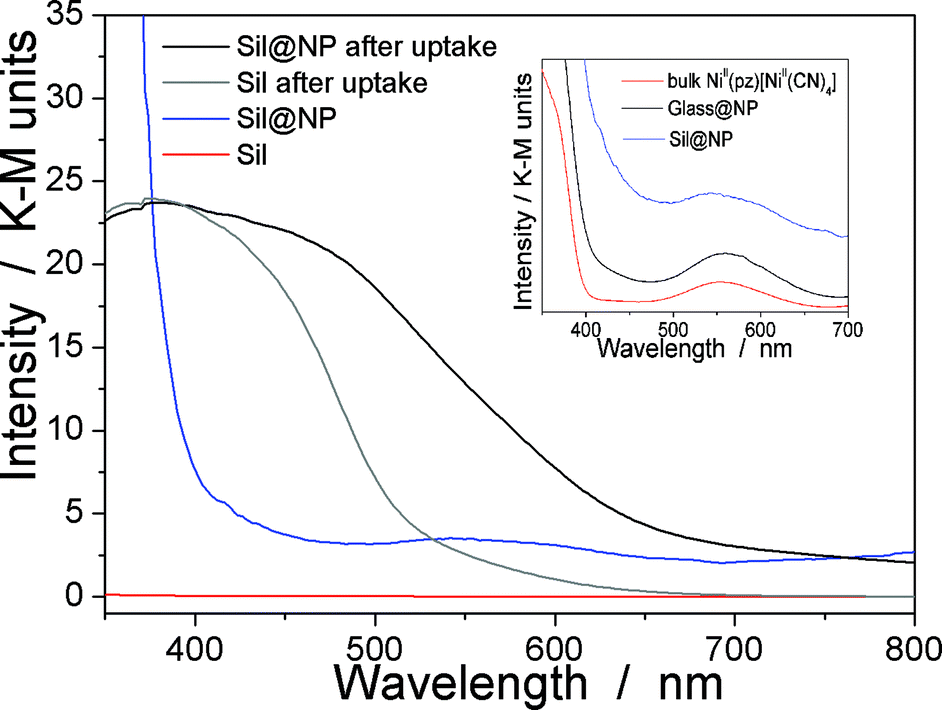 | ||
| Fig. 5 UV-Vis spectra in the solid state performed for the grafted silica Sil, Sil@NP nanocomposite and the bulk NiII(pz)[NiII(CN)4] before and after iodine uptake. Inset: magnification of the UV-Vis spectra in the range 350–700 nm for the Sil@NP and Glass@NP nanocomposites and bulk NiII(pz)[NiII(CN)4]. | ||
where Q (mmol g−1) is the amount of entrapped iodine at time t (min), Qe is the entrapped amount at equilibrium (t → ∞) and k (g mmol−1 min−1) is the kinetic constant. The I2 sorption kinetic curves show that the process is quite rapid for the bulk NiII(pz)[NiII(CN)4] with equilibrium being reached after 2 h (Fig. 6).13 The equilibrium time for the Sil@NP and Glass@NP samples is equal to around 14 h, whereas the process is much longer for the nanoparticles-free Sil and Glass samples (48 h) (Fig. 6 & S6, ESI†).
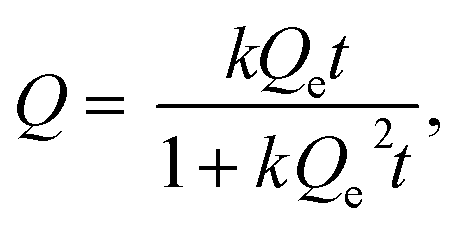
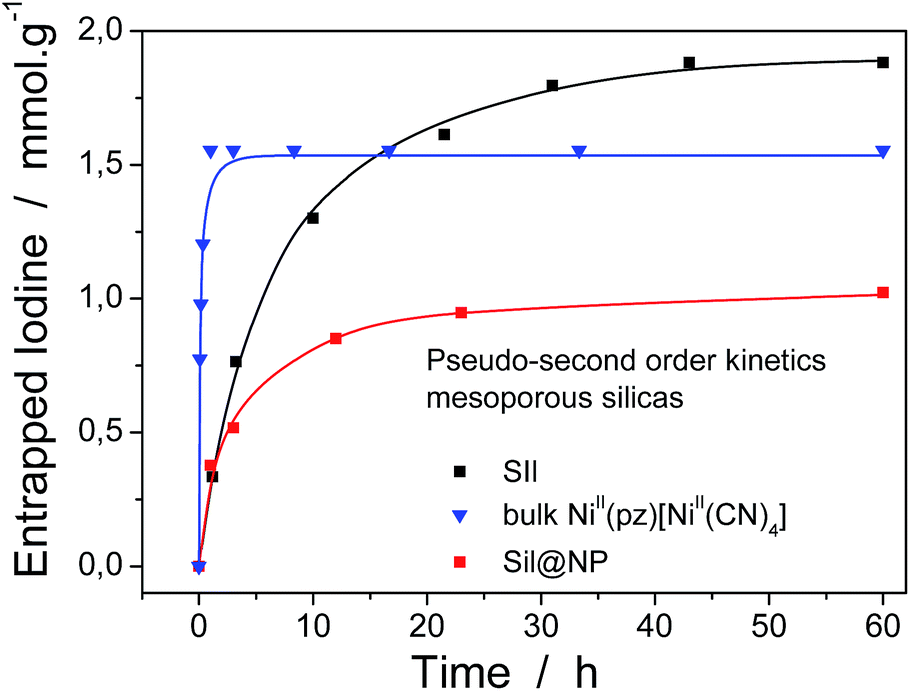 | ||
| Fig. 6 Kinetic curves for the grafted silica, the Sil@NP nanocomposite and the bulk NiII(pz)[NiII(CN)4] material in a cyclohexene solution. The solid lines represent the fit with a pseudo-second order model. | ||
For a complete characterization of the iodine uptake by these materials, sorption isotherms were obtained using iodine capture in a solution of cyclohexane with increasing iodine concentrations. The isotherm curves were fitted with the Langmuir model, which is described by the following formula:
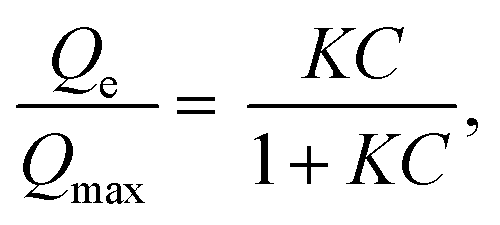
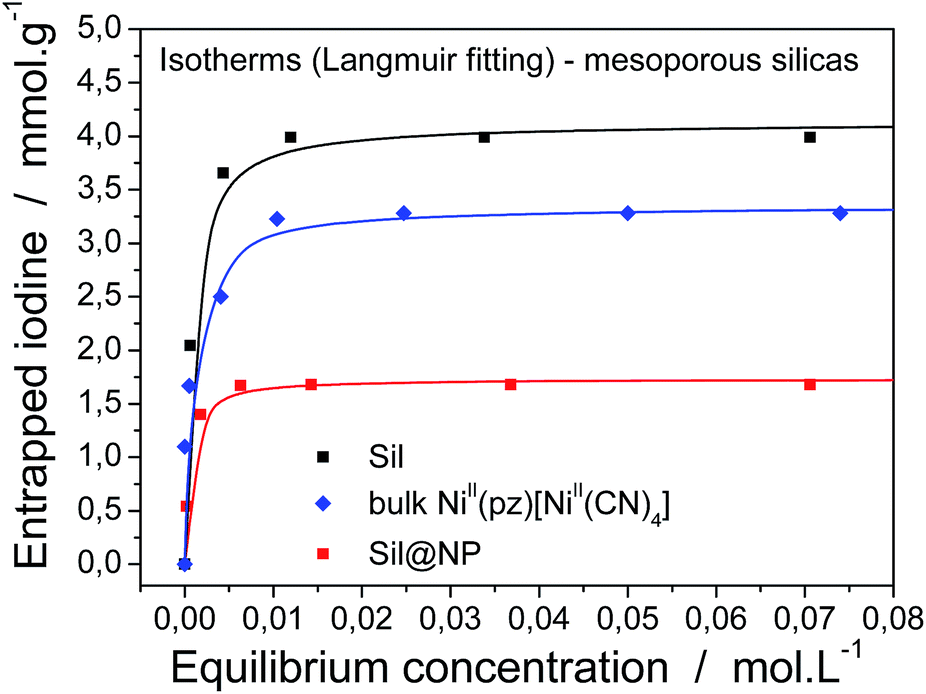 | ||
| Fig. 7 Iodine absorption isotherms in a cyclohexane solution for the grafted silica, the Sil@NP nanocomposite and the bulk NiII(pz)[NiII(CN)4] material. The solid lines represent the fit with the Langmuir model. | ||
Firstly, both grafted matrices are able to capture iodine from solution due to the presence of the diamine groups. The maximal sorption capacity of 4.1 ± 0.1 mmol g−1 (99 wt%) for Sil and 2.1 ± 0.1 mmol g−1 (53 wt%) for Glass were found (Table 1). The smaller amount of absorbed iodine by the glass matrix in comparison with silica is explained by the smaller quantity of grafted diamino functionality (17 wt% for Sil and 9 wt% for Glass).
Secondly, the nanocomposite materials show maximum absorption capacities equal to 1.7 ± 0.1 mmol g−1 (46 wt%) for Sil@NP and 1.1 ± 0.1 mmol g−1 (28 wt%) for Glass@NP (Table 2). This apparently lower capacity in comparison with those observed for the bulk Hofmann-type structure (3.28 mmol g−1) is explained by the fact that the nanocomposites contain only 39 and 18 wt% of NiII(pz)[NiII(CN)4] in the Sil and Glass matrices, respectively. Neglecting the iodine captured by residual diamines (see after), iodine sorption capacity of the nanocomposites were recalculated in mmol per gram of inserted Hofmann clathrate giving the capacity of 3.77 and 5.96 mmol g−1 of clathrate for Sil@NP and Glass@NP, respectively. In both cases, the iodine uptake by clathrate nanoparticles is higher in comparison to those obtained for the bulk materials (3.28 mmol g−1; one molecule of I2 per unit cell) demonstrating the higher efficiency of the nanoparticles in comparison with the corresponding bulk material, which is due to the higher available surface of the nanoparticles for physisorption.
In order to understand the iodine capture mechanism, the materials with a maximum iodine loading were characterized. First of all, the nanoparticles-free functionalized matrices (Sil and Glass) were investigated. Both matrices present similar features over the iodine capture characteristic of the triiodide (I3−) formed through the strong charge transfer occurring between the iodine and the σ-electron donor diamine functionality.36–38 Note also that the formation of an additional pentaiodide (I5−) species was observed for the glass pearls as a result of the interactions between I2 and I3−,39–41 but pentaiodide formation was not observed in silica. Indeed, the Raman spectrum of the Sil sample performed in the range 50–300 cm−1 shows a large peak at 150 cm−1 and a peak at 103 cm−1 classically assigned to the symmetric and the asymmetric stretching of I3− (Fig. 8, Table 2).41,42 In contrast, the spectrum of the Glass sample shows three bands related to the I5− species: two bands for I3− (at 110 cm−1 and 150 cm−1) and an additional band at 160 cm−1 for interacting I2 (Fig. 8, Table 2).41,42 This conclusion is also confirmed by the solid state UV-Vis spectra in diffuse reflectance mode showing a maximum absorption band at 360 nm for the I3− species, characteristic of the large HOMO and LUMO separation for the iodine molecule occurring during the charge transfer in Sil (Table 2, Fig. 5 and S5, ESI†).43 But when iodine is confined in the grafted glass pearls, two bands at 360 nm (I3−) and 450 nm (interacted I2) are visible in the spectrum (Table 2, Fig. S4, ESI†). This fact explains the yellow color of Sil and the red color of Glass sample observed after the iodine capture (Fig. 1). Therefore, as described in the literature,44 when iodine interacts with σ-electron donors, such as the diamine functionality grafted to the silica wall, a strong charge transfer occurs. The σ-electrons are given to the anti-bonding orbital 5σ* of iodine bringing about ionic dissociation:
| N: + 2I2 → N: I+ + I3− |
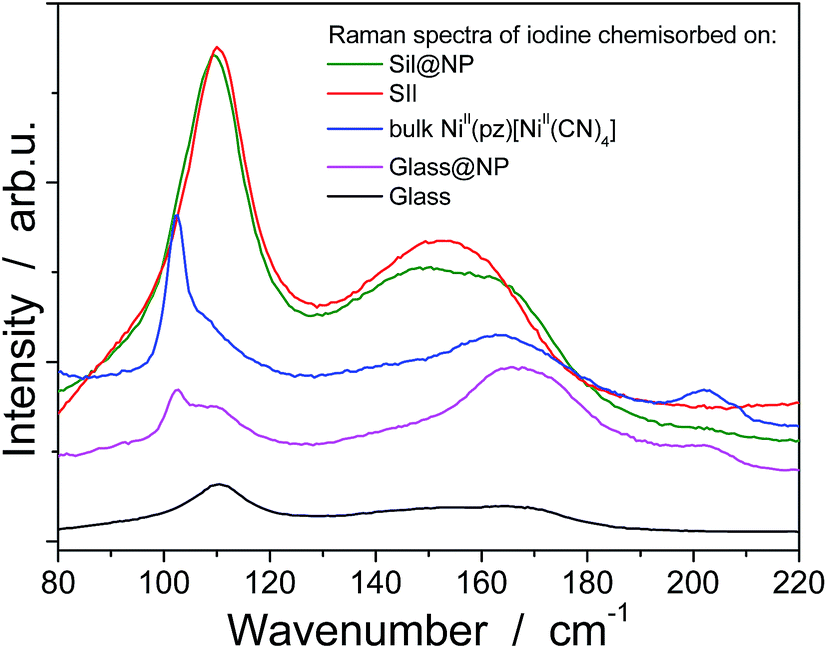 | ||
| Fig. 8 RAMAN spectra in the range 80–220 cm−1 for the functionalized matrices (Sil, Glass), bulk NiII(pz)[NiII(CN)4] and the nanocomposites (Sil@NP, Glass@NP) after I2 loading. | ||
The Sil@NP and Glass@NP nanocomposite materials obtained after growth of the NiII(pz)[NiII(CN)4] nanoparticles inside the pores of the functionalized matrices show different behaviour regarding the iodine uptake due to the presence of the nanoparticles. Previously, we demonstrated that the physisorption of iodine by the bulk Hofmann-type structure NiII(pz)[NiII(CN)4] is mainly due to the insertion of I2 molecules within the network cages through synergetic interactions with both the pyrazine pillar ligand and the cyanometallate moieties. A small amount of pentaiodides may also be present due to few lattice defects in the Hofmann-type network.15 When nanoparticles are introduced in the matrix pores, both the nanoparticles and the residual free diamine functionalities not coordinated to the nanoparticles are involved in the iodine trapping. This hypothesis is supported by the spectroscopic measurements of the nanocomposites after iodine loading. The Raman spectrum of Sil@NP shows, in addition to the peaks at 110 cm−1 and 150 cm−1 associated to I3− stretching vibrations observed for Sil matrix, an additional peak around 165 cm−1 assigned to the presence of confined solid state iodine (I2) found in the spectrum of the iodine loaded within the bulk Hofmann-type structure (Fig. 8, Table 2).15 Similarly, the peaks of the glass matrix (110, 150, 160 cm−1), as well as the one of the Hofmann-type structure (larger band at 165 cm−1) may be observed in the spectrum of Glass@NP (Fig. 8, Table 2). Moreover, the IR spectra of both nanocomposites after iodine uptake indicate a shift of the cyanide stretching vibration from 2172 to 2167 cm−1, which is indicative of an increase in the back-donation d–π* caused by an interaction between the iodine and the tetracyanonickelate moiety observed for the bulk Hofmann-type structure (Fig. S5, ESI†).15 The appearance of the band in the 500–520 nm range in UV-Vis spectra of the nanocomposites, characteristic of the confined I2 inside the pores of the Hofmann-type structure nanoparticles confirms this feature (Fig. 5 & S4, ESI†). XRPD for Sil@NP after uptake is in accordance with modifications as previously described for the bulk phase NiII(pz)[NiII(CN)4]·I2, since the iodine insertion triggers an increase in the lattice parameters. Relevantly, the peak at 25° for the Miller plane (200) shifts to lower 2θ values and it increases its intensity with respect to the plane (002) (Fig. 4).15
The iodine absorption kinetics curves also indirectly translate the presence of both the physisorption and the chemisorption mechanisms giving rise to an intermediate equilibrium time observed for nanocomposite materials; the presence of the Hofmann-type structure nanoparticles seems to speed up the iodine uptake.
However, as previously mentioned, nanoparticles show lower thermal stability in comparison with the bulk materials. For this reason, the iodine desorption executed at 250 °C for 4 h under a continuous flux of argon for the nanocomposites was investigated. The nanocomposite integrity after heating was confirmed by spectroscopy measurements as well as by EDX analyses. The obtained atomic ratio I/Ni = 0.20 for Sil@NP means that 1.49 mmol g−1 of I2 was desorbed off and 0.25 mmol g−1 of iodine was still chemisorbed on the residual free diamines in the pores. Similarly, the Glass@NP sample shows an atomic ratio I/Ni equal to 0.12 indicating desorption of 1.03 mmol g−1 of iodine, retaining 0.07 mmol g−1 after thermal treatment. FT-IR spectra for both nanocomposites show that the ν (CN) band at 2167 cm−1 was back to the original value of 2172 cm−1 meaning that confined iodine was fully evacuated off the clathrate nanoparticles (Fig. S8, ESI†), as already demonstrated in our previous work for the bulk material.15 In addition, a complete vanishing of the band in the range 500–520 nm in the UV-Vis spectra associated with the presence of the confined I2 is observed. However, the characteristic bands for the presence of residual I3− and I5− species, respectively for Sil@NP and Glass@NP, are still observed in the spectra of these desorbed samples (Fig. 9 & S9, ESI†). This result indicates that the heating of the nanocomposites at 250 °C permits the selective desorption of the iodine confined in the Hofmann-type structure nanoparticles, but the materials retain a small quantity of polyiodides in the residual diamine functionality. These moieties are lost simultaneously with the decomposition of the organic groups when heating to a higher temperature (Fig. S8, ESI†).
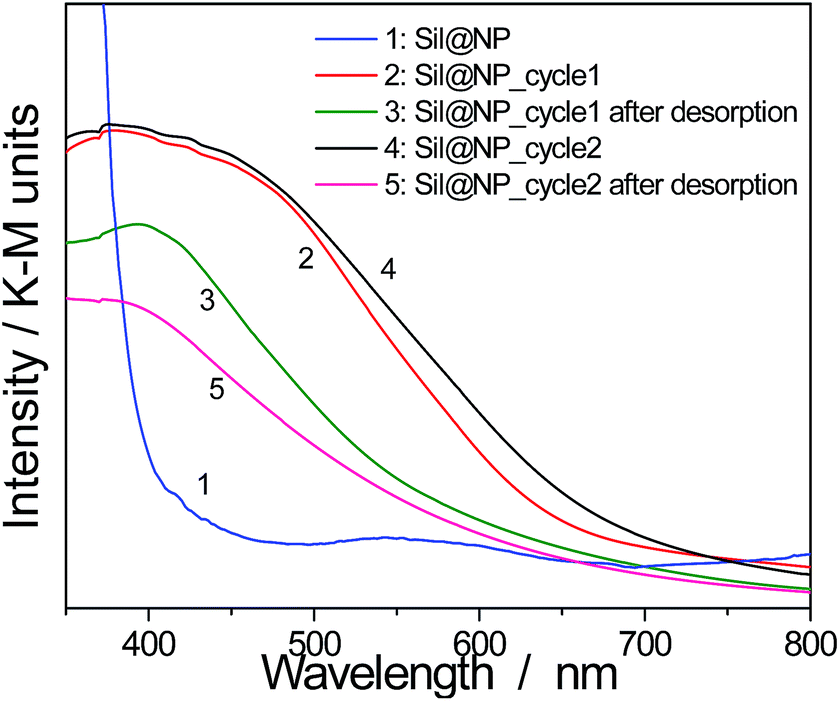 | ||
| Fig. 9 Solid state UV-Vis spectra in diffuse reflectance mode for the cycling process of Sil@NP (2 cycles of iodine uptake and desorption). | ||
Finally, in order to verify the possible cycling ability of the obtained nanocomposites, two sorption/desorption cycles were achieved indicating a perfect reproducibility in the iodine uptake. Note that UV-Vis spectroscopy shows an appearance and then a disappearance of the band at 500–520 nm associated with the presence of the confined I2 during the second iodine loading/desorption cycle (Fig. 9 & S9, ESI†). However, the iodine sorption on the functionalized support is not reversible and the departure of the iodine with heating occurs with the simultaneous decomposition of the organic anchors.
Conclusions
In the present work, we synthesized for the first time, two nanocomposites based on Hofmann-type structure nanoparticles NiII(pz)[NiII(CN)4] grown directly into the pores of mesostructured silica and porous glass pearls. These materials were obtained by successive coordination of nickel salts, pyrazine and cyanonickelate precursors into the pores through the diamine groups that functionalize the silica walls. Small spherical clathrate nanoparticles of 2–3 nm were obtained inside the matrix pores and were characterized by IR and Raman spectroscopy, UV-Vis spectroscopy, X-ray diffraction and TEM analysis. The obtained nanocomposites contain 39 and 18 wt% of nanoparticles for silica and glass pearls, respectively.The as-obtained nanocomposites were employed for iodine capture in cyclohexane solutions and their iodine sorption behavior was compared with the one of the functionalized parent matrix and the corresponding bulk Hofmann-type clathrate.
The first point to note is that the iodine sorption kinetics for the nanocomposites is four times faster than that compared to the ones of the parent amino functionalized matrix. Indeed, for the latter a strong charge transfer process arises between iodine and the σ-electron donor diamine functionality, i.e. a chemisorption process leading to the formation of I3− and/or I5− species. However, a physisorption process of molecular iodine into the Hofmann-type network cavities takes place for the nanocomposites, speeding up the iodine sorption. Still, the presence of the residual amino groups in the nanocomposites slows down the equilibrium sorption time when compared to the bulk Hofmann-type structure.
Secondly, both nanocomposite materials present interesting iodine sorption capacity of 1.7 mmol g−1 and 1.1 mmol g−1, respectively. The iodine capture occurs mostly on the nanoparticles, but a small quantity of iodide is also retained in the pores through the formation of adducts with the residual diamine functionality. The sorption capacity calculated by mmol g−1 of the Hofmann clathrate nanoparticles is higher in comparison with the capacity of the corresponding bulk Hofmann clathrate highlighting the efficiency of such supported nanoparticles.
Thirdly, thermal treatments at 250 °C show the reversible desorption of the iodine captured by nanoparticles and cycling ability is clearly observed for both nanocomposites.
In summary, the use of nanocomposite materials containing Hofmann-type structure nanoparticles covalently linked into the pores of supported matrices appears to be a promising approach for efficient and reversible iodine capture, which opens interesting perspectives in relation to the many possible variations in chemical composition of these materials for iodine decontamination and further waste storage.
Acknowledgements
The authors thank the ANR (ANR-AA-RMNP-003-01), University Montpellier 2 and CNRS for financial support. We also thank Mme D. Granier, (PAC Chimie Balard, Montpellier) for XRPD measurements, M. José-Marie Ruiz for Raman spectra, and Mme Veronique Richard and M. Franck Godiard for the preparation of the TEM grids.Notes and references
- G. Audi, O. Bersillon, J. Blachot and A. H. Wapstra, Nucl. Phys. A, 2003, 729, 3–128 CrossRef PubMed.
- D. R. Haefner and T. J. Tranter, Report INL/EXT-07-12299, Idaho National Laboratory, 2007.
- N. R. Soelberg, T. G. Garn, M. R. Greenhalgh, J. D. Law, R. Jubin, D. M. Strachan and P. K. Thallapally, Sci. Technol. Nucl. Install., 2013, 13, 702496 Search PubMed.
- (a) K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed; (b) T. Nenoff, M. A. Rodrigez, N. R. Soelberg and K. W. Chapman, Microporous Mesoporous Mater., 2014, 200, 297 CrossRef CAS PubMed.
- S. Matsuoka, H. Nakamura, T. Tamura, T. Takano and Y. Ito, J. Nucl. Sci. Technol., 1984, 21, 862–870 CrossRef CAS.
- C. Falaise, C. Volkringer, J. Facqueur, T. Bousquet, L. Gasnot and T. Loiseau, Chem. Commun., 2013, 49, 10320–10322 RSC.
- W. Guo, D. Wang, J. Hu, Z. K. Tang and S. Du, Appl. Phys. Lett., 2011, 98, 043105–043107 CrossRef PubMed.
- E. J. Doskocil, S. V. Bordawekar, B. G. Kaye and R. J. Davis, J. Phys. Chem. B, 1999, 103, 6277–6282 CrossRef CAS.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS PubMed.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256–16259 CrossRef CAS PubMed.
- P. Cui, L. Ren, Z. Chen, H. Hu, B. Zhao, W. Shi and P. Cheng, Inorg. Chem., 2012, 51, 2303–2310 CrossRef CAS PubMed.
- D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS PubMed.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filincuck, D. Popov, C. Rieckel, G. Ferey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- G. Massasso, J. Long, J. Haines, S. Devautour-Vinot, G. Maurin, A. Grandjean, B. Onida, B. Donnadieu, J. Larionova, C. Guérin and Y. Guari, Inorg. Chem., 2014, 53(9), 4269–4271 CrossRef CAS PubMed.
- J. Larionova, J. Long, Y. Guari, G. Massasso, A. Grandjean, Y. Barré, A. Tokarev and J. Causse, patent FD13715, SP52533FG, 2013.
- R. Turgis, G. Arrachart, C. Delchet, C. Rey, Y. Barré, Y. Pellet-Rostaing, Y. Guari, J. Larionova and A. Grandjean, Chem. Mater., 2013, 25, 4447–4453 CrossRef CAS.
- C. Delchet, A. Tokarev, X. Dumail, G. Toquer, Y. Barre, Y. Guari, C. Guerin, J. Larionova and A. Grandjean, RSC Adv., 2012, 2, 5707–5716 RSC.
- J. Causse, A. Tokarev, C. Ravaux, M. Moloney, Y. Barré and A. Grandjean, J. Mater. Chem., 2014, 2, 9461–9464 RSC.
- (a) S. Dourdain, X. Deschanels, G. Toquer, C. Grygiel, I. Monnet, S. Pellet-Rostaing and A. J. Grandhean, J. Nucl. Mater., 2012, 427, 411–414 CrossRef CAS PubMed; (b) S. Klaumunzer, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 225, 136–153 CrossRef CAS PubMed.
- (a) M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed; (b) P. Makowski, X. Deschanels, A. Grandjean, D. Meyer, G. Toquer and F. Goettmann, New J. Chem., 2012, 36, 531 RSC.
- (a) J. Larionova, L. Salmon, Y. Guari, A. Tokarev, K. Molvinger, G. Molnar and A. Bousseksou, Angew. Chem., Int. Ed., 2008, 47, 8236–8240 CrossRef CAS PubMed; (b) A. Tokarev, J. Long, Y. Guari, J. Larionova, F. Quinard, F. Agulhon, M. Robitzer, G. Molnar, L. Salmon and A. Bousseksou, New J. Chem., 2013, 37, 3420–3432 RSC.
- Y. Raza, F. Volatron, S. Moldovan, O. Ersen, V. Huc, C. Martini, F. Brisset, A. Gloter, O. Stephan, A. Bousseksou, L. Catala and T. Mallah, Chem. Commun., 2011, 11501–11503 RSC.
- B. Das, R. Carlin and A. Osteryoung, Inorg. Chem., 1989, 28, 421–426 CrossRef CAS.
- L. Cao, T. Man and M. Kruk, Chem. Mater., 2009, 21, 1144–1153 CrossRef CAS.
- J. S. Beck, K. D. Vartuli, W. J. Roth, M. E. Leonowitz, C. T. Kresge, K. D. Schmitt, C. T. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- P. Yang, D. Zhao, B. F. Chmelka and G. D. Strucky, Chem. Mater., 1998, 10, 2033–2036 CrossRef CAS.
- Z. Dongyuan, Q. Huo, J. Feng and B. F. Chmelka, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef.
- L. Wang, T. Qi, T. Y. Zhang and J. Chu, Microporous Mesoporous Mater., 2006, 91, 156–160 CrossRef CAS PubMed.
- T. Sangvanich, V. Sukwarotwat, R. J. Wiacek, R. M. Grudzien, G. E. Frywell, R. S. Addleman, C. Timchalk and W. Yantasee, J. Hazard. Mater., 2010, 182, 225–231 CrossRef CAS PubMed.
- G. Clavel, Y. Guari, J. Larionova and C. Guerin, New J. Chem., 2005, 29, 275–279 RSC.
- B. Folch, Y. Guari, J. Larionova, C. Luna, C. Sangregorio, C. Innocenti, A. Caneschi and C. Guerin, New J. Chem., 2008, 32, 273–282 RSC.
- R. W. Hutchinson, S. Kleinberg and F. P. Stein, J. Phys. Chem., 1973, 77, 870–875 CrossRef CAS.
- (a) C. Bartual-Murgui, L. Salmon, A. Akou, N. A. Ortega-Villar, H. J. Shepherd, M. C. Munoz, G. Molnar, J. A. Real and A. Bousseksou, Chem.–Eur. J., 2012, 18, 507–516 CrossRef CAS PubMed; (b) V. Niel, J. M. Martinez-Agudo, M. C. Munoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2001, 40, 3838–3839 CrossRef CAS.
- (a) W.-W. He, S.-L. Li, G.-S. Yang, Y.-Q. Lan, Z.-M. Su and Q. Fu, Chem. Commun., 2012, 48, 10001 RSC; (b) Z.-J. Zhang, W. Shi, Z. Niu, H.-H. Li, B. Zhao, P. Gheng, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2011, 47, 6425 RSC.
- P. Deplano, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, Coord. Chem. Rev., 1999, 188, 71–75 CrossRef CAS.
- S. Moulay, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, J. Polym. Eng., 2012, 33, 389–443 Search PubMed.
- M. Chorro, G. Kane, L. Alvarez, J. Cambedouzou, E. Paineau, A. Rossberg, M. Kociak, R. Aznar, S. Pascarelli, P. Launois and J. L. Bantignies, Carbon, 2013, 52, 100–108 CrossRef CAS PubMed.
- S. Kobinata and S. Nagakura, J. Am. Chem. Soc., 1966, 88, 3905–3909 CrossRef CAS.
- J. T. Ye, Y. Iwasa and Z. K. Tang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 193409 CrossRef.
- C. Pei, T. Ben, S. Xu and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179–7187 CAS.
- (a) L. Alvarez, J. L. Bantignies, R. Le Parc, R. Aznar, J. L. Sauvagol, A. Merlen, D. Machon and A. San Miguel, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205403 CrossRef; (b) P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef.
- B. Camarota, Y. Goto, S. Inagaki, E. Garrone and B. Onida, J. Phys. Chem., 2009, 113, 20396–20400 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental part (supplementary), pictures of glass pearl based samples (Fig. S1), TGA curves for the obtained materials (Fig. S2), nitrogen adsorption isotherms at 77 K (Fig. S3), UV-Vis spectra before and after iodine uptake (Fig. S4), FT-IR spectra before and after iodine uptake (Fig. S5), kinetic curves for the glass-based materials (Fig. S6), iodine absorption isotherms for the glass-based materials (Fig. S7), FT-IR spectra at sorption/desorption (Fig. S8), UV-Vis spectra demonstrating the cycling ability for the Glass@NP nanocomposite (Fig. S9). See DOI: 10.1039/c4ta04855e |
| This journal is © The Royal Society of Chemistry 2015 |