
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(N-isopropylacrylamide)–clay based hydrogels controlled by the initiating conditions: evolution of structure and gel formation†
Beata
Strachota
,
Libor
Matějka
*,
Alexander
Zhigunov
,
Rafał
Konefał
,
Jiří
Spěváček
,
Jiří
Dybal
and
Rudolf
Puffr
Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: matejka@imc.cas.cz; Tel: +420 296 809 281
First published on 15th September 2015
Abstract
The formation of the hydrogel poly(N-isopropylacrylamide)–clay (LAPONITE®) by redox polymerization was investigated, and the main factors governing the gel build-up were determined. The significant effect of the redox initiating system ammonium peroxodisulfate (APS) and tetramethylethylenediamine (TEMED) on gel formation and structure was established, making it possible to control the structure of the gel. Moreover, the pre-reaction stage involving the quality of the clay exfoliation in an aqueous suspension and the interaction of reaction components with the clay play a role in controlling the polymerization and gel structure. The molecular and phase structure evolution during polymerization was followed in situ by the following independent techniques: Fourier transform infrared spectroscopy (FTIR), chemorheology, small-angle X-ray scattering (SAXS) and ultraviolet-visible spectroscopy (UV/Vis). The combination of these methods enabled us to describe in detail particular progress stages during the gel formation and determine the correlation of the corresponding processes on a time and conversion scale. The mechanism of gel formation was refined based on these experimental results.
1. Introduction
Stimuli-responsive polymer hydrogels have attracted increasing attention as very promising soft materials because of their ability to undergo significant changes in volume in response to external stimuli. The most typical thermosensitive poly(N-isopropylacrylamide) (PNIPA)-based hydrogels are used for various applications. However, the PNIPA hydrogels, with covalently cross-linked network structures, are fragile materials and possess a low degree of swelling at higher crosslinking densities. Therefore, for practical applications, the mechanical properties of the soft materials should be improved.Soft PNIPA-based hydrogels showing extraordinary tensile properties and mechanical toughness were reported by Haraguchi et al.1–3 The gels were formed by in situ radical polymerization in the presence of clay. The low-temperature polymerization was initiated by the redox system potassium peroxodisulfate and N,N,N′,N′-tetramethylethylenediamine (TEMED). The clay platelets exfoliated in an aqueous solution act as a polyfunctional crosslinker of flexible polymer chains. The generally accepted gel formation mechanism in the PNIPA–clay hybrid4–7 involves the formation of the “clay-brush particles”, i.e., clay aggregates with polymer chains anchored on the surface of clay platelets, in the early polymerization stage. This process is accompanied by a reduction of optical transmittance. Linking of these clay brushes takes place to form large assemblies8–10 and microgel clusters followed by gelation of the system. Continuation of the polymerization and formation of long grafted chains result in a gradual homogenization and an increase in the transparency of a system. The model of the gelation mechanism was based on measurements of viscosity4 in the early pregel stage, changes in transparency during polymerization and dynamic light scattering (DLS) and small angle neutron scattering (SANS)8 data analysis. However, the mechanism is still a subject of discussion. The questions arise as to the nature of the interaction between the clay platelets and polymerization components, the way of system crosslinking, and other issues. In particular, the effect of the redox initiating system on the formation and properties of a polymer/clay gel is not understood.
The aim of this paper was to develop a better understanding of the formation process of PNIPA–clay gels. To gain complex insight into the mechanism of gel build-up, we followed the evolution of both molecular and phase structures during polymerization. The novelty of the investigation is based on the time and conversion correlation of all stages of gel formation determined in situ by different independent experimental techniques. The structure evolution and characterization of particular polymerization stages, including the determination of the gel point, was followed by in situ monitoring using Fourier transform infrared spectroscopy (FTIR), rheology, small-angle X-ray scattering (SAXS) and ultraviolet-visible spectroscopy (UV/Vis). In addition to reaction kinetics, the molecular, supramolecular and morphological structures were evaluated. To date, no complex in situ time correlation of the processes involved in the corresponding gel formation has been described in the literature. Okay and Oppermann11,12 studied the formation of a polymer–clay nanocomposite gel by chemorheology. However, the phase structure evolution or conversion correlation of the proposed polymerization stages was not followed. A detailed investigation of gelation was reported by Shibayama et al.8 using an in situ method: time-resolved DLS in addition to contrast-variation SANS measurements.
In our study, we investigated the factors governing the formation and properties of the PNIPA–clay gel to determine the formation–structure–property relationships and fine-tune the gel synthesis and properties. The importance of the pre-reaction state of the aqueous clay suspension is discussed, including the quality of the clay dispersion and the interactions of components of the reaction mixture with the clay. Substantial attention is paid to the effect of initiating conditions on the structure build-up and the role of the redox initiating system ammonium peroxodisulfate–N,N′-tetramethylethylenediamine (APS–TEMED). Usually, gel formation and properties are controlled by the clay and monomer content, while the effect of the initiators has been followed only in few papers. Thermal initiations with AIBN or photopolymerization were reported by Wang et al.13 and Ferse et al.,7 respectively. However, as to the redox system only recently Nigmatullin et al.14 demonstrated using reaction mixtures with high monomer and low clay concentrations that the properties of polyacrylamide–clay hydrogels significantly vary, depending on the initiating conditions during the synthesis. They found that the composition of the redox initiating system affects the polymerization kinetics and the mechanical properties of the hydrogels. The observed trend was attributed to the increase in the degree of self-crosslinking of polyacrylamides due to chain transfer, which was influenced by the composition of the initiating system. In our study, however, we determined a very strong effect of the redox initiator even in solutions of low monomer concentration and relatively high clay concentrations, and we suggest an alternative interpretation of the phenomenon. The effect of the initiator system described in this work makes it possible to efficiently control the polymerization process and thus tune the structure and properties of the final product in a relatively wide range.
Haraguchi et al. have mainly used the LAPONITE® XLG clay in their studies. This type of clay, however, is prone to aggregation in water at higher concentrations due to clay platelets assembling to form the so-called “house of cards” structures. This phenomenon is a result of attractive interactions between oppositely charged edges and surfaces of the platelets. To prepare aqueous dispersions of high clay content, which would not aggregate, we used pyrophosphate-modified LAPONITE® XLS clay. This approach provided better clay exfoliation and reduced the viscosity of the reaction mixtures.
2. Experimental
2.1 Materials
N-Isopropylacrylamide (NIPA), ammonium peroxodisulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED) and hydrofluoric acid were purchased from Aldrich and used as received. The synthetic hectorite clay, LAPONITE® XLS Na0.7+[(Si8Mg5.5Li0.3)O20(OH)4]0.7−, consisting of approximately circular platelets (diameter ∼30 nm, thickness ∼1 nm) and modified with pyrophosphate ions (P2O7)4− was donated by Rockwood Ltd. The XLS powder contained 10 wt% water, which was taken into account in the calculation of reactants and solvent amounts for the syntheses of hydrogels.2.2 Preparation of nanocomposite hydrogels
In the first synthesis step, LAPONITE® XLS (“XLS”) was put into a glass vial and homogeneously dispersed in water via mechanical stirring. During this step, the XLS/water mixture was cooled in an ice-water bath at +4 °C, and the stirring time was 1 h for standard samples. To investigate the exfoliation process, the duration of the XLS dispersion step was varied between 2 min and 48 h for selected samples. In the second step, the monomer (NIPA) was added and stirred until dissolution (ca. 5 min) and the reaction mixture was purged with argon to remove oxygen. Then, TEMED was added, and the mixture was purged again with Ar. As the last component, the radical initiator APS was added in the form of a 1% aqueous solution cooled to +4 °C, and the mixture was purged with Ar. Finally, the reaction mixture was transferred into a cuvette or fixture, in which the structure development was investigated at 25 °C (using SAXS, NMR, FTIR, and rheology). Solid gel specimens were prepared by transferring the reaction mixtures in argon-filled 5 mL ampoules with an inner diameter of 1 cm.The concentration of NIPA in solution was 0.75 mol L−1, corresponding to 8.5 wt% in the reaction mixture. As an exception the concentrations 0.5 M and 1 M were also applied. Four clay concentrations, CXLS, were used: 2, 3, 5, and 10 wt% in the reaction mixture, corresponding to approximately 20, 30, 40, and 60 wt% clay in dry poly(NIPA)/clay nanocomposites. The initial clay dispersions, prepared prior to the addition of the other reaction components, had concentrations of 2.6, 4, 6.5 and 13 wt%, respectively. The concentrations of the initiator APS and the activator TEMED were characterized with respect to the monomer content as a molar ratio [APS]/[NIPA] = [APS]r and [TEMED]/[NIPA] = [TEMED]r, respectively. The sample designation was generated using clay concentration in dry gels and relative concentrations of TEMED and APS. For example, “30X–0.028–0.0087” describes the sample with 30 wt% XLS in the dry nanocomposite with [TEMED]r = 0.028 and [APS]r = 0.0087.
2.3 Methods
| G = F/S0(λ−2 − λ) |
| [η] = KMav |
FTIR. Fourier transform infrared (FTIR) spectra were recorded on a Thermo Nicolet NEXUS 870 spectrometer with a DTGS TEC detector. The spectra with a resolution of 4 cm−1 were recorded every 10 s, and 9 scans were averaged. The reaction solution just after initiation was placed in a demountable liquid-cell between two CaF2 windows separated by a 20 μm teflon spacer. The integrated intensity of the band at ∼1417 cm−1 assigned to the C–H in-plane-bending vibration of the vinyl group was used in the quantitative analysis of the decrease of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.
C double bonds.
Chemorheology. Rheological properties were investigated at 25 °C on a strain-controlled ARES-G2 rheometer (TA Instruments, USA) using a parallel plate fixture with a diameter of 40 mm. Silicone oil was applied on the edge of the plates to prevent water evaporation. Dynamic shear storage modulus G′(t) and loss modulus G′′(t) were measured during polymerization by time sweep oscillatory measurements at a frequency of 1 Hz and 0.1% strain amplitude. The strain was kept low enough to be in the linear deformation region and to prevent breakage of the growing structure.
The gel point was determined using the multifrequency sweep measurement ranging from 1 to 64 rad s−1 and by applying the Winter–Chambon criterion19 for a power-law rheological behaviour at the critical state, G′ ∼ G′′ ∼ ωn. This criterion implies that the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curves obtained at different frequencies during polymerization intersect in the point of gelation.
δ curves obtained at different frequencies during polymerization intersect in the point of gelation.
Small-angle X-ray scattering (SAXS). The experiments were performed using a pinhole camera (Molecular Metrology SAXS System) attached to a microfocused X-ray beam generator (Osmic MicroMax 002) operating at 45 kV and 0.66 mA (30 W). The camera was equipped with a multiwire, gas-filled area detector with an active area diameter of 20 cm (Gabriel Design). Two experimental setups were used to cover the q range of 0.004–1.1 Å−1, where q = (4π/λ)sin
θ (λ is the wavelength and 2θ is the scattering angle). The scattering intensities were put on an absolute scale using a glassy carbon standard.
UV/Vis spectrometry. Changes in transparency during the polymerization were detected using a UV/Vis spectrophotometer (Perkin-Elmer Lambda 20 UV-Vis) at 600 nm wavelength at 25 °C.
3. Results and discussion
3.1 Clay dispersion in water
The properties of nanocomposite hydrogels crosslinked with the clay platelets are dependent on, among other factors, the initial state of the reaction mixture. The quality of a clay dispersion in water, the extent of exfoliation of clay particles and the stability of a clay dispersion prior to the polymerization are crucial for the reproducible synthesis and the final hydrogel properties. We used LAPONITE® XLS instead of LAPONITE® XLG, which does not flocculate to form a gel at low clay content, but still the quality of a clay dispersion is an important factor. For the preparation of a clay dispersion, the clay LAPONITE® XLS was suspended in water and stirred for a time tmix.The dispersion of clay in water requires hours or even days.11,20 We investigated the dispersion process of the clay powder in water and the stability of the dispersion by SAXS and measurement of viscosity as well as ζ-potential. Fig. 1a illustrates SAXS profiles of the clay powder before and during dispersion in water. The “dry” powder containing 10% water (see Experimental) shows an interference maximum at qmax = 0.50 Å−1 corresponding to the interlayer distance d = 13 Å. The figure displays a very quick exfoliation of the particles in water. After only 2 min of stirring, the maximum disappeared, and the individual clay platelets were detected in the 4 wt% suspension (sample 30X before addition of solution of APS). The SAXS profiles of dilute suspensions follow a power law I(q) ∼ q−x, with x = 2 suggesting thin layer shaped objects. The particles' exfoliation, however, is not complete in 2 minutes of stirring. The Guinier analysis of the SAXS profiles in the early mixing stage provides the molecular weight of the silicate nanoparticles of Mw = 3.9 × 106. Such a value corresponds to stacks of 2 or 3 clay layers, taking into account the molecular weight of the single platelet Mw = 1.7 × 106 reported in the literature.10 LAPONITE® clay particles containing two or three tightly bound platelets were previously demonstrated.21–23 The SAXS profiles in Fig. 1a show a decrease in the scattered intensity (Io) during mixing which is assigned to a decrease of molecular weight. Fig. 1b illustrates the gradual lowering of the determined molecular weight of silicate clusters, implying a disintegration of the stacks of layers upon further stirring. A complete exfoliation of clay in water is achieved in 24–48 h. The molecular weight then approaches the value corresponding to a single platelet. The SAXS results are corroborated by viscosity measurements in Fig. 2, showing a drop in the viscosity of clay suspensions during the mixing. The exfoliation process of clay particles depends on the concentration of an aqueous dispersion and, as expected, it takes a longer time in a more concentrated system of higher viscosity. However, even in the case of the highest clay content in the sample 60X (13 wt% – before addition of solution of APS) the clay particles were completely exfoliated in 48 h. In the literature, the stable dispersion of RDS LAPONITE® (5 wt%) was reported to be formed in 1 day11 or even in 1 h.10 The development of a clay dispersion over time was also followed by measurement of the ζ-potential. The exfoliation of clay particles is a result of clay wetting and hydration of Na+ interlayer ions that are released, thus leading to an increase in the negative charge of the ζ-potential as shown in Table 1. The platelet repulsion then prevails, and viscosity decreases. The clay dispersion and exfoliation were also proved by transmission electron microscopy (TEM). The TEM micrograph of the dried gel 30X in Fig. S1 (ESI†) shows that the gel is homogeneous and the clay platelets are exfoliated well.
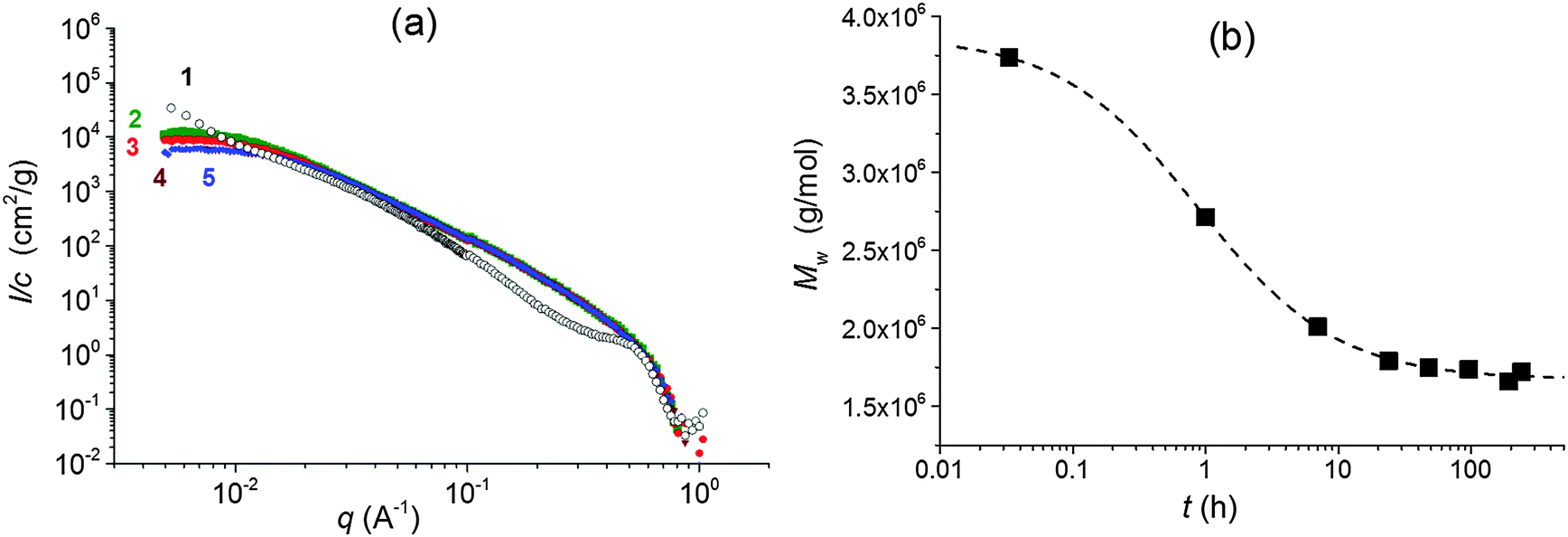 | ||
Fig. 1 (a) Evolution of SAXS profiles during mixing of clay suspensions; sample 30X curves (1; ○) – dry, t(mixing) = 0 (2;  ) – 2 min, (3; ) – 2 min, (3;  ) – 1 h, (4; ) – 1 h, (4;  ) – 24 h, and (5; ) – 24 h, and (5;  ) – 48 h; (b) evolution of the molecular weight Mw of clay particles/platelets during mixing, sample 30X. ) – 48 h; (b) evolution of the molecular weight Mw of clay particles/platelets during mixing, sample 30X. | ||
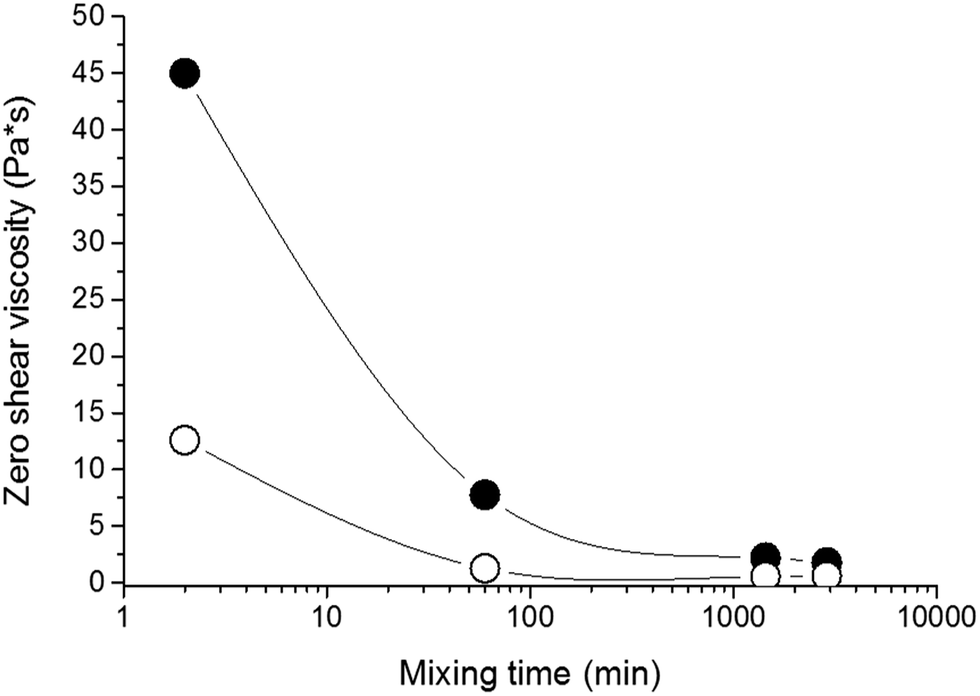 | ||
| Fig. 2 Zero shear viscosity of clay suspensions as a function of mixing time; curves ○ 30X and ● 60X. | ||
| Mixing time of aqueous clay dispersion | ζ-Potential (mV) |
|---|---|
| 2 min | −23 |
| 1 h | −55 |
| 2–14 days | −50 |
| 1 h + NIPA | −31 |
The time for the preparation of a clay suspension and the corresponding degree of clay exfoliation and dispersion was found to be a crucial parameter in hydrogel synthesis. The moduli of hydrogels significantly depend on the mixing time, mainly in systems with high clay concentration as shown in the next paper.24 For instance, the modulus of the gel 60X is more than 50% higher when the reaction mixture was mixed for 24 h prior to the polymerization compared to that undergoing mixing for only 1 h.
3.2 Interaction of clay with reaction constituents
The clay dispersion is also affected by the reaction mixture components due to their mutual interaction. The monomer NIPA is known to adsorb onto clay surfaces due to H-bonding and dipolar interactions which prevent the clay aggregation and improve the clay dispersion. On the contrary, Haraguchi et al.4 reported that both initiators ammonium persulfate and TEMED promote aggregation of the clay XLG. Both are located near the clay surface due to strong ionic interactions of persulfate or tertiary amine–clay interactions that are not well specified in the case of TEMED.We checked the effect of the individual reaction mixture agents on dispersion of the XLS in comparison with XLG and the effect of clay concentration and time of mixing on the reaction suspension. The clay XLS used in this paper exhibited a better dispersion in aqueous solution with respect to XLG clay. Fig. 3 displays the viscosity of different mixtures containing low (4 wt%) and high (13 wt%) amounts of clay, corresponding to the samples 30X and 60X, respectively, prepared by mixing for 1 h. In both systems, 30X and 60X, the viscosity of the clay suspension decreased after the addition of NIPA, due to the diminishing extent of clay–water interaction. The initiator APS only slightly augmented the viscosity in contrast to a dramatic increase reported in the case of the XLG suspension. However, the TEMED effect on viscosity growth at high clay content was very significant. The interpretation of this phenomenon is given below.
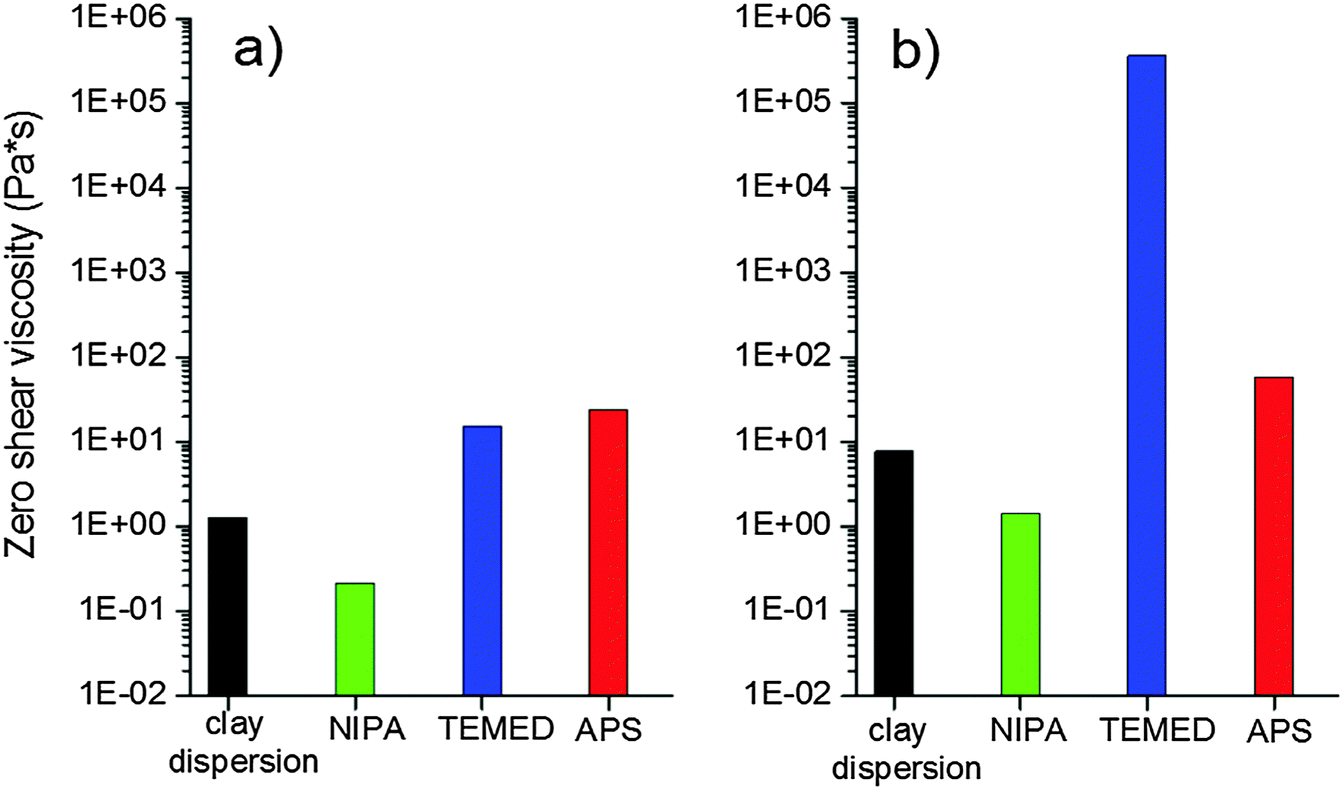 | ||
| Fig. 3 Effect of reaction components on the viscosity of clay dispersions for different clay contents, mixing time = 1 h: (a) 30X and (b) 60X. | ||
The interaction of the reaction mixture components NIPA, TEMED and polymer PNIPA with clay was studied by 1H NMR analysis. The analysis proved a very strong interaction for TEMED–clay resulting in immobilization of the TEMED molecule. This effect is demonstrated in Fig. 4, which shows the disappearance of the peaks corresponding to TEMED in the presence of clay. The signals at 2.50 and 2.22 ppm assigned to CH2 and CH3 groups of TEMED (see Fig. 4a) were also observed and found to be sharp in the mixture with NIPA (Fig. 4b) and even in the polymerization mixture containing the polymer PNIPA (Fig. 4c). The position of the corresponding peaks was slightly shifted to 2.75 and 2.40 ppm in the polymer solution in contrast to the mixture with monomer only. This effect is a result of an interaction of the TEMED molecule in solution with a polymer, likely due to adsorption. The TEMED fragment covalently bound to a polymer as an initiating end group was not detectable because the molecular weight of the polymer was too high (see below).
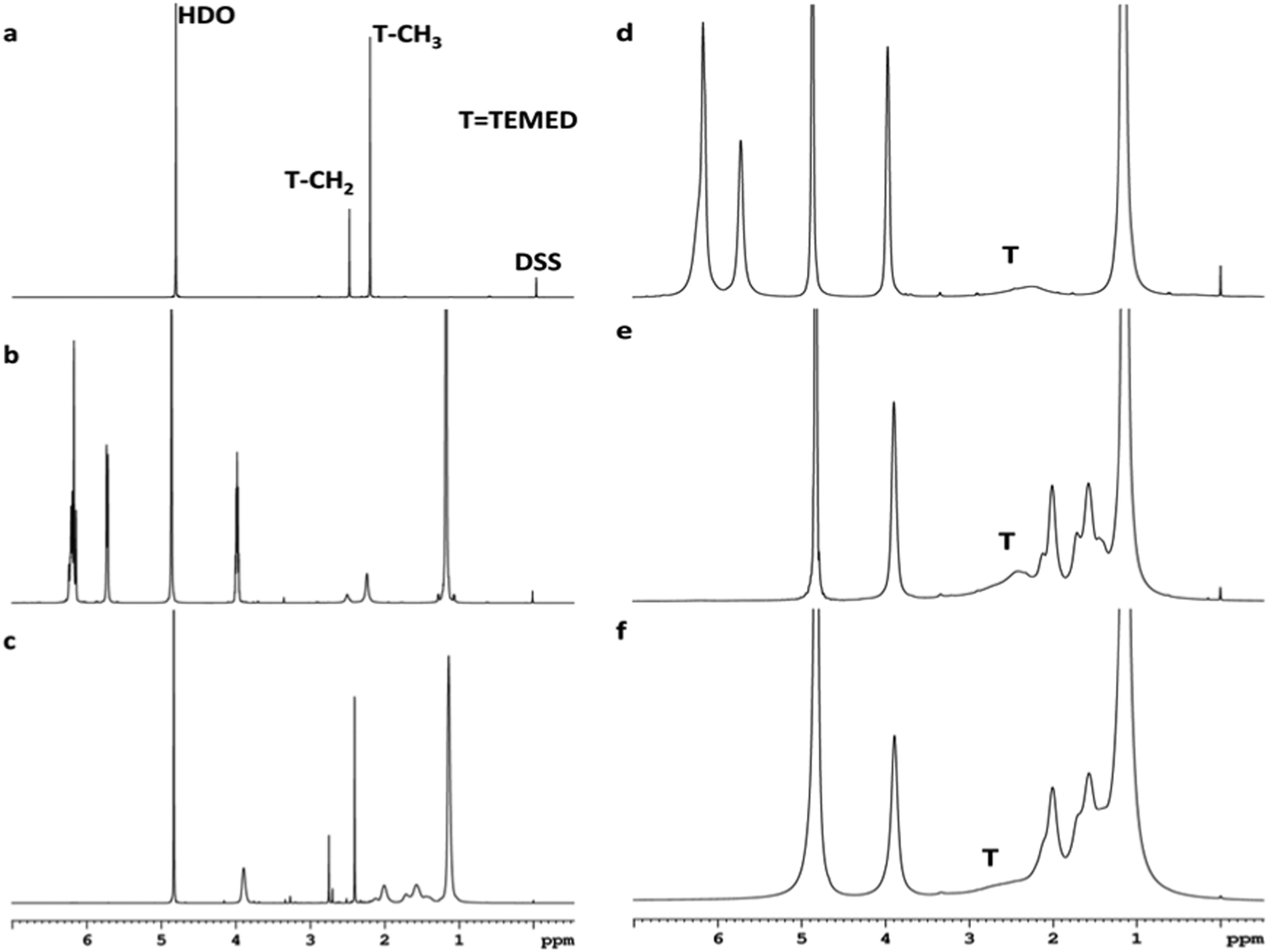 | ||
| Fig. 4
1H NMR spectra of TEMED (a), NIPA–TEMED (b), PNIPA–TEMED (c), reaction mixture 30X, clay–NIPA–TEMED/APS (3:1), t(reaction) = 0 (d), reaction mixture t = 20 min (e), and gel 30X (f). All spectra were recorded in D2O at room temperature. | ||
In the reaction suspension involving clay, however, the TEMED signals were very broad (Fig. 4d). During polymerization in the presence of clay (Fig. 4e) the situation was the same; in the gel, the TEMED signals were almost not detectable (Fig. 4f). The position of these broad bands appears to be shifted during polymerization by interaction with the polymer in the same way as in the absence of clay.
The disappearance of the peaks in the presence of clay is due to immobilization of the TEMED molecule by strong adsorption onto a clay surface. On the contrary, the peaks corresponding to both NIPA (vinylCH and CH2 at 5.73 ppm and 6.18 ppm, respectively, as well as isopropyl CH and CH3 at 3.98 ppm and 1.17 ppm, respectively) and PNIPA (main chain CH and CH2 at 2.01 ppm and 1.57 ppm, respectively, as well as isopropyl CH and CH3 at 3.89 ppm and 1.14 ppm, respectively) were still present in the spectra of the clay containing systems (Fig. 4d–f) in spite of NIPA and PNIPA also being adsorbed onto clay. The NMR results thus prove that the clay–TEMED interaction was much stronger compared to clay–NIPA and clay–PNIPA.
For a quantitative evaluation of the TEMED immobilization and thus the interaction strength with the clay in comparison with the interactions clay–NIPA and clay–PNIPA we determined the T2 relaxation times characterizing the mobility of the corresponding CH2 and CH3 protons. Table 2 shows that T2 values of TEMED were reduced by almost 3 orders of magnitude in the clay suspension and even more in the polymerized clay suspension and in the gel. The relaxation times of NIPA and PNIPA in the presence of clay were at least 10 times longer than that of TEMED revealing a higher mobility because of a weaker interaction with the clay. Moreover, the ratio of relaxation times corresponding to the TEMED molecule T2(NCH3)/T2(NCH2) for the clay–TEMED mixture was appreciably higher (∼2) than that for free TEMED (<1). This finding implies a stronger slowing down of the mobility of CH2 protons with respect to that of CH3 by adsorption onto clay. TEMED is often used as a bidentate ligand for metal ions, and the redox initiation is also supposed to proceed through a cyclic intermediate including both TEMED nitrogens (see below). Therefore, we assume that both nitrogens are also involved in the interaction with a silicate layer by N-coordination to exchangeable cations or by H-bonds through a H2O bridge on an oxygen containing clay surface to form a stable coordination cycle with mainly the CH2 groups being severely immobilized. The compound dimethylethyleneamine (DMEA) was used as a mononitrogen model of TEMED to test this hypothesis. The higher T2 values of DMEA in Table 2 compared to TEMED demonstrate a weaker bonding through one nitrogen only and may support the idea of a strong cyclic-type TEMED interaction on the clay surface.
| Sample | T 2 [ms] | |||||
|---|---|---|---|---|---|---|
| TEMED (or DMEA) | NIPA (or PNIPA) | |||||
| NCH2 | NCH3 | CH2 |
CH |
NCH | CH3 | |
| a Determined after deconvolution from the linewidth Δν using the relation T2 = (πΔν)−1. b One broad band for both NCH3 and NCH2 protons. c PNIPA. | ||||||
| TEMED | 945 | 868 | ||||
| NIPA | 805 | 648 | 1343 | 722 | ||
| TEMED–NIPA | 26 | 24 | 111 | 104 | 287 | 71 |
| TEMED–clay | 1.1a | 2.2a | ||||
| DMEA–clay | 3.0a | 3.8a | ||||
| Reaction mixture, t = 0 TEMED–NIPA–clay | 0.9a,b | 10 | 9 | 24 | 15 | |
| Polymerization, t = 20 min TEMED–NIPA–PNIPA–clay | 0.8a,b | 7c | ||||
| Final gel | 0.5a,b | 7c | 7c | |||
Another possibility of the interaction involving both nitrogens is an interplatelet bridge giving rise to the formation of plate doublets. Such an increase in doublet formation explains the dramatic increase in viscosity of a clay suspension after the addition of TEMED as shown in Fig. 3b. This phenomenon occurs only at a high clay content of 60X (13 wt%), where the platelets are close to each other and could be bridged by the TEMED molecule. No significant increase in viscosity was observed at a lower clay amount, 30X (4 wt%), as shown in Fig. 3a.
Unfortunately, it was not possible to follow the interaction of the initiator APS in a similar way as the behaviour of TEMED because in 1H NMR spectra the signal of APS was overlapped by the signal of the solvent (residual HDO). Moreover, due to low sensitivity of the 15N nuclei signal, APS was undetectable in the 15N NMR spectra.
3.3. Hydrogel formation
Gelation is the crucial step of network formation. Moreover, in the multiphase hybrid systems the relative rate of gelation with respect to phase structure evolution, such as aggregation or reaction induced microphase separation, governs the structure and morphology of a hybrid network. Therefore, determination of a correlation between molecular and phase structure development is a way to understand the mechanism of formation and the structure of the hybrid gel. We followed the formation of the PNIPA–clay hybrid hydrogel in all the process complexity using several complementary experimental methods that allowed us to monitor in situ both molecular and phase structures and evaluate the corresponding correlation.(i) the kinetics of polymer growth,
(ii) the molecular weight Mw of the polymer, and
(iii) crosslinking, gelation and the postgel stage.
In situ FTIR and chemorheology measurements were the techniques used to gain the corresponding information.
Polymerization. Polymerization of NIPA is initiated by the redox system involving APS as the initiator and TEMED as an activator promoting the decomposition of APS into free radicals. TEMED decreases the activation energy Ea of polymerization making the reaction possible at low temperatures. While Ea = 62 kJ mol−1 in the polymerization initiated with APS, the initiating system APS/TEMED reduces the activation energy to Ea = 22 kJ mol−1.25 The initiation mechanism proposed by Feng et al.26 is shown in Scheme 1. The reaction scheme is assumed to proceed via a charge transfer complex and a cyclic transient state to form two main types of primary initiating radicals.
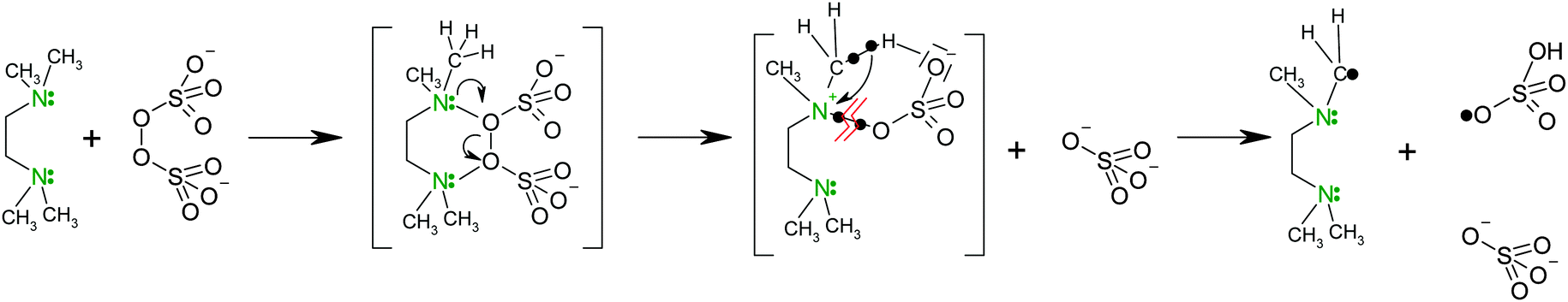 | ||
| Scheme 1 Mechanism of the primary radical formation in redox initiation by APS and TEMED.26 | ||
In addition, hydroxyl radicals formed by hydrolysis (*OSO3H + H2O → H2SO4 + HO*) are considered to be responsible for initiation.
In the presence of clay minerals, the polymerization process is affected and inhibition27 as well as catalysis28,29 of free radical polymerization by clay have been described in the literature.
The reaction kinetics of NIPA polymerization were monitored in situ by FTIR spectroscopy. Fig. 5 shows the progress of conversion of the NIPA monomer during the reaction evaluated using the peak at 1417 cm−1 attributed to the presence of CC bonds in vinyl groups. Polymerization initiated by the redox system at 25 °C was relatively fast under the used reaction conditions. Seventy percent conversion was reached in 15 min and the complete conversion in approximately 40 min in the case of the reaction mixture with the following composition: 30X–0.028–0.0043, i.e., [NIPA] = 0.75 M, CXLS = 3 wt%, [TEMED]r = 0.028, and [APS]r = 0.0043 (see Fig. 5a).
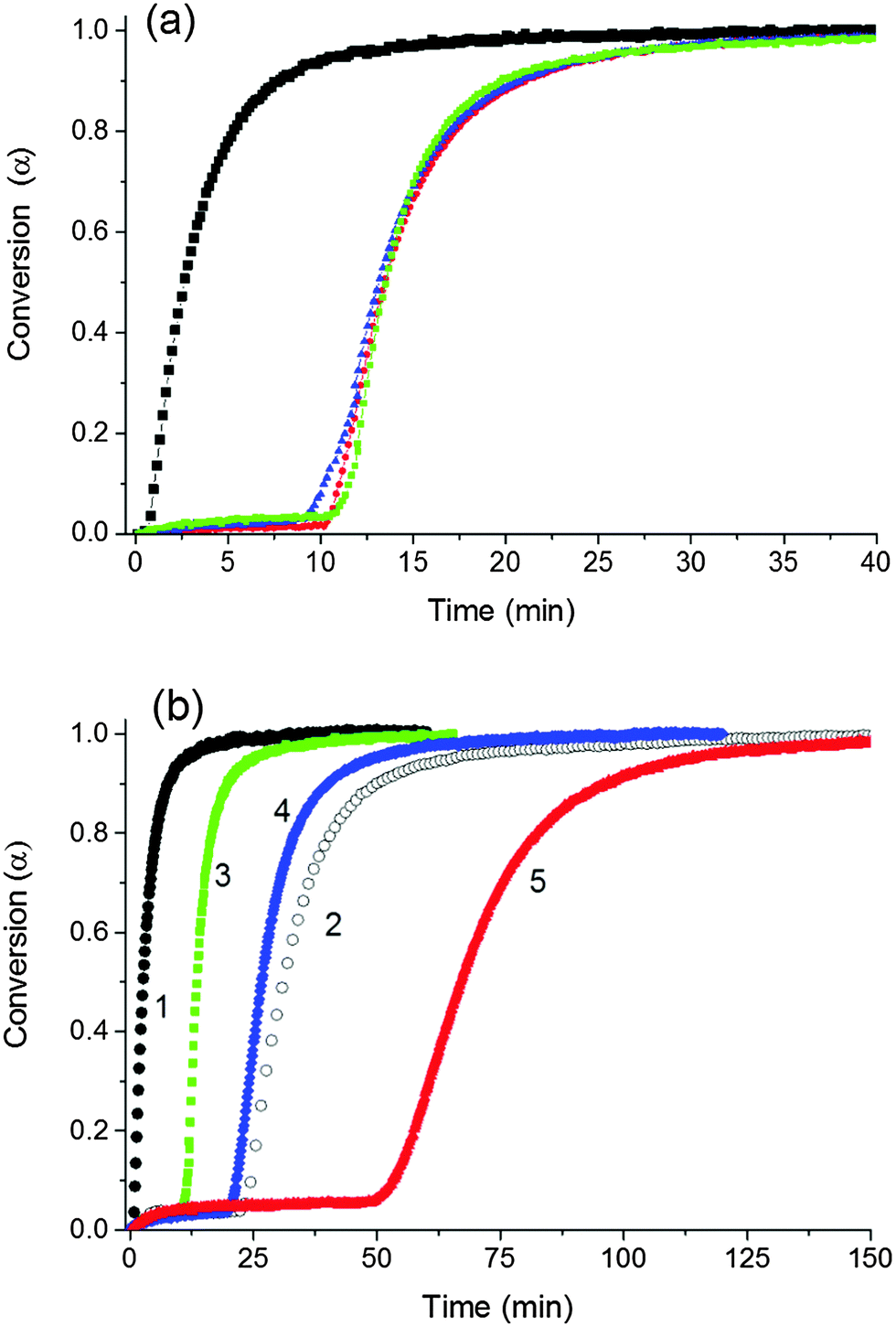 | ||
Fig. 5 Kinetics of NIPA conversion during polymerization in systems with a different content of clay and initiators, T = 25 °C (a) the effect of clay content; (■) 0X neat PNIPA, ( ) 30X, ( ) 30X, ( ) 40X, ( ) 40X, ( ) 60X [TEMED]r = 0.028, [APS]r = 0.0043; (b) the effect of TEMED and APS content; 1(●) 0X–0.028–0.0043, 2 (○) 0X–0.01–0.0014, 3 ( ) 60X [TEMED]r = 0.028, [APS]r = 0.0043; (b) the effect of TEMED and APS content; 1(●) 0X–0.028–0.0043, 2 (○) 0X–0.01–0.0014, 3 ( ) 60X–0.028–0.0043, 4 ( ) 60X–0.028–0.0043, 4 ( ) 60X–0.028–0.0014, and 5 ( ) 60X–0.028–0.0014, and 5 ( ) 60X–0.01–0.0014. ) 60X–0.01–0.0014. | ||
Initiation stage. The reaction exhibited an induction period that was lengthened by the decreasing content of initiators APS and TEMED (Fig. 5b). A comparison of the curves 1 and 2 shows the effect of the initiators on the neat NIPA polymerization, and curves 3–5 illustrate the polymerization in the presence of clay. Curves 3 and 4 prove the influence of a decreasing content of APS and curve 5 shows the polymerization under diminished concentration of both APS and TEMED. Fig. 5a reveals that the polymerization was slowed down and the induction period was longer in the presence of clay. This period corresponds to an initiation stage that is considered to involve two steps: generation of primary radicals by decomposition of the initiator and production of the first monomer radical by the reaction of the monomer and the primary radical. A slight consumption of the monomer up to 5% can be observed during this induction period.
The initiation is the rate determining step in free radical polymerization. It is dependent on the concentration of the initiator [I], the rate of decomposition kd and the efficiency of initiation fi characterizing an effective radical concentration.
| Ri ∼ 2kdfi[I], | (1) |
In the redox polymerization, usually the initiator and the activator are applied in a constant ratio of concentrations, r = [activator]/[initiator]. In particular, in the case of PNIPA–clay gel preparation, the ratio r = [TEMED]:[APS] = 3:1 is often used in the literature. We have thus determined the rate of initiation as a function of the initiators' concentration [TEMED, APS]r=3 while keeping the ratio constant at r = 3. The rates of initiation were evaluated as being inversely proportional to the corresponding induction period (tind); Ri ∼ 1/tind. The following dependencies were established for polymerization of neat NIPA and the PNIPA–clay system 60X:
| Ri(PNIPA) ∼ [TEMED, APS]r=31.2 | (2a) |
| Ri(60X) ∼ [TEMED, APS]r=30.8 | (2b) |
Propagation stage. After primary radical formation, the polymerization of nearby adsorbed NIPA monomers begins, and the slow initiation step is followed by a fast propagation, the rate of which is less dependent on the clay and initiators (see Fig. 5). The growing polymer chains are physically bound to clay platelets by coordination and dipolar interactions of the chain end groups from the initiation radicals. Moreover, adsorption of chain segments onto the clay surface also occurs. The molecular weight of a polymer generally increases with increasing monomer concentration [M] and decreasing initiator content as follows from the general expression,
| Pn ∼ kp[M]/2(ktfikd[I]init[I]act)1/2 | (3) |
Haraguchi et al.30 proved that the Mw of PNIPA during the free NIPA polymerization corresponds to a theoretical dependence on the monomer content with the power equal to 0.98. The theoretical prediction as to the effect of the initiator was proved by Feng et al.26 who determined a decrease in the molecular weight of poly(acrylamide) with increasing TEMED content.
The effect of clay in reducing the amount of initiating radicals should give rise to the generation of fewer growing chains of higher length. Haraguchi, however, determined that clay does not affect the Mw of the polymer which is constant regardless of the clay concentration. We investigated the effect of clay and the effect of initiators upon polymerization in the presence of clay. Our experiments revealed substantially higher values of molecular weight of PNIPA in the gel compared to the linear PNIPA particularly at higher clay content in accordance with a lower efficiency of initiation in the presence of clay. Fig. 6 displays the Mv of PNIPA formed during the linear free NIPA polymerization as well as the Mv of the chains in the hybrid network as a function of concentration of TEMED or APS initiators. The molecular weights of the network chains were determined by a technique used previously by Haraguchi et al.17 (see Experimental). The Mv in Fig. 6 decreased with increasing amount of initiators. At low APS and TEMED content ([APS]r < 0.0087, [TEMED]r < 0.028), the molecular weight reached the value 5 × 106, while it is lower by two orders of magnitude, Mv = 4.6 × 104, at high concentrations ([APS]r = 0.087, [TEMED]r = 0.28). The power dependencies determined from double logarithmic plots, however, were smaller compared to the theoretical power 1/2 in eqn (3).
| Mv(PNIPA) ∼ [TEMED]−0.4[APS]−0.2 | (4a) |
| Mv(60X) ∼ [TEMED]−0.35[APS]−0.1 | (4b) |
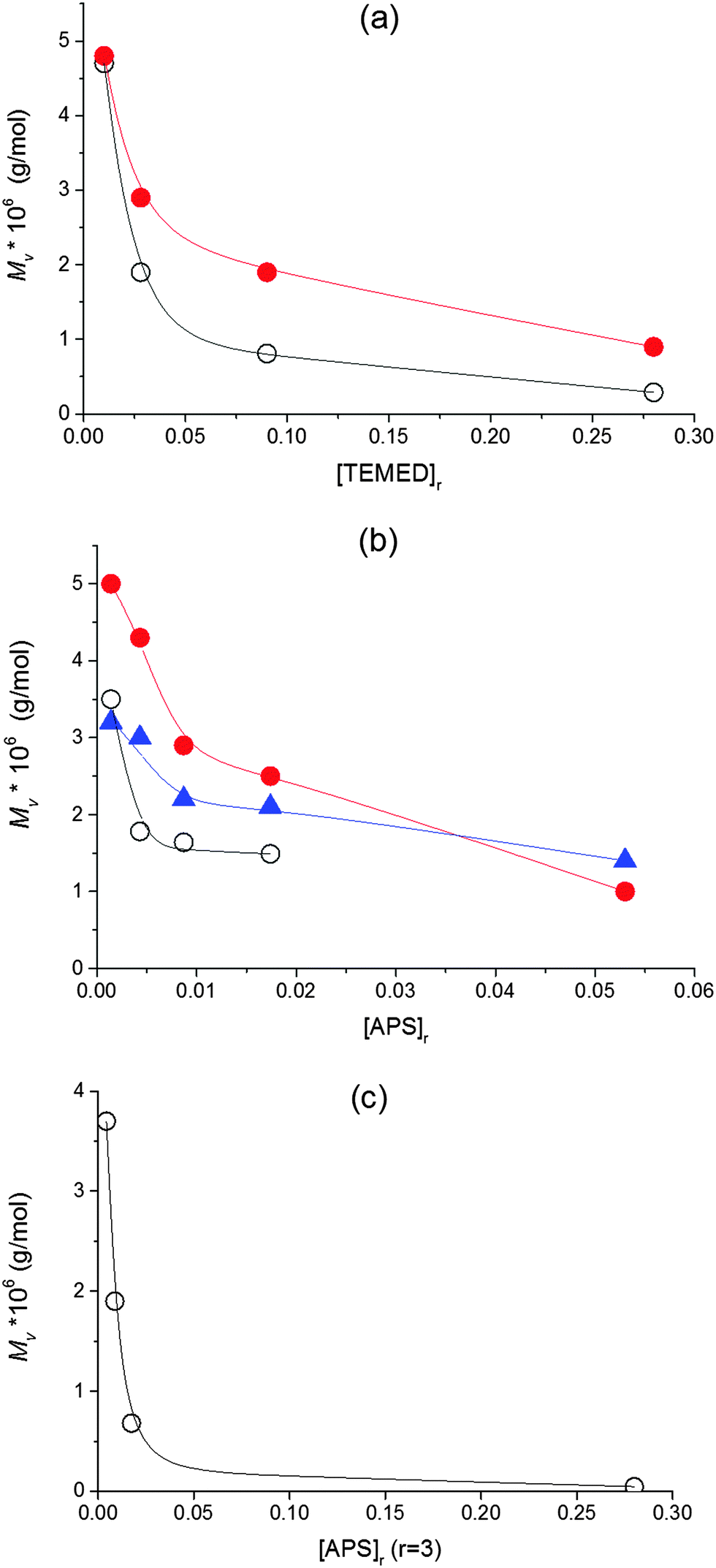 | ||
Fig. 6 Molecular weight Mv of PNIPA during linear NIPA polymerization and in the clay based gel as a function of (a) the content of TEMED at constant [APS]r = 0.0087, (b) the content of APS at constant [TEMED]r = 0.028, and (c) the content of APS and TEMED at a constant ratio r(=[TEMED]/[APS]) = 3. (○) free PNIPA, ( ) PNIPA in gel 30X, and ( ) PNIPA in gel 30X, and ( ) PNIPA in gel 60X. ) PNIPA in gel 60X. | ||
In the literature the redox initiators' ratio r is often considered to be crucial in affecting the redox polymerization and the final gel properties, while, typically, the constant ratio is used in the preparation of polymer–clay gels. Adrus et al.32 found that increasing the ratio (r > 1) decreases gel times in nonlinear PNIPA polymerization while the low ratio results in a long induction period. Also Nigmatullin et al.14 reported the effect of the ratio on the rate of polymerization/gelation of the polyacrylamide–clay system. They observed a faster polymerization with increasing r at a constant initiator APS concentration, while the polymerization speed was decreased with an increasing r at constant activator content (fastest at r < 1). In all these cases, however, the polymerization was accelerated by an increasing overall concentration of the initiator or activator regardless of the ratio of their content. We found, however, that the optimum ratio r = 3 promotes the formation of the PNIPA–clay gels displaying the best properties.24 Taking this into account, we followed the effect of the concentration of initiators while keeping the ratio constant at r = 3. Fig. 6c shows that such a dependence of Mv is very dramatic, thus revealing that the overall concentration of both initiators is important for the initiation stage and control of the forming polymer structure. The Mv decreases mainly if the content of both the initiator and activator is high. In this case, termination of primary radicals is favoured.33
The molecular weight of the polymer chains between crosslinks, i.e., the length of elastically active chains between clay platelets in our case, is an important structural parameter of a gel. This factor determines the structure as well as mechanical and swelling properties of gels. Therefore, the initiators affect not only the rate of polymerization but also the polymer structure. Consequently, the initiating conditions, i.e., the concentration of initiators or their composition, can be used to efficiently control the polymer structure and the final gel properties.24
Similar to the initiators, clay also exhibits an effect on the polymerization process and polymer structure. A lower efficiency of the initiation during NIPA polymerization in the presence of clay results in a decrease of the polymerization speed with respect to the free NIPA reaction and higher molecular weights of polymer chains in a gel.
Crosslinking and gelation. Crosslinking in the PNIPA–clay system could occur by several different ways. The most typical is termination by the recombination of two chains from different clay platelets thus forming a bridge between the platelets that acts as an elastically active network chain. The formation of physically trapped entanglements of two chains anchored on different platelets is another and a very important way of crosslinking. This type of physical bridging of the clay sheets is indirectly experimentally proved by the perfect self-healing properties of these clay–polymer gels.34 Another possibility is a chain termination in a “radical centre” on another or the same platelet to create an elastically active chain or inactive cycle. In the later stage, however, the access of the chain end to a primary radical on another platelet is limited due to an absorbed polymer layer on the surface of the majority of clay platelets (see below). An unlikely possibility is termination by disproportionation at room temperature polymerization due to the high temperature dependence of this reaction. The self-crosslinking by radical transfer to a chain is typical of free acrylate polymerization.35,36 Nigmatullin et al.14 reported self-crosslinking at a high monomer concentration even in the presence of a low clay amount. Haraguchi et al., however, excluded such a crosslinking in the systems containing clay above a certain critical concentration.17 In addition to elastically active chains inactive loops and dangling chains are formed.
Chemorheology, i.e., dynamic shear rheology of a system performed during a chemical reaction, is a reliable and applicable method to follow the in situ evolution of a molecular structure, crosslinking and gelation during polymerization. The determined time of gelation tgel is the most important parameter characterizing network formation. However, only few papers have described the chemorheology investigation of PNIPA–clay and similar hydrogel formation. Okay and Oppermann11 monitored the development of complex shear moduli G′(t) and G′′(t) during redox polymerization of acrylamide in an aqueous dispersion of LAPONITE® RDS. They observed that the crosslinking reaction, characterized by a fast growth of the G′ modulus, is promoted by an increasing content of LAPONITE®. However, the overall kinetics of structure evolution, including the pregel stage and gelation times, were not determined because the initial period of the reaction exhibiting low moduli below the detection level was not taken into account. In the case of NIPA–XLS polymerization, Okay et al.12 showed a very unusual progress during the crosslinking reaction consisting of an initial abrupt increase in the modulus followed by a continuous decrease with polymerization time. This behaviour was interpreted by inhomogeneity of the gel and the existence of clusters of clay platelets. Nigmatullin et al.14 studied by means of rheology the rate of polymerization of acrylamide in the presence or absence of montmorillonite clay. The rate was characterized by the duration of the initial induction period to reach a certain value of modulus (G′ = 3 Pa). This period is not equivalent to the polymerization induction period corresponding to the initiation stage because it includes the rheological behaviour of the polymer. The systems were also characterized by dynamics of the storage modulus evolution in the gelation and postgelation stages.
We monitored the in situ crosslinking and gelation of the PNIPA–XLS system by following the evolution of the complex moduli G′(t) and G′′(t). None of the abnormalities reported by Okay et al.12 were observed. During polymerization both moduli increased and the system classically progressed through the three stages illustrated in Fig. 7. In the pregel stage G′′ > G′, in the postgel stage G′ > G′′ and at the gel point G′ ≅ G′′. The precise determination of the gel times tgel was based on the Winter–Chambon criterion of the critical state19 (see Experimental). The gel times are given in Table 3 together with induction periods of polymerization for the selected systems for comparison. For the sake of simplicity, however, only the evolution of the dynamic storage modulus G′(t) is displayed in the following figures. The increase of G′(t) during polymerization of a series of reaction mixtures with different contents of clay (20X–60X), APS and TEMED is shown in Fig. 8. The pregel stage includes an induction period of the polymerization governed by the initiation process (see above) as well as propagation of a polymer chain. An increase in the modulus was relatively small in this stage and was detected under our experimental conditions only during the slow reactions showing a high enough pregel modulus. This enhancement of the modulus is a result of the increasing fraction of a polymer and probably also a result of bridging of the platelets by the growing polymer, thereby forming polymer–clay clusters. The steep increase in the modulus corresponds to the onset of gelation. Gelation is accelerated by an increase in the monomer NIPA concentration (Table 3) and concentrations of the initiators APS and TEMED (Fig. 8) as expected due to the faster kinetics (cf.Fig. 5). Moreover, Fig. 8a reveals that gelation was slowed down by the increasing content of clay.
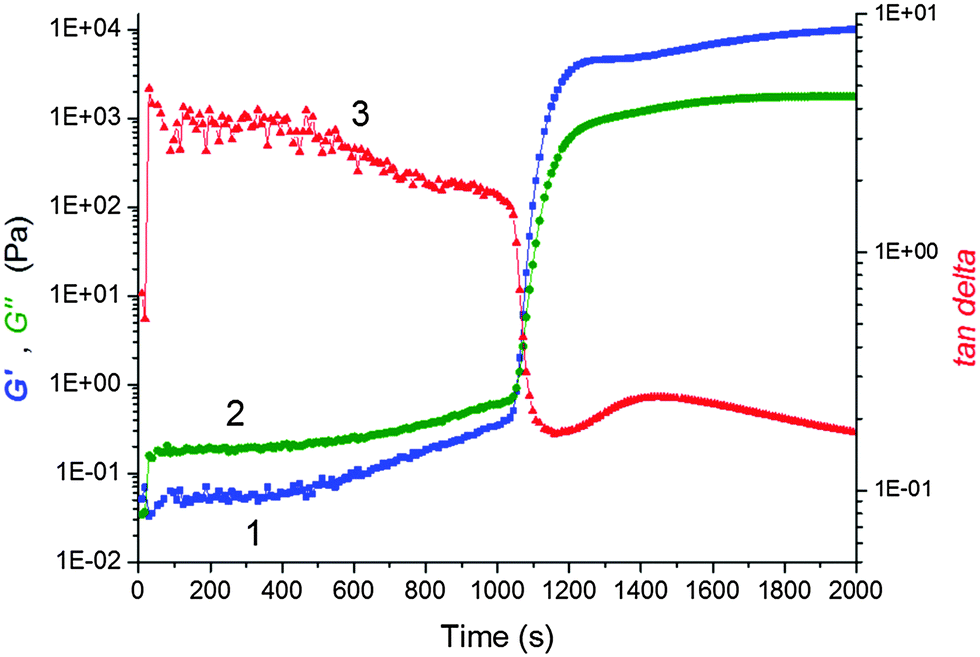 | ||
Fig. 7 Evolution of complex moduli G′(t) and G′′(t) and the loss factor tanδ during polymerization of the system 60X–0.01–0.0087; 1-(blue  ) storage modulus, 2-(green ) storage modulus, 2-(green  ) loss modulus, and 3-(red ) loss modulus, and 3-(red  ) loss factor tanδ. ) loss factor tanδ. | ||
| [NIPA] [mol L−1] | Sample | t ind [min] | t gel [min] |
|---|---|---|---|
| 0.75 | 20X–0.028–0.0087 | 3 | |
| 30X–0.028–0.0087 | 4 | 4 | |
| 40X–0.028–0.0087 | 5 | 6 | |
| 60X–0.028–0.0087 | 5 | 9 | |
| 20X–0.028–0.0043 | 14 | ||
| 30X–0.028–0.0043 | 10 | 18 | |
| 40X–0.028–0.0043 | 9 | 22 | |
| 60X–0.028–0.0043 | 10.5 | 25 | |
| 0.5 | 30X–0.028–0.0087 | 11 | |
| 1 | 30X–0.028–0.0087 | 2.5 | |
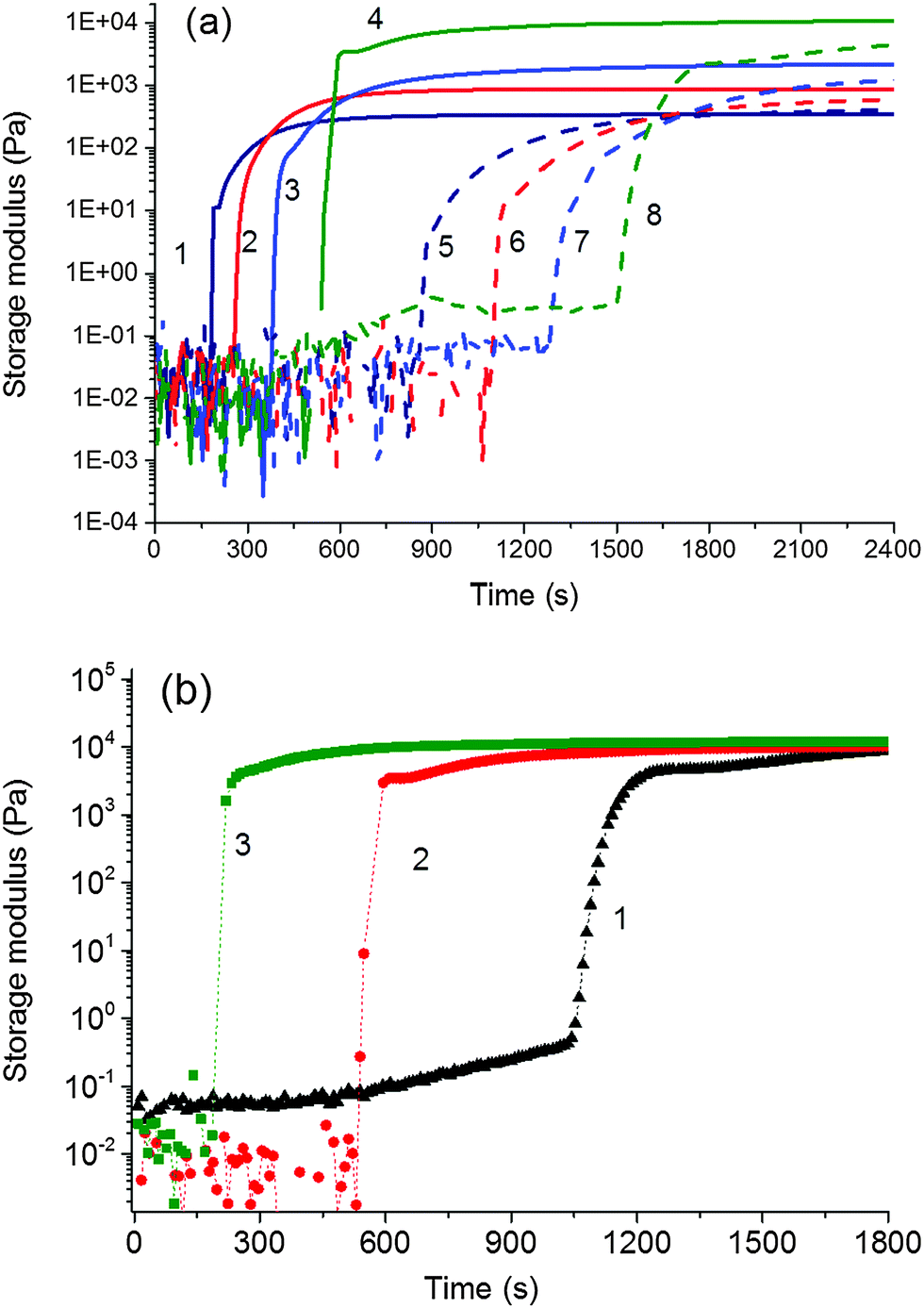 | ||
Fig. 8 Evolution of the storage modulus G′(t) during polymerization of the system NIPA–clay at 25 °C, [NIPA] = 0.75 mol L−1 (a) effect of clay and APS content, [TEMED]r = 0.028; curves 1 – 20X, [APS]r = 0.0087; 2 – 30X, [APS]r = 0.0087; 3 – 40X, [APS]r = 0.0087; 4 – 60X, [APS]r = 0.0087; 5 – 20X, [APS]r = 0.0043; 6 – 30X, [APS]r = 0.0043; 7 – 40X, [APS]r = 0.0043; 8 – 60X, [APS]r = 0.0043; (b) the effect of TEMED content, [APS]r = 0.0087, 60X; curves 1(▲) [TEMED]r = 0.01, 2( ) [TEMED]r = 0.028, and 3( ) [TEMED]r = 0.028, and 3( ) [TEMED]r = 0.09. ) [TEMED]r = 0.09. | ||
The sharp increase in the modulus at the gel point is typical for fast reacting chemical systems mainly at low viscosity reaction mixtures. In contrast, during the gelation of LAPONITE® without polymer, Tanaka et al. found that the complex viscosity changed much more smoothly at the gelation point.37 This difference in rheology evolution reflects the different types of gelation.38,39 While LAPONITE® undergoes aggregation, physical gelation, the PNIPA–XLS gelation, is the chemical process, mainly the recombination of PNIPA chains. Colloidal gelation occurring via noncovalent bonds is often controlled by diffusive transport, whereas the chemical gelation is reaction limited and realized by covalent bond percolation.40 Diffusion control could lead to a smooth change in viscosity at the gel point of LAPONITE®. On the contrary, the PNIPA–clay gelation is reaction rate limited resulting in a faster evolution of rheology close to the gel point. The PNIPA–clay is a physical gel because of physical interactions of clay–PNIPA. However, the crosslinking proceeds by combination of chemical reactions and physical interactions. In the early stage and during gelation, the chemical reactions dominate resulting in the steep increase in the modulus at the gel point. Consequently, the structure growth and gelation of the PNIPA–clay system can be treated as chemical crosslinking and gelation.
The rate of gel formation is generally determined mainly by the kinetics of the crosslinking reaction and the content and functionality (f) of a crosslinker governing the critical conversion at the gel point αc = 1/(f − 1). At high functionality the gelation occurs earlier at a low conversion. In our case two opposite factors played a role. The clay serves as a multifunctional crosslinker and increasing its amount should accelerate the gelation of a system. On the other hand, the clay slows down the polymerization of NIPA, thus delaying gelation. The kinetics are decelerated in the presence of clay; however, this effect is independent of clay content (Fig. 5 and Table 3) contrary to gelation (Fig. 8 and Table 3). Hence, another factor must be responsible for the deceleration of gelation with increasing clay content. The polymerization is initiated at the clay surface by TEMED (or APS) radical fragments. At a constant TEMED concentration and increasing content of clay, the average number of initiating sites on the clay platelet, i.e., a theoretical functionality of the plate fth(platelet) (∼[TEMED–APS]/CXLS under full initiating efficiency) is reduced (see Scheme 2). Gelation of the system, i.e., the time of gelation, depends on an average theoretical functionality of a clay platelet. Therefore, tgel ∼ 1/fth(platelet) = CXLS/[TEMED–APS]. Consequently, at a constant TEMED (and APS) concentration and increasing content of clay, the gelation speed is decreased. This hypothesis is supported by the plot in Fig. 9. The master curve of the dependence of gel times on theoretical functionality of the platelet describes relatively well the gelation of all studied systems with different contents of clay, TEMED and APS. The ratio CXLS/[TEMED–APS] in different systems is considered relatively with respect to the standard mixture 30X–0.028–0.0087 (see Experimental).
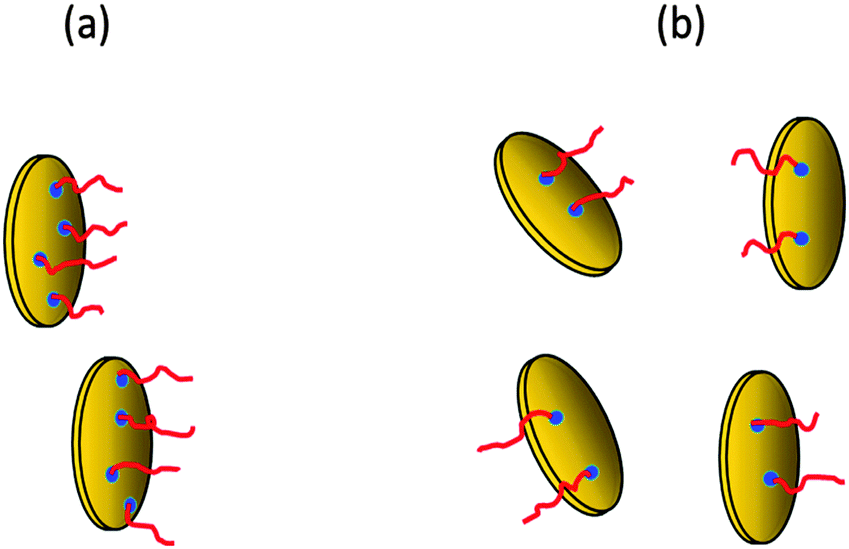 | ||
| Scheme 2 Functionality of clay platelets corresponding to the number of issuing polymer chains at (a) low clay content and (b) high clay content, for polymerization at a constant concentration of initiators. | ||
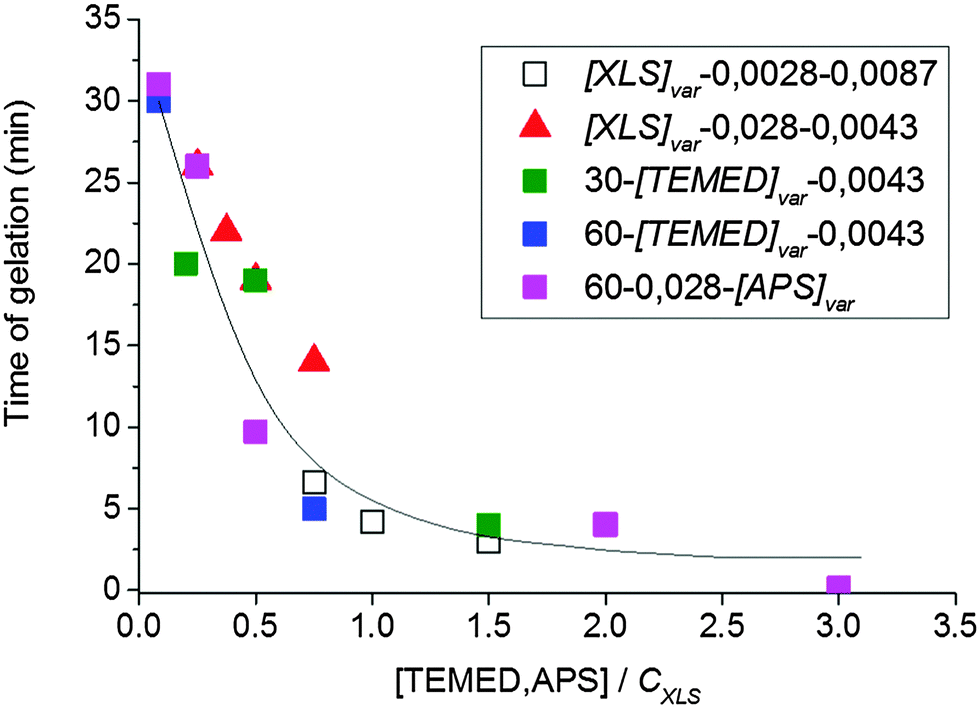 | ||
| Fig. 9 Gel time as a function of the ratio of concentrations [TEMED–APS]/CXLS corresponding to the theoretical functionality of the platelet. | ||
The ratio is determined relatively with respect to the standard mixture 30X–0.028–0.0087.
Postgel stage. In the postgel stage the modulus continues to grow as the crosslinking density gradually increases and finally reaches a plateau. Fig. 8a shows the complexity of the gel formation. While the time of gelation is delayed with increasing clay content, the modulus in the postgel stage is enhanced. The interpretation of this fact and discussion of the mechanical properties are given in the next paper.24 The chemorheology measurements of hydrogels, however, are not well suited for a long monitoring of the polymerization because of water evaporation and a corresponding change in the degree of swelling. Therefore, a long time evolution of the modulus in the postgel stage, displayed in Fig. 10, was determined by measuring the separate samples polymerized ex situ in closed vials for a particular reaction time. The majority of modulus enhancement occurred within 1 h during the polymerization of the 60X sample. A slight increase, however, continued up to 24 h despite the fact that the radical polymerization was finished, i.e., the monomer completely reacted in less than 30 min. We suggest that this increase of modulus was brought about by a slow formation of trapped entanglements thus generating additional physical crosslinking.
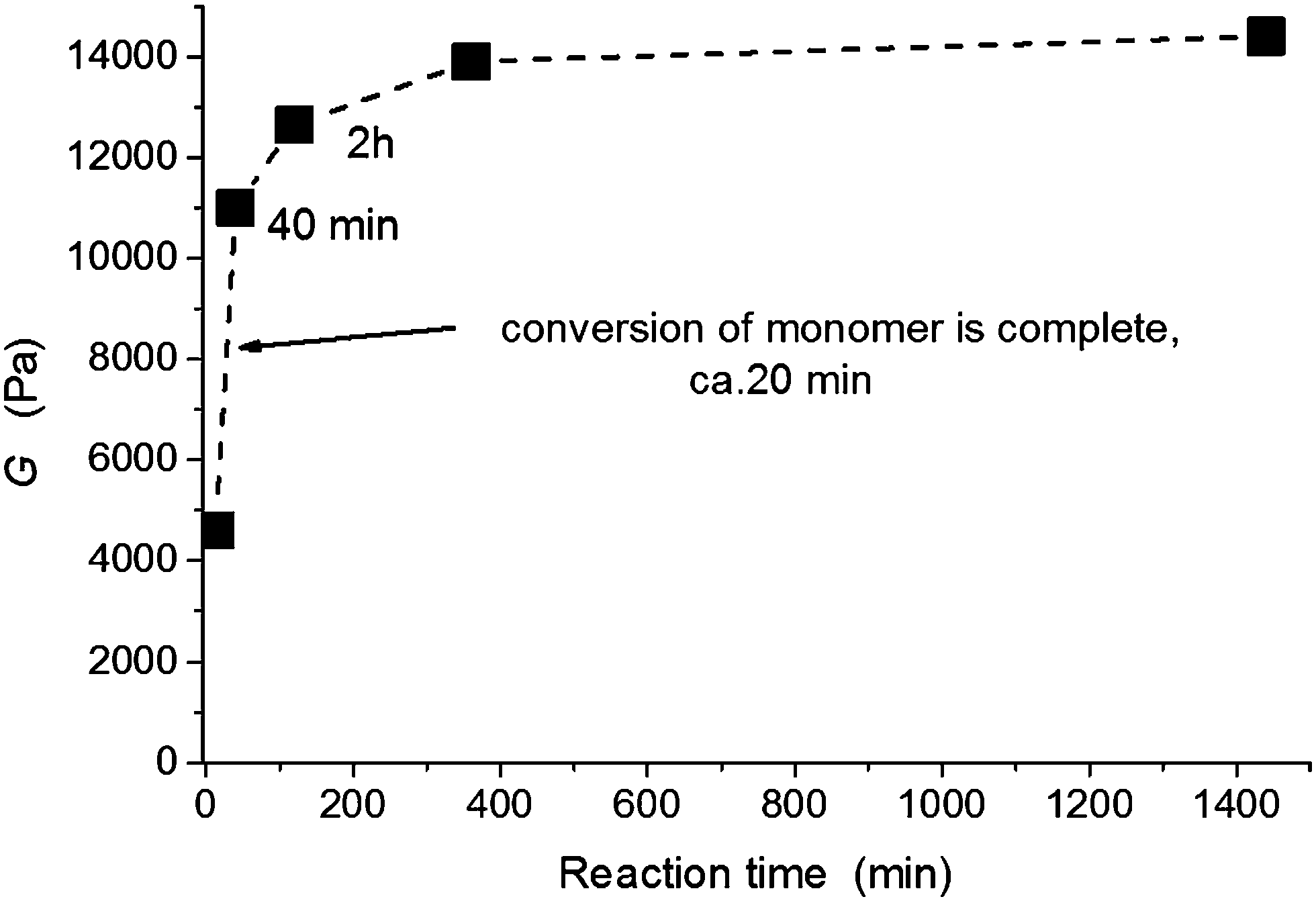 | ||
| Fig. 10 Evolution of the modulus in the postgel stage during polymerization of the system 60X, the content of initiators [APS]r = 0.0087, [TEMED]r = 0.028 (T = 25 °C). | ||
The development of the phase structure during polymerization followed by in situ time resolved SAXS is illustrated in Fig. 11. One can see the appearance and growth of a shoulder at high q values (∼0.5 Å−1) of the SAXS profiles in the early polymerization stage. This shoulder is attributed to the formation of a polymer layer on clay platelets. The radius of gyration of the adsorbed polymer determined by the function of a generalized Gaussian coil was found to be Rg = 6.3 Å, which roughly corresponds to the polymer layer thickness D = 16 Å [geometrical diameter of a particle D = (5/3)1/22Rg under an assumption of a spherical shape, which reflects an upper limit of the layer thickness]. This finding is in agreement with literature data as Shibayama et al.8 reported the polymer layer with a thickness of ca. 10 Å and Nelson et al.41 determined the monolayer of polyethers on the clay surface of 11–16 Å thickness. Rg of the layer is approximately constant for all the time, thus proving that no further polymer adsorption takes place after saturation of the clay surface by a monolayer. The gradual increase in the SAXS intensity in the shoulder region is in accordance with an increase in the number of clay platelets covered with a polymer layer. In approximately 30 min, the intensity reaches a plateau, likely revealing that all the accessible platelets are covered as in the case of the sample 0.75N–60X–0.028–0.0043. This phase structure evolution was compared with the molecular structure evolution determined by chemorheology. Fig. 8a shows that the corresponding system gelled at approximately 25 min, which approximately corresponds to the time of coverage of the majority of the clay platelets.
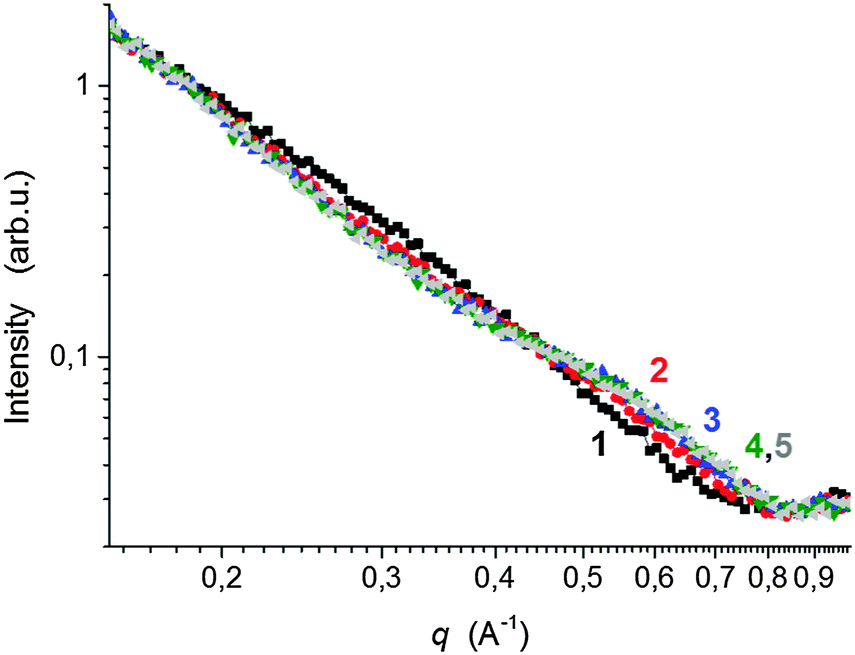 | ||
Fig. 11 SAXS profiles during polymerization of PNIPA–clay at 25 °C, (60X–0.028–0.0043). The curves are indicated according to the reaction time; 1 (■) 10 min,  – ( – ( ) 20 min, ) 20 min,  – ( – ( ) 30 min, ) 30 min,  – ( – ( ) 40 min, and ) 40 min, and  – ( – ( ) 100 min. ) 100 min. | ||
In situ UV/Vis spectrometry measurements were conducted during polymerization to examine the turbidity in the PNIPA–clay system as described in the literature7 and correlate it with molecular structure evolution. Fig. 12 illustrates the evolution of the absorbance at 600 nm of the reaction dispersions during polymerization of four systems with different clay contents (20X–60X). The initially homogeneous transparent reaction mixtures exhibited a sharp drop in transparency followed by a period of turbidity and the recovery of a partly transparent state revealing a final homogenization. The evolution of turbidity was compared with the gel points of the systems determined by chemorheology. The mixtures became cloudy shortly before the gel point. The dashed lines in the figure display gelation. The increasing clay amount in the mixtures shifted the appearance of turbidity to longer reaction times in agreement with the corresponding shift of gelation. The cloudy period was relatively short, being only 1.5 min for the sample 20X, however, the period was lengthened with increasing clay content in the gel. Also the transparency of the finally cured gels decreases at a higher amount of clay. Moreover, very cloudy systems were formed in the case of high concentration of the APS initiator.
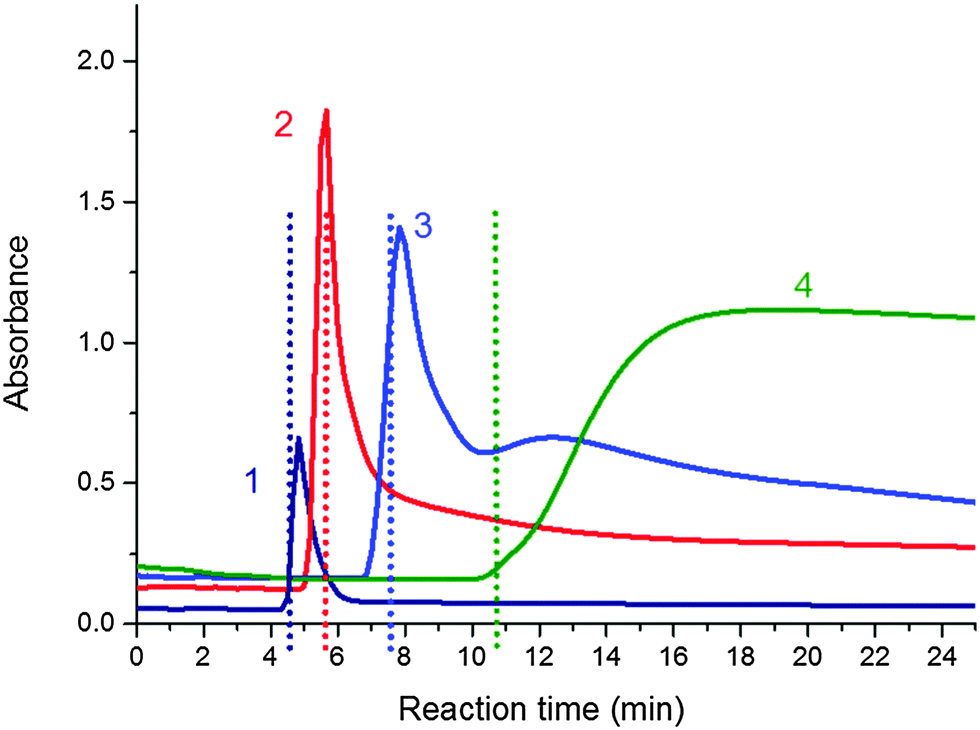 | ||
| Fig. 12 Evolution of absorbance at 600 nm measured by UV-Vis spectrometry during polymerization of the PNIPA–clay system at 25 °C, [APS]r = 0.0087, [TEMED]r = 0.028; curves: 1 – 20X, 2 – 30X, 3 – 40X, and 4 – 60X. Dashed lines show the gelation point. | ||
These results related to the phase structure are in agreement with literature data4,8 generated using DLS and SANS. The novelty of our investigation consists in the precise time correlation of these phenomena with molecular structure development, such as the initiation of polymerization, growth of the polymer fraction, gelation and postgel increase in the crosslinking density as determined by independent techniques.
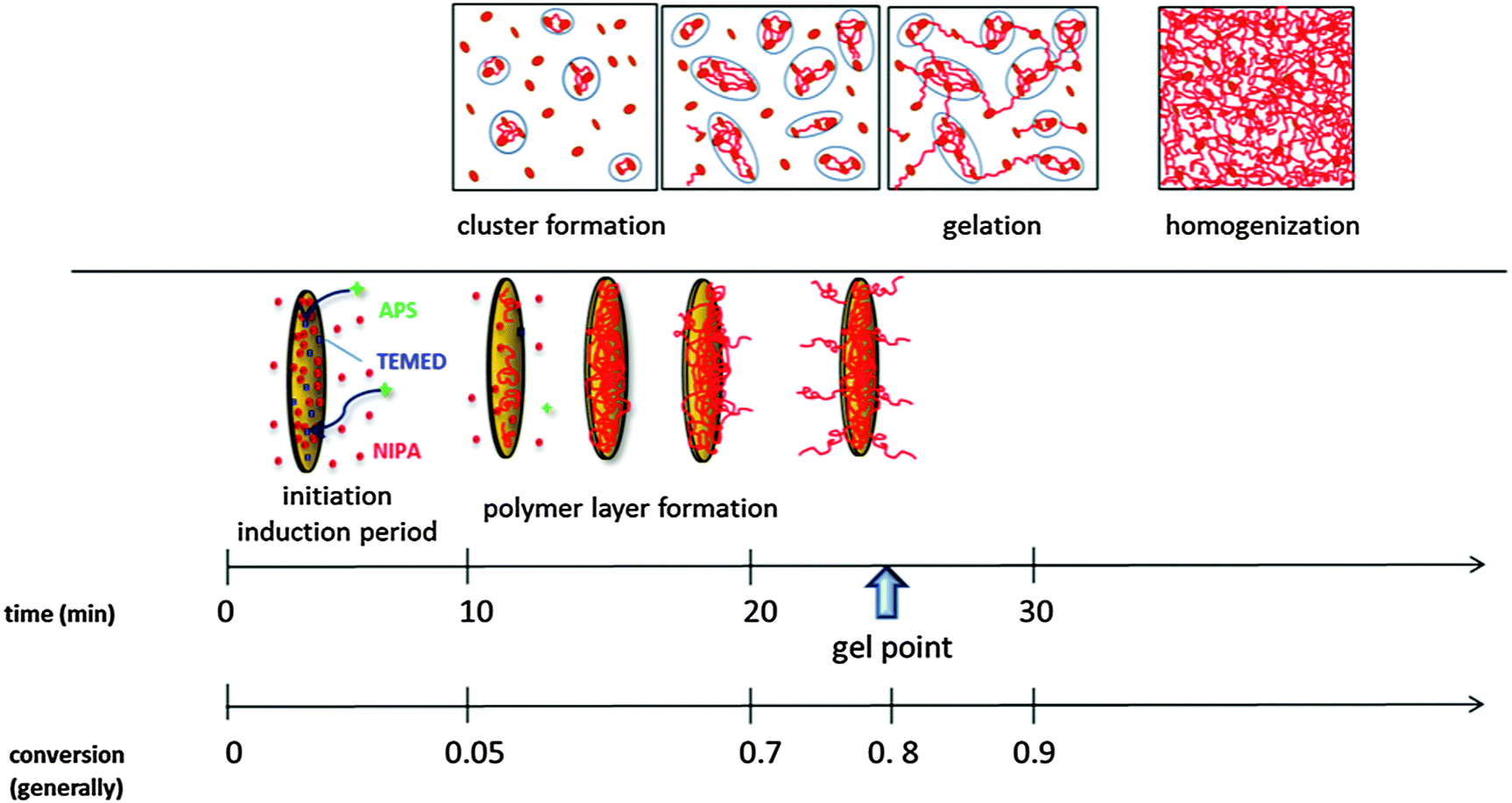 | ||
| Scheme 3 Progress of gel formation during polymerization. The time and conversion scales determined for polymerization of the sample 0.75N–60X–0.028–0.0043 at T = 25 °C. | ||
(a) Pregel stage. The initial slow reaction stage is determined by the formation of primary radicals and the initiation of the NIPA polymerization in the aqueous suspension of well-dispersed and exfoliated clay platelets. The redox type initiation is manifested by the induction period, which is the rate determining step of the process. An increasing content of initiators, APS and TEMED, reduces the induction period and accelerates the polymerization thus enabling control of the polymerization rate. On the contrary, the presence of clay in a reaction mixture results in inhibition of polymerization and lengthening of the induction period, tind (Fig. 5) due to the mobility and steric restrictions to form the active redox complex APS–TEMED (see Scheme 1). The strong adsorption of TEMED onto a clay surface and a high medium viscosity could play a role.
The induction period is followed by a fast polymerization and polymer fraction growth (Fig. 5). The formed polymer chains adsorb onto the clay platelets and create a polymer monolayer of a constant thickness of ∼1.5 nm on their surface. The fraction of the covered clay sheets gradually increases until all accessible sheets are grafted with a polymer layer (tlayer) (cf.Fig. 11).
After saturation of the clay platelet surface, the grafted chains continue to grow outward from the platelet into the solution interplatelet space. Only then can the polymer chains issuing from different platelets crosslink and form a bridge between platelets, most likely by a chemical reaction, such as recombination and formation of entanglements. The bridging of the clay sheets gives rise to the formation of clusters and results in system heterogeneity due to the irregular density of growing crosslinked domains (clusters). This phenomenon is manifested by a slight increase in the modulus in the pregel stage (Fig. 8) (also an increase in the polymer fraction contributes to this modulus enhancement) and the appearance of turbidity (Fig. 12). The gradual increase of the modulus, likely characterizing the formation and growth of clusters (one of the possible interpretations), sets early in the pregel stage (tclustering) immediately after the induction period (cf.Fig. 5 and 8) and continues until gelation. The process occurs in the conversion region of the monomer α ∼ 0.1–0.8. The turbidity, however, is observed only just before gelation in the conversion of ∼0.7 when large clusters detectable by UV/Vis spectrometry are created (tturbidity) (cf.Fig. 8 and 12). In the case of XLG clay, Shibayama and Haraguchi reported4,8 a distinct reduction in transparency already in the early stage and a monomer conversion α = 0.07 at a minimum transparency. Both crosslinking and the appearance of turbidity are delayed by increasing the content of clay. This finding is contrary to the results of Chen and Xu42 who observed an earlier turbidity period in systems with a higher concentration of clay LAPONITE® RD. This difference could be related to the clay type as RD LAPONITE® is prone to form “house of cards” structures at higher clay content unlike the XLS LAPONITE®. The heterogeneity is more pronounced in gels prepared with a high content of initiators. In this case, the extremely fast polymerization leads to excessive clustering and build-up of a microgel-like structure, which is manifested by strong turbidity. This phenomenon is discussed in the next paper.24
(b) Gelation. Gelation of the system sets in at a relatively high conversion of the monomer, i.e., the fraction of the polymer being ∼0.7–0.9 (cf.Fig. 5 and 8) (this is not the conversion of the crosslinking reaction).
A large fraction of polymer first covers the clay platelets, and only after the surface saturation by the polymer layer does the crosslinking and clustering occur. The clusters are gradually interconnected, mainly by the reaction (recombination) of PNIPA chains, and gelation of the system occurs by bond percolation (tgel) as in the typical chemical gelation. The delay of gelation by clay is a result of the inhibition effect of clay and a slower polymerization, as well as by decreasing the functionality of clay platelets with increasing clay concentration.
(c) Postgel stage. Gradually all clay sheets are covered by a polymer layer (Fig. 11) and issue polymer chains forming bridges between platelets. They are thus incorporated into the gel and enhance the gel fraction. The increasing crosslinking density in the postgel stage and continuous bridging of the clusters within the gel results in homogenization of the system and recovery of transparency (Fig. 12). The recovery is delayed by a high clay content. In the late postgel stage, even after termination of the radical polymerization, the crosslinking density is slowly enhanced by the formation of trapped entanglements (cf.Fig. 5 and 10).
The particular progress stages were characterized by the reaction times tx and conversion α during polymerization. While the time scale is applicable for a particular system only, the conversion scale could be more general. The experimentally determined development during polymerization of the sample 0.75N–60X–0.028–0.0043 at T = 25 °C is as follows:
t ind = 11 min, αind = 0–0.05 – induction period (FTIR)
t layer = 11–30 min, αlayer = 0.05–0.8 – formation of a polymer layer on the clay platelet up to all covered platelets (SAXS)
t clustering = 11–25 min, αclustering = 0.1–0.8 – bridging of platelets, formation of clusters (chemorheology)
t turbidity = 22.5 min (maximum at 30 min), αturbidity = 0.8–0.85 – formation of large clusters (UV/Vis)
t gel = 25 min, αgel = 0.8–0.9 – gelation of the system (chemorheology)
t transparency > 35 min, αtransparency > 0.9 – a gradual increase in transparency due to homogenization of the gel (UV/Vis)
t ent ≥ 60 min, αent = 1 – formation of entanglements dominates this stage (DMA).
Scheme 3 illustrates the progress in gel formation involving time and conversion correlations of the experimentally determined processes.
4. Conclusions
In situ monitoring of the gel formation by several independent methods (FTIR, chemorheology, SAXS, and UV/Vis) enabled the detailed time and conversion correlation of molecular and phase structure evolution during gel formation. Such a correlation has not been performed to date. Using these complementary methods, 7 progress stages of the gel formation were experimentally determined during polymerization and were then characterized and correlated to each other: (1) induction period (FTIR), (2) polymer propagation and coverage of clay platelets by a polymer monolayer (FTIR and SAXS), (3) saturation of the clay surface followed by crosslinking and bridging of clay platelets resulting in clustering (SAXS and chemorheology), (4) large cluster generation giving rise to the formation of an inhomogeneous structure and the appearance of turbidity (UV/Vis), (5) gelation by interconnection of clusters and bond percolation, including precise determination of the gel point (chemorheology), (6) postgel stage structure homogenization (UV/Vis) and (7) postgel growth of crosslinking density by entanglement formation (FTIR and DMA).The crucial factors determining structure evolution are discussed, and their effect on the formation and properties of PNIPA–clay hydrogel is clarified. The gel formation and properties are known to be determined by the content of clay and monomer in an aqueous suspension. In this and the next paper we show, in addition, very efficient control of both gel build-up (this work) and the gel properties (next paper)24 by reaction conditions, including the redox initiator system and the prereaction stage of clay suspension preparation. Moreover, the complex effect of clay on NIPA polymerization and gel formation was clarified.
Dispersion of the XLS clay powder in the prereaction stage is fast; however, stacks of 2 or 3 clay layers are present for a long time in the clay suspension mainly at high clay content. A perfect exfoliation takes time, and a careful preparation of a clay suspension is therefore necessary as the degree of exfoliation prior to polymerization strongly determines the final gel properties.24
The process of polymerization and gel formation is significantly affected by the redox initiating system, the initiator APS and the activator TEMED. They control not only the rate of polymerization but also the polymer structure. Their initiation efficiency is manifested in the early stage of the polymerization by the induction period corresponding to the formation of primary radicals. Reducing the concentration of the initiators leads to a decrease in the speed of polymerization. However, on the other hand, this reduction also leads to the generation of higher molecular weight chains. This finding implies the possibility of controlling the chain length between crosslinks in the gel independently of the crosslinking density determined by the clay content.
The clay acts in the gel formation not only as a crosslinker and reinforcing nanofiller but also influences the polymerization process as well. The presence of clay promotes the inhibition of polymerization by lowering the efficiency of the initiators. This effect is attributed mainly to sterical and mobility restrictions for the formation of primary radicals in a high viscosity medium. A very strong interaction of the activator TEMED with the clay platelets of a coordination type likely plays a role. Moreover, increasing the clay content at a constant initiator concentration results in a delay of gelation due to the decreasing functionality of the clay platelet despite an improvement in the mechanical properties.
Acknowledgements
The authors acknowledge the financial support of the Grant Agency of the Czech Republic (Projects P108/12/1459 and 13-23392S). B.S. and R.K. acknowledge the Charles University Faculty of Science for providing the opportunity to pursue their PhD studies.References
- K. Haraguchi, T. Takehisa and S. Fan, Macromolecules, 2002, 35, 10162 CrossRef CAS.
- K. Haraguchi and H. J. Li, Macromolecules, 2006, 39, 1898 CrossRef CAS.
- K. Haraguchi, H. J. Li, H. Y. Ren and M. Zhu, Macromolecules, 2010, 43, 9848 CrossRef CAS.
- K. Haraguchi, H. J. Li, K. Masuda, T. Takehisa and E. Elliott, Macromolecules, 2005, 38, 3482 CrossRef CAS.
- K. Haraguchi, Colloid Polym. Sci., 2011, 289, 455 CAS.
- K. Haraguchi and Y. J. Xu Colloid, Polym. Sci., 2012, 290, 1627 CrossRef CAS.
- B. Ferse, S. Richter, F. Eckert, A. Kulkarni, C. M. Papadakis and K. F. Arndt, Langmuir, 2008, 24, 12627 CrossRef CAS PubMed.
- S. Miyazaki, H. Endo, T. Karino, K. Haraguchi and M. Shibayama, Macromolecules, 2007, 40, 4287 CrossRef CAS.
- S. Abdurrahmanoglu, V. Can and O. Okay, J. Appl. Polym. Sci., 2008, 109, 3714 CrossRef CAS.
- J. Nie, B. Du and W. Oppermann, Macromolecules, 2005, 38, 5729 CrossRef CAS.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378 CrossRef CAS.
- S. Abdurrahmanoglu and O. Okay, J. Appl. Polym. Sci., 2010, 116, 2328 CAS.
- Y. Wang, J. Ma, S. Yang and J. Xu, Colloids Surf., A, 2011, 390, 20 CrossRef CAS.
- R. Nigmatullin, M. Bencsik and F. Gao, Soft Matter, 2014, 10, 2035 RSC.
- T. C. Farrar and E. D. Becker, Pulse and Fourier transform NMR, Academic Press, New York, 1971, p. 27 Search PubMed.
- M. Ilavský, Adv. Polym. Sci., 1993, 109, 173 CrossRef.
- K. Haraguchi, Y. Xu and G. Li, Macromol. Rapid Commun., 2010, 31, 718 CrossRef CAS PubMed.
- S. Fujishige, Polym. J., 1987, 19, 297 CrossRef CAS.
- H. H. Winter and M. Mours, Adv. Polym. Sci., 1997, 134, 165 CrossRef CAS.
- C. Martin, F. Pignon, A. Magnin, M. Meireles, V. Lelièvre, P. Lindner and B. Cabane, Langmuir, 2006, 22, 4065 CrossRef CAS PubMed.
- J. Fripiat, J. Cases, M. Francois and M. Letellier, J. Colloid Interface Sci., 1982, 89, 378 CrossRef CAS.
- L. Rosta and H. R. von Gunten, J. Colloid Interface Sci., 1990, 134, 397 CrossRef CAS.
- J. M. Saunders, J. W. Goodwin, R. M. Richardson and B. Vincent, J. Phys. Chem. B, 1999, 103, 9211 CrossRef CAS.
- B. Strachota, J. Hodan and L. Matějka, submitted.
- X. Guo, K. Qiu and X. Feng, Chin. J. Polym. Sci., 1989, 7, 165 CAS.
- X. D. Feng, X. Q. Guo and K. Y. Qiu, Makromol. Chem., 1988, 189, 77 CrossRef CAS.
- D. H. Solomon and J. D. Swift, J. Appl. Polym. Sci., 1967, 11, 2567 CrossRef CAS.
- P. Bera and S. K. Saha, Polymer, 1998, 39, 1461 CrossRef CAS.
- A. S. Badran, A. A. Abdelhakim, A. B. Moustafa and M. A. Abdelghaffar, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 609 CrossRef CAS.
- Y. Xu, G. Li and K. Haraguchi, Macromol. Chem. Phys., 2010, 211, 977 CrossRef CAS.
- H. Mauroy, Z. Rozynek, T. S. Plivelic, J. O. Fossum, G. Helgesen and K. D. Knudsen, Langmuir, 2013, 29, 371 CrossRef CAS PubMed.
- N. Adrus and M. Ulbricht, React. Funct. Polym., 2013, 73, 141 CrossRef CAS.
- G. S. Misra and U. D. N. Bajpai, Prog. Polym. Sci., 1982, 8, 61 CrossRef CAS.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253 CrossRef CAS PubMed.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5212 CrossRef CAS.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5217 CrossRef CAS.
- H. Tanaka, S. Jabbari-Farouji, J. Meunier and D. Bonn, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 021402 CrossRef PubMed.
- A. Zaccone, J. J. Crassous and M. Ballauff, J. Chem. Phys., 2013, 138, 104908 CrossRef PubMed.
- A. Zaccone, H. H. Winter, M. Siebenburger and M. Ballauff, J. Rheol., 2013, 58, 1219 CrossRef.
- S. Coresszi, D. Rioretto and F. Sciortino, Soft Matter, 2012, 8, 11207 RSC.
- A. Nelson and T. Cosgrove, Langmuir, 2004, 20, 2298 CrossRef CAS PubMed.
- Y. Chen and W. Xu, J. Mater. Res., 2014, 29, 820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM micrograph of the dried gel PNIPA-XLS, 30X. See DOI: 10.1039/c5sm01996f |
| This journal is © The Royal Society of Chemistry 2015 |