
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorous ‘ponytails’ lead to strong gelators showing thermally induced structure evolution†
Harshita
Kumari
*a,
Sarah E.
Armitage
b,
Steven R.
Kline
c,
Krishna K.
Damodaran
d,
Stuart R.
Kennedy
 b,
Jerry L.
Atwood
*e and
Jonathan W.
Steed
*b
b,
Jerry L.
Atwood
*e and
Jonathan W.
Steed
*b
aJames L. Winkle College of Pharmacy, University of Cincinnati, 3225 Eden Avenue, Cincinnati, OH, USA. E-mail: kumariha@ucmail.uc.edu
bDepartment of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk
cNIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD, USA
dDepartment of Chemistry, Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavík, Iceland
eDepartment of Chemistry, University of Missouri – Columbia, 601 South College Avenue, Columbia, MO 65211, USA
First published on 3rd September 2015
Abstract
Appending perfluoroalkyl substituents to bis(urea) gelators results in significantly decreased inter-chain interactions with markedly thinner fibres and hence more cross-linked and more transparent gels with potential applications in the crystallisation of fluorinated pharmaceuticals. Gel structure has been probed by detailed SANS measurements which indicate a surprising structure evolution on thermal cycling, not seen for hydrocarbon analogues. The SANS data are complemented by the single crystal X-ray structure of one fluorinated gelator.
Introduction
Bis(urea) derivatives are well-known low molecular weight gelators (LMWG) capable of gelling a variety of solvents at around 0.1–10 percent by weight of the total mixture.1–10 A wide variety of solvents including both water and less polar media can be immobilised by self-assembled fibrillar networks (SAFINs) based on the self-association of these small molecules, depending on the substituent groups on the gelator and hence its solubility characteristics. Bis(urea) hydrogels have been reported, for example, bearing hydrophilic carboxylic acid functionalities.11 Hydrophobic solvents such as aliphatic and aromatic hydrocarbons, esters, ketones, and alcohols are also gelled by bis(ureas) bearing alkyl, aryl or ester substituents.2 Bis(ureas) have not been significantly explored as gelators of fluorous media such as perfluorocarbons (PFCs) despite the interest in gels of these solvents as wound protecting creams, as lubricants and for in vivo oxygen delivery.12 Thus far, gelation of PFCs has predominantly been achieved by fluorous surfactants in the presence of 1–20% water. A range of amide-derived LMWG bearing fluorinated substituents (generally multiple perfluoroalkyl chains) have been used to gel some fluorinated solvents with some success.13–17 However, due to their low surface tensions and the problems associated with solubilising organic compounds in a lipophobic medium, there are very few compounds which possess the ability to gel pure PFCs.13–16,18,19 The presence of hydrophobic and lipophobic fluorous chains in LMWG compounds results in peculiar chemical behaviour and also means that these types of compounds cannot usually gel organic solvents.13 In 1999 an extraordinary example of the gelation ability of fluorinated LMWGs was reported by Beckman et al.20 who were able to gel supercritical carbon dioxide by increasing the “CO2-philicity” of a bis(urea) derived compound by addition of perfluoroalkyl chains. The adaptation of LMWG to gel fluorous media or act as crystal growth media for fluorous drugs represents a novel direction in supramolecular gel chemistry.While the primary supramolecular structure of LMWG fibres is reasonably well understood, the evolution of a one-dimensional hydrogen bonded motif to give an interconnected, entangled three-dimensional sample-spanning framework capable of immobilising a contiguous fluid phase is still the topic of speculation.21–24 The problem is exacerbated by the inability of many ‘molecular level’ techniques to give information on extended supramolecular structure. Moreover, sample preparation methods such as drying a gel to give a xerogel suitable for analysis by X-ray powder diffraction may well result in changes to the structure and degree of crystallinity of the material. Ultimately in situ techniques capable of probing supramolecular structure on a variety of length scales are best placed to provide insight into questions of higher order supramolecular structure and the process by which a gelator is deposited from solution into an interconnected fibrous morphology. In this regard small angle neutron scattering (particularly using isotopic substitution to enhance contrast) is of particular benefit in combination with complementary techniques such as SAXS, WAXS, SEM and single crystal and powder X-ray diffraction. We now present a detailed comparison of the gelation behaviour of bis(urea) gelators bearing perfluoroalkyl ‘ponytails’25 with their hydrocarbon analogues. It was anticipated that ‘insulating’ the urea hydrogen bonded motif within a fluorous ‘sheath’ may result in novel gelation behaviour while retaining the compatibility of the gelator with a range of solvents. A detailed SANS analysis has been undertaken of the structure of the gels which shows very different thermal evolution and SAFIN morphology for the two classes of materials.
Gel preparation and properties
The lipophilic gelators 1a–d and their fluorinated analogues 2a–b were readily prepared in yields ranging between 79–92% in THF from the corresponding diisocyanate and aniline derivatives (see ESI,† pages S1–S4). The gelation propensity of all compounds was screened in a total of 31 solvents with a range of polarity by dissolving the gelator in the hot solution in concentrations of (0.2–10) wt% with sonication26 and allowing the resulting sol to cool. All compounds were insoluble in water. The hydrocarbon compounds of type 1 gel the majority of the solvents tested (20 in the case of compound 1a, 26 for 1b, 21 for 1c and 17 for 1d) with minimum gelation concentration (mgc) (1.5–9) wt%, suggesting relatively weak gelation. All four gelators of type 1 gel polar solvents such as cyclohexanone and 1-butanol as well as apolar media such as toluene and xylenes. The gels are thermoreversible but tend to break down on mechanical agitation at the mgc. The appearance of the gels is opaque (Fig. 1), suggesting relatively large, aggregated fibres.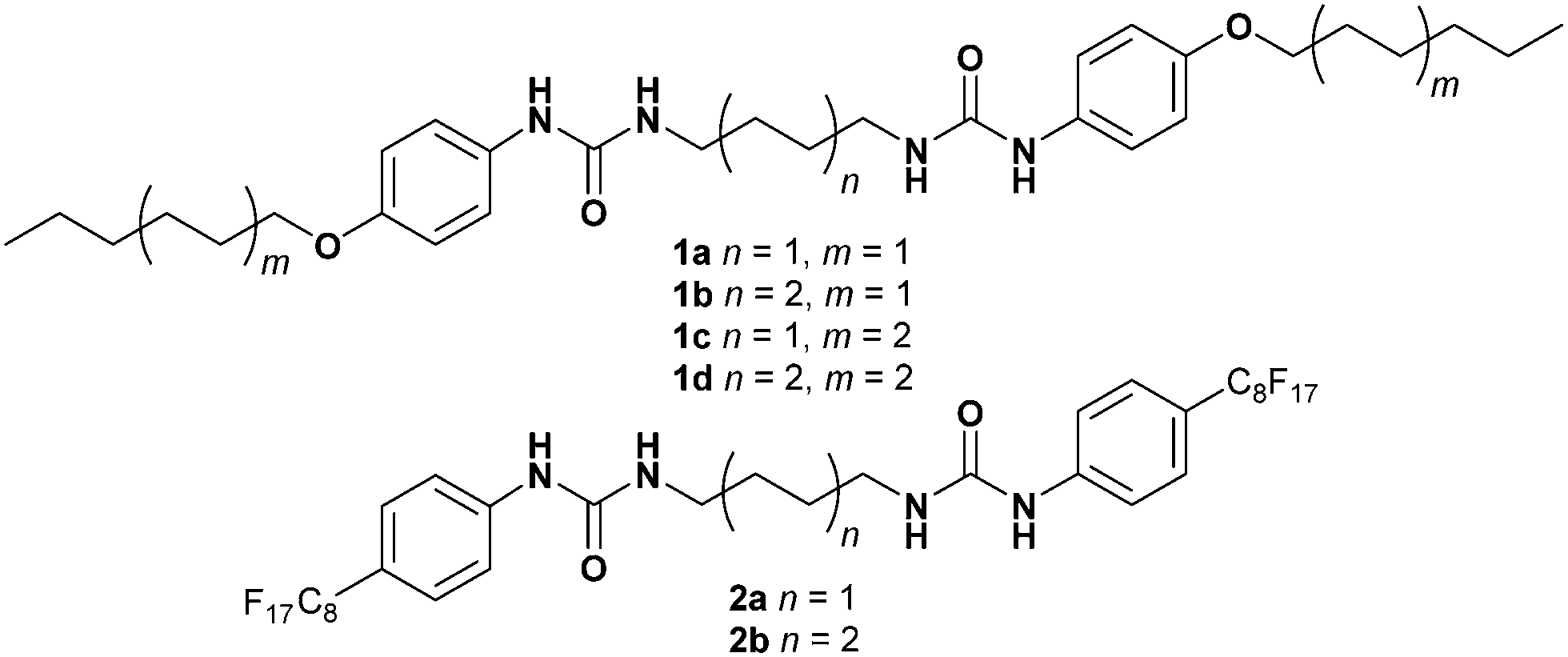
 | ||
| Fig. 1 Gels in DMSO at the mgc: from left to right 1d (3 wt%), 2b (0.5 wt%), 1b (7 wt%), 1c (2.5 wt%), 1a (3 wt%) and 2a (0.2 wt%). The fluorinated gels have a markedly more transparent appearance. | ||
In contrast, the fluorous gelators of type 2 gel a narrower range of solvents than their hydrocarbon analogues (12 and 14 of the 31 solvents tested for 2a and 2b, respectively). Compounds 2 are more effective in immobilising more polar solvents such as DMSO, alcohols and ketones. While the mgc required varied widely from (0.2–10) wt%, the fluorous gelators generally exhibit much lower mgc than their hydrocarbon analogues in the same solvent. Gels of compounds of type 2 are generally much more robust, resistant to mechanical agitation and transparent in appearance, suggesting a denser network of narrower fibres. While most gels formed by thermal cycling, gels of 2b in DMF, DMSO and cyclopentanone displayed the unusual ability to gel at room temperature after around 15 minutes following 30 s of sonication. Room temperature gelation requires a fine balance between solubility and aggregation rate, and relatively few examples have been reported.27
Gelation experiments were also undertaken for compounds of type 2 in fluorinated solvents. Fluorous phases are generally very challenging to gel and most successful gelators for these media possess multiple perfluoroalkyl substituents.12 Compounds 2a and 2b were designed to achieve a fluorous ‘sheath’ around a lipophilic core rather than as efficient gelators of fluorous solvents. While these compounds are indeed ineffective at gelling the majority of fluorous solvents examined, surprisingly compound 2b proved to gel perfluorodecalin at 10 wt% to give an opaque, thermoreversible gel.
The elastic properties of the DMSO gels of compounds 1 and 2 were probed by stress sweep and frequency sweep rheometry using a rough, parallel plate geometry. Experiments were conducted both at the mgc and at higher concentrations. For all gels the plateau G′ value proved to be around one order of magnitude above the viscous modulus (G′′) and invariant to frequency, confirming the gels solid-like nature.28,29 Gels with the shorter butylene spacer all proved to have a larger G′ and yield stress at a given concentration. This greater strength may be due to the more rigid nature of the shorter spacer, greater compatibility with the polar DMSO solvent and in the case of the fluorinated gels of type 2, less incompatibility between hydrocarbon and fluorocarbon regions. For compounds of type 1 gels with the shorter hexyl termini proved to be weaker than those bearing longer octyl groups. The perfluoroalkane gels of type 2 proved stronger still, consistent with the expectation that the fluorinated tail sheaths the urea hydrogen bonded region from solvent competition.
Gel strength was also probed by measurement of Tgel using the dropping ball method.23 For the hydrocarbon terminated gelators of type 1Tgel increases with concentration from 3 to 7 wt% suggesting that relative high concentrations are needed to reach maximum stability in the highly competitive DMSO medium. As suggested by the rheological data, compound 1c forms the most thermally stable gels (Tgel = 93 °C at 7 wt%). Across the whole series of compounds of type 1 and 2, the least temperature-stable gel at 7 wt% is compound 2b, consistent with the lower G′ and yield stress for this compound. This factor may reflect incompatibility between the hydrocarbon and fluorocarbon regions of the gel fibres. For the fluorinated compound of type 2 gel thermal stability increases from the mgc to 3.0 wt% whereupon a plateau is reached, consistent with the generally stronger nature of these gels. For example, DMSO gels of 2a exhibit a Tgel of 50 °C at 0.2 wt% rising to 77 °C at 1 wt% and reaching a plateau value of 80–82 °C at 3 wt% and above. In DMF the Tgel values for compounds of type 1 and 2 are significantly lower. For compound 2a at 2 wt% the Tgel is 52 °C rising to 62 °C at 5 wt%. For 1d in DMF 2 wt% gels do not support the test ball, whereas at 5 wt% the same Tgel of 62 °C is obtained.
The morphology of the DMSO and ethanol xerogels of compounds of type 1 and 2 were probed by SEM using chromium coating. The micrographs reveal networks of anisotropic fibres with relatively high aspect ratios in all gels investigated, however, fibre morphology differs greatly between xerogels of compounds with perfluorinated and hydrocarbon end groups (Fig. 2). The xerogel fibres of the hydrocarbon gelators are more crystalline, linear, flat and ribbon-like in morphology with fibre widths ranging from 0.9–5 μm. The xerogel fibres of the perfluorinated gelators are more amorphous, less linear and more cylindrical in morphology, and are much narrower with fibre diameters of 50–130 nm. Fibres formed from the perfluorinated compounds have much higher aspect ratios than their hydrocarbon analogues leading to highly entangled fibrous networks which is likely to be a key reason for the ability of compounds of type 2 to gel at such low mgc compared to their hydrocarbon analogues. The narrower fibre width in turn is likely to be related to the lower mutual affinity of the fluorinated chains and hence slower growth perpendicular to the fibre axis.
 | ||
| Fig. 2 SEM micrographs of ethanol xerogels (a) large, flat ribbons of compound 1d typical of compound of type 1, (b) narrow cylindrical fibres of 2b typical of compounds of type 2. | ||
Gel structure
Significant evidence exists to suggest that bis(urea)s form gels by hydrogen bonded urea α-tape motifs along the fibre axis, resulting in the formation of high aspect ratio fibrils, that bunch and interlink to give the gel network.30–32 In order to obtain structural insight, extensive crystallization tests were undertaken with a view to isolating diffraction quality crystals of 2a. Crystals were eventually isolated from a 1.0 wt% gel of 2a in 2-butanone which was found to convert over several hours to give small, needle shaped crystals (see ESI,† page S11). A needle morphology is common for gelators and is consistent with rapid growth along the urea α-tape hydrogen bonded axis.33 The single crystal structure of 2a was obtained using beamline I19 at the diamond synchrotron source is shown in Fig. 3.‡ The structure comprises two independent gelator molecules and one disordered molecule of 2-butanone solvent. Specifically the asymmetric unit has one molecule of gelator 2a in a general position, a half molecule of 2a lying about an inversion centre and a half-occupancy 2-butanone molecule lying disordered about another inversion centre. Each independent gelator takes part in an independent antiparallel arrangement of urea α-tape motifs involving somewhat unsymmetrical six-membered hydrogen bonded rings. Each hydrogen bonded tape exhibits distinct hydrocarbon and fluorocarbon regions with the fluorous region in the periphery of the tape, as anticipated. Surprisingly, however, the fluorous region of the second independent tape is situated adjacent to the hydrocarbon region of the first. This arrangement appears to maximise shape complementarity and hence minimise void space despite the incompatibility of the different regions of the molecules. A small residual lattice void is occupied by the 2-butanone solvent molecule. This system thus seems to be consistent with the premise that awkwardly shaped molecule (dumbbells in this instance) can give rise to multiple crystallographically independent motifs.34 Thermogravimetric analysis on the crystals gave a weight loss of 2.07% between 75–140 °C corresponding to the loss of 2-butanone (confirmed by TGA-MS). The desolvated material decomposes at 200 °C. | ||
| Fig. 3 (a) One of the two independent antiparallel double urea α-tape hydrogen bonded motifs found in the X-ray crystal structure of 2a, showing and separated fluorous and hydrocarbon regions. (b) Unexpected offset stacking of fluorous and hydrocarbon regions in the two independent stacks. NH⋯O hydrogen bond lengths: 2.863(3)–3.126(3) Å. | ||
Analysis of the dried DMSO xerogel and the as-synthesised bulk solid 2a by X-ray powder diffraction (XRPD) showed that both have the same crystal packing arrangement, which also matches the calculated XRPD data from the X-ray crystal structure, albeit with lower crystallinity in the case of the xerogel in particular (see ESI,† Fig. S2). This suggests that the crystal packing arrangement can tolerate a variety of included solvents in addition to 2-butanone. While it is always uncertain to extrapolate diffraction data to the structure of the gel itself, the observation of only a single solid form with the expected hydrogen bonding arrangement isolated from a gelled sample suggests that this X-ray structure is indeed representative of the structure of the gel fibres themselves. Further support for this suggestion comes from SAXS and WAXS data on the DMSO gels (10 wt%) and xerogels of 2a and 2b which give similar results for both gel and xerogel. SAXS data indicate a characteristic spacing of 40.13 Å and 40.65 Å for the gel and xerogel of 2b, respectively, with a shorter distance of 37.84 Å and 37.58 Å, respectively for compound 2a, consistent with the shorter length of the compound. WAXS data gives a spacing of 4.2 Å for both xerogels, increasing to 4.4 Å in the gels themselves. This appears to correlate with the molecular thickness and suggests perhaps some shrinkage of the fibres on drying.
SANS study
SANS has been used to probe the structures, the effect of temperature, the effect of concentration and the thermo-reversibility of bis-urea based gels of fluorinated gelator 2a and non-fluorinated gelator 1d. Measurements were conducted on the NG7 30 m SANS instrument at the NIST Center for Neutron Research (NIST-NCNR) in Gaithersburg, MD35 using neutrons of wavelength λ = 6 Å with full width half-maximum Δλ/λ = 15%. The instrument covers the q range of 0.00116 Å−1 < q < 0.522 Å−1. Gels were studied at 25 °C, and then cycled to 80 °C and back to 25 °C in DMSO-d6 and DMF-d7 solution at 2 wt% and 5 wt%. The temperature of 80 °C was chosen since it is the Tgel of 2a in this solvent. Deuterated solvents improve the scattering contrast and thus the coherent scattering of neutrons that contain structural information. The low q data were modelled using both spherical and flexible cylinder fractal models. Factors such as instrumental geometry, apertures, wavelength spread, effect of gravity on neutron trajectory, etc. were accounted for by employing resolution-smearing as part of the model. The spherical fractal model describes the scattering from fractal-like aggregates built from spherical building blocks. The model calculates the number density, aggregation number, and other parameters based on the volume fraction and polydisperse particle volume. The spherical building blocks aggregate to form fractal-like clusters. The clusters have a correlation length corresponding to their overall size, and self-similarity dimension. Alternatively, the fractal flexible cylinder model uses a flexible cylinder as the “building block”. The block radius of this model corresponds to the radius of gyration of this flexible cylinder. The high q data for each scenario primarily exhibited two peaks. Each of the peaks was fitted separately using a restricted Gaussian fit to yield ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 between q position and a D-spacing (Table 1 and Fig. 4).
:2 between q position and a D-spacing (Table 1 and Fig. 4).
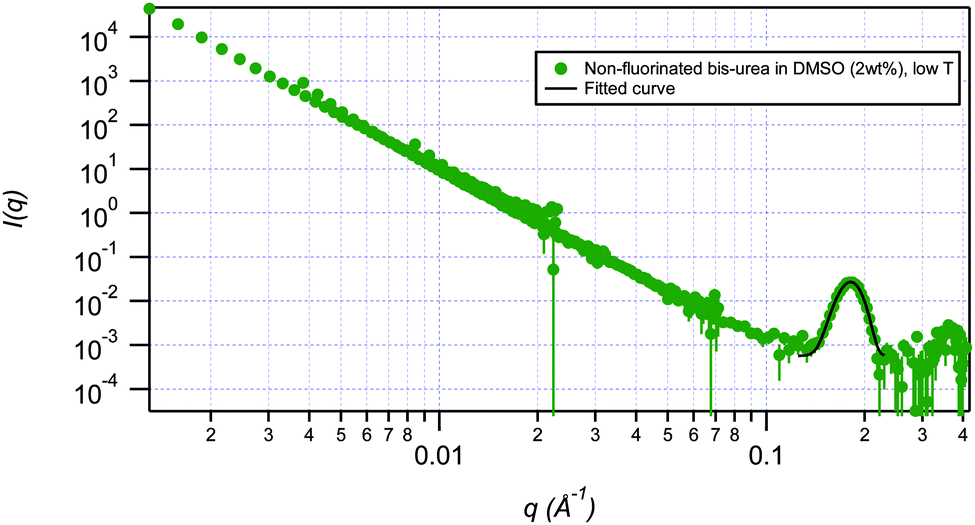 | ||
| Fig. 4 Gaussian fit of the first peak of non-fluorinated bis-urea gelator (1d). | ||
The ratio of peak positions at high q (1:2) reflects the presence of lamellar structures for both fluorinated and non-fluorinated bis-urea gelators (Tables 1 and 2). The overall lamellar structure is unaffected with change in temperature in all cases for both gelators with an exception of fluorinated gelator 2a in DMF which breaks apart when subjected to high temperature conditions. Gaussian fitting of high q data for the fluorinated gelator yields lamellar structures with a D-spacing of ≈39 Å. In contrast, the D-spacing for the non-fluorinated gelator is ≈35 Å. The higher D-spacing in fluorinated gelator correlates with the greater elasticity and the transparent appearance of the fluorinated gels.
| Conc. (wt%) | Solvent | Temp. (°C) | D spacing (Å) | Peak position ratio |
|---|---|---|---|---|
| 2 | DMSO | 25 | 38.7 | 2 |
| 2 | DMSO | 80 | 39.1 | 2.1 |
| 2 | DMSO | Cool | 38.5 | 2.2 |
| 5 | DMSO | 25 | 38.9 | 2 |
| 5 | DMSO | 80 | 38.8 | 2 |
| 5 | DMSO | Cool | 39 | 2 |
| 2 | DMF | 25 | 39.7 | 2 |
| 2 | DMF | 80 | Structure broken | |
| 2 | DMF | Cool | 38.9 | 1.9 |
| 5 | DMF | 25 | 39.4 | 2 |
| 5 | DMF | 80 | Structure broken | |
| 5 | DMF | Cool | 39.6 | 2 |
| Conc. (wt%) | Solvent | Temp. (°C) | D spacing (Å) | Peak position ratio |
|---|---|---|---|---|
| 2 | DMSO | 25 | 34.8 | 2 |
| 2 | DMSO | 80 | 35.2 | 2 |
| 2 | DMSO | Cool | 34.7 | 2 |
| 5 | DMSO | 25 | 34.6 | 2 |
| 5 | DMSO | 80 | 35.1 | 2 |
| 5 | DMSO | Cool | 34.6 | 2 |
| 2 | DMF | 25 | 34.7 | 2 |
| 2 | DMF | 80 | 35.3 | 1.9 |
| 2 | DMF | Cool | 34.7 | 2 |
| 5 | DMF | 25 | 34.7 | 2 |
| 5 | DMF | 80 | 35.1 | 2 |
| 5 | DMF | Cool | 34.6 | 2 |
Fitting the experimental data for the fluorinated gelator 2a in DMSO-d6 to the smeared spherical fractal model gives a correlation length of 365 ± 4 Å at 25 °C. This length increases markedly to 530 ± 6 Å at 80 °C. The correlation length may be thought of as representing the average distance between gel nodes or the ‘mesh size’ of the SAFIN. Interestingly, the correlation length remains unaltered when the gel is cooled from 80 °C back to 25 °C (523 ± 7 Å). This data suggests that that the gel structure changes irreversibly upon heating with individual fibres growing longer and more ordered. This may be interpreted as a phenomenon analogous to Ostwald ripening36,37 of the gelator component of the solid and suggests that the initially formed network is metastable with initial fibre growth arrested by rapid depletion of the local supersaturation giving a relatively disordered, defective network. The observation is in contrast to the reversible thinning observed in bis(urea)s by Bouteiller et al.38 The temperature of 80 °C corresponds to the gel–sol transition temperature (Tgel) and indicates that at the point of mechanical breakdown the fibrous network persists but the less stable nodes are lost and redissolved, resulting in an ‘annealed’ network upon re-cooling.
In addition to the correlation length, the gel is characterized by its block radius and fractal dimension. The fractal dimension provides a statistical measure of the complexity of the gel network whereas the block radius represents the local dimensions of the gel fibres. The block radius changes from 6 Å to 10 Å upon heating from 25 °C to 80 °C, and remains at 10 Å upon cooling back to 25 °C further confirming the annealed nature of the fibres. The fractal dimension (df = 2.9 to 3.0) remains unaltered as a function of temperature. However, the fit to the SANS data with this model does not adequately describe the low q scattering. This region corresponds to longer length scales and hence gives information about the bulk fibrous structure and trends in its non-uniformity. To address the low q region the data for 2a in DMSO was re-fitted using only the low q data. To focus only on the trends in the correlation length the block radius, corresponding to the local (high q) structure was held constant. Using this model the trend in correlation length remained the same, albeit with a larger value obtained for the initial low temperature data of 425 Å at 25 °C rising to 526 Å at 80 °C and remaining unchanged on cooling. The fractal dimension, remained unaltered suggesting the density or complexity of the network stays the same at all temperatures and hence, no significant differences in correlation lengths and no significant variation was observed as a function of fixed block radius.
The same SANS data for 2a in DMSO-d6 (2% mass fraction) was also fitted to a smeared fractal flexible cylinder model particularly to address the flexibility of cylindrical fibres of gelators. Using this more sophisticated description a similar trend in correlation length is observed for both full q and low q SANS data as the temperature is cycled, further supporting the fractal model. In addition to establishing the non-thermoreversible nature of gels, the cylindrical gel fibres are revealed to be quite rigid. The rigidity is determined by comparing the difference in the contour and Kuhn lengths (Table 3). The Kuhn length represents the average length over which the flexible cylinder building blocks can be considered to be rigid while the contour length is the end-to-end length to the polymer chain if it were fully stretched out (i.e. length of one monomer times the degree of polymerisation). The larger the difference in the two lengths the more flexible the gel fibres are. In this case the contour length is the total length of the flexible cylinder and if the Kuhn length is similar to the contour length, then is the cylinder can be considered rigid over its entire span. For 2a in DMSO-d6, the contour length is never more than twice the Kuhn length, indicating nearly rigid fibers.
| Conc. (wt%) | Solvent | T (°C) | Contour length | Kuhn length |
|---|---|---|---|---|
| 2 | DMSO | 25 | 58.3 | 52.1 |
| 2 | DMSO | 80 | 67.6 | 48.4 |
| 2 | DMSO | Cool | 59.7 | 51.7 |
| 5 | DMSO | 25 | 41.6 | 59.6 |
| 5 | DMSO | 80 | 52 | 49.5 |
| 5 | DMSO | Cool | 62 | 40.7 |
| 2 | DMF | 25 | 39.7 | 24.6 |
| 2 | DMF | 80 | Structure broken | |
| 2 | DMF | Cool | 30.5 | 20.8 |
| 5 | DMF | 25 | 40.4 | 25.1 |
| 5 | DMF | 80 | Structure broken | |
| 5 | DMF | Cool | 42.4 | 20.5 |
The aliphatic gelator 1d in DMSO-d6 is far better described by the fractal flexible cylinder model and this was used exclusively. In contrast to 2a (correlation lengths: 300 Å to 550 Å), gels of 1d give correlation lengths approximately an order of magnitude higher. The correlation length of the initially formed gel at 25 °C is 2735 ± 139 Å and decreases to 1806 ± 57 Å upon heating to 80 °C. Upon subsequent re-cooling the correlation length recovers to 3262 Å. The contour length is again less than twice the Kuhn length at all three temperatures. This indicates that the rigidity of the fibres is similar in case of 2a and 1d and is unaffected by the change in temperature. The cylindrical radius of fibres in 1d reduces by about 13 Å (from 49 to 36 Å) upon heating and expands back to 52 Å, upon cooling. For 1d 80 °C is above the Tgel of the material at 2 wt% (52 °C) and it is interesting that fibrous material remains in the sample even after the collapse of the 3D gel network.
SANS data for 2a in DMF-d6 (2% mass fraction) fitted using a smeared spherical fractal model shows a distinct contrast to the data in DMSO-d6 and provides evidence for a drastic structural transition as a function of temperature (Fig. 5). At 25 °C, the fibers have a correlation length of 231 Å, but are completely disrupted upon heating to 80 °C (Fig. 6). The high temperature SANS data fits to a power law instead of smeared spherical fractal model showing the presence of small spheres at high q and a weak power law at low q. Power law fitting indicates that the gel has completely broken up and there are simply “primary” aggregates (spheres of average radius 9.1 Å), with some small aggregates remaining. Interestingly, the thermally disrupted structure reforms to give a fibrous network upon re-cooling to 25 °C with the same correlation length (full q fitting). This result contrasts sharply with the increase in correlation length upon heating and retention of the annealed fibrous network upon cooling that is observed for the same gelator in DMSO-d6. In this case the DMF gel is significantly beyond the measured Tgel of 52 °C and has reverted to a sol at high temperature. The fractal dimension in DMF, remains unchanged on re-cooling (it ranges between 2.9 Å to 3.0 Å) highlighting the thermoreversibility of the DMF gel. The smeared fractal flexible cylindrical fits give similar qualitative results as a function of temperature.
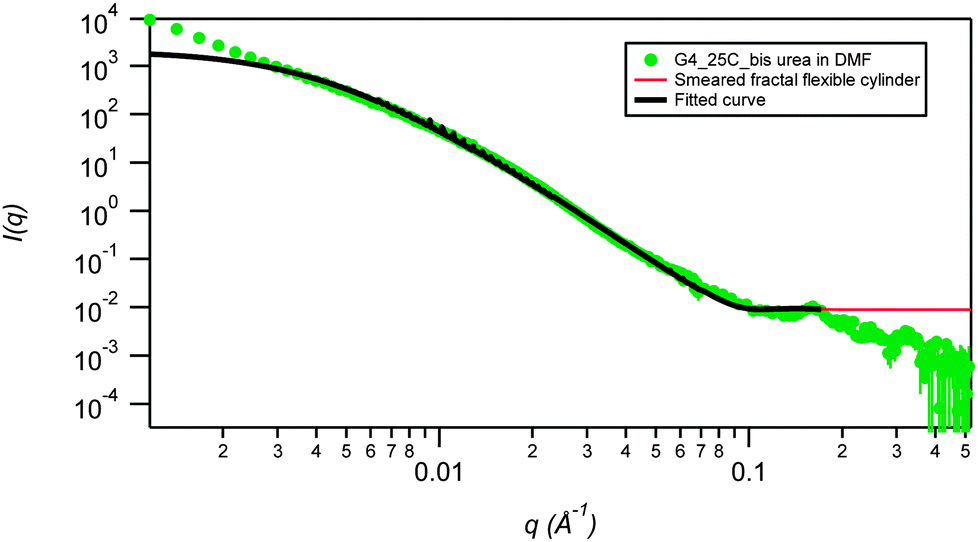 | ||
| Fig. 5 SANS smeared fractal flexible cylinder fit (low q) for the fluorinated bis(urea) gelator 2a in DMF at 2 wt%, measured at 25 °C. Errors on contour and Kuhn lengths are ±0.05. | ||
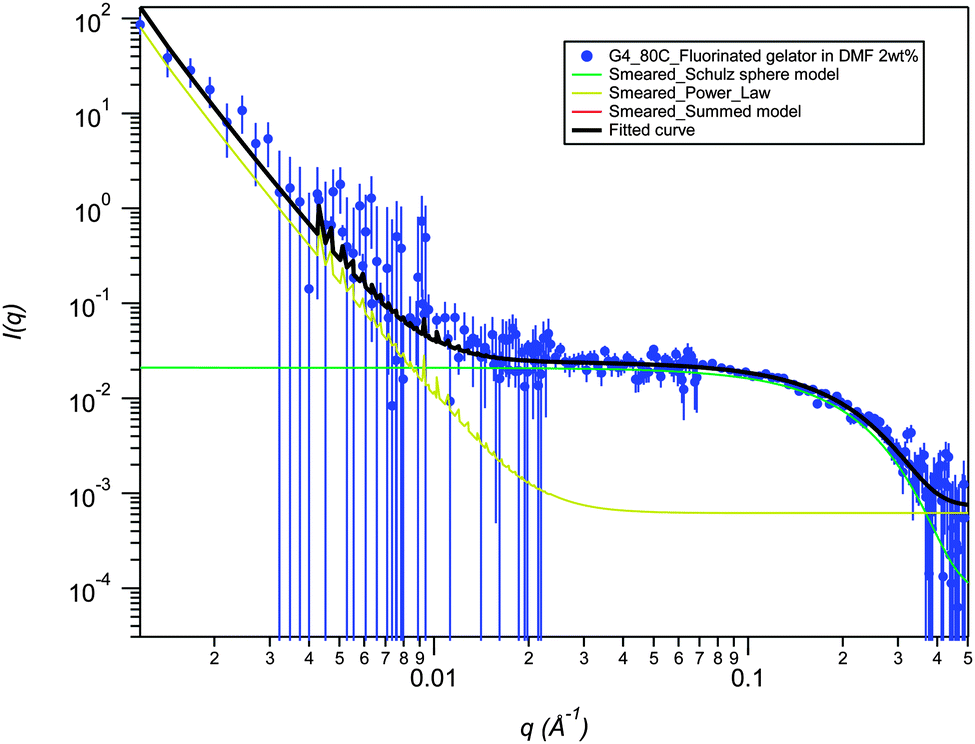 | ||
| Fig. 6 SANS smeared summed model (red line beneath black fitted curve line) of Porod_law fit and Schulz sphere fit for the fluorinated bis-urea gelator 2a in DMF at 2 wt%, measured at 80 °C, showing breaking of the gel structure. | ||
In contrast, gels of 1d in DMF at 2 wt% do not show any disruption of gel fibres upon heating. However, the cylindrical radius shrinks from 50 Å to about 6.5 Å before recovering to a very similar value upon re-cooling. This suggests that the fluorinated bis-urea can disrupt and reform its gel network whereas the non-fluorinated gelator are stable, with the fibres thinning out upon heating and thickening back to the original structure upon cooling. For 1d in DMF at all three temperatures the contour lengths and Kuhn lengths are similar, suggesting that the fibres are very rigid and remain rigid upon heating (Table 4). The length of fibres in 1d in DMF is again an order of magnitude higher (400 Å vs. 1800 Å) than 2a. However, the length of fibres of 1d is smaller in DMF (1100–1800 Å) than in DMSO (1805–3262 Å). In both solvents, the correlation length of fibres of 1d decreases upon heating and subsequently increases again upon cooling.
| Conc. (wt%) | Solvent | T (°C) | Contour length | Kuhn length |
|---|---|---|---|---|
| 2 | DMSO | 25 | 37.4 | 40.1 |
| 2 | DMSO | 80 | 27.5 | 33.4 |
| 2 | DMSO | Cool | 40.8 | 20.6 |
| 5 | DMSO | 25 | 39.0 | 38.2 |
| 5 | DMSO | 80 | 22.4 | 38.8 |
| 5 | DMSO | Cool | 31.3 | 36.0 |
| 2 | DMF | 25 | 20.2 | 59.6 |
| 2 | DMF | 80 | 26.5 | 40.7 |
| 2 | DMF | Cool | 26.1 | 53.3 |
| 5 | DMF | 25 | 34.6 | 35.7 |
| 5 | DMF | 80 | 36.1 | 34.0 |
| 5 | DMF | Cool | 34.3 | 45.2 |
The opacity and consistency of these gels can vary significantly as a function of gelator concentration, suggesting variations in gel structure. Thus, in addition to the effect of temperature and solvent, the effect of concentration was also investigated. Gels of 2a were studied using SANS at 5% mass fractions in both DMSO-d6 and DMF-d7.
SANS data fit to smeared spherical fractal model of this concentrated gel of 2a in DMSO shows an increase in correlation length from 392 Å at RT to 474 Å upon heating to 80 °C and retention of the fibrous correlation length (473 Å) upon cooling to 25 °C in a similar way to the 2% mass fraction sample, although the change is less significant for the more concentrated sample. The block radius for 2a at 2% mass fraction changes from 6 Å at RT to 10 Å upon heating to 80 °C and remains similar upon re-cooling (9.7 Å). At 5% mass fraction it changes from 7.3 Å at RT to 11.5 Å upon heating to 80 °C and back to 8.2 Å upon re-cooling. The fractal dimension is very similar to the lower concentration sample suggesting that although the complexity of the fibrous network is insensitive to concentration the local dimension of fibers shows significant variation at different concentrations during the temperature cycling.
The cylindrical radius of fibers at 5% mass fraction (44 Å to 45 Å to 59 Å) and at 2% mass fraction (44 Å to 45 Å to 61 Å) are similar to one another. However, the correlation lengths are slightly shorter than that for 2% mass fraction. Overall, a change in concentration for DMSO gels of 2b causes a difference in local dimension of block radius and correlation lengths but the qualitative aspects (cylindrical radius and flexibility) remain unaltered.
For gels of 2a in DMF at 5% mass fraction the smeared spherical fractal model shows the presence of a longer fibrous network at 25 °C. The correlation length of 348 ± 2 Å at 5% mass fraction is 115 Å longer than that for 2% mass fraction sample. However, in both cases the structure disrupts at 80 °C, evidenced by fitting to a power law. The resultant spherical aggregates of 11 Å for 5% mass fraction are about 2 Å larger than those at 2% mass fraction. Upon cooling, the gel reforms in DMF with slightly a longer correlation length of 387 Å but similar block radius of 6.5 Å. In contrast, an increment of 1 Å in block radius is observed in DMF at 2% mass fraction. The fractal dimensions are again unaffected by the concentration, solvent and temperature changes. Again, in both cases an increase in the correlation length is observed before and after heating, but the effect is less marked at higher concentration (difference of 250 Å vs. 60 Å). The gel also shows a longer initial gel fibre length at higher concentration (5 wt%).
Conclusions
In conclusion, this work has demonstrated that appending perfluoroalkyl substituents to a bis(urea) gelator markedly modifies the properties of the resulting gel, making it stronger and more transparent, apparently as a result of the decreased inter-chain interactions in fluorous compounds. Even a single fluorinated substituent at each end of the molecule is sufficient to impart interesting gelation properties including the ability to get a fluorous solvent. The simple molecular structure of unbranched bis(ureas) allows ready prediction of the basic features of their self-assembled fibres, and the urea α-tape structure is conserved in both the gel and crystalline state. The structure of 2a is surprising in that fluorinated and non-fluorinated regions stack on top of one another in a fashion that maximises crystal packing efficiency rather than separating fluorous and hydrocarbon regions.The SANS results demonstrate an interesting annealing effect on the fluorinated gels which is absent for the non-fluorinated analogues, and may be analogous to Ostwald ripening. The data show that in DMSO the gel structure is retained right up to the Tgel of the materials (80 °C) but that fully reversible collapse to spherical aggregates occurs if the Tgel is exceeded (DMF data). The aliphatic gelator 1d shows thermo-reversible behavior in both DMF and DMSO. A change in concentration for 2a, shows a change in the local structure but qualitative aspects of the network such as fibre rigidity and cylinder radius remain unaltered. The SANS data reveal that the physical appearance of a gel is not a good indicator of its structure and demonstrates that SAFINs can anneal from disordered, defect-filled metastable structures to more ordered networks, or may exhibit full thermoreversibility. Moreover the gel structure behaves as a solid network even while undergoing significant changes in fibre rigidity, connectivity, and size. Interestingly, the metastable nature of the initially formed fluorinated gels and their short contour length does not adversely affect their elastic properties. These SANS results address, for the first time, some of the questions pertaining to the self-assembly and maturation phenomena of SAFINs. This kind of study lends unique insight into the formation, growth and ripening in nanoassemblies and related self-assembled complex networks.
Supramolecular gels have recently been shown to have novel applications as pharmaceutical crystal growth media. Gel phase crystal growth limits convection effects and can result in control of polymorphism and crystal habit. Supramolecular gels allow tailored access to non-aqueous solvents and can be removed under mild conditions by the introduction of competitor hydrogen bond acceptors such as anions.39–46 The low mgc values and transparency of the gels formed by compounds 2a and 2b makes them good candidates for the crystallisation of drug compounds. Moreover, fluorination is often used as a site protection strategy in drug design,47 suggesting that some fluorinated drugs such as lansoprazole and fluoxetine might be compatible with fluorous gelators. Work is ongoing on the application of compounds of type 2 in this regard and further SANS studies probing the simultaneous, orthogonal self-assembly of gels networks and included pharmaceutical crystals will be reported in due course.
Acknowledgements
We thank the Engineering and Physical Sciences Research Council for funding (grant reference EP/J013021/1). We are grateful to The Diamond Light Source for the award of instrument time on I19 (grant reference MT 6749) and we thank Dr Andrei Batsanov and the instrument scientists for support. This work utilized facilities supported in part by the National Science Foundation under Agreement No. DMR-0944772 (S.R.K.). Certain trade names and company products are identified to adequately specify the experimental procedure. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products are necessarily best for the purpose.Notes and references
- J. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron Lett., 1997, 38, 281–284 CrossRef CAS.
- Y. Li and M. Liu, Chem. Commun., 2008, 5571–5573, 10.1039/b812567h.
- F. S. Schoonbeek, J. H. van Esch, R. Hulst, R. M. Kellogg and B. L. Feringa, Chem. – Eur. J., 2000, 6, 2633–2643 CrossRef CAS.
- F. Fages, F. Vögtle and M. Žinic, Top. Curr. Chem., 2005, 256, 77–131 CrossRef CAS.
- M. George, G. Tan, V. T. John and R. G. Weiss, Chem. – Eur. J., 2005, 11, 3243–3254 CrossRef CAS PubMed.
- Y. Jeong, K. Hanabusa, H. Masunaga, I. Akiba, K. Miyoshi, S. Sakurai and K. Sakurai, Langmuir, 2005, 21, 586–594 CrossRef CAS PubMed.
- C. Wang, D. Q. Zhang and D. B. Zhu, Langmuir, 2007, 23, 1478–1482 CrossRef CAS PubMed.
- D. Mangin, F. Puel and S. Veesler, Org. Process Res. Dev., 2009, 13, 1241–1253 CrossRef CAS.
- M. Yamanaka and H. Fujii, J. Org. Chem., 2009, 74, 5390–5394 CrossRef CAS PubMed.
- R. G. Ellis-Behnke, Y.-X. Liang, K. C. David, P. W. F. Kau, G. E. Schneider, S. Zhang, W. Wu and K.-F. So, Nanomedicine, 2006, 2, 207–215 CrossRef CAS PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of porous solids and powders: surface area, pore size and density, Springer, The Netherlands, 2006 Search PubMed.
- J. G. Riess, Tetrahedron, 2002, 58, 4113–4131 CrossRef CAS.
- J. Loiseau, M. Lescanne, A. Colin, F. Fages, J. B. Verlhac and J. M. Vincent, Tetrahedron, 2002, 58, 4049–4052 CrossRef CAS.
- M. George, S. L. Snyder, P. Terech, C. J. Glinka and R. G. Weiss, J. Am. Chem. Soc., 2003, 125, 10275–10283 CrossRef CAS PubMed.
- M. George, S. L. Snyder, P. Terech and R. G. Weiss, Langmuir, 2005, 21, 9970–9977 CrossRef CAS PubMed.
- J. Gan, M. El Bakkari, C. Belin, C. Margottin, P. Godard, J.-L. Pozzo and J.-M. Vincent, Chem. Commun., 2009, 5133–5134 RSC.
- E. Faggi, R. Maria Sebastian and A. Vallribera, Tetrahedron, 2010, 66, 5190–5195 CrossRef CAS PubMed.
- P. Terech, V. Rodriguez, J. D. Barnes and G. B. McKenna, Langmuir, 1994, 10, 3406–3418 CrossRef CAS.
- E. Krieg, H. Weissman, E. Shimoni, A. Bar On and B. Rybtchinski, J. Am. Chem. Soc., 2014, 136, 9443–9452 CrossRef CAS PubMed.
- C. Shi, Z. Huang, S. Kilic, J. Xu, R. M. Enick, E. J. Beckman, A. J. Carr, R. E. Melendez and A. D. Hamilton, Science, 1999, 286, 1540–1543 CrossRef CAS.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- J. Zhang, H. F. Chow, M. C. Chan, G. K. Chow and D. Kuck, Chem. – Eur. J., 2013, 19, 15019–15025 CrossRef CAS PubMed.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- J. Gladysz and M. Jurisch, in Fluorous Chemistry, ed. I. T. Horváth, Springer, Berlin, Heidelberg, 2012, ch. 282, vol. 308, pp. 1–23 Search PubMed.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- Y. Li, T. Wang and M. Liu, Tetrahedron, 2007, 63, 7468–7473 CrossRef CAS PubMed.
- C. Chassenieux and L. Bouteiller, in Supramolecular Chemistry from Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, Chichester, 2012, vol. 2, pp. 517–528 Search PubMed.
- T. G. Mezger, The Rheology Handbook, William Andrew Publishing, Norwich, NY, USA, 2nd edn, 2006 Search PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS PubMed.
- J. H. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem. – Eur. J., 1999, 5, 937–950 CrossRef CAS.
- C. E. Stanley, N. Clarke, K. M. Anderson, J. P. Lenthall and J. W. Steed, Chem. Commun., 2006, 3199–3201 RSC.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- C. J. Glinka, J. G. Barker, B. Hammouda, S. Krueger, J. J. Moyer and W. J. Orts, J. Appl. Crystallogr., 1998, 31, 430–445 CrossRef CAS.
- IUPAC, Compendium of Chemical Terminology, (the “Gold Book”), 2nd edn, 1997, online corrected version: (2006–).
- W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9607 CrossRef CAS PubMed.
- L. Bouteiller, O. Colombani, F. Lortie and P. Terech, J. Am. Chem. Soc., 2005, 127, 8893–8898 CrossRef CAS PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- Y. Diao, K. E. Whaley, M. E. Helgeson, M. A. Woldeyes, P. S. Doyle, A. S. Myerson, T. A. Hatton and B. L. Trout, J. Am. Chem. Soc., 2012, 134, 673–684 CrossRef CAS PubMed.
- F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 48, 5757–5759 RSC.
- D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154–5156 RSC.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS PubMed.
- R. I. Petrova, R. Patel and J. A. Swift, Cryst. Growth Des., 2006, 6, 2709–2715 CAS.
- M. Pauchet, T. Morelli, S. Coste, J.-J. Malandain and G. Coquerel, Cryst. Growth Des., 2006, 6, 1881–1889 CAS.
- L. A. Estroff, L. Addadi, S. Weiner and A. D. Hamilton, Org. Biomol. Chem., 2004, 2, 137–141 CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic information in CIF format for the structure of 2a (CCDC 1412542). Experimental details covering synthesis of new compounds, rheology, Tgel measurement, XRPD and SANS analysis. See DOI: 10.1039/c5sm01865j. The underlying research data for this paper is available in accordance with EPSRC open data policy from http://dx.doi.org/10.15128/73666481z |
‡ Crystal data: C106H68N12O7F102; MW = 3559.58 g mol−1; T = 120 K; λ = 0.68890 Å; triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 13.287(3) Å; b = 14.229(3) Å; c = 18.870(4) Å; α = 70.476(2)°; β = 72.879(2)°; γ = 85.688(2)°; V = 3212.4(12) Å3; Z = 3; R1 = 0.0543; wR2 = 0.1567. ; a = 13.287(3) Å; b = 14.229(3) Å; c = 18.870(4) Å; α = 70.476(2)°; β = 72.879(2)°; γ = 85.688(2)°; V = 3212.4(12) Å3; Z = 3; R1 = 0.0543; wR2 = 0.1567. |
| This journal is © The Royal Society of Chemistry 2015 |