
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anomalous and anisotropic nanoscale diffusion of hydration water molecules in fluid lipid membranes†
Laura
Toppozini
a,
Felix
Roosen-Runge
b,
Robert
I. Bewley
c,
Robert
M. Dalgliesh
c,
Toby
Perring
c,
Tilo
Seydel
b,
Henry R.
Glyde
d,
Victoria
García Sakai
c and
Maikel C.
Rheinstädter
*a
aDepartment of Physics and Astronomy, McMaster University, Hamilton, Ontario, Canada. E-mail: rheinstadter@mcmaster.ca
bInstitut Laue-Langevin, Grenoble, France
cISIS, Rutherford Appleton Laboratory, Didcot, UK
dDepartment of Physics and Astronomy, University of Delaware, Newark, Delaware, USA
First published on 19th August 2015
Abstract
We have studied nanoscale diffusion of membrane hydration water in fluid-phase lipid bilayers made of 1,2-dimyristoyl-3-phosphocholine (DMPC) using incoherent quasi-elastic neutron scattering. Dynamics were fit directly in the energy domain using the Fourier transform of a stretched exponential. By using large, 2-dimensional detectors, lateral motions of water molecules and motions perpendicular to the membranes could be studied simultaneously, resulting in 2-dimensional maps of relaxation time, τ, and stretching exponent, β. We present experimental evidence for anomalous (sub-diffusive) and anisotropic diffusion of membrane hydration water molecules over nanometer distances. By combining molecular dynamics and Brownian dynamics simulations, the potential microscopic origins for the anomaly and anisotropy of hydration water were investigated. Bulk water was found to show intrinsic sub-diffusive motion at time scales of several picoseconds, likely related to caging effects. In membrane hydration water, however, the anisotropy of confinement and local dynamical environments leads to an anisotropy of relaxation times and stretched exponents, indicative of anomalous dynamics.
1 Introduction
Diffusion is the fundamental mechanism of motion for lipids, proteins and water molecules throughout a membrane. While diffusion describes a stochastic, intrinsically non-directed motion, many biological processes are related to highly directed motions. The incoherent interaction between active processes in the cytoplasm was recently shown to lead to random fluctuation forces,1 which in return may change the character of a directed motion into a thermally-induced diffusion process.In biology, diffusion often occurs in crowded media, which for instance, have been found to lead to anomalous diffusion of proteins.2–4 The dynamics of the atoms in protein or lipid molecules are more complex than those in ordinary liquids. In a simple liquid, atoms move ballistically at short times, followed by a crossover to Brownian diffusion. In dense fluids a “caging” effect is observed, where the atoms can be trapped momentarily by their neighbours. The motions of protein and lipid atoms are further complicated by their polymeric structure, characterized by connectivity and flexibility.
Sub-diffusion of lipid molecules in membranes has been observed in computer simulations.8–10 The experimental observation of sub-diffusive lipid or hydration water motion, however, has turned out to be more difficult and a conclusive observation has yet to be reported.11,12
While numerous studies addressed nanoscale lipid dynamics, see, e.g.,5–7,13–21 significantly less work has been done to study dynamics of membrane hydration water, most likely due to the corresponding computational and in particular experimental challenges. However, hydration water was shown to have distinct properties as compared to bulk water, such as a slower diffusion,22 reduced freezing temperature23,24 and glassy dynamics.25
Properties of hydration water play an important role for the dynamics of biological systems up to the point that their corresponding dynamics have been suggested to be “slaved” by the solvent's fluctuations.26 Protein dynamics and folding were reported to strongly depend to hydration water dynamics.27–29 Lipid structural dynamics have a complex relationship with hydration water30 and coupling of membrane and hydration water dynamics has been reported.24,31
From computer simulations of lipid bilayers, sub-diffusive and ballistic regimes of hydration water motions have been predicted on short time scales and correspondingly small distances.32,33 Water molecules are expected to move anisotropically when diffusing along the membranes or approaching and penetrating the bilayers, as reported from nuclear magnetic resonance imaging34 and are shown to slow further from the introduction of proteins by sum frequency generation spectroscopy.35 Time-resolved vibrational spectroscopy has shown that water in DOPC membranes is distributed anisotropically and forms nano-clusters36 for a range of hydration levels, supported by molecular dynamics simulations.37
While water molecules can penetrate lipid bilayers,38 the perpendicular diffusion, D⊥, is typically at least an order of magnitude lower than lateral diffusion along the bilayers. Water motion through a bilayer is not a simple diffusion process as free energy and diffusion rate strongly depend on the position in the bilayer.39
In this work we combined neutron diffraction, incoherent quasi-elastic neutron scattering and computer modelling to study nanoscale diffusion of hydration water molecules throughout lipid bilayers on length scales of ∼3–100 Å. By preparing oriented membranes, the experimental results give unambiguous access to the anisotropic local molecular confinement geometry to which the diffusive processes are restricted. Large, 2-dimensional detector arrays further enabled us to differentiate between motions of water molecules along and across the membranes separately.
Hydration water diffusion was also modelled using Brownian dynamics in a coarse-grained simulation of the trajectory of water molecules represented by center of mass coordinate for 10 μs (in time steps of 0.1 ps). Diffusion constants and potentials for water molecules are specific to the region, such as hydrophobic core and hydration water layer, with friction included as an effective term. From the computer model, we find that the anisotropy of confinement and local dynamical environments of membrane hydration water lead to an anisotropy of relaxation times and stretched exponents, indicative of anomalous diffusion.
2 Materials & methods
2.1 Experimental sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform and 2,2,2-trifluoroethanol (TFE) was prepared. The Si wafers were cleaned by alternate 12 minute sonications in ultra pure water and methanol at 40 °C (313 K). This process was repeated twice. The cleaned wafers were placed on a heated sample preparation surface, which was kept at 35 °C (308 K). This temperature is well above the main phase transition for DMPC that occurs at 23.4 °C (296.4 K), thus the heated substrates ensured that the lipids were in the fluid phase during deposition and the self-assembly of the lipids. An aliquot of 1.2 mL of the lipid solution was deposited on each Si wafer and allowed to dry. The wafers were kept under vacuum overnight to remove all traces of solvent. Following this protocol, ∼3000 highly-oriented, stacked membranes were supported on each wafer with a total thickness of ∼10 μm.
:1 chloroform and 2,2,2-trifluoroethanol (TFE) was prepared. The Si wafers were cleaned by alternate 12 minute sonications in ultra pure water and methanol at 40 °C (313 K). This process was repeated twice. The cleaned wafers were placed on a heated sample preparation surface, which was kept at 35 °C (308 K). This temperature is well above the main phase transition for DMPC that occurs at 23.4 °C (296.4 K), thus the heated substrates ensured that the lipids were in the fluid phase during deposition and the self-assembly of the lipids. An aliquot of 1.2 mL of the lipid solution was deposited on each Si wafer and allowed to dry. The wafers were kept under vacuum overnight to remove all traces of solvent. Following this protocol, ∼3000 highly-oriented, stacked membranes were supported on each wafer with a total thickness of ∼10 μm.
Deuterated lipids, DMPC-d54, hydrated with H2O, were used for the incoherent experiment to emphasize the incoherent scattering of the hydration water molecules. In order to study water dynamics from the quasi-elastic neutron scattering experiments, eighteen Si wafers were stacked with two 0.3 mm aluminum spacers placed in between each wafer to allow for the membranes to be properly hydrated. Wafers and spacers were stacked into an aluminum sample can, as shown in Fig. 1(a)–(c). Aluminum foil between the wafers and sample can lid provided tension to keep the wafers in place. Samples were then hydrated with ultra pure H2O and incubated at 30 °C (303 K) for 24 hours before the measurements. Energy resolution was determined by cooling a sample with a large incoherent scattering contribution consisting of protonated lipids hydrated with H2O down to 15 K to freeze all molecular dynamics, such that the observed scattering was considered to be purely elastic.
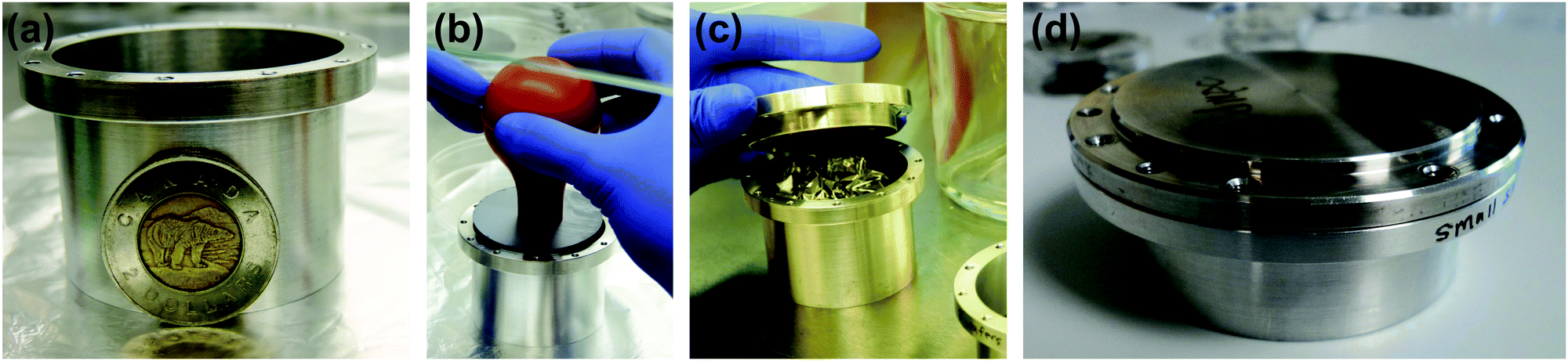 | ||
| Fig. 1 Sample cans used for the neutron scattering experiments. (a) A cylindrical sample can was used for the incoherent neutron scattering experiments. (b) 18 2′′, 300 μm thick Si wafers were loaded into the aluminium cans. (c) Wafers were separated by 0.3 mm thick aluminum spacers to allow for hydration and the remaining space was filled with aluminum foil. (d) Sample can used for the neutron reflectivity where a single 2′′ wafer was mounted. | ||
In this experiment, the bilayers were hydrated from water vapor to avoid high absorption and background signals from bulk solvent hydration.40 Solvent-saturated filter paper was placed above loosely packed balls of aluminum foil and cadmium shields were mounted carefully to avoid scattering of the filter paper and bulk water. The can was mounted inside of a temperature controlled cryostat with a temperature stability of ±0.01 K. Data were taken while the membranes were fully hydrated and in their fluid phase, at a temperature of T = 30 °C (303 K). Due to the typically low scattering of the sample (∼10%), we did not observe evidence of multiple scattering events.
The membrane structure was determined via neutron diffraction in which a single Si wafer was mounted in an aluminum can, as shown in Fig. 1(d) and aligned in a temperature controlled chamber on a neutron reflectometer. Temperature was set to T = 30 °C (303 K) and controlled by a circulating water bath to better than ±0.1 K. Samples were hydrated with ultra pure H2O or heavy water (D2O), and incubated at 30 °C (303 K) for 24 hours before the measurements.
2.2 Elastic and quasi-elastic neutron scattering
We used OFFSPEC to probe the structure of the lamellar bilayers perpendicular to the plane of the bilayers. The thickness of the hydration water layer and water distribution throughout the bilayers, bilayer head group and tail region thickness were determined from these measurements. Neutron scattering has the advantage of scattering differently from hydrogen and deuterium atoms due to their vastly different scattering cross sections. This difference allows measurements to highlight the structure of particular atoms and functional groups through deuterium labelling. The corresponding reflectivity profiles are shown in Fig. 2: (red) chain-deuterated DMPC hydrated with H2O to emphasise the scattering contribution of the hydrophobic membrane core, (blue) DMPC hydrated with heavy water (D2O) to highlight contribution of the hydration water, and (green) DMPC and H2O to emphasise the head group in the bilayers.
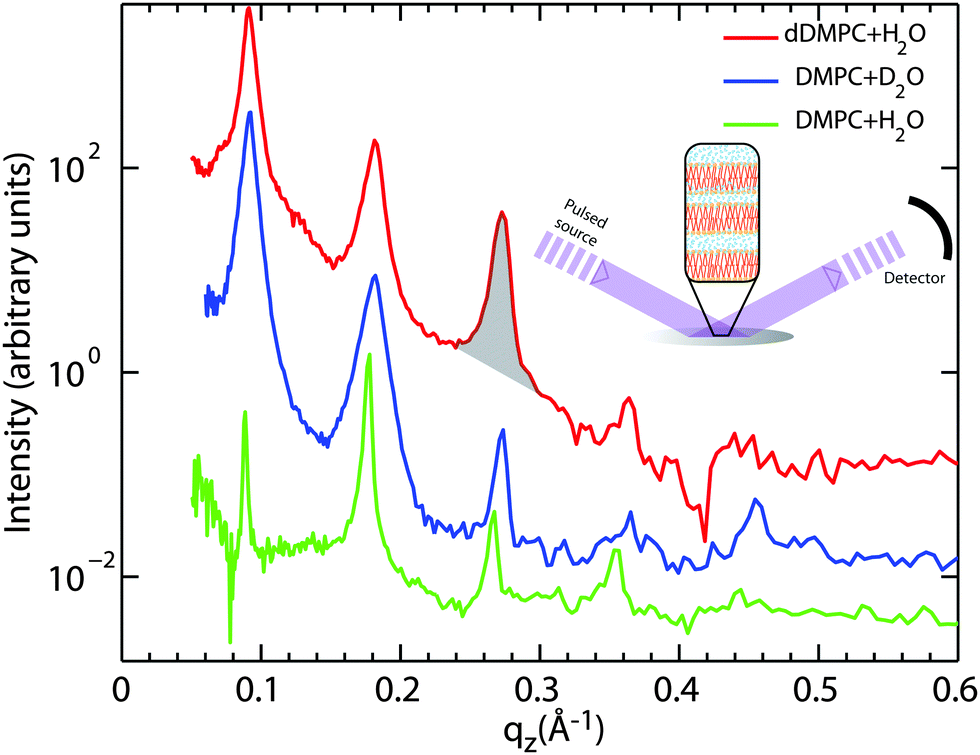 | ||
| Fig. 2 Neutron diffraction data collected at the OFFSPEC reflectometer are shown for three hydrated lipid bilayer samples: (red) deuterated DMPC hydrated by H2O, (blue) DMPC hydrated by D2O, and (green) DMPC hydrated by H2O. The neutron scattering length profiles in Fig. 5 were calculated from these data by using the location and areas (as shown by the shading under the peak) under each peak. All measurements were taken at T = 30 °C (303 K). | ||
The equally spaced Bragg peaks are indicative of a well organized lamellar membrane structure. While up to 12 diffraction orders can be observed in partially-hydrated DMPC membranes,43–46 thermal fluctuations in fully hydrated fluid lipid membranes usually strongly suppress higher diffraction orders.47 Four to five diffraction orders were observed in each diffractrogram in Fig. 2 and used for Fourier synthesis of the scattering length density.
The OFFSPEC measurements covered a qz range from 0.04 to 1.10 Å−1, however Bragg peaks could be observed only to a maximum of qz ∼ 0.50 Å−1. The out-of-plane and in-plane component of the total scattering vector, Q, are denoted qz and qxy, respectively, in what follows. Fitting of Lorentzian peak profiles was used to determine the location of each reflection. The peak areas were integrated by manual determination of the corresponding peak bounds. An example of such an area is marked in Fig. 2 in the third reflection of the DMPC-d54/H2O sample (red).
The neutron scattering length density (NSLD), ρ(z), can be approximated by a 1-dimensional Fourier analysis, where the different Fourier components are observed in the experiment as the integrated intensities of the reflectivity Bragg peaks:
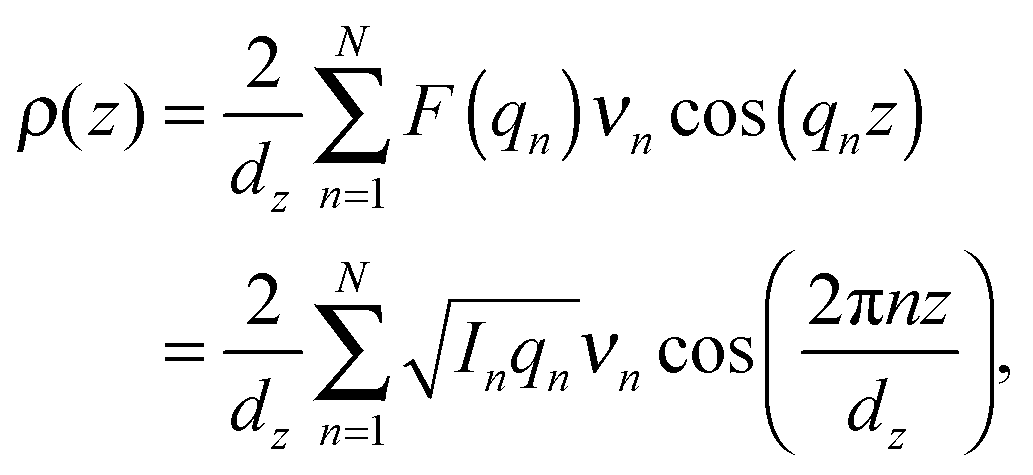 | (1) |
When the form factor F(qz) is measured at several qz values, T(qz), which is proportional to F(qz), can be fit:
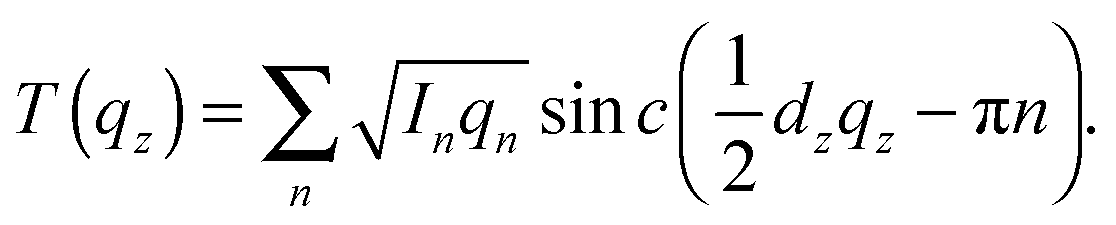 | (2) |
The lamellar spacings, dz, were determined from the peak spacing in Fig. 2 to be: DMPC-d54 hydrated with H2O: dz = 69.22 Å, DMPC hydrated with D2O: dz = 70.92 Å, and DMPC hydrated with H2O: dz = 68.98 Å. As the lamellar spacing includes the thickness of the bilayer plus the thickness of the water layer between the stacked bilayer, it is an established measure of the level of hydration reached. Values of dz as a function of hydration are well documented for DMPC by the Nagle group and dz for DMPC was reported to show a power-law dependence on membrane hydration.51dz values in excess of 63 Å denote a significant increase in the thickness of the water layer. The different membranes prepared for this study can, therefore, be considered to be fully hydrated with more than 25 water molecules per lipid molecule such that the results of each system can directly be compared.
In order to cover as large a range of momentum transfer, Q, and dynamical range as possible different incident neutron energies, Ei, were combined: 0.64, 0.96, 1.62, 3.28, and 9.8 meV corresponding to neutron wavelengths of λ = 11.3, 9.23, 7.11, 4.99 and 2.89 Å, respectively. Position- and time-sensitive 3He detectors collected scattered neutrons with an angular range of −10° to 70° (up to 140° for bulk water) horizontally and ±30° vertically.
A 3-dimensional spectrum (E, Q, S(Q, E)) for each incident energy was acquired in .nxspe (Nexus event-mode) format. A Matlab (Mathworks Inc.) script tailored to stacked membranes used a cylindrical geometry where the components of scattering vector, Q, are the membrane plane, qxy, and the cylindrical axis, qz.
Bulk water data were sliced using a powder geometry in DAVE (Data Analysis and Visualization Environment) software from NIST Center for Neutron Research.54 All data were cut into ten equal slices in qz and up to 22 equal slices in qxy to make a grid of 220 squares in reciprocal space. For Eis of 0.64, 0.96, 1.62, 3.28, 9.80 meV the widths in qz and qxy were 0.027, 0.033, 0.043, 0.062, and 0.106 Å, respectively.
Each square results in a spectrum of S(q(xy,z), E) vs. E and was fit with functions that will be described in detail in the next section (Section 2.2.3). Only data from detector positions of greater than ∼10° (the sixth slice and greater) in the lateral direction were used in the analysis due to contributions of the super mirror guides. The super mirror guides cause a beam divergence, which in turn affects the lower scattering angles.
As the membranes were disordered in the plane of the bilayers (lateral average), the lateral Q-range was scaled by 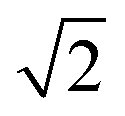 to convert the lateral scale from
to convert the lateral scale from 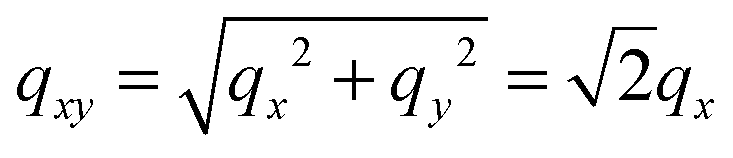 to qx. The qx-range for each of the incident energies was as follows: 0.10–0.34 Å−1 (0.64 meV), 0.12–0.42 Å−1 (0.96 meV), 0.15–0.54 Å−1 (1.62 meV), 0.22–0.79 Å−1 (3.28 meV), and 0.37–1.35 Å−1 (9.80 meV). The qz-ranges for each of the incident energies were: 0–0.27 Å−1 (0.64 meV), 0–0.33 Å−1 (0.96 meV), 0–0.43 Å−1 (1.62 meV), 0–0.62 Å−1 (3.28 meV), and 0–1.06 Å−1 (9.80 meV).
to qx. The qx-range for each of the incident energies was as follows: 0.10–0.34 Å−1 (0.64 meV), 0.12–0.42 Å−1 (0.96 meV), 0.15–0.54 Å−1 (1.62 meV), 0.22–0.79 Å−1 (3.28 meV), and 0.37–1.35 Å−1 (9.80 meV). The qz-ranges for each of the incident energies were: 0–0.27 Å−1 (0.64 meV), 0–0.33 Å−1 (0.96 meV), 0–0.43 Å−1 (1.62 meV), 0–0.62 Å−1 (3.28 meV), and 0–1.06 Å−1 (9.80 meV).
Resolution measurements for the membrane system were taken using a ∼15 K DMPC/H2O sample and bulk H2O resolution measurements using a vanadium sample. Resolutions and quasi-elastic spectra were cut to the same dimensions in reciprocal space. The fits are based on the Matlab nonlinear least-squares minimization algorithm “lsqnonlin”. Fits were a numerical convolution of the resolution (Gaussian function(s)) with the δ-shaped function and the model for the QENS, with the addition of a background. The resolution function of Let depends on the incident energy and scattering angle and may deviate from a simple Gaussian function. Therefore, the resolution spectra were fit with one Gaussian function (Ei = 0.64, 0.96, 1.62 meV) or two Gaussian functions (Ei = 3.28, 9.80 meV). A Toeplitz matrix was constructed from each resolution spectrum, forming a square matrix dependent on the size of the scattering function spectrum. Convolution was carried out by multiplication of the Toeplitz matrix with the sum of the δ-shaped function and broadening model.
| 〈[δr(t)]2〉 = 2Dt, | (3) |
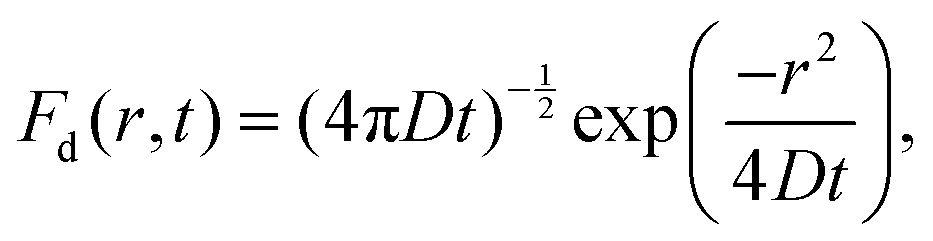 | (4) |
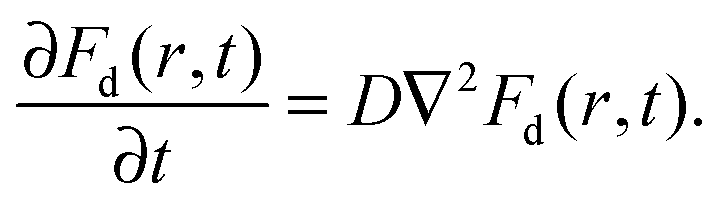 | (5) |
The corresponding intermediate scattering function decays exponentially,
| Id(Q, t) = exp(−Q2Dt), | (6) |
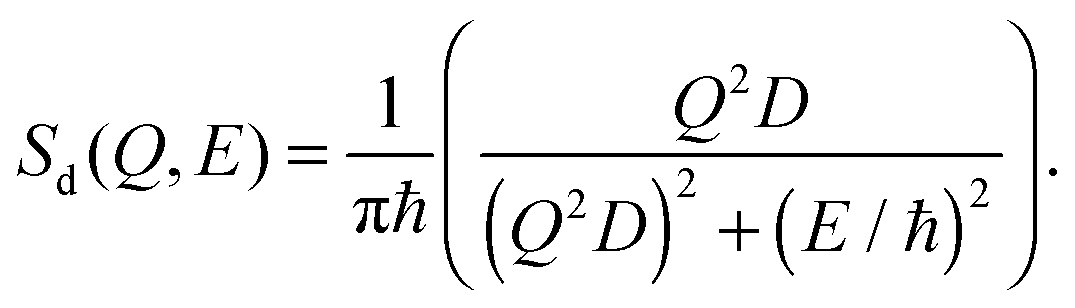 | (7) |
Water dynamics are more complex than that of ordinary liquids. In a simple liquid56 atoms move ballistically at short times. The ballistic MSD 〈[δr(t)]2〉 ∝ 〈(vt)2〉 ∝ t2 of the single molecule is eventually followed by a crossover to Brownian diffusion, characterized by 〈[δr(t)]2〉 ∝ t, for long times. In dense or strongly interacting fluids, the caging effect leads to a plateau in 〈[δr(t)]2〉. The motion in this regime is sub-diffusive with 〈[δr(t)]2〉 ∝ tβ with an exponent β < 1. This anomalous regime of hydration water dynamics was observed in computer simulations of lipid bilayers.32,33
The models used to fit the incoherent neutron data incorporate a constant background, a resolution function, R(t) (in the time domain) or R(E − E′) (in energy domain), which is numerically convolved with the sum of a δ-like function and a function that describes the quasi-elastic broadening.
In the first model, motion of the water molecules is modelled by a single relaxation time, τ:
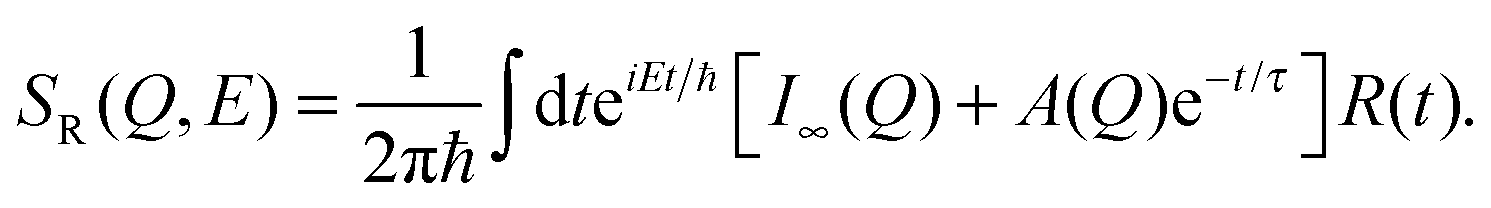 | (8) |
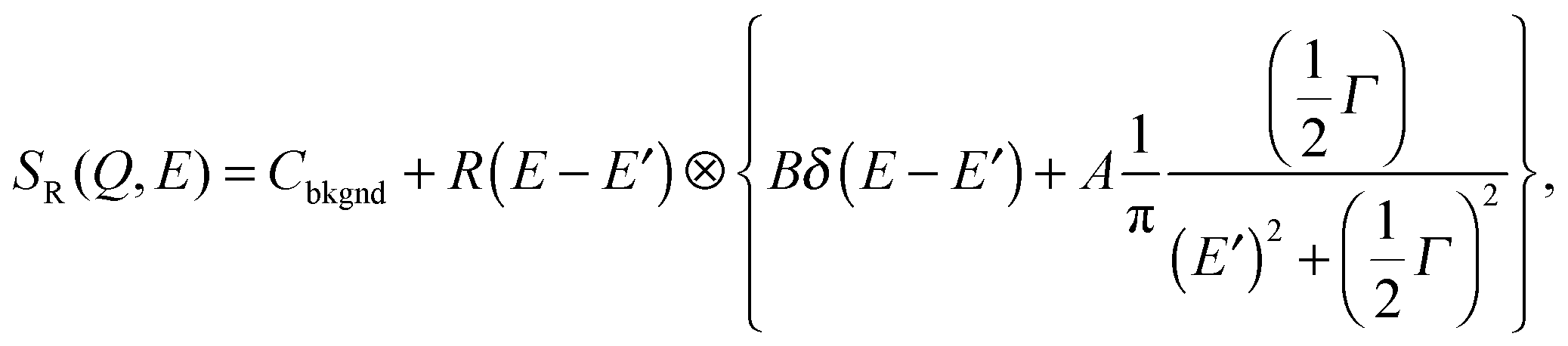 | (9) |
In the second model, the observed dynamics are described by two Brownian diffusion processes,
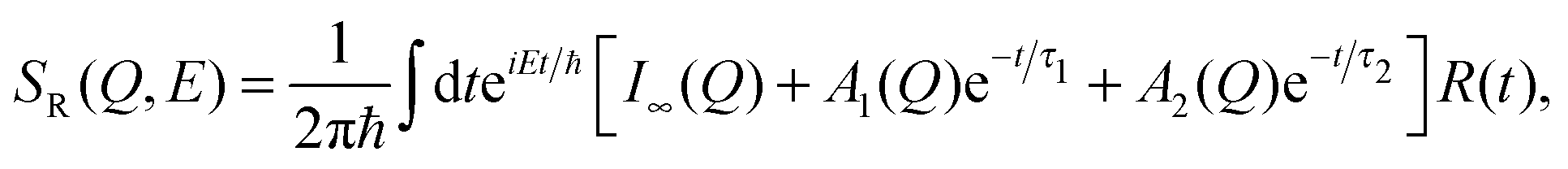 | (10) |
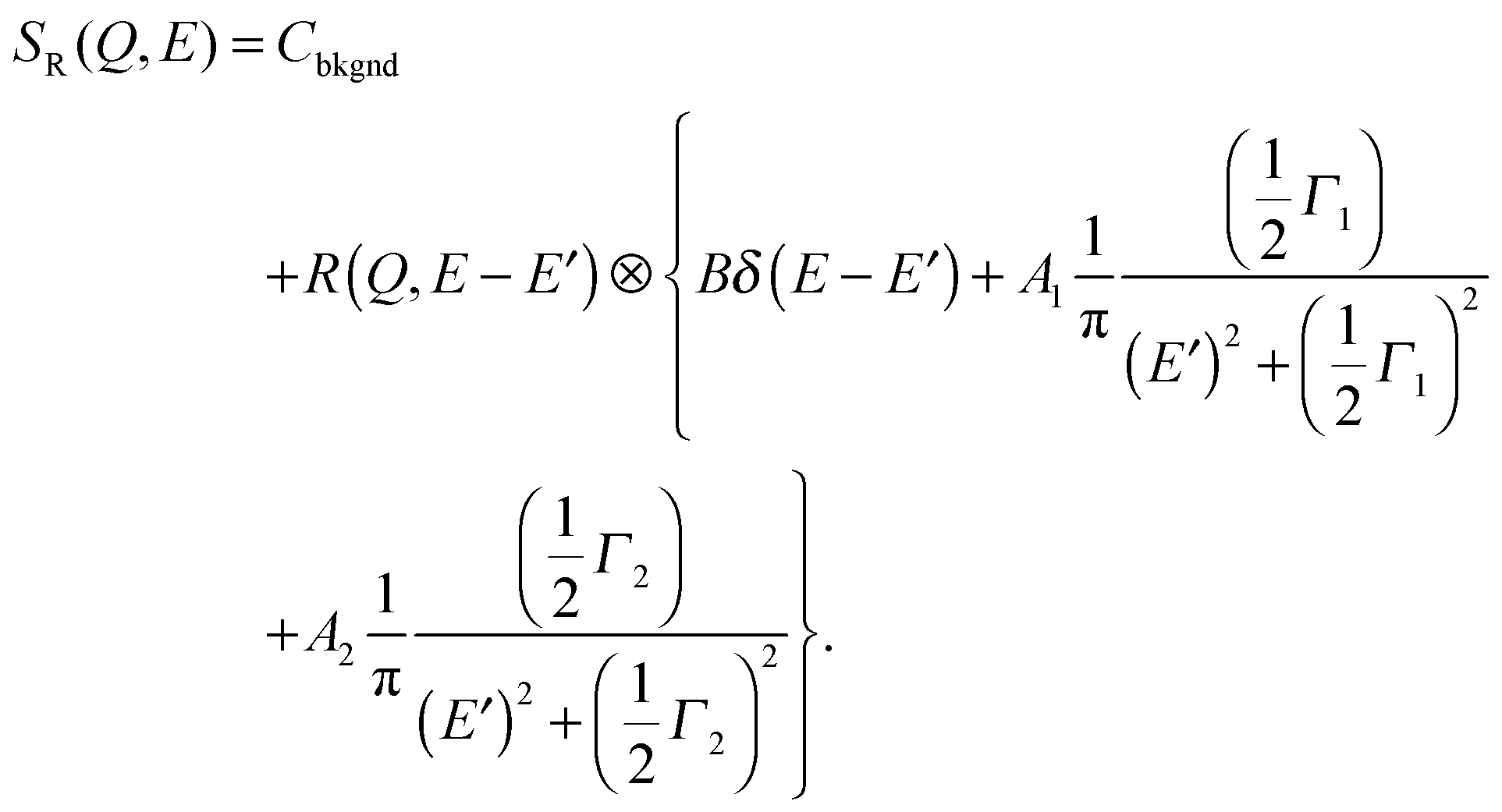 | (11) |
In the third model, we used the Kohlrausch–Williams–Watts (KWW) function to describe motion of the water molecules.59 This function describes a distribution of relaxation processes characterized by relaxation time τ and stretching exponent β. Values of β less than unity signify a sub-diffusive regime, where a stretching exponent of unity is indicative of Brownian diffusion.
Using this model one obtains
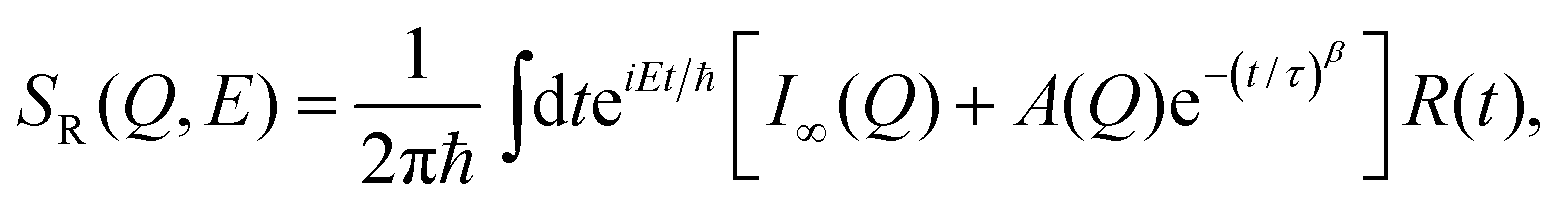 | (12) |
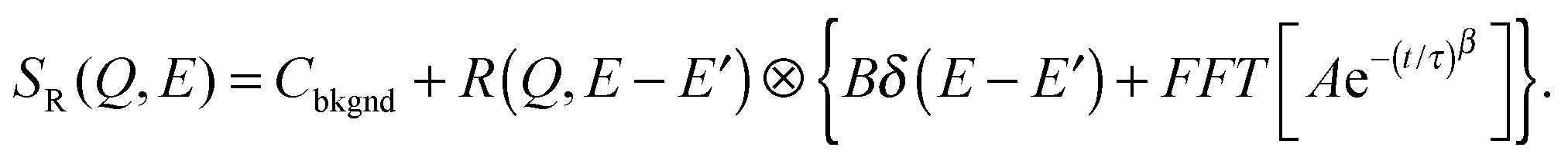 | (13) |
The choice of which of the above functions to use to model incoherent quasi-elastic neutron scattering data is not a purely mathematical problem. The use of a single Lorentzian peak implies a relaxation process with a single relaxation time τ. Two Lorentzian peaks imply two well defined dynamical processes. Stretched exponential correlation functions are often more appropriate to describe dynamics in heterogeneous systems to take into account the heterogeneous local structure in which water molecules move. β exponents of less than 1 describe a distribution of relaxation times. The dynamics are then “glassy” and the corresponding diffusion dynamics slower than a Brownian diffusion. This regime is referred to as “sub-diffusive”. The difference in the distribution of relaxation times between a single and stretched exponentials, is pictured in Fig. 3 for different values of β.
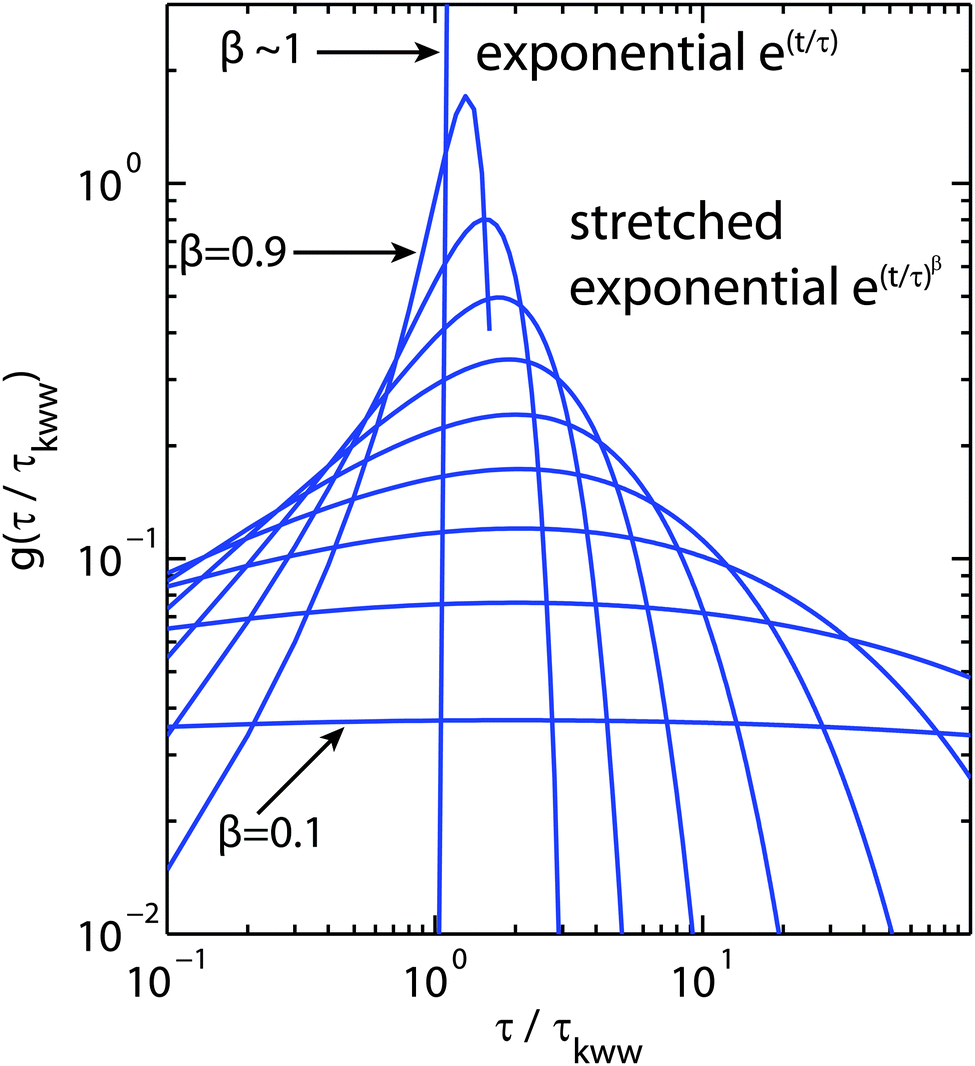 | ||
| Fig. 3 Plot of the relaxation time probability density function (pdf) as a function of τ/τKWW where τKWW is the relaxation time introduced in eqn (12) and (13).60β values range from 0.1 to ∼1 to illustrate the range of relaxation distributions for sub-diffusive systems as opposed to the δ-like distribution for Brownian diffusion (approximated by β = 0.9999). | ||
A clear distinction between the different models often turns out to be difficult due to low statistics in experiments. Examples of different models fit to quasi-elastic scattering are shown in Fig. 4. While the one-Lorentzian fit shown in parts (a)–(c) fits the data reasonably well, fitting with two Lorentzians (parts (d–f)) or the Fourier transform of the KWW function (in parts (g–i)) provides a significantly better fit.
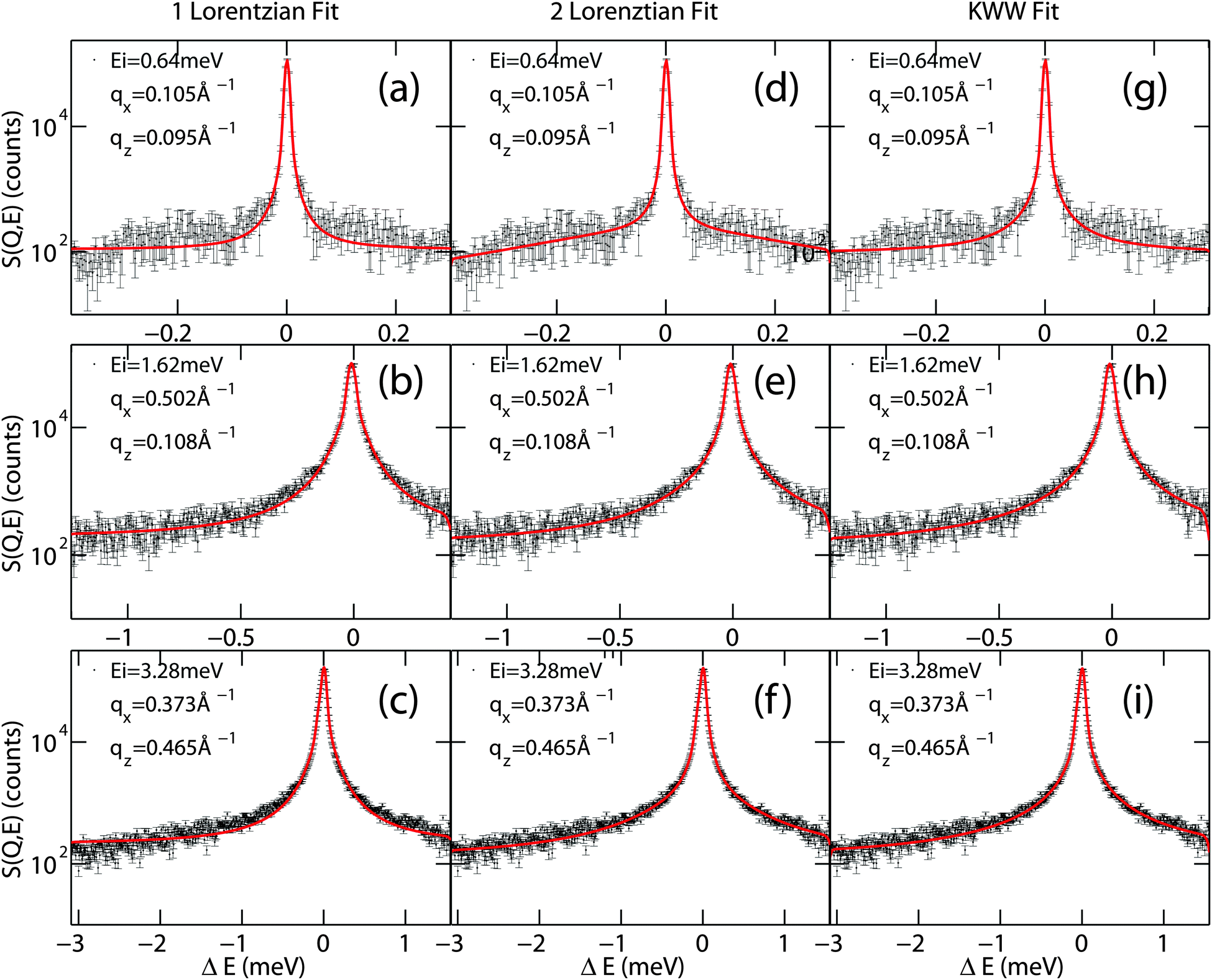 | ||
| Fig. 4 Comparison between a one-Lorentzian peak fit, two-Lorentzian peak fit and Fourier transform of KWW fit of quasi-elastic broadening spectra of water diffusing in a lipid membrane. Values for qx and qz are indicated in each plot. | ||
In order to quantitatively compare the different models, the reduced chi-squared values (![[small chi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e11b.gif) 2) of the different models were calculated using61
2) of the different models were calculated using61
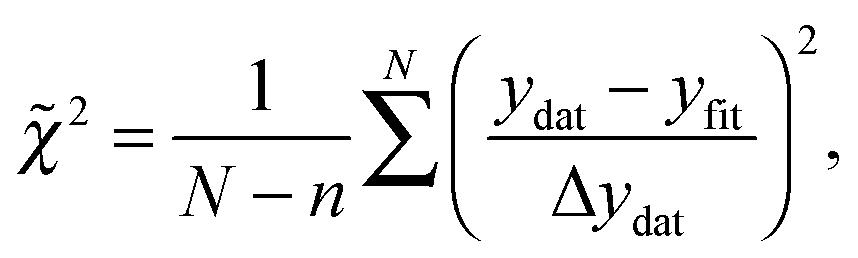 | (14) |
2 = 1, the fit is within the error bars of the spectrum. For 2 > 1, the spectrum is under-fitted, i.e., there is more information in the spectrum that has not been reproduced in the fit. For 2 < 1, the spectrum is over-fitted.
Each spectrum was fit with all three models and the corresponding reduced χ2 calculated. The results are listed in Table 1. A detailed list is given for the qx-averaged 2 for an incident energy of 3.28 meV to show that the fit quality also depends on qz. Mean 2s were calculated and are given for all energies. It is observed that the fit of a single Lorentzian provides a significantly poorer fit while two Lorentzian peaks and a KWW function seem to adequately describe the quasi-elastic data with almost identical 2s.
2 values (mean x2). All qz values are listed for an incident neutron energy of 3.28 meV; averages are shown for other energies, only. Mean 2s are calculated from fits to one Lorentzian (eqn (9)), two Lorentzians (eqn (11)) and the KWW function (eqn (13)) of each spectrum. Exemplary fits are shown in Fig. 4
| Energy (meV) | q z (Å−1) | Mean 2 |
||
|---|---|---|---|---|
| 1 Lorentzian | 2 Lorentzians | KWW | ||
| 0.64 | 1.21 | 1.07 | 1.15 | |
| 0.96 | 2.14 | 2.06 | 2.03 | |
| 1.62 | 5.58 | 5.03 | 5.15 | |
| 3.28 | 0.0310 | 16.5 | 9.73 | 10.6 |
| 0.0930 | 15.6 | 8.56 | 8.85 | |
| 0.155 | 14.6 | 7.41 | 7.73 | |
| 0.217 | 15.2 | 8.13 | 8.25 | |
| 0.279 | 13.6 | 6.49 | 6.58 | |
| 0.341 | 12.2 | 5.34 | 5.37 | |
| 0.403 | 12.7 | 5.93 | 5.86 | |
| 0.465 | 11.5 | 5.67 | 5.47 | |
| 0.527 | 12.5 | 7.48 | 7.30 | |
| 0.589 | 13.7 | 7.99 | 8.17 | |
| Mean 2 |
13.7 | 7.27 | 7.41 | |
| 9.80 | 71.0 | 30.7 | 41.7 | |
|
Total
2
|
18.7 | 9.21 | 11.5 | |
We note that this problem is not unique to incoherent quasi-elastic neutron scattering experiments. Lipid motion determined by fluorescence correlation spectroscopy (FCS), for instance, was reported to be equally well described by anomalous diffusion or two Brownian diffusion processes.11
Following eqn (9), (11) and (13), the corresponding fitting functions involve 2, 4 and 3 free parameters. While a larger number of free parameters typically leads to a better fit, the fundamental question is if motion of molecules in anisotropic and heterogeneous environments is more reasonably described by a series of Brownian diffusion processes or rather by a distribution of relaxation times, as described by the KWW function. We will present evidence that the KWW provides the correct description of hydration water dynamics based on results from the neutron experiment and computer simulations further below. In the remaining sections, the quasi-elastic broadening is characterized by the Fourier transform of a KWW function to describe anomalous dynamics, and τ and β parameters are determined.
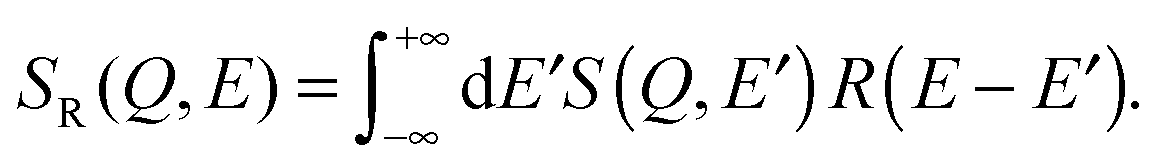 | (15) |
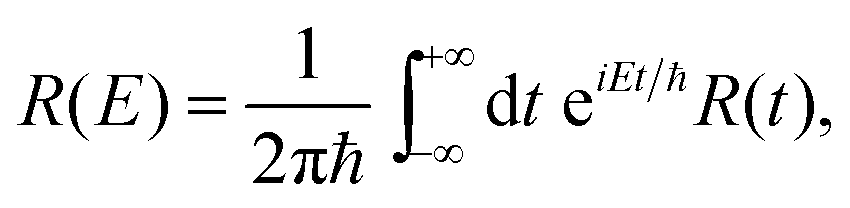 | (16) |
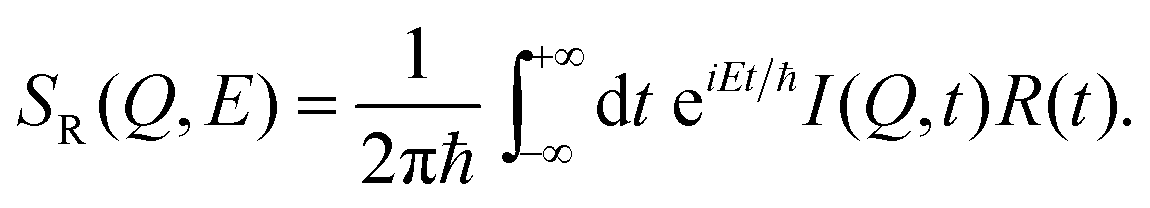 | (17) |
When assuming a Gaussian resolution function, common in quasi-elastic incoherent neutron scattering experiments, R(E) can be written as
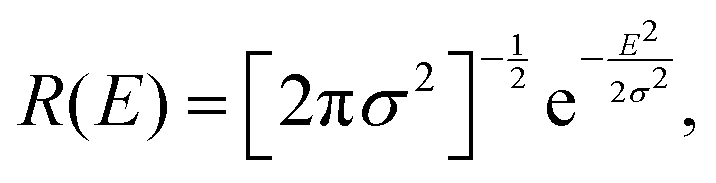 | (18) |
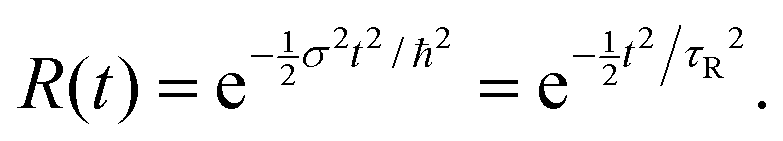 | (19) |
When R(t) is substituted into R(E), the integral in eqn (17) is cut off after a time τR. For a Gaussian R(t), the “cut-off” time (the experimental observation time τR) is related to the FWHM of R(E) by
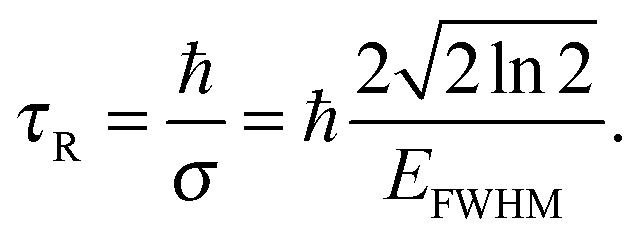 | (20) |
Experimental observation time windows, τR, for the bulk water sample were calculated from the average σ of the resolution peaks of 4.3, 6.8, 12.7, 20.0, 96.4 μeV to be 154.2, 97.2, 51.7, 23.5, 6.6 ps, respectively. The membrane samples have average σ resolution peaks of 4.3, 6.8, 12.6, 25.8, 96.0 μeV corresponding to observation time windows of 151.6, 97.4, 52.3, 25.5, 6.9 ps, respectively. The slight difference in the experimental resolution and observation time for the two samples is a purely geometrical effect as the experimental resolution depends on the size (mainly the diameter) of the sample used.
Molecular motions slower than the experimental observation time can still be accessed in experiments, however potential additional statistical noise in a spectrum can lead to a systematic error, which tends to overestimate the actual relaxation time, τ, as will be discussed below.
2.3 Simulations
In particular, we choose:
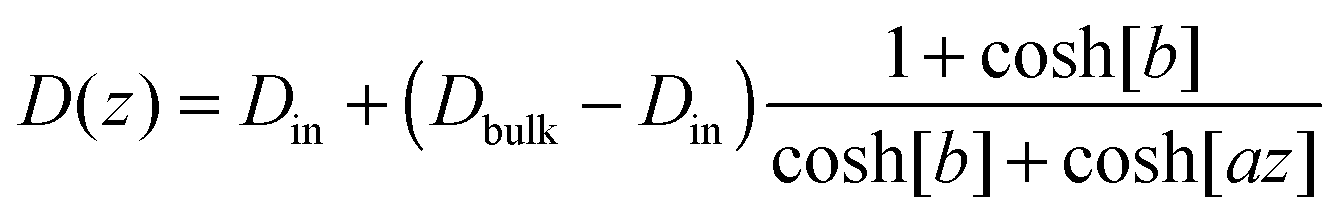 | (21) |
| V(z) = cz2.75, | (22) |
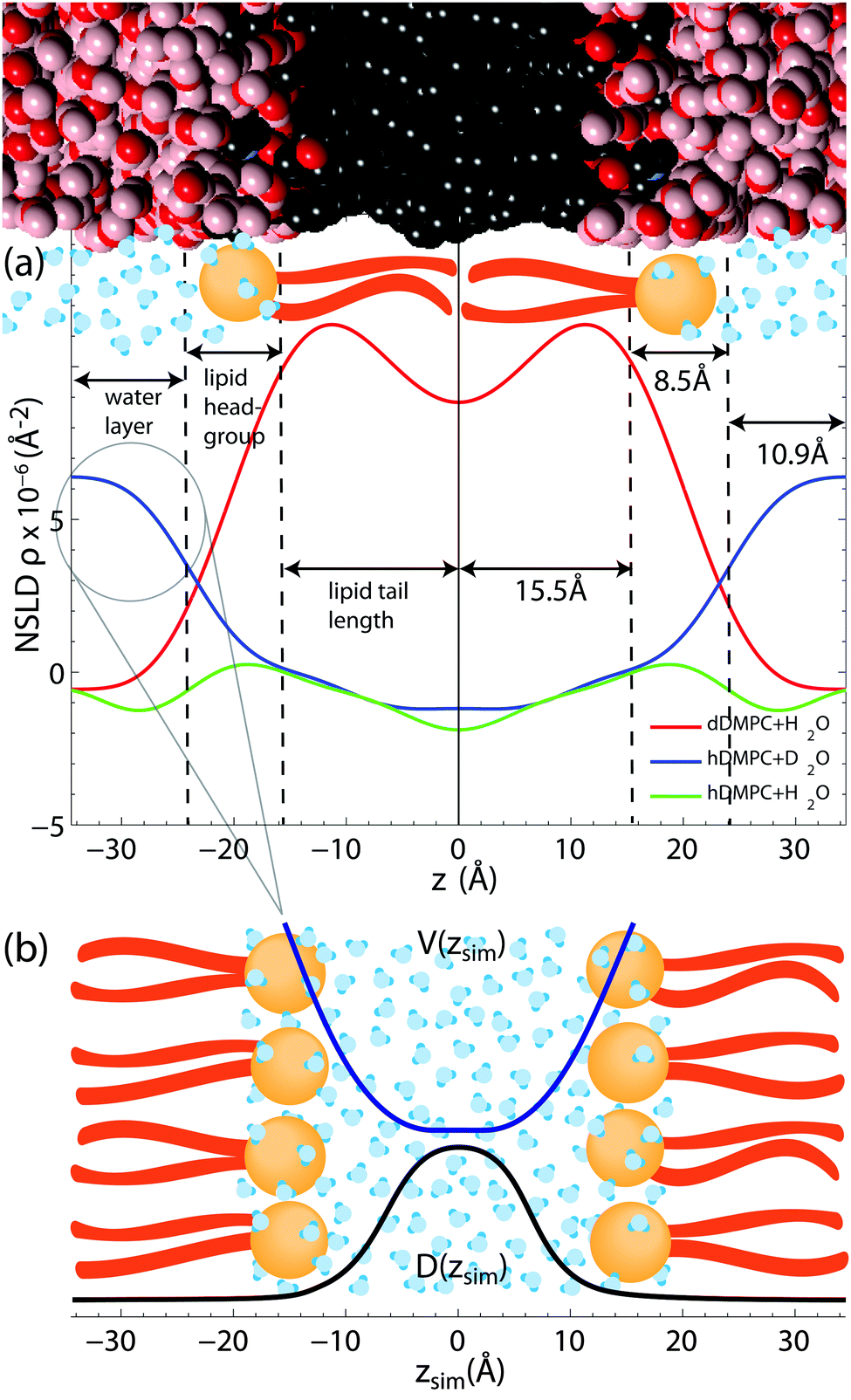 | ||
| Fig. 5 (a) Scattering length density profiles as synthesized through Fourier transformation of the reflectivity curves in Fig. 2. Selective labelling of hydration water and lipid tails highlights the hydration water layer and the hydrophobic membrane core. Fully protonated membranes enable the determination of the lipid head groups as phosphate and carbonyl groups have larger coherent neutron scattering cross section. (b) The shape of the water segment of the D2O/DMPC sample was used to model the shape of the potential, V(z), in the simulations. | ||
The displacement Δr of the center-of-mass of one water molecule within a time step Δt is calculated via
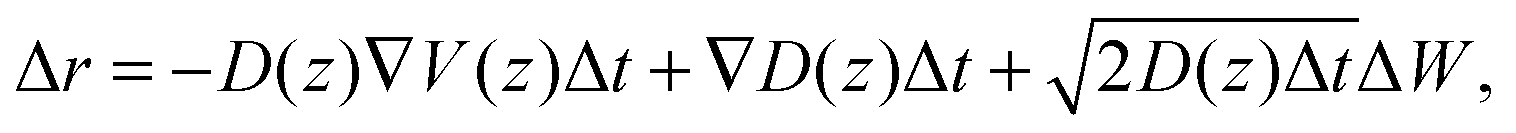 | (23) |
The trajectory for one water molecule is simulated with Δt = 0.1 ps for a time of 10 μs. As starting condition we choose the center of the water layer, in agreement with the experimental data in Fig. 5.
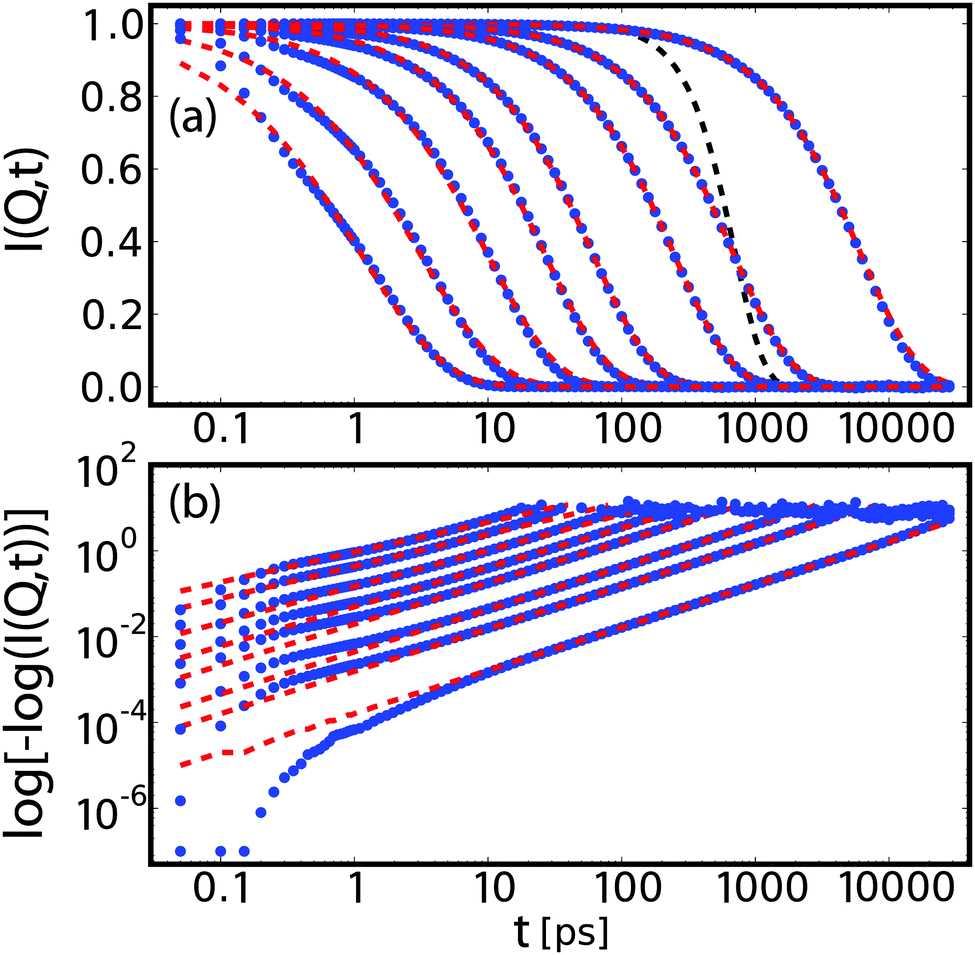 | ||
| Fig. 6 Intermediate scattering function for bulk water at qx = 0 and qz = 0.025, 0.075, 0.125, 0.25, 0.375, 0.6, 1.0, 1.5 Å−1 in lin–log (a) and log–log (b) representation. These data (blue dots) are fitted corresponding to eqn (12) with a stretched exponential plus constant (red dashed line). The linear scaling over decades in time in the log–log representation supports the applicability of a stretched exponential as fit function, although deviations are observed at low times. The artificial resolution function is plotted as a black dashed line. | ||
The intermediate scattering functions were fitted with a stretched exponential plus a constant (non-zero only for qx = 0), consistent with the experimental analysis as laid out in eqn (13). To render the fitting more robust, we employed the following fit procedure: first, for the case of hydration water and qx = 0, I∞(qx, qz) is fixed to the mean of the last 3 points of the correlation function. For all other cases, I∞(qx, qz) is set to zero. Second, we fit a stretched exponential to the re-scaled data.
In order to investigate conditions closer to the experiment, we include a hypothetical Gaussian experimental resolution via fitting weights R(t) = exp(−½t2/τR2) with τ = 500 ps. Fig. 6 shows a set of I(Q, t) profiles with corresponding fits and artificial resolution function. Except for very small correlation times, the model of a stretched exponential provides good fits over several decades in times.
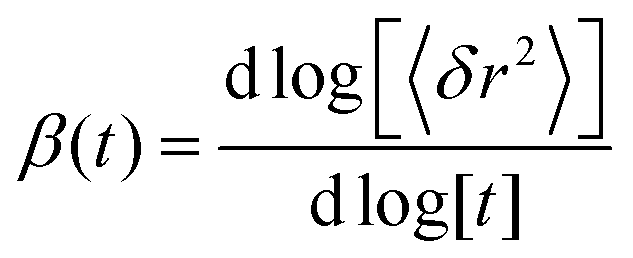 | (24) |
3 Results & discussion
3.1 Membrane structure
In order to confirm fully hydrated bilayers neutron reflectivity measurements were done to determine the bilayer structure. Fig. 5 shows the scattering length density profiles, as determined from the reflectivity curves in Fig. 2.The DMPC/H2O sample allowed us to determine the extent of the head group, the water layer and lipid tail regions. A combination of X-ray scattering, neutron scattering and simulations have been used to determine bilayer structures of a variety of phospholipids (PC).64–66 Since these lipids have the same PC head group, these publications were instructive in finding the extent of our head group region. We define the extent of the head group to be the center of the distributions of its most interior/exterior molecules: carbonyl/choline groups.
The probability distributions for the head group components65,66 show the head group/water boundary at approximately the centre of the choline distributions. The head group/water boundary should be approximately at the inflection point of the head group peak bordering the water at z = 24 Å, as shown in Fig. 5.
We assumed the maximum of our NSLD peak is centered between the phosphate and carbonyl groups. As the distribution of the carbonyl64–66 has a half-width at half-maximum of ∼3 Å, we approximate the head group/tail boundary at 15.5 Å. Half the average lamellar repeat distance minus the head group/water boundary (34.9–24.0 Å) results in half a water layer of 10.9 Å. Based on this analysis, the membrane core was found to have a thickness of 31 Å (15.5 Å per leaflet), the head group region was 8.5 Å thick and the hydration water layer had a total thickness of 21.8 Å (10.9 Å on each side of the density profile). These values were used in the simulations and confirm that the phospholipid bilayers were in their fully hydrated fluid phase. We note that the values for the bilayer dimensions are in excellent agreement with reference values for DMPC by the Nagle group51 of DHH = 35.3 Å, DW = 19.2 Å and 9 Å for the head group thickness.
The raw NSLDs for each sample were scaled in the following way: the profile edges were scaled to the density of bulk water (ρH2O = −5.6 × 10−7 Å−2 or ρD2O = +6.39 × 10−6 Å−2) and bilayer centers were scaled to a CH3 (or CD3) group (ρCH3 = −1.83 × 10−6 Å−2) or (ρCD3 = +8.89 × 10−6 Å−2) plus the NSLD of 0.10 water molecule. The center of the bilayer in the NSLD profiles are typically scaled to a CH3 group, however, differences between tail regions of DMPC/H2O and DMPC/D2O profiles point to non-zero water content at the center of the hydrophobic core.
We determine the water content at the center of the hydrophobic core to be 0.10 solvent molecules (H2O or D2O) per lipid such that the NSLD profiles of the two DMPC samples overlap from ∼6 Å to 10 Å.
Though this technique cannot determine the absolute number of water molecules in the entire tail group region, our experiments provide evidence for ∼1 water molecule for every 10 lipids. The presence of water in the center of lipid bilayers is supported by IR spectroscopy experiments,36 as well as simulations.37,39 While the density of water molecules in the hydrophobic core is low compared to the hydration water layer, the center of the bilayer is a region of low lipid density, with a larger free volume fraction39 and consequently lower free energy and higher probability for water molecules to be present.
The NSLD profiles in Fig. 5 are in very good agreement with results from recent computer simulations in the same system by Hansen et al.33 and Yang, Calero and Marti.67 Both our experiment and simulations agree that water molecules penetrate the bilayer into head group and tail region, however the simulations do not provide evidence for an increased water density in the center of the bilayers. We note that the simulations used a potential to implicitly model the membrane where water was not explicitly accounted for within the membrane core.
3.2 Water dynamics from experiments
Fig. 7(a) and (b) show the results for bulk water. A sketch of the scattering geometry is shown in Fig. 7(c). Stretching exponent (Fig. 7(a)) and relaxation time (part (b)) are displayed in 2-dimensional maps as a function of parallel and perpendicular momentum transfer, qx and qz. τ is plotted in units of picoseconds and on a logarithm scale due to its multiple orders of magnitude range. The map covers motions of water molecules from ∼3 Å to ∼100 Å in the x-direction and from ∼6 Å to ∼60 Å in the z-direction.
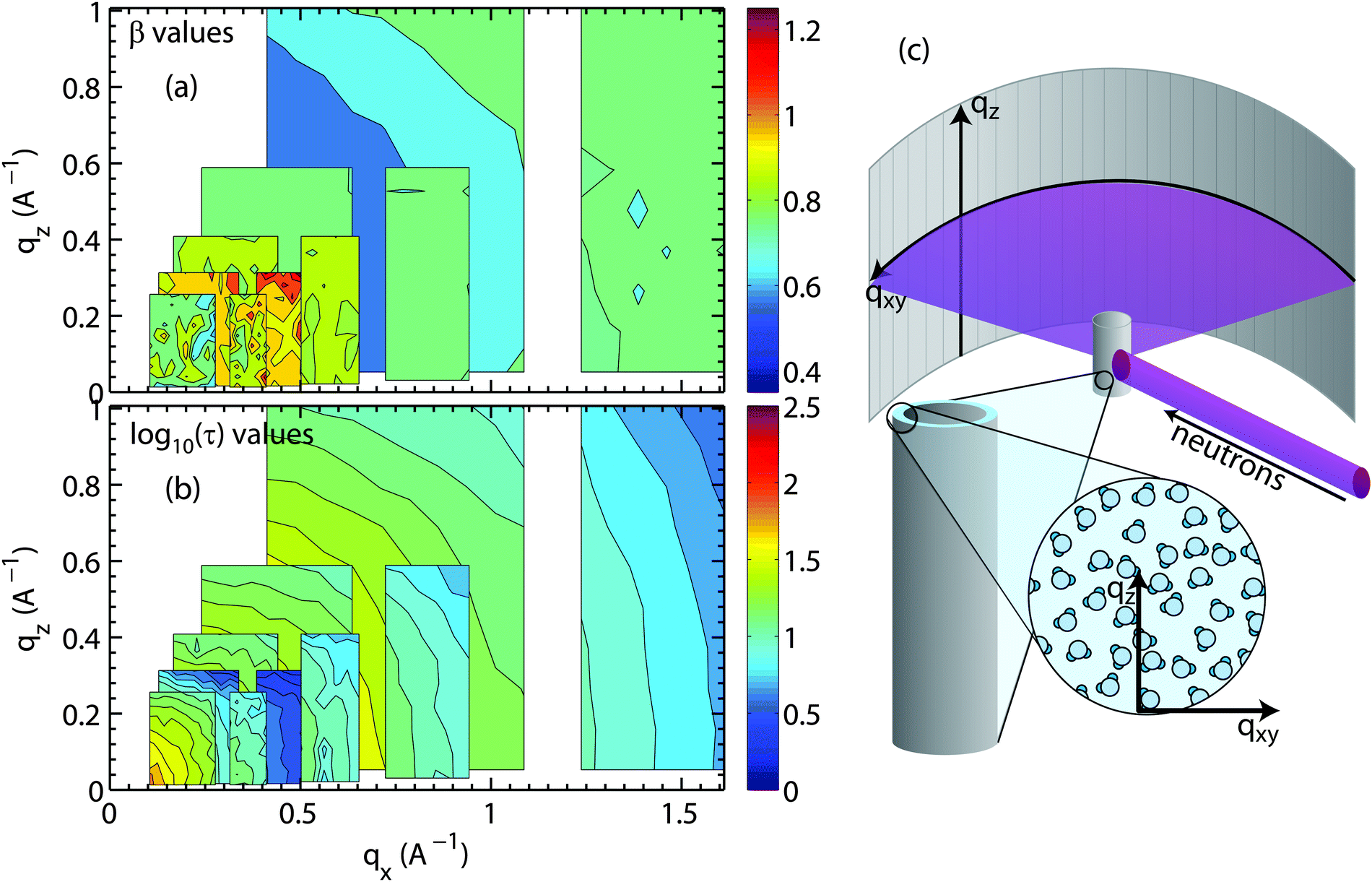 | ||
| Fig. 7 Values of (a) β and (b) log10(τ) (τ in ps), fit from each bulk water spectrum, are plotted as a function of qz and qx. Measurements were made with five different energy resolutions. The isotropic nature of the bulk water dynamics can be seen in the concentric rings in (b). (c) Sketch of the scattering geometry. We note that the 17th slice in each measurement was taken out because the detector array at this scattering angle was, unfortunately, malfunctioning during the experiment. | ||
Motions over longer distances (small qx,z-values) are slower and require longer observation times (higher energy resolution or longer neutron wavelength, λ) as compared to short-range motions at larger qx,z-values. Following Bragg's equation (q = 4πsin(θ)/λ or 2dsin(θ) = λ with d = 2π/q), the accessible qx and qz-range strongly depends on the incident neutron wavelength. The larger λ, the larger the observation time and the higher the energy resolution, however, the smaller the qx,z-range covered. In order to cover the largest range of length and time scales, results from different energy resolutions must, therefore, be combined, as has been done in Fig. 7. The pattern of concentric rings of relaxation times in Fig. 7(b) is indicative of an isotropic motion of water molecules with longer relaxation times at longer length scales.
The values for β in Fig. 7(a) between 0.85 and 1 indicate that the corresponding nanoscale dynamics of water molecules is slightly sub-diffusive. Sub-diffusive dynamics is often found in supercooled water. Simulations of supercooled bulk water68–70 show sub-diffusive behaviour with exponents in the range of β = 0.85–1 for our parameter ranges. Chen et al.69,70 found that β stayed close to 1 for low Q values and low temperatures T < 225 K, however Sciortino et al.68 reported that supercooled bulk water at 285 K at Q ∼ 1.5 Å−1 reached β = 0.85. Experiments using light scattering71 and neutron scattering71,72 observed 1 ≥ β ≥ 0.6 for bulk water in our temperature range (293–313 K72) for Q ∼ 0.25–1.5 Å−1.
The bulk water measurements prove that diffusion can be studied by analyzing different areas on the large 2-dimensional detector array. We note that it is not trivial to determine a diffusion constant in the sub-diffusive regime as the standard approach is to use eqn (7) and plot the quasi-elastic broadening as a function square of the scattering vector, Q2; this approach fails in the case of sub-diffusion and non-Lorentzian fit functions.
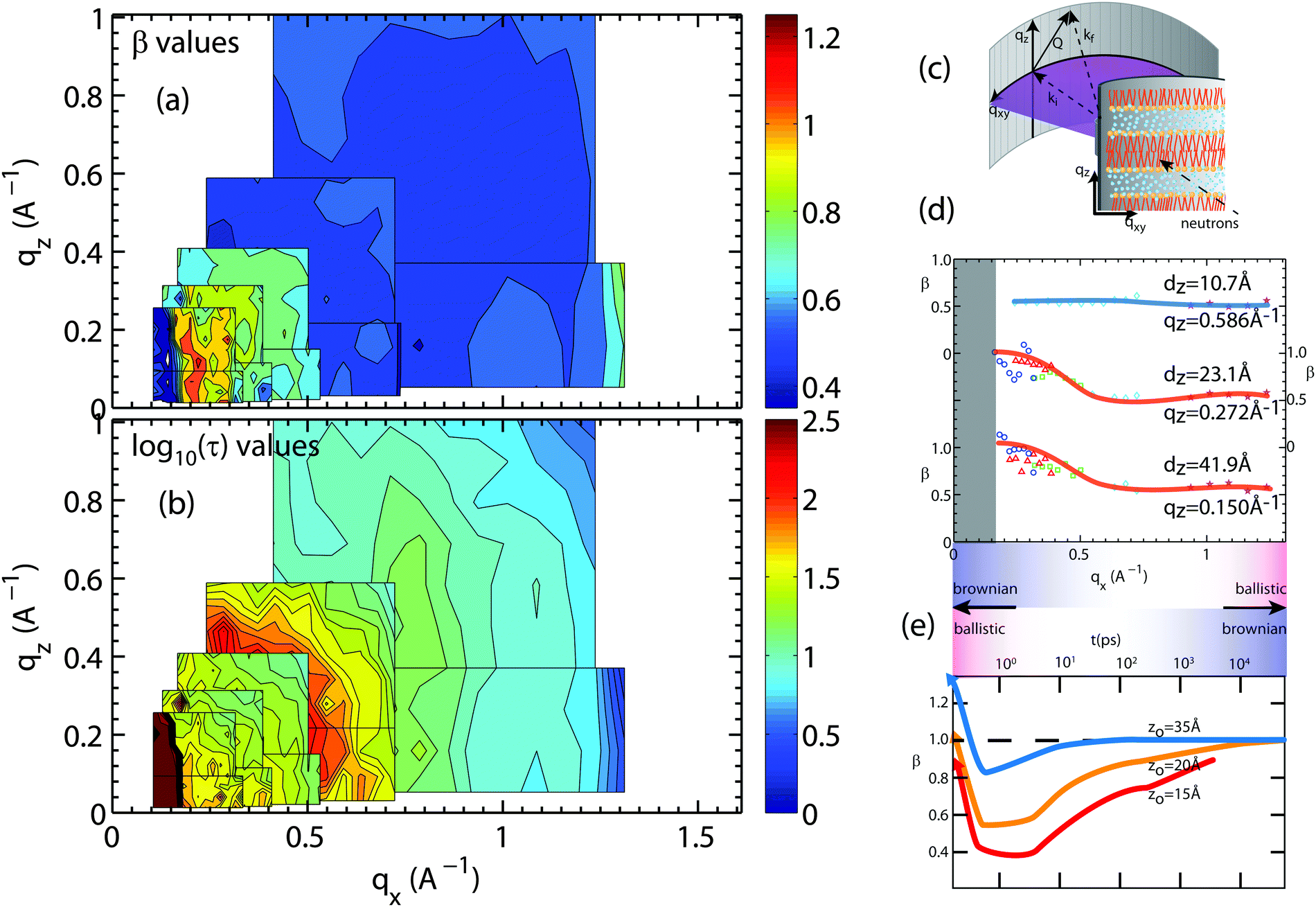 | ||
| Fig. 8 Plots of the (a) stretching exponent, β, and (b) the logarithm of the relaxation time log10(τ), τ in ps, from fits of hydration water spectra that have been integrated over qz and qx. The smallest map corresponds to the highest resolution measurement (Ei = 0.64 meV) while the largest map corresponds to the lowest resolution measurement (Ei = 9.80 meV). The experimental error on determining the β- and τ-values depends on the actual Q-value and the resolution of the spectrometer. Relative standard error values for β and τ were between 5% to 10%. (c) A schematic of the membrane system in the Let spectrometer. (d) β values for slices in qz of 0.15, 0.272, and 0.586 Å−1 for water motion varying in qx. (e) β values adapted from simulations done by von Hansen et al.32 plotted as a function of time. | ||
The relaxation times in Fig. 8(b) show fast hydration water dynamics at large qx, which continuously slows down towards longer length scales (smaller q-values). In contrast to the bulk water results in Fig. 7, the relaxation times along qx and qz are anisotropic with clear deviations from the circular pattern. The most striking feature is a localized region with slow relaxation times at qx ∼ 0.15 Å−1 (equivalent to a length scale of 40 Å). The feature is also seen in the β data in part (a) and exhibits small values of β of less than 0.40. Shape and position of this feature agrees well with the feature observed in the computer model of the stacked bilayers and is a result of confinement, as will be discussed below.
Fig. 8(d) shows qx-slices at different qz-values, which allow access to different water molecules. Based on the structural results in Fig. 5, the highest density of water molecules in the hydrated bilayer stack is found in the center of the hydration water layer. Different values of the transverse momentum transfer, qz, then correspond to the spread of z-values that the molecules explore when diffusing. qz = 0.586 Å−1 corresponds to a vertical distance of dz = 2π/0.586 = 10.7 Å, within the hydration water layer. The slice centered at qz = 0.272 Å−1 contains water molecules that penetrate the head group region of the bilayers; qz = 0.15 Å−1 molecules, which migrate to the lipid acyl chains. Based on Fig. 8(d), water molecules, which diffuse laterally in the hydration water layer show a sub-diffusive behaviour with a β exponent of ∼0.50. This β-value is significantly lower than the value that we measured for bulk water, indicative that hydration water motion is significantly anomalous. Water molecules in contact with lipid head groups and hydrophobic core show a sub-diffusive regime at small distances with a transition to Brownian motion at larger distances of ∼14 Å. We note that ballistic motion of water molecules with β-values of 2 is expected to occur at larger qx-values, outside of the experimentally accessible range in this experiment.
The results are in qualitative agreement with a recent computer simulation study by von Hansen, Gekle and Netz,32 which reports a sub-diffusive regime for hydration water dynamics, however in the time domain, as shown in Fig. 8(e) (data adapted from ref. 32). The simulation data were evaluated for water molecules at different distances from the bilayer center, z0 = 15 Å being located in the lipid head group region and z0 = 35 Å, at the farthest distance from the bilayer. We note that simulations were conducted in a single hydrated bilayer, where water dynamics away from the bilayer are more likely Brownian, as compared to the confined water layer in our experiment. Water molecules diffusing along the head group region or within the lipid tails show a distinct sub-diffusive regime, with β values of 0.50, in good agreement with the experimental results in Fig. 8(d).
Corresponding cuts of the relaxation time, τ, at qz-values of 0.15 Å−1 and 0.586 Å−1 (corresponding to z motions over 10.7 Å and 41.9 Å, respectively) are shown in Fig. 9. Again, slices were centered in the hydration water layer and qz-values are a measure of the corresponding slice width. The relaxation time for hydration water molecules diffusing laterally with small z motions (large qz), inside the water layer, was found to be almost qx independent. A slight slowing down of the water molecules at a qx-value of ∼0.70 Å−1 is likely related to the presence of the bilayer interface as a disturbance caused by the lipid head groups. This feature corresponds to the slight decrease in β-values in Fig. 8(e) in the computer simulations by von Hansen, Gekle and Netz32 at z0 = 35 Å.
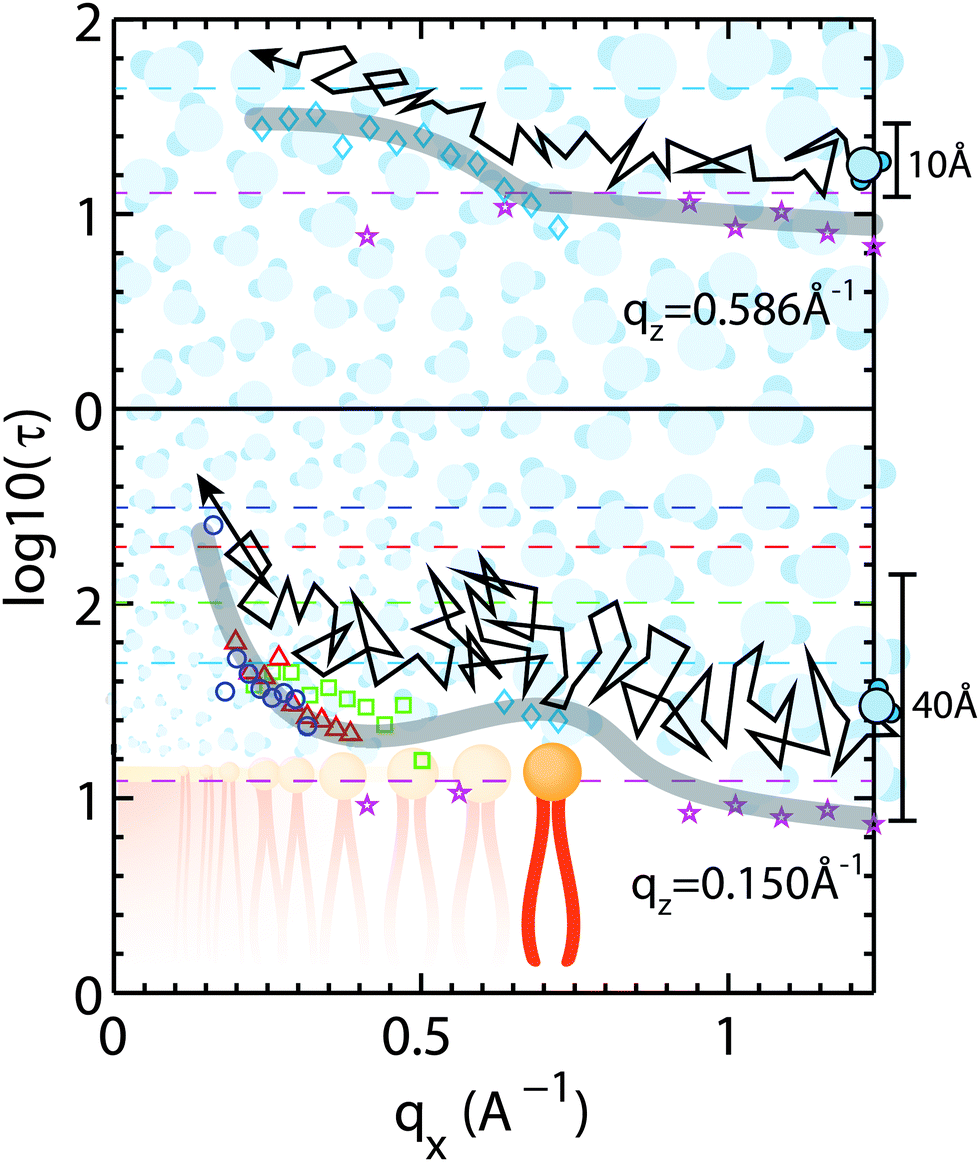 | ||
| Fig. 9 Lateral diffusion of membrane water molecules as shown by relaxation time τ (τ in ps). Slices along qx are shown for two values of qz. The lower curve occurs at qz = 0.15 Å−1 corresponding to length scale of ∼40 Å. The lateral motion of a water molecule is slowed significantly for a lateral length scale of ∼10 Å denoted by an increase in τ at qx = 0.6 Å−1; most likely due to the water's interaction with the head group, where hydrogen bonding occurs. The upper curve occurs at qz = 0.586 Å−1 corresponding to ∼10 Å. This signal is most likely dominated by water molecules diffusing in the hydration water layer. Dashed lines correspond to maximum viable τ; lines and symbols have corresponding colors. | ||
A hydration water molecule that moves about a range of z = 40 Å while diffusing laterally penetrates the bilayer to the lipid acyl chains. Its motion appears to slow down at a qx value corresponding to the distance between two lipid head groups indicated by the peak in the relaxation time. The relaxation times then increase significantly towards larger length scales.
Fig. 10 illustrates the effect: Fig. 10 (a) shows τ distributions of the fit function to data measured at the same (qx, qz) position in reciprocal space from the data in Fig. 8 (which should have the same relaxation time), however, measured at different experimental resolution times, τR. τR-values are given in the figure and marked by the shaded areas. For some resolutions, the peak in the time distribution is outside of the experimentally covered window and the corresponding relaxation time was extrapolated by the fit. As a consequence, when the quasi-elastic broadening was too small to be properly resolved by the instrumental resolution, the relaxation time was found to be systematically over-estimated.
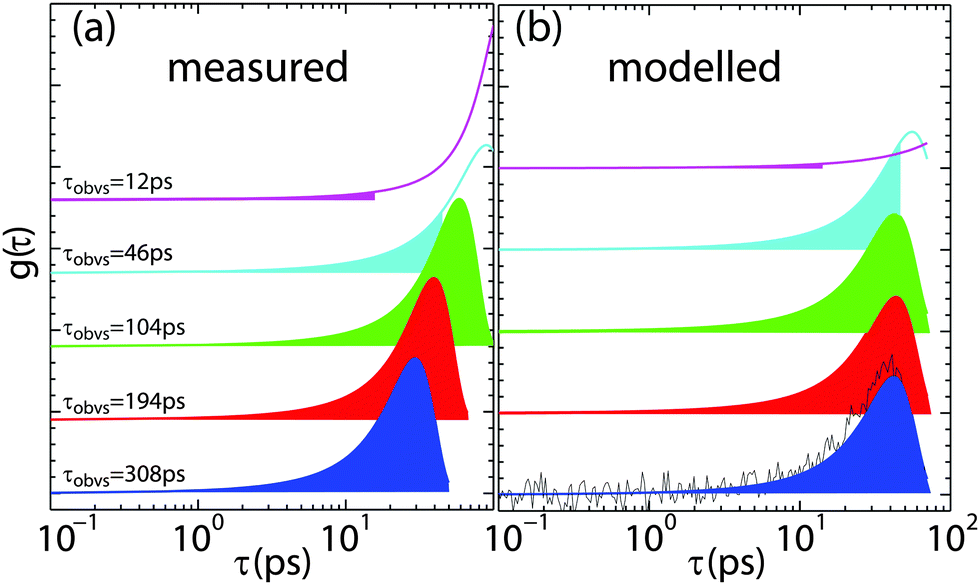 | ||
| Fig. 10 (a) Plot of relaxation time probability density functions using β and τKWW values obtained from fits of QENS data for an area of reciprocal space bounded by Δqz = 0.2–0.1 Å−1 and Δqx = 0.4–0.3 Å−1 for each resolution probed. (b) Plot of fits to relaxation time pdfs with β = 0.8 with noise. Values of τKWW and amplitude were obtained for fits to the noisy pdf within each resolution range. Blocks of color represent the observation time window for that particular resolution setup. Observation times (τR) are given. | ||
Though counterintuitive, this behaviour is a systematic effect that can be modelled. If there is little or no noise or error, fit functions can typically be determined correctly even when the function is not fully covered by the experimentally accessible range of data. This is the case when the relaxation time is outside the experimental resolution window, as defined in eqn (20). The combination of experimental noise and partial coverage, however, may cause a systematic error in the determination of relaxation times in quasi-elastic incoherent neutron scattering experiments. In Fig. 10(b), the τ-distribution from part (a) was generated and a window equivalent to the experimentally accessible time window was selected. Statistical noise was added to the generated data to mimic experimental conditions. The resulting data were then fitted again with the original distribution that was used to create the distribution (and should reproduce the original data). The results are shown as solid lines in part (b) and well reproduce the systematic deviation: the relaxation time tends to be overestimated when the distribution is outside of the experimental window and experimental noise is present.
3.3 Water dynamics from computer modelling
In order to consolidate the picture of anisotropy and anomaly of water dynamics obtained by the neutron scattering experiments, we employed two sets of computer modelling. First, using full-atomic detail molecular dynamics, we studied the intrinsically anomalous character of isotropic, bulk water diffusion at picosecond time scales.For hydration water dynamics, we used a coarse-grained Brownian dynamics simulation to investigate the effects of geometrical confinement and locally varying dynamical environments of water molecules interacting with lipid bilayers. The corresponding systems are inherently larger in size and the number of molecules involved, therefore, making full atomistic simulations challenging.
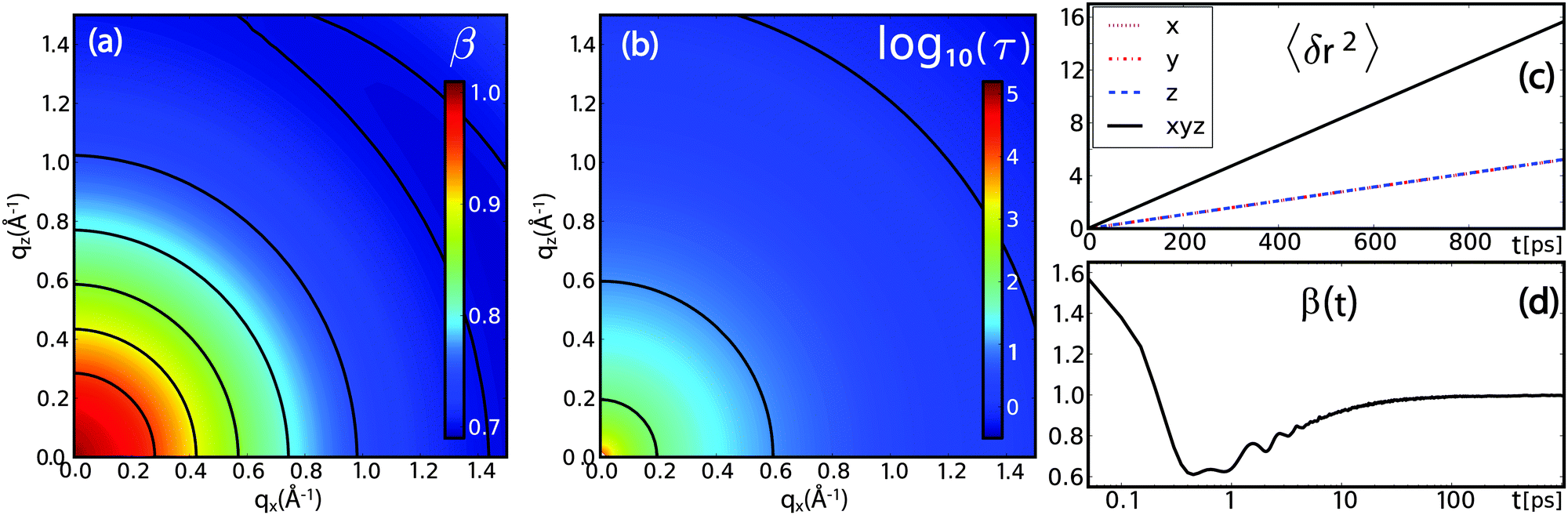 | ||
| Fig. 11 Results from full atomistic molecular dynamics simulations of bulk water using the SPCE model: (a) intrinsic anomaly of water diffusion: the stretch exponent β indicates a transition from normal diffusion at low qx-value to predominantly sub-diffusive behaviour at larger qx. (b) The relaxation time τ (plotted in units of 1 ps) shows no anisotropic behaviour, as expected for bulk water. (c) and (d) The time-dependent stretch exponent as extracted from the MSD 〈δr2〉 viaeqn (24) changes from a super-diffusive, ballistic regime at times below 0.5 ps to a sub-diffusive regime for several ps before approaching the normal diffusive regime for longer time scales. | ||
The MSD and the derived time-dependent stretch exponent β are shown in Fig. 11(c) and (d). The displacements along different directions (〈δx2〉, 〈δy2〉 and 〈δz2〉) coincide, indicative of an isotropic motion. After a ballistic regime at sub-picosecond time scales, a sub-diffusive regime at time scales of several picoseconds follows, and, finally, normal diffusion is reached at nanosecond time scales, in agreement with computer simulations by von Hansen et al.32 Thus, both experiments and simulations consistently indicate that water diffusion has an intrinsically sub-diffusive character on picosecond time scales, also under ambient conditions.
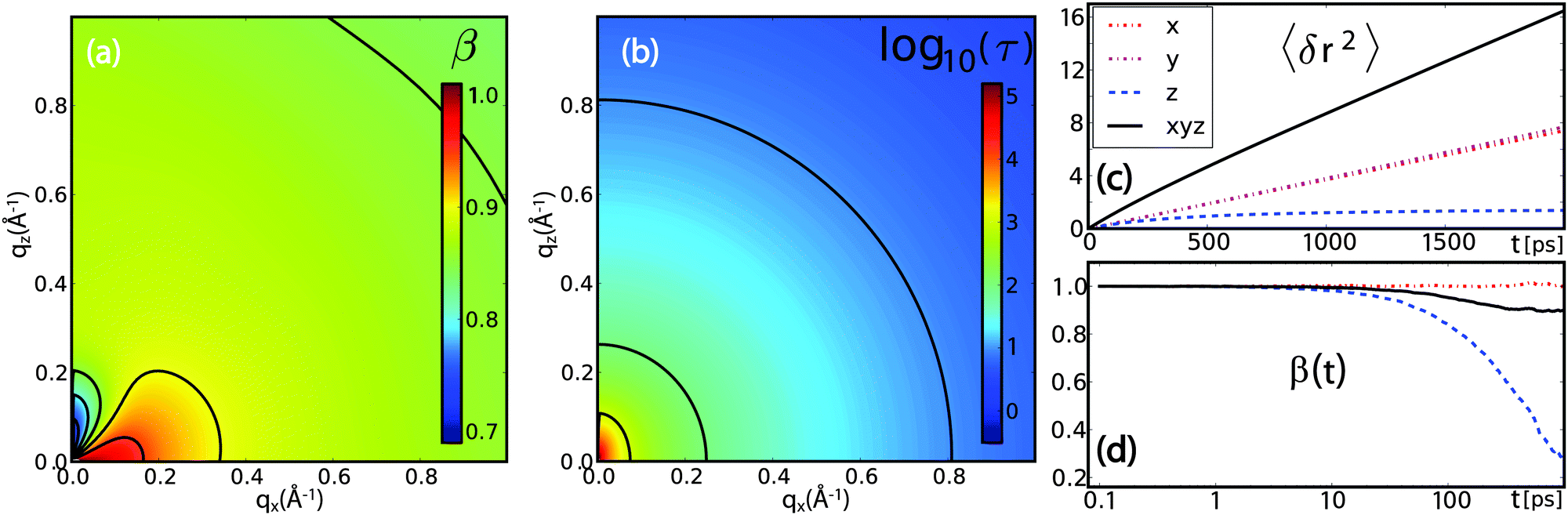 | ||
| Fig. 12 Results from coarse-grained Brownian dynamics simulation for water confined in a membrane system: (a) anomaly of water diffusion due to dynamical heterogeneity and confinement: the stretch exponent β shows sub-diffusive behaviour at all visible qx values, with a trend of stronger sub-diffusive motion at higher qx. Anisotropic behaviour is observed at low qx due to geometrical confinement of the membranes. (b) The relaxation time τ (plotted in units of 1 ps) shows a slight anisotropic behaviour at low qx. Water diffusion is overall slower than in bulk (see Fig. 11(a)). (c) and (d) Mean-squared displacements 〈δr2〉 and time-dependent stretch exponents β(t) (see eqn (24)) for different coordinate directions evidence a confined motion in z-direction (blue dashed), while the MSD in xy-direction (red dash-dotted) resembles that of normal diffusion. The resulting overall MSD (black solid) has an apparent sub-diffusive character emerging solely from the confinement of hydration water molecules. | ||
However, the stretch exponent β in Fig. 12(a) shows sub-diffusive behaviour at all qx values, with a trend of stronger sub-diffusive motion at higher qx. Pronounced anisotropic behaviour is observed at low qx. The constant I∞(Q) is zero except for qx = 0 Å−1 and qz ≲ 0.2 Å−1, indicating geometrical confinement due to the membrane on a length scale of ∼30 Å. The relaxation time τ (plotted in units of 1 ps in Fig. 12(b)) shows anisotropic behaviour at low qx. Water motion is overall slower than in bulk water in Fig. 11(b).
The significantly longer τ results from the slowing down of dynamics close to the membrane, and a longer average relaxation time. The value of β indicates a distribution of local dynamical environments, and thus a distribution of relaxation times. This finding is consistent with the observed trend of β approaching 1 with lowering qx,z, since the molecule explores a larger space and thus performs a temporal average of dynamical environments, finally reaching normal diffusion with the average diffusion coefficient.
With increasing correlation time, the molecules experience confinement due to the membrane, which induces an apparent sub-diffusive character for the diffusion displacement. We note that this factor is only caused by the z coordinate, as shown in the MSD in Fig. 12(c), while diffusion along x and y is normal diffusion, as expected from the simulation framework.
At low qx,z, distinct anisotropic features appear in τ, β and I∞, mirroring the features observed in experiments. In particular, when cutting along the qx axis for small qz, a non-monotonous behaviour is observed for β and τ. The evolution of β as a function of time is plotted in Fig. 12(d). This non-monotonicity seems to be directly related to confinement effects, since its appearance coincides with a non-zero I∞, i.e., a non-vanishing correlation at long times.
This relation of geometrical confinement and dynamical effects can be understood when considering the role of Q in the calculation of the intermediate scattering function if one coordinate is confined:
I(Q, t) = 〈exp[−i![[Q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0051_20d1.gif) ·( ·(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) (t) − (0))]〉 ∼ exp[−qx2〈δx2〉 − qy2〈δy2〉 − qz2〈δz2〉] +…, (t) − (0))]〉 ∼ exp[−qx2〈δx2〉 − qy2〈δy2〉 − qz2〈δz2〉] +…, | (25) |
3.4 Motion of water molecules
Lipid diffusion has been studied for decades and it is commonly accepted that the Brownian motion of lipid molecules over long, micrometer length scales is characterized by a continuous diffusion process.13–17,21 Although this is a well studied fundamental process, our understanding of nanoscale diffusion in membranes is still being challenged by new results from experiments and simulations.Experimental evidence that lipids move coherently in loosely bound clusters, rather than as independent molecules was presented.7,18–20 A “hopping” diffusion of lipids into nearest neighbour sites was observed in single supported bilayers.6 Ballistic lipid motion in fluid membranes was recently reported.5 It has also been suggested that there is a flow-like component to the motion of the lipid molecules over long length scales.7
Significantly less work has been done to study dynamics of hydration water molecules, most likely due to the experimental and computational challenges. We find that lateral water diffusion is affected at all distances from the bilayers and motions of hydration water molecules differs significantly from the isotropic diffusion of bulk water.
From the quasi-elastic neutron scattering experiments and computer simulations, we obtain a consistent picture of the phenomenology of water diffusion in membrane systems, as well as different origins of anomaly and anisotropy. As a first origin of anomaly, water shows an intrinsic anomalous diffusion on picosecond time scales, as also observable in bulk water. The second origin of anomalous diffusion is related to the distribution of local dynamical environments of hydration water molecules, which leads to a distribution of relaxation times and sub-diffusive behaviour. The anisotropic effects of confinement and local dynamical environments on the motions in different directions give rise to direction-dependent relaxation times and stretch exponents due to the varying sampling time of the system along the unconfined coordinates.
A clear distinction of which model to use is often difficult in QENS data because of the limited statistics in the data, such that different models fit the data equally well. We note that FCS suffers from the same issue and cannot unambiguously determine the basic character of lipid dynamics in membranes.11 However, we argue that motion of water molecules in anisotropic and heterogeneous environments is physically better described by a distribution of relaxation times (a KWW function) rather than a sum of individual Brownian processes (several Lorentzian functions). This interpretation is supported by the computer simulations, i.e., by the linear scaling of intermediate scattering function in the log–log representation over several decades which is indicative of a single stretched exponential.
We further tested the hypothesis of Lorentzian vs. KWW function in the experiment by using different instrumental resolutions. If the processes fitted by the 2 Lorentzian peak broadenings were actual physical processes, their corresponding width should be constant and they should be independent of the instrumentally available energy window. However, each energy resolution was well fit by a narrow and a broad Lorentzian peak, with no correlation between the broad peaks at different resolutions. This is strong evidence that the broad Lorentzian is likely a fitting artefact in the case of hydration water. We can, of course, not make a statement about lipid dynamics as this was not part of the current study.
Measurements at different hydrations could possibly be used to distinguish between Lorentzian and KWW models in QENS experiments in the future. One may expect that de-hydration leads to a more heterogeneous environment of water molecules and correspondingly smaller values of β, possibly up to the point where Lorentzians no longer describe the data.
4 Conclusion
Diffusion of hydration water molecules in hydrated phospholipid bilayers was studied using 2-dimensional incoherent quasi-elastic neutron scattering combined with computer modelling. By aligning the membranes horizontally in the spectrometer, lateral and transverse molecular motions can be studied separately, but simultaneously, by analyzing different positions on the 2-dimensional detector array. The dynamics were described in the energy domain by KWW functions, corresponding to stretched exponentials in the time domain.The nanoscale dynamics of hydration water molecules is anisotropic, and shows a sub-diffusive behaviour on nanometer length scales. As a first origin of the anomalous character of hydration water we have identified the intrinsic sub-diffusion of water at picosecond time scales, as also observed in bulk water. Secondly, a dynamically heterogeneous environment causes a distribution of relaxation times that finally appear as a sub-diffusive motion. These 2-dimensional data show a distinct regime of very slow dynamics at small Q-values, related to the geometry of the stacked bilayers used for the experiment.
Diffusion experiments using 2-dimensional detectors will in the future be used to precisely measure penetration and permeability of water molecules across bilayers as a function of bilayer composition,40,73 and to study the interaction of water with membrane inclusions, such as proteins and peptides.
Acknowledgements
Experiments were funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada, the National Research Council (NRC), the Canada Foundation for Innovation (CFI), and the Ontario Ministry of Economic Development and Innovation. L.T. is the recipient of an NSERC Canada Graduate Scholarship, M.C.R. is the recipient of an Early Researcher Award from the Province of Ontario. H.R.G. was supported by the United States DOE, Office of Basic Energy Sciences under Contract No. 46680. We profited from discussions with Jean-Louis Barrat (Grenoble) on the general interpretation.References
- M. Guo, A. J. Ehrlicher, M. H. Jensen, M. Renz, J. R. Moore, R. D. Goldman, J. Lippincott-Schwartz, F. C. Mackintosh and D. A. Weitz, Probing the stochastic, motor-driven properties of the cytoplasm using force spectrum microscopy, Cell, 2014, 158, 822–832 CrossRef CAS PubMed.
- F. Höfling and T. Franosch, Anomalous transport in the crowded world of biological cells, Rep. Prog. Phys., 2013, 76, 046602 CrossRef PubMed.
- D. S. Banks and C. Fradin, Anomalous diffusion of proteins due to molecular crowding, Biophys. J., 2005, 89, 2960–2971 CrossRef CAS PubMed.
- F. Roosen-Runge, M. Hennig, F. Zhang, R. M. J. Jacobs, M. Sztucki, H. Schober, T. Seydel and F. Schreiber, Protein self-diffusion in crowded solutions, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11815 CrossRef CAS PubMed.
- C. L. Armstrong, M. Trapp, J. Peters, T. Seydel and M. C. Rheinstädter, Short range ballistic motion in fluid lipid bilayers studied by quasi-elastic neutron scattering, Soft Matter, 2011, 7, 8358–8362 RSC.
- C. L. Armstrong, M. D. Kaye, M. Zamponi, E. Mamontov, M. Tyagi, T. Jenkins and M. C. Rheinstädter, Diffusion in single solid supported lipid bilayers studied by quasi-elastic neutron scattering, Soft Matter, 2010, 6, 5864–5867 RSC.
- S. Busch, C. Smuda, L. C. Pardo and T. Unruh, Molecular mechanism of long-range diffusion in phospholipid membranes studied by quasielastic neutron scattering, J. Am. Chem. Soc., 2010, 132, 3232–3233 CrossRef CAS PubMed.
- E. Flenner, J. Das, M. C. Rheinstädter and I. Kosztin, Sub-diffusion and lateral diffusion coefficient of lipid atoms and molecules in phospholipid bilayers, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 011907 CrossRef PubMed.
- G. R. Kneller, K. Baczynski and M. Pasenkiewicz-Gierula, Communication: consistent picture of lateral subdiffusion in lipid bilayers: molecular dynamics simulation and exact results, J. Chem. Phys., 2011, 135, 141105 CrossRef PubMed.
- J.-H. Jeon, H. M.-S. Monne, M. Javanainen and R. Metzler, Anomalous diffusion of phospholipids and cholesterols in a lipid bilayer and its origins, Phys. Rev. Lett., 2012, 109, 188103 CrossRef PubMed.
- P. Schwille, J. Korlach and W. W. Webb, Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes, Cytometry, 1999, 36, 176–182 CrossRef CAS PubMed.
- D. S. Martin, M. B. Forstner and J. A. Käs, Apparent subdiffusion inherent to single particle tracking, Biophys. J., 2002, 83, 2109–2117 CrossRef PubMed.
- S. König, W. Pfeiffer, T. Bayerl, D. Richter and E. Sackmann, Molecular dynamics of lipid bilayers studied by incoherent quasi-elastic neutron scattering, J. Phys. II, 1992, 2, 1589–1615 CrossRef.
- S. König, E. Sackmann, D. Richter, R. Zorn, C. Carlile and T. M. Bayerl, Molecular dynamics of water in oriented dppc multilayers studied by quasielastic neutron scattering and deuterium-nuclear magnetic resonance relaxation, J. Chem. Phys., 1994, 100, 3307–3316 CrossRef.
- S. König, T. M. Bayerl, G. Coddens, D. Richter and E. Sackmann, Hydration dependence of chain dynamics and local diffusion in l-alpha-dipalmitoylphosphatidylcholine multilayers studied by incoherent quasi-elastic neutron scattering, Biophys. J., 1995, 68, 1871–1880 CrossRef.
- W. Pfeiffer, Th. Henkel, E. Sackmann and W. Knorr, Local dynamics of lipid bilayers studied by incoherent quasi-elastic neutron scattering, Europhys. Lett., 1989, 8, 201–206 CrossRef CAS.
- W. Pfeiffer, S. König, J. F. Legrand, T. Bayerl, D. Richter and E. Sackmann, Neutron spin echo study of membrane undulations in lipid multibilayers, Europhys. Lett., 1993, 23, 457–462 CrossRef CAS.
- E. Falck, T. Róg, M. Karttunen and I. Vattulainen, Lateral diffusion in lipid membranes through collective flows, J. Am. Chem. Soc., 2008, 130, 44–45 CrossRef CAS PubMed.
- M. C. Rheinstädter, J. Das, E. J. Flenner, B. Brüning, T. Seydel and I. Kosztin, Motional coherence in fluid phospholipid membranes, Phys. Rev. Lett., 2008, 101, 248106 CrossRef PubMed.
- C. L. Armstrong, L. Toppozini, H. Dies, A. Faraone, M. Nagao and M. Rheinstädter, Incoherent neutron spin-echo spectroscopy as an option to study long-range lipid diffusion, ISRN Biophys., 2013, 2013, 9 Search PubMed.
- U. Wanderlingh, G. D'Angelo, C. Branca, V. C. Nibali, A. Trimarchi, S. Rifici, C. Finocchiaro, D. ad Crupi, J. Ollivier and H. D. Middendorf, Multi-component modeling of quasielastic neutron scattering from phospholipid membranes, J. Chem. Phys., 2014, 140, 174901 CrossRef CAS PubMed.
- S. R. Wassall, Pulsed field gradient-spin echo NMR studies of water diffusion in a phospholipid model membrane, Biophys. J., 1996, 71, 2724 CrossRef CAS PubMed.
- M. Weik, U. Lehnert and G. Zaccai, Liquid-like water confined in stacks of biological membranes at 200 K and its relation to protein dynamics, Biophys. J., 2005, 89, 3639–3646 CrossRef CAS PubMed.
- L. Toppozini, C. L. Armstrong, M. D. Kaye, M. Tyagi, T. Jenkins and M. Rheinstädter, Hydration water freezing in single supported lipid bilayers, ISRN Biophys., 2012, 2012, 520307 Search PubMed.
- M. C. Rheinstädter, T. Seydel, F. Demmel and T. Salditt, Molecular motions in lipid bilayers studied by the neutron backscattering technique, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 061908 CrossRef PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Slaving: solvent fluctuations dominate protein dynamics and functions, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047–16051 CrossRef CAS PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and R. D. Young, Bulk-solvent and hydration-shell fluctuations, similar to α- and β-fluctuations in glasses, control protein motions and functions, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14408–14413 CrossRef CAS PubMed.
- H. Frauenfelder, P. W. Fenimore, G. Chen and B. H. McMahon, Protein folding is slaved to solvent motions, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15469–15472 CrossRef CAS PubMed.
- G. Zaccai, The effect of water on protein dynamics, Philos. Trans. R. Soc., B, 2004, 359, 1269–1275 CrossRef CAS PubMed.
- O. Kel, A. Tamimi, M. C. Thielges and M. D. Fayer, Ultrafast structural dynamics inside planar phospholipid multibilayer model cell membranes measured with 2D IR spectroscopy, J. Am. Chem. Soc., 2013, 135, 11063–11074 CrossRef CAS PubMed.
- K. Wood, M. Plazanet, F. Gabel, B. Kessler, D. Oesterhelt, D. J. Tobias, G. Zaccai and M. Weik, Coupling of protein and hydration-water dynamics in biological membranes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18049–18054 CrossRef CAS PubMed.
- Y. von Hansen, S. Gekle and R. R. Netz, Anomalous anisotropic diffusion dynamics of hydration water at lipid membranes, Phys. Rev. Lett., 2013, 111, 118103 CrossRef PubMed.
- F. Y. Hansen, G. H. Peters, H. Taub and A. Miskowiec, Diffusion of water and selected atoms in DMPC lipid bilayer membranes, J. Chem. Phys., 2012, 137, 204910 CrossRef CAS PubMed.
- P. Wästerby, G. Orädd and G. Lindblom, Anisotropic water diffusion in macroscopically oriented lipid bilayers studied by pulsed magnetic field gradient NMR, J. Magn. Reson., 2002, 157, 156–159 CrossRef.
- A. Ghosh, R. Kramer Campen, M. Sovago and M. Bonn, Structure and dynamics of interfacial water in model lung surfactants, Faraday Discuss., 2009, 141, 145–159 RSC.
- L. Piatkowski, J. de Heij and H. J. Bakker, Probing the distribution of water molecules hydrating lipid membranes with ultrafast förster vibrational energy transfer, J. Phys. Chem. B, 2013, 117, 1367–1377 CrossRef CAS PubMed.
- S. M. Gruenbaum and J. L. Skinner, Vibrational spectroscopy of water in hydrated lipid multi-bilayers. iii. water clustering and vibrational energy transfer, J. Chem. Phys., 2013, 139, 175103 CrossRef CAS PubMed.
- J. Andrasko and S. Forsén, NMR study of rapid water diffusion across lipid bilayers in dipalmitoyl lecithin vesicles, Biochem. Biophys. Res. Commun., 1974, 60, 813–819 CrossRef CAS PubMed.
- S.-J. Marrink and H. J. C. Berendsen, Simulation of water transport through a lipid membrane, J. Phys. Chem., 1994, 98, 4155–4168 CrossRef CAS.
- M. D. Kaye, K. Schmalzl, V. C. Nibali, M. Tarek and M. C. Rheinstädter, Ethanol enhances collective dynamics of lipid membranes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 050907 CrossRef PubMed.
- J. R. P. Webster, S. Langridge, R. M. Dalgliesh and T. R. Charlton, Reflectometry techniques on the second target station at ISIS: methods and science, Eur. Phys. J. Plus, 2011, 126, 1–5 Search PubMed.
- R. Dalgliesh, S. Langridge, J. Plomp, V. O. de Haan and A. A. van Well, Offspec, the ISIS spin-echo reflectometer, Phys. B, 2011, 406, 2346–2349 CrossRef CAS.
- M. A. Barrett, S. Zheng, G. Roshankar, R. J. Alsop, R. K. R. Belanger, C. Huynh, N. Kučerka and M. C. Rheinstädter, Interaction of aspirin (acetylsalicylic acid) with lipid membranes, PLoS One, 2012, 7, e34357 CrossRef CAS PubMed.
- M. A. Barrett, S. Zheng, L. A. Toppozini, R. J. Alsop, H. Dies, A. Wang, N. Jago, M. Moore and M. C. Rheinstädter, Solubility of cholesterol in lipid membranes and the formation of immiscible cholesterol plaques at high cholesterol concentrations, Soft Matter, 2013, 9, 9342–9351 RSC.
- R. J. Alsop, M. A. Barrett, S. Zheng, H. Dies and M. C. Rheinstädter, Acetylsalicylic acid (ASA) increases the solubility of cholesterol when incorporated in lipid membranes, Soft Matter, 2014, 10, 4275–4286 RSC.
- R. J. Alsop, C. Armstrong, A. Maqbool, L. Toppozini, H. Dies and M. C. Rheinstadter, Cholesterol expels ibuprofen from the hydrophobic membrane core and stabilizes lamellar phases in lipid membranes containing ibuprofen, Soft Matter, 2015, 11, 4756–4767 RSC.
- K. Hristova and S. H. White, Determination of the hydrocarbon core structure of fluid dioleoylphosphocholine (dopc) bilayers by x-ray diffraction using specific bromination of the double-bonds: effect of hydration, Biophys. J., 1998, 74, 2419–2433 CrossRef CAS PubMed.
- S. Tristram-Nagle, Y. Liu, J. Legleiter and J. F. Nagle, Structure of gel phase DMPC determined by X-ray diffraction, Biophys. J., 2002, 83, 3324–3335 CrossRef CAS PubMed.
- J. F. Nagle and M. C. Wiener, Relations for lipid bilayers, Biophys. J., 1989, 55, 309–313 CrossRef CAS PubMed.
- J. F. Nagle, R. Zhang, S. Tristram-Nagle, W. Sun, H. I. Petrache and R. M. Suter, X-ray structure determination of fully hydrated lα phase dipalmitoylphosphatidylcholine bilayers, Biophys. J., 1996, 70, 1419–1431 CrossRef CAS PubMed.
- N. Kučerka, Y. Liu, N. Chu, H. I. Petrache, S. Tristram-Nagle and J. F. Nagle, Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles, Biophys. J., 2005, 88, 2626–2637 CrossRef PubMed.
- R. I. Bewley, J. W. Taylor and S. M. Bennington, Let, a cold neutron multi-disk chopper spectrometer at ISIS, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 637, 128–134 CrossRef CAS.
- R. I. Bewley, J. W. Taylor and S. M. Bennington, Let: a low energy multiple chopper spectrometer at ISIS, Notiziario Neutroni e Luce di Sincrotrone, 2011, 16, 4–13 Search PubMed.
- R. T. Azuah, L. R. Kneller, Y. Qiu, P. L. W. Tregenna-Piggott, C. M. Brown, J. R. D. Copley and R. M. Dimeo, Dave: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data, J. Res. Natl. Inst. Stand. Technol., 2009, 114, 341–358 CrossRef CAS PubMed.
- M. Bée, Quasielastic Neutron Scattering: Principles and Applications in Solid State Chemistry, Biology and Materials Science, Taylor & Francis, 1988 Search PubMed.
- J. Hansen and I. McDonald, Theory of Simple Liquids, Elsevier, Oxford, 2006 Search PubMed.
- D. Vural and H. R. Glyde, Intrinsic mean-square displacements in proteins, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 011926 CrossRef PubMed.
- D. Vural, L. Hong, J. C. Smith and H. R. Glyde, Long-time mean-square displacements in proteins, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 052706 CrossRef PubMed.
- G. Williams and D. C. Watts, Non-symmetrical dielectric relaxation behaviour arising from a simple empirical decay function, Trans. Faraday Soc., 1970, 66, 80–85 RSC.
- C. P. Lindsey and G. D. Patterson, Detailed comparison of the Williams–Watts and Cole–Davidson functions, J. Chem. Phys., 1980, 73, 3348 CrossRef CAS.
- J. R. Taylor, A Introduction to Error Analysis: the study of uncertainties in physical measurements, University Science Books, 1997 Search PubMed.
- D. Vural, L. Hong, J. C. Smith and H. R. Glyde, Motional displacements in proteins: the origin of wave-vector-dependent values, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 052705 CrossRef PubMed.
- J. M. Sancho, Brownian colloidal particles: ITO, stratonovich, or a different stochastic interpretation, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 062102 CrossRef CAS PubMed.
- N. Kučerka, J. F. Nagle, J. N. Sachs, S. E. Feller, J. Pencer, A. Jackson and J. Katsaras, Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data, Biophys. J., 2008, 95, 2356–2367 CrossRef PubMed.
- H. Petrache, S. Feller and J. Nagle, Determination of component volumes of lipid bilayers from simulations, Biophys. J., 1997, 70, 2237–2242 CrossRef.
- J. F. Nagle and S. Tristram-Nagle, Structure of lipid bilayers, Biochim. Biophys. Acta, 1469, 2000, 159–195 Search PubMed.
- J. Yang, C. Calero and J. Marti, Diffusion and spectroscopy of water and lipids in fully hydrated dimyristoylphosphatidylcholine bilayer membranes, J. Chem. Phys., 2014, 140, 104901 CrossRef CAS PubMed.
- F. Sciortino, P. Gallo, P. Tartaglia and S.-H. Chen, Supercooled water and the kinetic glass transition, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 54, 6331–6343 CrossRef CAS.
- F. Y. Chen, W. C. Hung and H. W. Huang, Critical swelling of phospholipid bilayers, Phys. Rev. Lett., 1997, 79, 4026–4029 CrossRef CAS.
- S.-H. Chen, C. Liao, F. Sciortino, P. Gallo and P. Tartaglia, Model for single-particle dynamics in supercooled water, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 59, 6708–6714 CrossRef CAS.
- V. Crupi, D. Majolino, P. Migliardo and V. Venuti, Diffusive relaxations and vibrational properties of water and h-bonded systems in confined state by neutrons and light scattering: state of the art, J. Phys. Chem. A, 2000, 104, 11000–11012 CrossRef CAS.
- V. Crupi, D. Majonlino, P. Migliardo and V. Venuti, Neutron scattering study and dynamic properties of hydrogen-bond liquids in mesoscopic confinement. 1. The water case, J. Phys. Chem. B, 2002, 106, 10884–10894 CrossRef CAS.
- L. Toppozini, C. L. Armstrong, M. A. Barrett, S. Zheng, L. Luo, H. Nanda, V. Garcia Sakai and M. Rheinstädter, Partitioning of ethanol into lipid membranes and its effect on fluidity and permeability as seen by X-ray and neutron scattering, Soft Matter, 2012, 8, 11839–11849 RSC.
Footnote |
| † 87.16.D-, 87.15.Vv, 83.85.Hf, 87.15.A- |
| This journal is © The Royal Society of Chemistry 2015 |