
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Grafted biomembranes containing membrane proteins – the case of the leucine transporter†
Vivien
Jagalski‡
ab,
Robert D.
Barker‡
*c,
Mikkel B.
Thygesen
b,
Kamil
Gotfryd§
d,
Mie B.
Krüger
d,
Lei
Shi
e,
Selma
Maric¶
f,
Nicolas
Bovet
a,
Martine
Moulin
c,
Michael
Haertlein
c,
Thomas
Günther Pomorski
f,
Claus J.
Loland
d and
Marité
Cárdenas
*ag
aDepartment of Chemistry and Nano-Science Center, University of Copenhagen, Universitetsparken 5, DK 2100, Copenhagen, Denmark. E-mail: cardenas@nano.ku.dk
bDepartment of Chemistry and Nano-Science Center, University of Copenhagen, Thorvaldsensvej 40, DK-1871 Frederiksberg C, Denmark
cInstitute Laue Langevin, 71 avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France. E-mail: barker@ill.eu
dDepartment of Neuroscience and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, DK-2200, Copenhagen, Denmark
eDepartment of Physiology and Biophysics and Institute for Computational Biomedicine, Weill Cornell Medical College, New York 10065, USA
fCentre for Membrane Pumps in Cells and Disease (PUMPKIN), Department of Plant and Environmental Sciences, University of Copenhagen, Thorvaldsensvej 40, DK-1871 Frederiksberg C, Denmark
gMalmo University, Biofilm – Research Center for Biointerfaces and Department of Biomedical Science, Health & Society, 20506 Malmoe, Sweden. E-mail: marite.cardenas@mah.se
First published on 20th August 2015
Abstract
Here, we bind the sodium dependent amino acid transporter on nitrilotriacetic acid/polyethylene glycol functionalized gold sensors in detergents and perform a detergent–lipid exchange with phosphatidylcholine. We characterize the LeuT structure in the adsorbed film by magnetic contrast neutron reflection using the predicted model from molecular dynamic simulations.
Today, novel methodologies to study membrane proteins are much sought to develop biotechnological sensing applications for diagnostics. This is mainly due to the highly amphipathic character of membrane proteins that turns them into biomolecules that are extremely difficult to study. This is demonstrated by the fact that only ∼1% of the known total protein structures (101, 218 protein crystal structure appeared on the protein data bank on May 20th, 2015) belong to the membrane protein category, yet they are among the most important drug targets and potential biomarkers. In this communication we outline how we control membrane protein tethering on surfaces specifically by using self-assembled monolayers (SAMs) made from a nitrilotriacetic acid/polyethylene glycol (NTA–PEG) anchor and a polyethylene glycol (PEG) spacer (Fig. 1A).
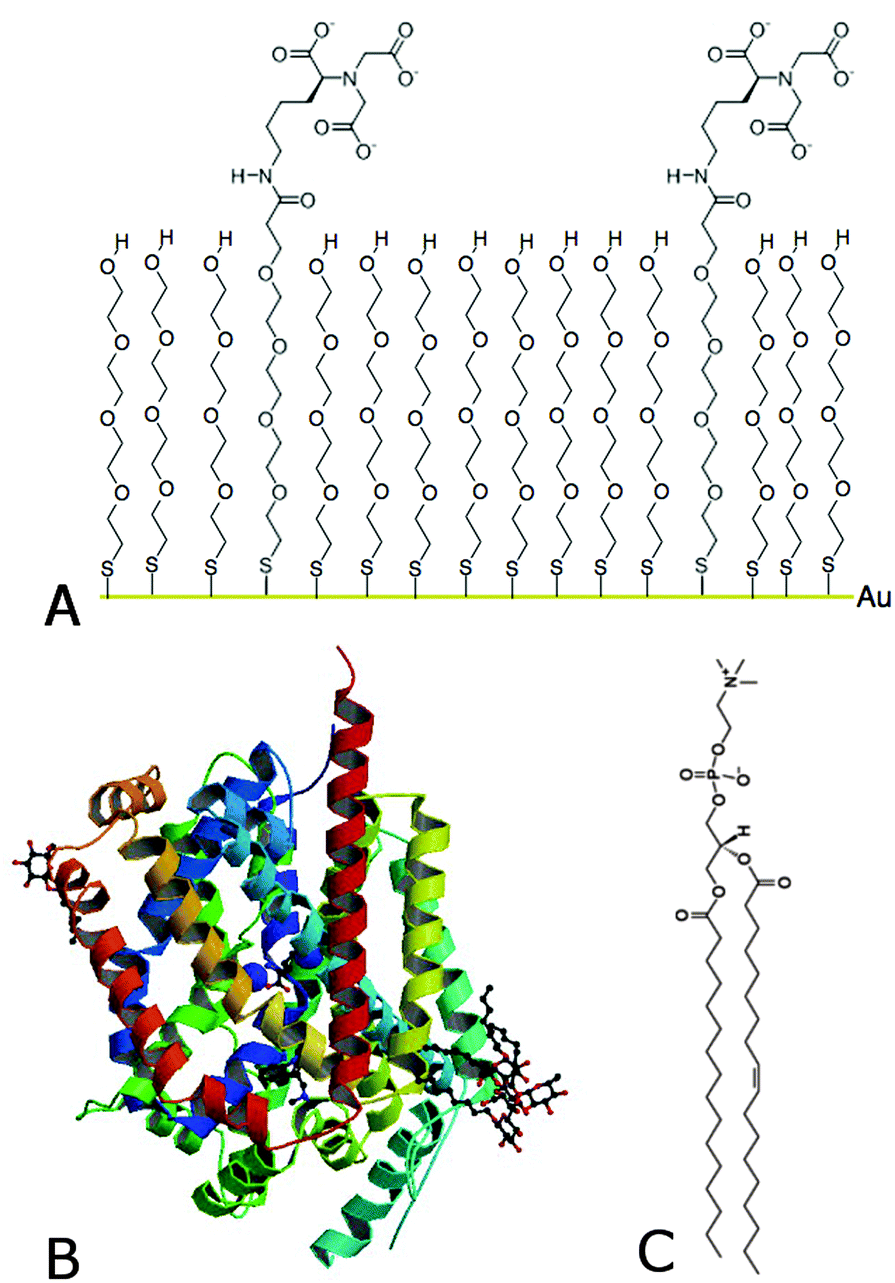 | ||
Fig. 1 Schematic drawing and structure of (A) NTA–PEG (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) SAM on a gold surface via a covalently bound sulfur atom, (B) membrane protein LeuT1,2 and (C) the lipid POPC. :9) SAM on a gold surface via a covalently bound sulfur atom, (B) membrane protein LeuT1,2 and (C) the lipid POPC. | ||
We describe the reconstitution of a lipid environment surrounding a neurotransmitter:sodium symporter (NSS) family member, the hydrophobic amino acid transporter, LeuT1,2 (Fig. 1B), on surfaces. LeuT is a secondary active transporter utilizing the sodium gradient across membranes to drive the transport of the solute against its chemical gradient. Crystal structures of LeuT in detergents showed a compact protein with twelve transmembrane α-helices1 while molecular dynamics (MD) simulations predict that this conformation is preserved in a native-like environment.3 On the other hand, protein adsorption to surfaces constitutes a robust approach for sensing applications. However, protein binding at surfaces may induce changes in the protein structure and thus affects the protein function.4 Particularly, membrane protein tethering and the reconstitution of a native-like lipid environment on surfaces represent a major challenge. This is an important step though for fully assessing membrane protein function. There are many strategies for selective protein tethering on surfaces that include irreversible binding (covalent binding via sulfur bridges in cysteine groups to Au substrates5), and reversible binding through (1) the specific avidin–streptavidin binding6 or (2) chelators such as complexes between the divalent metal ions and nitrilotriacetic acid (NTA)7 and others.8 The high affinity (nM range) his-tag–metal–NTA bonds can be easily activated with Cu+2 or Ni+2, for example,7 while the protein can be eluted with competing chelating agents such as EDTA or imidazole.9 However, a major limitation is the high percentage of non-specific binding on these surfaces, where electrostatic and van der Waals forces play an important role. The high affinity of the metal–NTA technology together with the protein-repelling properties of PEG10 constitutes a valid approach to minimize non-specific protein adsorption to surfaces.11,12
In this communication we show that NTA–PEG coated Au surfaces can be used to promote the specific immobilization of LeuT in detergents. Moreover, our neutron reflectivity (NR) data confirm the importance of the detergent choice for effective lipid replacement and we present a method that exploits trajectories from MD simulations to analyze NR data using only three parameters. For this we used previously published MD simulations of LeuT in POPC under similar conditions to this work.3 We synthesized NTA–PEG and PEG thiols using a similar procedure to that reported earlier12 and produced mixed NTA–PEG SAM by exposing Au coated silica crystals to 1 mM thiol solutions containing different molar proportions of NTA–PEG anchor and PEG thiols in ethanol (Experimental details for synthesis and SAM formation are found in the SI). High resolution X-ray photoelectron spectroscopy (XPS) data confirmed the quality and determined the ratio of NTA–PEG and PEG on the surface (ESI,† Fig. S1), which showed a higher NTA–PEG content than that of the nominal mixture. We then determined the optimal content of NTA–PEG linkers within the SAM in order to obtain minimal non-specific protein binding. This was done by measuring quartz crystal microbalance with dissipation (QCM-D) signals before and after imidazole and detergent washes of pre-adsorbed LeuT in detergents (ESI,† Fig. S2 and S3) as well as for the soluble bovine serum albumin that is often used to assess non-specific binding on surfaces13 (ESI,† Fig. S4). As for previous results,5,12 non-specific binding increased with the NTA–PEG linker content until non-specific binding dominates protein binding to pure NTA–PEG SAM. We observed that 10% NTA–PEG and 90% PEG SAM gave similar non-specific binding to that of a SAM lacking the NTA–PEG linker. Thus, we continued with this NTA–PEG composition in all following experiments. Interestingly, a non-linear relationship is found between the adsorbed amount of protein and the NTA–PEG content (ESI,† Fig. S3 and S4) suggesting that the NTA–PEG anchor preferentially binds the Au surface in agreement with our XPS results.
We then proceeded to replace the detergent with lipids using the Tiberg (or mixed lipid/detergent micelle) approach,14 which is based on the higher aqueous solubility of the detergent with respect to the lipid. This method was proposed earlier for biomembrane reconstitution of a membrane protein immobilized via the Ni-NTA technology on non-PEGylated linkers.15 Initially, two different detergents, n-dodecyl β-D-maltopyranoside (DDM) or maltose-neopentyl glycol (MNG-3), were used for the preparation of LeuT. Both detergents are known to preserve the stability and activity of LeuT.16 QCM-D was used to follow the changes in frequency and dissipation as a function of time and in this way follow the LeuT immobilization process and the reconstitution of a lipid environment in situ (Fig. 2). For adsorption processes in general, the QCM-D signals typically show a decrease in frequency and an increase in dissipation while the opposite is true for desorption processes. First, the detergent was introduced to the liquid flow cell to avoid the dilution of the detergent upon addition of LeuT leading to reversible detergent adsorption (ESI,† Fig. S5). Secondly, LeuT in detergents was added and, as expected, protein binding occurred within minutes. The QCM-D data show an interfacial process in which mass adsorption is followed by desorption prior to stabilization, and the extent of excess adsorption seems to depend on the detergent choice. This signal is a signature for the adsorbed layer formation from soft self-assembled aggregates where adsorption is accompanied by the release of water from aggregates.17 The equilibrium QCM-D signal, on the other hand, did not depend on the detergent type. Moreover, controls show minimal protein binding on pure PEG surfaces or detergent binding on NTA–PEG surfaces (ESI,† Fig. S3 and S5).
 | ||
| Fig. 2 QCM-D frequency and dissipation of overtone 7 for LeuT adsorption in detergents (blue trace: DDM; black trace: MNG-3) onto a NTA–PEG SAM at 14 °C. The NTA–PEG SAM was first equilibrated in a detergent-free buffer. Then, the detergent containing buffer was flushed to the surface followed by LeuT in a detergent buffer. Next, the surface was flushed with a detergent containing buffer to remove all unbound protein from the liquid flow cell. Then, POPC was added to the detergent (molar ratio detergent:POPC of 5:1) followed by a final rinse (30 min at 10 μL min−1) with a detergent and lipid free buffer. During the membrane re-lipidation, significant changes in the QCM-D signals were observed upon lipid addition and detergent removal for DDM only. | ||
Subsequently, we removed LeuT from the bulk solution by flushing with a detergent-rich buffer followed by rinsing with a buffer containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) in detergents at a molar ratio of 1:5. The change in frequency and dissipation was in this case detergent type dependent: a slight decrease in the frequency and constant dissipation was observed for LeuT in MNG-3 while a considerable decrease in frequency and an increase in dissipation occurred for DDM. Moreover, a major increase in frequency occurred upon rinsing with a detergent-free buffer suggesting a mass loss when DDM was used. Thus, POPC clearly induces changes in the structure of the LeuT–detergent layer for DDM only. This dramatic change may be a consequence of restructuring and decoupling (swelling) the adsorbed layer from the surface that leads to (i) water incorporation on the surface upon addition of lipids and (ii) lipid/detergent when diluting DDM. Since the QCM-D signals remained stable upon POPC addition and after MNG-3 removal by rinsing with a detergent-free buffer, we conclude that the use of MNG-3 is key to stabilize the adsorbed LeuT–lipid layer. This is, to our knowledge, the first reported study to demonstrate that an adsorbed LeuT–lipid layer can be stabilized by the choice of the detergent.
To establish whether this layer stabilization is due to lipid–detergent exchange on the surface, we performed magnetic contrast neutron reflection (NR) using a polarized neutron beam and D2O-matched phosphatidylcholine (dPC).18 In this way, the lipid–detergent exchange process could be followed due to the isotopic contrast between the hydrogen-rich detergent and the deuterium-rich PC. We monitored the reflectivity during incubation with both dPC and MNG-3 (ESI,† Fig. S6), and after extensive rinsing with a detergent free buffer (Fig. 3A). The NR data showed a shift in the positioning of the fringe minimum towards a higher momentum transfer vector (Q) and a slight decrease in the reflectivity around this minimum. Upon rinsing with a detergent-free solution, there was no further change in the reflection profile even after 4 h of rinsing at 20 μL min−1 (ESI,† Fig. S6). This suggests that dPC exchanges the detergent in the presence of MNG-3. Moreover, the data suggest that the lipid–detergent exchange occurs within the first 10 min of incubation (duration of the first measurement) as no more changes were observed even after 4 h rinsing with detergent free buffer. This is to our knowledge the first time that lipids are shown to effectively exchange detergents surrounding a membrane protein prior to rinsing with a detergent free buffer. We cannot though – at this stage – completely disregard the presence of some hydrogenated detergent in the adsorbed layer.
 | ||
| Fig. 3 Magnetic contrast neutron reflection for adsorbed layers of LeuT in detergents or lipids. (A) NR profiles for the intermediate Q-range in detergents and after addition of dPC and extensive rinsing (4 h at 20 μL min−1) with a detergent-free buffer. (B) NR profiles (symbols) and best fits (lines) for LeuT–dPC membranes after removal of MNG-3 by extensive rinsing in D2O based buffer (circles) and H2O based buffer (crosses). (C) Scattering length density profiles for the best fit obtained by applying the density profiles obtained by MD simulations combined with a pre-fitted substrate. | ||
We then measured NR profiles after the lipid exchange with surfactant-free H2O and a D2O-based buffer over a complete Q-range (Fig. 3B). Due to the high complexity of the interfacial structure, we created a combined scattering length density (SLD) profile for the whole interface by taking the SLD obtained using the traditional box model to fit the substrate (NTA–PEG–Au–Ni/Fe–SiO2–Si, ESI,† Fig. S1) added to the density profiles obtained from atomistic MD simulations of LeuT in POPC (biomembrane) under similar conditions.3 The detailed MD trajectories used for this analysis were extracted from simulations published earlier.3 This composite modeling approach allowed the molecular structure of the protein in the adsorbed layer to be used to constrain our fit to the NR data. By separating the lipid and solvent densities from the protein (normalized by their volume and number ratio) from the MD simulations we could analyze the NR data using only three parameters: the biomembrane coverage, number of lipids per protein and the water ratio in the biomembrane. Best fits were obtained for 45% biomembrane area coverage composed of 83 lipids per LeuT molecule, and by increasing the water contribution by a factor of 2.5. The latter affects mainly the headgroup region as the water contribution in the lipid core is close to zero.3 The outstanding quality of the fit (Fig. 3B) suggests that the protein retains the conformation predicted by the MD simulation. Such fit quality could not be obtained using a standard box approach (ESI,† Fig. S7).
The fitted number of lipids per protein is low. However, the higher water content (suggested by the increased water contribution) around the intra- and extracellular parts of the protein can in part compensate for this lower ratio. In fact, 52 ± 3 lipids/LeuT are necessary to cover one molecular shell of lipids around the LeuT as calculated by using a 3.9 Å cut off for the C–C distance in the MD simulations.3 Thus, the adsorbed layer (with ∼45% surface coverage) is probably mainly composed of LeuT surrounded by smaller lipid patches on the surface around the active NTA–PEG sites. An alternative approach to create a continuous biomembrane is to include lipids that can bind to the NTA–PEG anchor as the protein does.19
Finally, we compared the expected (simulated) NR profiles for LeuT with and without bound leucine as predicted by the MD simulations published earlier3,20,21 (ESI,† Fig. S8). The modeled differences in NR profiles between these two states are at the limits of detection, but could be distinguished by following the changes in spin-asymmetry using polarized NR, combined with specific deuteration of the protein to offer additional contrast. Furthermore, the change in LeuT conformation upon leucine binding needs to be produced experimentally by sodium-mediated ligand transport (and thus requires the formation of a tight biomembrane) or via the use of a binding site mutant that does not bind leucine.
Here, we have shown for the first time that it is possible to constrain the analysis of NR data more realistically, with fewer free parameters, by parameterizing the density profiles obtained for membrane protein containing biomembranes from atomistic MD simulations (rather than forcing a structural model using boxes/layers22,23). Previously, density profiles from MD simulations of lipid monolayers were compared to those fitted to NR data.24
Experimental section
Neutron reflectivity (NR)
In a specular (mirror like) NR experiment, the intensity of reflected neutrons (R) is measured as a function of Q perpendicular to the interface25,26 giving the averaged composition and structure of an interface (perpendicular to the surface). The reflectivity profiles for the substrates were analyzed by fitting a simulated reflectivity curve of a model structure of the system to the experimental data using the software Motofit.27 The density profiles were extracted by MD simulations performed on the same system using the VMD plugin28 and combined with the characterized lower surface layers using custom procedures within the RasCAL29 software (see ESI,† for further details). Polarized NR experiments were performed on the POLREF instrument at ISIS, Oxfordshire, UK. The Au coated Si-permalloy surfaces were first characterized before exposure over 6 h to NTA-PEG (10 mol%) and PEG thiol (90 mol%) solutions to a total concentration of 150 mM in ethanol, rinsed in ethanol, MilliQ water, CuSO4 solution, and 50 mM Tris buffer pH 8 containing 199 mM KCl and 1 mM NaCl prior to characterization of the NTA–PEG layer in D2O and H2O contrast. LeuT (300 nM) was then injected in MNG-3 (0.05 wt%) containing buffer and characterized by NR. Then, 0.0083 mol% lipids in MNG-3 containing buffer were added and NR collected prior to rinsing at 100 μL min−1 with a detergent-free buffer. The final NR characterization was performed both in D2O and H2O rich buffer solutions. QCM-D experiments were performed using the same detergent, lipid and protein concentrations.Quartz crystal microbalance with dissipation monitoring (QCM-D)
A Q-Sense E4 system was used. Au sensors were modified with NTA-PEG SAMs as described in SI. Sensors were dried under nitrogen flow and mounted into the flow cells. Then they were equilibrated in water until a stable signal was observed prior to use. The flow rate was maintained throughout the whole experiment at 50 μL min−1 until the point of detergent dilution where the flow rate was reduced to 10 μL min−1. Experiments were performed at 20 °C.Conclusions
In this communication we have demonstrated that (1) the choice of detergent is crucial for the stabilization of a LeuT–lipid layer on surfaces, (2) lipids effectively exchange with detergents in a LeuT adsorbed layer prior to detergent removal, with no major changes in the adsorbed biomembrane structure taking place upon detergent removal, and (3) the number of free parameters for fitting NR data can be reduced by parameterizing the density profiles obtained from atomistic MD simulations. This approach opens up many possibilities to model complex biointerfaces including transmembrane and membrane bound proteins. For example, the NTA–PEG approach could be used as an alternative for larger membrane bound proteins such as the CYP450s that have been shown to change their conformational equilibration in nanodisc films using NR.30Acknowledgements
We thank ISIS for beam time allocation and Timothy Chalton and Christy Kinane for local contact. We thank Thierry Bigault (ILL) for providing coating of our Si blocks and Andrew Wildes (ILL) for initial discussions. We gratefully acknowledge the access to the Deuteration Laboratory (D-Lab) PSB platform within ILL's Life Sciences Group. We acknowledge funding from the Swedish Research Council (MC), DANSCATT Centre (Danish government), the Research Centre ‘bioSYNergy’ funded by the UCPH Excellence Programme for Interdisciplinary Research and the Danish National Council for Independent Research – Sapere Aude program (CJL). VJ and MC also thank funding from Plant Power: Light-driven synthesis of complex terpenoids using cytochrome P450s (12-131834) funded by Innovation Fund Denmark (previously the Danish Council for Strategic Research, Programme Commission on Strategic Growth Technologies).Notes and references
- A. Yamashita, S. K. Sight, T. Kawate and E. Gouaux, Nature, 2005, 437, 215–223 CrossRef CAS PubMed.
- C. J. Loland, Biochim. Biophys. Acta, 2015, 1850, 500–510 CrossRef CAS PubMed.
- S. Stolzenberg, M. Quick, C. Zhao, K. Gotfryd, G. Khelashvili, U. Gether, C. J. Loland, J. A. Javitch, S. Noskov, H. Weinstein and L. Shi, J. Biol. Chem., 2015, 290, 13992–14003 CrossRef CAS PubMed.
- M. Wahlgren and T. Arnebrant, Trends Biotechnol., 1991, 9, 201–208 CrossRef CAS.
- Y. Okada, T. Y. Takano, N. Kobayashi, A. Hayashi, M. Yonekura, Y. Nishiyama, T. Abe, T. Yoshida, T. A. Yamamoto, S. Seino and T. Doi, Bioconjugate Chem., 2011, 22, 887–893 CrossRef CAS PubMed.
- P. M. Wolny, J. P. Spatz and R. P. Richter, Langmuir, 2010, 26, 1029–1034 CrossRef CAS PubMed.
- E. Hochuli, H. Dobei and A. Schacher, J. Chromatogr., 1987, 411, 177–184 CrossRef CAS.
- D. L. Johnson and L. L. Martin, J. Am. Chem. Soc., 2005, 127, 2018–2019 CrossRef CAS PubMed.
- J. Schmitt, H. Hess and H. G. Stunnenberg, Mol. Biol. Rep., 1993, 18, 223–230 CrossRef CAS.
- K. Reimhult, K. Petersson and A. Krozer, Langmuir, 2008, 24, 8695–8700 CrossRef CAS PubMed.
- G. B. Sigal, C. Bramdad, A. Barberis, A. Strominger and G. M. Whitesides, Anal. Chem., 1996, 68, 490–497 CrossRef CAS.
- A. Vaish, V. Silin, M. L. Walker, K. L. Steffens, S. Krueger, A. A. Yeliseev, K. Gawrisch and D. J. Vanderah, Chem. Commun., 2013, 49, 2685–2687 RSC.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS PubMed.
- F. Tiberg, I. Harwigsson and M. Malmsten, Eur. Biophys. J., 2000, 29, 196–203 CrossRef CAS.
- K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter and J. Heberle, J. Am. Chem. Soc., 2004, 126, 16199–16206 CrossRef CAS PubMed.
- P. S. Chae, S. G. F. Rasmussen, R. R. Rana, K. Gotfryd, R. Chandra, M. A. Goren, A. C. Kruse, S. Nurva, C. J. Loland, Y. Pierre, D. Drew, J.-L. Popot, D. Picot, B. G. Fox, L. Guan, U. Gether, B. Byrne, B. Kobilka and S. H. Gellman, Nat. Methods, 2010, 7, 1003–1008 CrossRef CAS PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- S. Maric, M. B. Thygesen, J. Schiller, M. Marek, M. Moulin, M. Haertlein, V. T. Forsyth, M. Bogdanov, W. Dowhan, L. Arleth and T. G. Pomorski, Appl. Microbiol. Biotechnol., 2015, 99, 241–254 CrossRef CAS PubMed.
- U. Rädler, J. Mack, N. Persike, G. Jung and R. Tampé, Biophys. J., 2000, 79, 3144–3152 CrossRef.
- D. P. Claxton, M. Quick, L. Shi, F. D. de Carvalho, H. Weinstein, J. A. Javitch and H. S. McHaourab, Nat. Struct. Mol. Biol., 2010, 17, 822–829 CAS.
- C. Zhao, S. Stolzenberg, L. Gracia, H. Weinstein, S. Noskov and L. Shi, Biophys. J., 2012, 103, 878–888 CrossRef CAS PubMed.
- J. C. Karst, R. Barker, U. Devi, M. J. Swann, M. Davi, S. J. Roser, D. Ladant and A. Chenal, J. Biol. Chem., 2012, 287, 9200–9212 CrossRef CAS PubMed.
- S. A. Holt, A. P. Le Brun, C. F. Majkrzak, D. J. McGillivray, F. Heinrich, M. Lösche and J. H. Lakey, Soft Matter, 2009, 5, 2576–2586 CAS.
- A. Dabkowska, L. E. Collins, D. J. Barlow, R. Barker, S. E. McLain, M. J. Lawrence and K. Lorenz, Langmuir, 2014, 30, 8803–8811 CrossRef CAS PubMed.
- G. Fragneto, Eur. Phys. J.: Spec. Top., 2012, 213, 327–342 CrossRef CAS PubMed.
- E. B. Watkins, R. D. Barker, S. Singh, M. J. Waltman, H. L. Smith, L. Pocivavsek and J. Majewski, Biointerphases, 2015, 10, 019014 CrossRef PubMed.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- T. Giorgino, http://multiscalelab.org/utilities/DensityProfileTool.
- A.V. Hughes, RasCAL, Sourceforge, 2013, downloaded from: http://sourceforge.net/projects/rscl/.
- M. Wadsater, T. Laursen, A. Singha, N. S. Hatzakis, D. Stamou, R. Barker, K. Mortensen, R. Feidenhans'l, B. L. Moller and M. Cardenas, J. Biol. Chem., 2012, 287, 34596–34603 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, synthesis of NTA–PEG thiols, formation and characterization of the NTA–PEG SAM, optimization of the NTA–PEG content for minimal non-specific protein binding and LeuT binding on inactivated NTA–PEG surfaces. See DOI: 10.1039/c5sm01490e |
| ‡ These authors contributed equally. |
| § Present address: Department of Biomedical Sciences Faculty of Health and Medical Sciences, University of Copenhagen, DK-2200, Copenhagen, Denmark. |
| ¶ Present address: Malmo University, Biofilm – Research Center for Biointerfaces and Department of Biomedical Science, Health & Society, 20506 Malmoe, Sweden. |
| This journal is © The Royal Society of Chemistry 2015 |