
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming inactivation of the lung surfactant by serum proteins: a potential role for fluorocarbons?
Marie Pierre
Krafft
Institut Charles Sadron (CNRS), University of Strasbourg, 23 rue du Loess, 67034 Strasbourg Cedex, France. E-mail: krafft@unistra.fr
First published on 18th June 2015
Abstract
In many pulmonary conditions serum proteins interfere with the normal adsorption of components of the lung surfactant to the surface of the alveoli, resulting in lung surfactant inactivation, with potentially serious untoward consequences. Here, we review the strategies that have recently been designed in order to counteract the biophysical mechanisms of inactivation of the surfactant. One approach includes protein analogues or peptides that mimic the native proteins responsible for innate resistance to inactivation. Another perspective uses water-soluble additives, such as electrolytes and hydrophilic polymers that are prone to enhance adsorption of phospholipids. An alternative, more recent approach consists of using fluorocarbons, that is, highly hydrophobic inert compounds that were investigated for partial liquid ventilation, that modify interfacial properties and can act as carriers of exogenous lung surfactant. The latter approach that allows fluidisation of phospholipid monolayers while maintaining capacity to reach near-zero surface tension definitely warrants further investigation.
1. Introduction
The native lung surfactant is a complex mixture comprising about 90% of lipids (mainly dipalmitoylphosphatidylcholine, DPPC) and about 10% of specific proteins (SP-A, SP-B, SP-C and SP-D) that forms a monolayer at the surface of the alveoli in mammals. This monolayer lowers the tension at the alveolus–air interface upon compression during expiration, thus reducing the work of breathing, and respreads quickly on expansion during inspiration. As the lung is ceaselessly submitted to compression–expansion cycles (∼15–20 cycles per minute) and undergoes a typical total volume variation of ∼20%, one easily understands that the viscoelastic properties of the monolayer of pulmonary surfactant is a critical determinant of the lung's functionality.There are many pulmonary diseases in which the lung surfactant is either absent or inactivated. The surfactant deficiency that results from lung immaturity in preterm babies leads to the neonatal respiratory distress syndrome (NRDS). In NRDS, the lack of surfactant results in progressive lung failure characterised by alveoli collapse (atelectasis), decreased lung compliance, systemic hypoxia and lung oedema. In new-borns, surfactant inactivation can be due to meconium inhalation immediately before or during delivery, as in the meconium aspiration syndrome (MAS). In children and adults, surfactant inactivation often occurs as a consequence of acute lung injury (ALI) and is a primary cause of acute respiratory distress syndrome (ARDS). ALI/ARDS may result from both direct lung injury (pneumonia, aspiration, drowning or toxic inhalation) and indirect lung injury (sepsis, nonthoracic trauma, multiple blood transfusions, major surgery, acute pancreatitis or shock).1–3
Three sequential pathological phases are commonly encountered:4 a first acute exudative phase is characterized by the influx of protein-rich oedema fluid into the air spaces as a consequence of increased permeability of the alveolar–capillary barrier; a second phase consists of proliferation of type II cells and accumulation of fibroblasts associated with collagen deposition in the extracellular matrix. This phase may result in a third fibrotic phase that leads to obliteration of alveolar spaces by deposition of extracellular matrix proteins. Although ARDS has a more complicated pathology than the simple deficiency of surfactant, it has similar symptoms than NRDS, including reduced lung compliance, restriction of lung volume and refractory hypoxemia, necessitating mechanical ventilation. ALI is diagnosed by PaO2/FiO2 ≤300 mmHg and ARDS by PaO2/FiO2 ratio ≤200 mmHg.5
The occurrence of NRDS is nowadays low in developed countries (current neonatal mortality rate is ∼4 per 1000 live births in the US).6 This is due to the fact that, since 1990, NRDS can be prevented or efficiently reverted by surfactant therapy (administration of exogenous surfactant), along possibly with other major innovations, such as antenatal corticosteroids and advanced care technologies (high-frequency oscillations).6 Neonatal mortality contributes however to up ∼40% to the mortality rate for children younger than 5 years worldwide. Several lung surfactant substitutes consisting of purified and complemented lung extracts from porcine or bovine tissues (e.g. Curosurf, Survanta, Alveofact and BLES) have proven effective in NRDS treatment.7,8
Where ALI/ARDS is concerned, the situation is different. Over a period of 25 years, the annual incidence of ALI/ARDS is predicted to be 335![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, with 147000 deaths per year.9 Although surfactant therapy has demonstrated some improvements in gas exchange and survival in animal ARDS models, its efficacy in human clinical trials is limited and mortality remains high.3,10,11 It has been recognised that surfactant inactivation is a major factor in ARDS and also that the lack of clinical inefficacy of the exogenous surfactant preparations results from such inactivation. The inhibitors responsible for inactivation of both native and exogenous lung surfactants include serum proteins (albumin, fibrinogen monomer, fibrinogen, immunoglobulin, C-reactive protein), red blood cell components (haemoglobin), pro-inflammatory cytokines (IL-1, TNF), lipopolysaccharides (LPS), and certain lipids such as cholesterol (a major component of meconium), fatty acids, bile salts and lysophosphatidylcholine. The surfactant components can also be hydrolysed by phospholipases or proteases (the concentration of which can increase strongly during microbial infection), or peroxidised by reactive oxygen species (ROS), or altered by various pollutants (acids, fumes, particles). In order to design lung substitute preparations that are more efficient against ARDS, it is necessary to better understand the biophysical mechanisms of lung surfactant inactivation, especially by the most damaging inhibitors.
000, with 147000 deaths per year.9 Although surfactant therapy has demonstrated some improvements in gas exchange and survival in animal ARDS models, its efficacy in human clinical trials is limited and mortality remains high.3,10,11 It has been recognised that surfactant inactivation is a major factor in ARDS and also that the lack of clinical inefficacy of the exogenous surfactant preparations results from such inactivation. The inhibitors responsible for inactivation of both native and exogenous lung surfactants include serum proteins (albumin, fibrinogen monomer, fibrinogen, immunoglobulin, C-reactive protein), red blood cell components (haemoglobin), pro-inflammatory cytokines (IL-1, TNF), lipopolysaccharides (LPS), and certain lipids such as cholesterol (a major component of meconium), fatty acids, bile salts and lysophosphatidylcholine. The surfactant components can also be hydrolysed by phospholipases or proteases (the concentration of which can increase strongly during microbial infection), or peroxidised by reactive oxygen species (ROS), or altered by various pollutants (acids, fumes, particles). In order to design lung substitute preparations that are more efficient against ARDS, it is necessary to better understand the biophysical mechanisms of lung surfactant inactivation, especially by the most damaging inhibitors.
This short review presents the strategies that have recently been proposed to overcome the inactivating effect of proteins on the pulmonary surfactant. In one approach, SP-B/SP-C protein analogues or simpler peptides have been designed that mimic the constituents of the native surfactant that are responsible for its innate resistance to inactivation. Another new and cost-effective approach consists of using hydrophilic polymers and electrolytes. A markedly different approach makes use of the unique properties of fluorocarbons (biological inertness, gas transport capacity, spreadability, low surface tension and high vapour pressure with regard to molecular weight). These water-insoluble compounds have been investigated in the 1990's in clinical trials of partial liquid ventilation and as injectable oxygen carriers.12 Fluorocarbon gases are routinely utilised as ophthalmic tamponades for repair of retina detachment and as stabilisers of the commercial gas microbubbles used as ultrasound diagnosis contrast agents. Fluorinated compounds also have potential as reporters for 19F magnetic resonance imaging (MRI).13 The review emphasises recent investigations that demonstrate the potential of fluorocarbons as components for lung surfactant therapy.
2. The native lung surfactant
2.1. A complex bio-interface
The respiratory pulmonary gas exchange takes place through a complex physical barrier that includes the thin aqueous film that lines up the alveolus (the alveolar lining layer), the alveolar epithelial cells, the interstitial layer, the endothelial cells that form blood capillaries, the blood plasma and the erythrocyte membrane (Scheme 1a).7,14,15 In mammals, the native lung surfactant typically contains a large proportion of lipids (∼90% by surfactant mass), primarily DPPC (∼40%), small fractions of polyunsaturated fatty-acid-based phospholipids, anionic phospholipids such as phosphatidylglycerols, neutral lipids such as diacyl- and triacylglycerols, free fatty acids such as palmitic acid, plasmalogens, and cholesterol (∼8%) (Scheme 1b).16,17 | ||
| Scheme 1 (a) Structure of the alveolus; from ref. 1. (b) Typical composition of the lung surfactant in mammals; from ref. 18. (c) Electron micrograph of a rat lung showing lamellar bodies forming tubular myelin (bar 1 mm); from ref. 19. | ||
The remaining 10% of the composition comprises four proteins, named surfactant proteins SP-A, SP-B, SP-C and SP-D.20 SP-A (monomeric, MW 26–38 kDa) and SP-D (43 kDa) are large hydrophilic glycoproteins that belong to the calcium-dependent carbohydrate-binding collectin family.21,22 SP-A and SP-D play important roles in pulmonary host defence and control of lung inflammation. These proteins bind to various microorganisms and pathogen-derived components and modulate leukocyte functions such as chemotaxis, cytokine function and phagocytosis.23 By associating with cell surface pattern recognition receptors, SP-A and SP-D regulate inflammatory cellular responses such as the release of lipopolysaccharides-induced pro-inflammatory cytokines.22 SP-A associates with surfactant membranes via its globular carbohydrate recognition domains and can promote surfactant vesicle aggregation in the presence of calcium.18 SP-A also facilitates formation of surfactant tubular myelin, thus regulating surfactant secretion and recycling. Contrary to SP-A, SP-D is not associated with surfactant membranes. SP-D participates in surfactant reuptake and recycling and has potent protective antioxidant properties. The smaller hydrophobic proteins SP-B and SP-C play important roles for determining the surface activity of the lung surfactant, including efficient surface adsorption, film stability and re-spreading processes of surfactant during the continuous compression–expansion breathing cycles.24 SP-B, a 79-residue polypeptide (MW 8.7 kDa) composed of amphiphilic α-helices connected by amino acids containing apolar loops, belongs to the saposin-like protein family.25,26 It exhibits a positive net charge, which results in interactions with anionic phospholipids. It was suggested that SP-B interact and connect surfactant bilayers and monolayers due to its amphiphilic character and presence of charged residues. SP-B is essential for the biogenesis of lamellar bodies, the organelles used by type II pneumocytes to assemble, store and secrete surfactant membranes (Scheme 1c). SP-C is a 35 residues monomer (4.2 kDa) that forms a single α-helix rich in branched aliphatic residues and has a trans-membrane orientation in surfactant bilayers.27
2.2. Formation of the surfactant monolayer and its role in respiration
The lipids and hydrophobic proteins SP-B and SP-C are synthesized mainly in the alveolar space by epithelial type II pneumocytes where they are stored in lamellar bodies (1–3 μm in diameter) before being secreted by exocytosis into the alveolus. The lipids of the lamellar bodies rearrange in the alveolus into an expanded membrane called tubular myelin, from which the monolayer is formed and spreads at the air-fluid interface just above the alveolar epithelium (Scheme 2a). SP-A and SP-D are secreted independently of the lamellar bodies and are associated with the surfactant lipids in the alveolar lumen. From the alveolus, the surfactant components are recycled into type II cells, where they are re-utilised or removed from the cycle by phagocytosis and degraded within alveolar macrophages.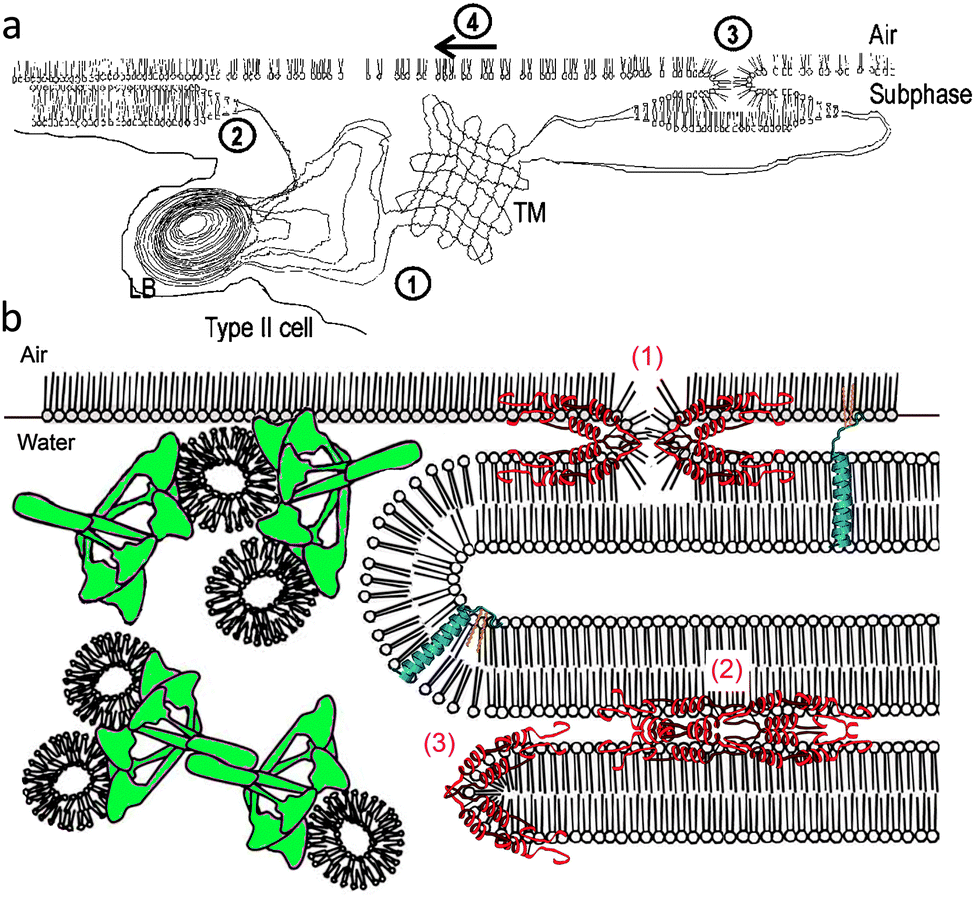 | ||
| Scheme 2 (a) A model illustrating surfactant secretion from the alveolar type II cells and the steps by which it forms a surface layer on the alveolar sub-phase, including (1) secretion of lamellar bodies from type II cells and formation of tubular myelin surfactant; (2) spreading of the tubular myelin surfactant as individual bilayers; (3) fusion and conversion of bilayers to monolayers; and (4) lateral spreading of surfactant monolayers; from ref. 2. (b) A model for surfactant reservoirs associated with the interfacial monolayer showing the possible roles of SP-A (green), SP-B (red) and SP-C (blue) in the dynamic folding and re-spreading of interfacial films. SP-B is represented in three hypothesized roles: (1) facilitating formation of the intermediate lipid structure between monolayer and bilayer; (2) generating stacks of bilayers via protein–protein interactions; and (3) promoting formation of bilayer disks; from ref. 18. | ||
One key role of the lung surfactant is to form a monolayer at the interface between air and the fluid hypophase that lines the alveoli. The surfactant monolayer is in charge of reducing the surface tension upon compression (i.e., during expiration), thus reducing the work of breathing, and must respread easily on expansion (i.e., during inspiration) with spreading rates compatible with respiration.
These conflicting requirements are achieved by a combination of specialised compounds that work synergistically to promote adsorption and spreading during dynamic compression–expansion cycling. DPPC alone can generate near-zero surface tension at the air–water interface during compression, but cannot respread fast enough upon expansion because it forms highly cohesive (semi-crystallised liquid-condensed) two-dimensional domains at the air–water interface due to strong lateral interactions. It is the presence of unsaturated lipids such as phosphatidylglycerols and phosphatidylcholines, and of the amphiphilic proteins SP-B and SP-C that allows reversion to the fluid liquid-expanded phase.7,28 On the other hand, fluid monolayers would not allow reaching the low surface tensions necessary for proper lung function. Therefore, in order to reach low tensions some minor components (pre-dominantly non-phosphatidylcholine compounds) are squeezed out from the monolayer upon compression, which results in an enrichment of the monolayer in DPPC and allows transition to the condensed phase. Surface tension is then reduced while alveolar collapse is prevented. A concept involving a “surface-associated reservoir” was developed to account for the fact that phospholipid uptake occurs too fast to be explained solely by de novo adsorption.7,18,29 In this model, layers of surfactant are folded in the sub-phase and remain in contact with the monolayer at the air/liquid interface, ready for rapid replenishment of the surfactant layer (Scheme 2b).
2.3. Lung surfactant substitutes
Surfactant therapy involves the administration of exogenous surfactant preparations in order to replace the absent or damaged native surfactant.7,8,18 Clinically used surfactant preparations consist mostly in two classes. A first class includes surfactants that are extracted from animal sources and are depleted in SP-A and SP-D, such as BLES (BLES Biochemicals, London, ON, Canada), Curosurf (Chiesi Farmaceutici, Parma, Italy), Infasurf (ONY, Amherst, NY, US), Survanta (Abbott Laboratories, North Chicago, IL, US), Surfacten (Tokyo Tanabe Co., Tokyo, Japan) and Surfacen (CENSA, Mayabeque, Cuba). Despite their intrinsic drawbacks, such as potential risks of pathogen transmission, variability in composition and high cost, the modified “natural” surfactants are generally more effective than the synthetic formulations (Table 1). Their efficacy is limited, however, to the treatment of NRDS. They were found to improve oxygenation, but did not reduce mortality in the case of surfactant inactivation such as in ARDS or MAS.3,8,30,31 A second class of more recently developed preparations includes synthetic surfactants that contain lipids and recombinant SP-C (Venticute, Altana Pharm. Konstanz, Germany),32 SP-B/SP-C protein analogues or simple peptides that mimic surfactant proteins (KL4 peptide, Surfaxin, Discovery Laboratories, Warrington, PA, US).33 Other exogenous surfactant preparations include combinations of lipids with Hel 13-5 peptide (mimic of SP-C)34,35 or with polymers (poly-L-lysine electrostatically complexed with poly-L-glutamic acid, Synsurf).36| Approaches | Development | Advantages | Limitations and challenges |
|---|---|---|---|
| Modified animal surfactants complemented with SP-B & C | Commercial In the clinic | • Available | • Transmission risks (lack of SP-A & D) |
| • Adequate surface activity | • Batch-to-batch variations | ||
| • Variable sensitivity to inactivation | |||
| • Variable results in ARDS/ALI treatment | |||
| Synthetic preparations with surfactant protein mimics or peptides | In the clinic or preclinical | • Batch-to-batch consistency | • Under development |
| • Large scale production | • Less effective than modified natural surfactants | ||
| • Cost effectiveness | |||
| Polymers | Preclinical | • Surfactant preparation adsorption enhancement | • Under development |
| • Resistance to inactivation | • Not yet defined formulations | ||
| • Versatility and cost-efficiency | • Need tests in animal models | ||
| Fluorocarbon gases & liquids | Basic research | • DPPC monolayer fluidisation & near-zero surface tension | • Need defined formulations, delivery systems, administration modes |
| • DPPC adsorption enhancement | • Need tests in animal models | ||
| • Resistance to inactivation (albumin) | |||
| • Biological acceptance of fluorocarbons demonstrated | |||
| • Versatility and cost efficiency | |||
| Fluorinated surfactants | Basic research | • DPPC adsorption enhancement | • Assessment of toxicity |
| • Resistance to inactivation (fibrinogen) | • Non defined formulations, delivery modes | ||
| • Need tests in animal models | |||
3. Biophysical mechanisms of surfactant inactivation by proteins
The various processes by which the normal activity of the pulmonary surfactant is reduced or suppressed are referred to as surfactant inactivation. Common conditions related to surfactant inactivation include ALI/ARDS, MAS and pulmonary oedema. Certain mechanisms of surfactant inactivation are related to chemical degradation of surfactant constituents or to alteration of the surfactant's aggregate structure and morphology. These impairments can occur at various time-points of the surfactant's life-time, including during secretion of the lamellar bodies into the alveolar liquid layer and unpacking in membrane vesicles, during formation of the monolayer, or during reuptake by type II cells or macrophages. Impairments can also occur during transport from the alveoli to the airways.14,18,37 All these abnormalities may prevent the phospholipids from reaching low surface tension values during monolayer compression or hamper their re-spreading during expansion. In most of these cases, surfactant inactivation is slow and permanent.18,37By contrast, in ARDS and ALI, inactivation is rapid and often correlated with increased concentration of various substances, including plasma proteins, unsaturated phospholipids, lysophospholipids, free unsaturated acids (e.g. oleic acid), bile salts, diacylglycerol and cholesterol, or meconium.7,38 Despite large variations, most studies consistently report an increase of albumin concentrations in the alveolar fluid during ARDS (∼25 mg mL−1 in ARDS patients versus ∼5 mg mL−1 in healthy patients).39 The protein concentration in the alveolar fluids of ARDS patients correlates actually with the severity of the disease.40 Leakage of plasma proteins into the alveolar space due to impaired function of the alveolar-capillary barrier was recognised as an early event in the pathogenesis of ARDS and may contribute substantially to surfactant alterations.8,41
Various in vitro studies have reported that different inhibitors, including plasma proteins and certain lipids, can interfere at different stages with phospholipid functions by hindering phospholipid adsorption and formation of a functional surfactant film, by preventing the film from reaching low interfacial tensions or by increasing film compressibility, and by affecting phospholipid re-spreading from surfactant reservoirs upon expansion.7
3.1. Proteins prevent access of surfactant to the alveolar interface by competitive adsorption
Soluble proteins such as fibrinogen, fibrinogen monomer, haemoglobin and albumin were found to interfere with surfactant phospholipid de novo adsorption.2,42–53 Surfactant adsorption was also strongly impaired by lysophosphatidylcholine, while supraphysiological concentrations of neutral lipids (monoacylglycerol, diacylglycerol, unsaturated fatty acids, cholesterol and cholesterol esters) showed little effect on adsorption. All serum proteins are surface active and can compete with surfactant membranes for adsorption at the air–water interface. Phospholipids, which are present in the form of large aggregates, adsorb and spread slowly at the interface through “unzipping” processes.7 Despite their lower surface tension, they adsorb more slowly than small amphiphilic proteins, which adsorb rapidly by molecular diffusion. Once adsorbed at the interface, the film of protein prevents phospholipids from reaching the interface by creating a steric and/or electrostatic (DLVO-type) energy barrier.37 It is reasonable to think that, at the surfactant/albumin concentration ratios that prevail in ARDS conditions, competitive adsorption plays a role in the inhibition of the surfactant. However, other mechanisms have been proposed that suggest interaction and/or binding of the proteins with the surfactant.54 It is important to note that albumin is a relatively weak inhibitor, as compared to other serum proteins, such as fibrinogen.3.2. Proteins prevent surfactant from reaching low surface tension and from re-spreading at the alveolar interface
The ability of the surfactant phospholipids to attain low surface tensions during compression is hampered by serum proteins, fibrinogen, fibrinogen monomer, and C-reactive protein, which bind to the polar phosphorylcholine group, and to a lesser extent, by albumin.53 Simultaneous increase in C-reactive protein and decrease in SP-A in the alveolar space were associated with ALI.55 It was suggested that C-reactive protein inhibits the surfactant by fluidising the surfactant film.56 Surfactant compressibility is markedly increased by supraphysiological concentrations of cholesterol. Surface tension reduction at end-expiration is also affected by high monoacylglycerol, diacylglycerol, unsaturated fatty acids, bile salts and cholesterol (a major component of meconium) and cholesterol esters.18 Respreading of surfactant phospholipids from surface-associated reservoirs is inhibited by CRP and, at high surface tensions, by albumin. Supraphysiological concentrations of cholesterol have also a deleterious effect on surfactant re-spreading.4. Optimising lipids and proteins in order to resist surfactant inactivation
The higher efficiency of modified natural surfactants over protein-free synthetic surfactants demonstrates the importance of the native proteins in overcoming surfactant inactivation (Table 1). In animal experiments, surfactant preparations containing SP-A showed higher resistance to inactivation than those without SP-A.57 Addition of SP-A increased the resistance of lipid extract surfactants to blood proteins46,47 and meconium.58 Both SP-B7,50 and SP-C59,60 have been reported to increase the resistance of lung surfactant to inactivation by various agents. These proteins have therefore been proposed for optimizing the surface behaviour of the surfactant.15 A preparation containing a synthetic C16:0 diether phospholipid (DEPN-8, structurally resistant to phospholipase degradation) and SP-B/C was found to be highly resistant to inhibition by serum proteins, phospholipase A2 and lysophosphatidylcholine.61 Addition of peptide analogues of SP-B and/or SP-C also improved the resistance to surfactant inactivation.62,63 Recently, a new generation of synthetic analogues, which incorporates recombinant proteins or SP-B/SP-C peptides, has been tested in vivo. CHF 5633 is a fully synthetic surfactant containing two phospholipids, DPPC and 1-palmitoyl-2-oleoyl-glycero-3-phospho-1-glycerol sodium salt (reported to inhibit lung inflammation) and two peptidic analogues of human SP-B and SP-C, designed to resist to oxidative injury.64 The SP-C analogue is a 33-amino acid protein that includes both the N-terminal and C-terminal helical segments found in native SP-C. The SP-B analogue is a 34-amino acid protein derived from the full-length natural SP-B. A study demonstrated superior oxygenation and lung compliance in ventilated preterm lambs treated with CHF 5633 as compared to animal-derived surfactant preparations.64 In another study led after surfactant inactivation in preterm lambs, CHF 5633 showed a survival benefit over the gold standard treatment Curosurf at the same doses.65
5. Using hydrophilic polymers and electrolytes
Introduced in the late 1990's, certain water-soluble polymers were found to enhance adsorption and increase the surface activity of various surfactant substitutes and to reverse or reduce inactivation by serum, meconium, albumin and other substances, both in vitro and in vivo.2,66–69 Many polymers were found effective, including non-ionic polymers (poly(oxyethylene) PEG 10000, dextran 9500 and 71000, polyvinylpyrrolidone PVP), anionic polymers (hyaluronan) and cationic polymers (chitosan).
5.1. Nonionic and anionic polymers—generating depletion forces
The enhancement of surfactant adsorption by addition of PEG 10000 or hyaluronan was found to be a function of both polymer concentration and molecular weight (Fig. 1, Table 1), which supported a model based on polymer-induced depletion-attraction, a non-specific polymer-induced entropic force.2,37,52 A quantitative theory of surfactant inactivation by charged surface-active serum proteins has been proposed.2,37,52 This theory uses Smolukowski's analysis of colloidal stability and suggests formation of a repulsive energy barrier by serum protein at the interface, with contributions of both steric effects and electrostatic repulsions.2,37 When two vesicles of lung surfactant are approaching each other, there is a point at which the polymer can no longer fit into the space available between these vesicles and will be excluded from this space. The resulting enrichment in polymer in other zones produces an increase in osmotic pressure that draws water from the polymer-depleted region, thus promoting vesicle fusion. A similar process occurs at the air/water interface. The resulting polymer depletion drives the vesicles towards this interface, thus decreasing the energy barrier for surfactant adsorption. The depletion attraction is purely entropic and independent of the chemical composition of the surfactant, protein and polymer, which explains that PEG, dextran and hyaluronan were all effective at enhancing adsorption of various surfactant substitutes (Survanta, Curosurf and Infasurf).2,37,52
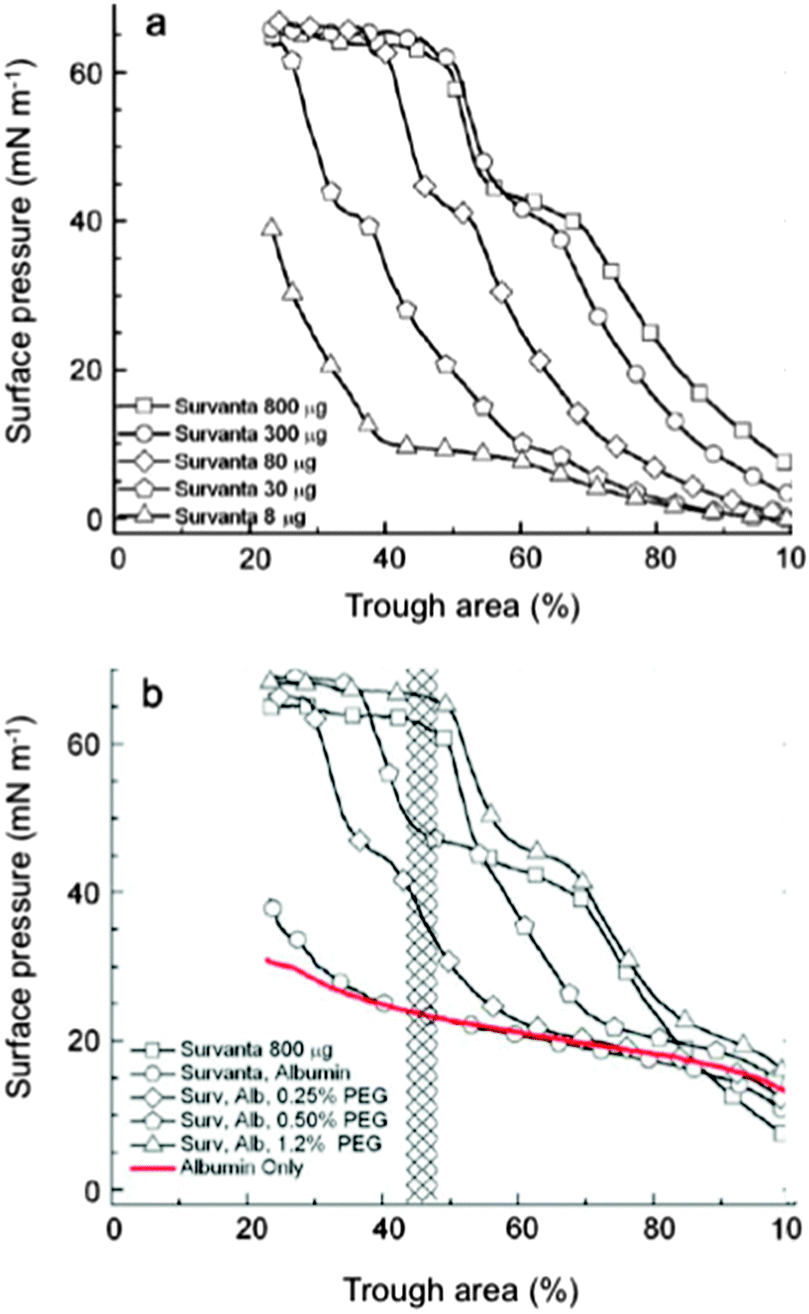 | ||
| Fig. 1 Compression isotherms of (a) Survanta on a buffer sub-phase, showing increased adsorption with increased concentration, and (b) Survanta (800 μg) on sub-phases containing albumin (2 mg mL−1) with increasing PEG concentrations. Albumin produces the same effect as decreasing Survanta concentration. Adding increasing amounts of PEG produces the same effect as increasing Survanta concentration. From ref. 71. | ||
This view has recently been confirmed by a study of the rheological dilational properties of surface films formed by Exosurf, Survanta, Curosurf or Alveofact in the presence of albumin and PVP, PEG or dextran. The hydrophilic polymers were found to restore the DPPC content of the surface film.70
In another recent study, the effect of hydrophilic polymers was investigated using thin (black) foam films.72 It was indeed suggested that the lung surfactant exists as an intrapulmonary foam of air bubbles filling the terminal lung unit.73 The contacts between bubbles, and between bubbles and cell surfaces consist of black foam films. The ability of the polymers to decrease the effect of albumin inhibition was expressed by their capacity of restoring stable and homogeneous black films (Fig. 2).
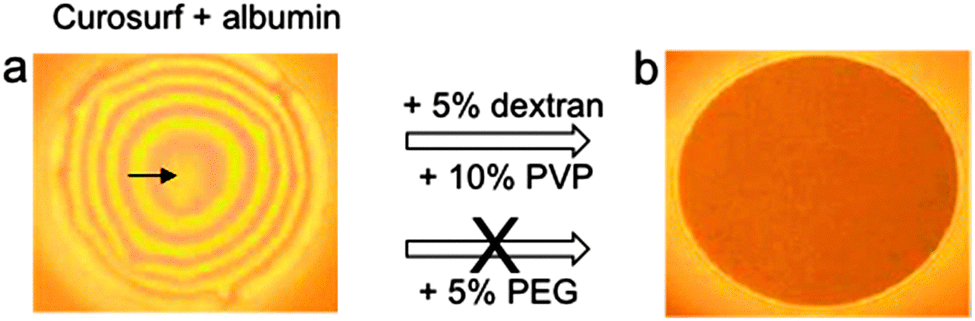 | ||
| Fig. 2 Inhibition of Curosurf by albumin in foam films, and effect of hydrophilic polymers (dextran, PVP or PEG) on the recovery of surfactant surface properties. Dextran and PVP were efficient, but not PEG. Foam films thicknesses were (a) ∼1133 nm and (b) ∼15 nm; from ref. 72. | ||
A microinterferometric method allowed direct visualisation of foam film morphology, evaluation of its stability and viscosity, and provided access to the disjoining pressure, a measure of the interactions (electrostatic, van der Waals, steric, etc.) between two interfaces. This study indicated that some specific interactions might take place, as certain polymers have an effect on certain surfactant substitutes, but not on all of them (e.g., Survanta recovered its effectiveness with PEG, PVP, and hyaluronan, while Curosurf recovered it with dextran, PVP, and hyaluronan) and suggested that both depletion attraction and specific interaction were operative.
5.2. Electrolytes—decreasing Debye length
Classical methods of manipulation of the double-layer repulsion in colloids using electrolytes were shown to have similar predictable effects on surfactant adsorption.37In vitro studies showed that increasing NaCl concentration above physiological levels decreases the Debye length and the magnitude and range of the double-layer repulsion, and restores surfactant adsorption in the presence of albumin (Fig. 3).71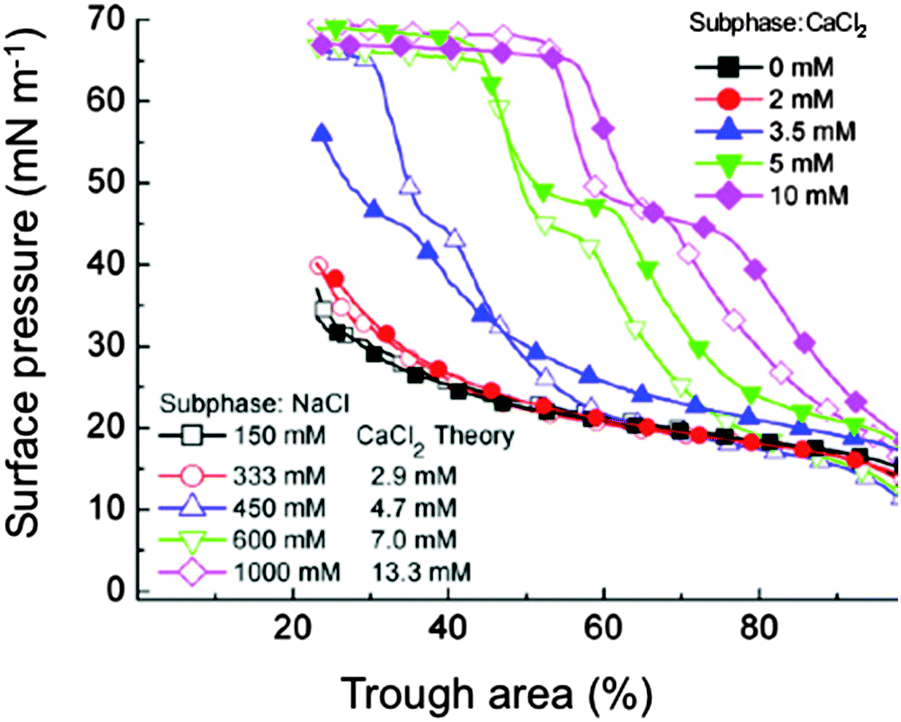 | ||
| Fig. 3 Compression isotherms of Survanta (800 μg) on a buffered saline sub-phase (0.2 mM NaHCO3, pH 7) containing albumin (2 mg mL−1). Various electrolyte concentrations are plotted. For each NaCl concentration, the theoretical CaCl2 concentration according to the Schulze-Hardy rule, is given; from ref. 71. | ||
It would obviously be impracticable to use high levels of saline because of their osmotic effect on lung tissue. However, it has been calculated (using the Schulze-Hardy rule) that divalent calcium would enhance surfactant adsorption at concentrations 2−6 lower than monovalent sodium. It was experimentally verified that CaCl2 concentrations of ∼15–20 mM would be sufficient to overcome albumin adsorption and would have minimal effects on the fluid balance in a rat lung.71
5.3. Cationic additives (polyelectrolytes and peptides)—flocculating surfactant aggregates
The interactions between a cationic polyelectrolyte, chitosan (612, 113 and 213 kD), or two cationic peptides (polylysine 50 kDa and polymyxin B), and BLES were studied using dynamic compression–expansion cycles of dilute BLES preparations.74,75 Extremely low concentrations of both chitosan and of the two peptides were found to improve the adsorption of the surfactant substitute at the air/water interface in the presence of albumin. The optimal additive/surfactant ratio was determined based on the minimal surface tension that could be reached upon dynamic compression carried out in a constrained sessile drop device in the presence of serum. The zeta potential of the lung surfactant aggregates in the sub-phase suggests that chitosan and the peptides bind to the anionic lipids (phosphatidylglycerols) in BLES, and that this binding is responsible for the changes in surface activity (elasticity and stability) observed for these surfactant–polyelectrolyte mixtures. However, unlike calcium or PEG, increasing chitosan concentration above optimal caused less surfactant to adsorb, and inactivation occurred again. The optimal formulation containing these peptides were able to reach low minimum surface tensions in systems containing 500 μL mL−1 of serum, thereby matching the effectiveness of the complete lung surfactant.6. Using fluorocarbon compounds in lung therapy
6.1. Relevant physicochemical properties of fluorocarbons and fluorinated surfactants
Fluorocarbons and perfluoroalkylated surfactants have properties of their own that distinguish them markedly from their alkylated analogues.76,77 Fluorocarbons (FCs) have been investigated for various biological applications due to their high biological inertness, remarkable ability to solubilize gases, extremely low solubility in water and low surface tension, including for intravascular oxygen transport12,78 and stabilization of gaseous microbubbles used as contrast agent in ultrasound imaging.79–82 The surface tension of lungs has been measured in situ by depositing small droplets of various fluorocarbons on excised lungs of rats.83 Interactions among fluorinated chains are weak, due to the lower polarisability of fluorine as compared to hydrogen (and hence, lower van der Waals forces), resulting in extremely low water solubility. Perfluoroalkyl chains are much more hydrophobic than alkyl chains, and have also a lipophobic character. Consequently, fluorinated surfactants can reduce air–water superficial tension more effectively than their hydrogenated counterparts, reaching values that cannot be obtained with the latter. Their capacity for segregation and self-association is also much more pronounced, fostering higher organization, including with phospholipids.78,84–866.2. Partial liquid ventilation with fluorocarbons: improvement of lung compliance, recruitment of lung alveoli and anti-inflammatory effects
At the end of the 1960's the discovery that rodents immersed in a liquid fluorocarbon can survive by breathing the oxygen solubilised in this fluid87,88 coincided with the initial description of ARDS.89 In the 1990's, there have been numerous studies aiming at assessing the utility of fluorocarbons in partial liquid ventilation (PLV) for the treatment of ARDS. Perfluorooctyl bromide (PFOB) in particular has been extensively investigated for its capacity to act as liquid positive end-expiratory pressure (“liquid PEEP”) to recruit collapsed alveoli and improve oxygenation and ventilation.PLV with FC has been described in many animal models (premature, full-term neonatal and adults) and a few human studies. Improved oxygenation and lung compliance were achieved in preterm animal models,90 as well as in premature infants.91,92 Adequate gas exchange could be maintained in healthy rabbits at airway pressures comparable to those used in conventional ventilation. A study on a saline lavage rabbit model of respiratory failure showed improvement in oxygenation with PFOB, effective gas exchange at relatively low airway pressures, and overall improvement in both pulmonary mechanics and lung histology as compared with conventional ventilation.93,94 In another study, severe respiratory failure was induced with oleic acid in young sheep.95 Improved pulmonary mechanics (increased compliance), improved oxygenation, and marked reduction in alveolar haemorrhage, lung fluid accumulation and inflammatory infiltration was demonstrated in the PFOB group, as compared to the gas ventilated animals. A human adult trial found significant improvement in physiological shunt and static pulmonary compliance.96 Studies were also conducted on human neonates that were impressive regarding oxygenation.91,97 Another study evaluated the use of PLV in trauma patients as compared with conventional mechanical ventilation.98 The alveolar inflammatory response was investigated, as measured by white cell infiltration and capillary leak. The sequence of pro-inflammatory and anti-inflammatory cytokines exhibited an anti-inflammatory effect in the alveolar environment with reduction of pro-inflammatory interleukins IL-1 and IL-6, white blood cell count, and protein capillary leak.98 PLV also reduced alveolar neutrophils independent of IL-8. Administration of PFOB reduced total white cell and neutrophil infiltration into the alveoli, capillary leak of proteins, and expression of the inflammatory cytokines IL-b and IL-6. The anti-inflammatory IL-10 was also reduced. All of these alveolar responses appeared to be beneficial when viewed individually, but did not result in significant clinical improvement. The fact that FCs attenuate the pro-inflammatory and pro-coagulatory responses of activated monocytes and of alveolar macrophages may contribute to the protective role of FCs in injuries associated with local activation of inflammatory processes.99 Delivery of vaporized FCs to oleic acid-injured ARDS sheep resulted in significant and sustained improvements of gas exchange and of lung compliance.100 PLV trials were abandoned when improved, less damaging ventilators became available.
6.3. Combined administration of exogenous surfactant and fluorocarbons
It was suggested that the low efficacy of the surfactant substitutes in the treatment of ARDS could be related, in addition to surfactant inactivation, to inhomogeneity in patient aeration due to adjacent atelectatic and overinflated areas.101 Since exogenous surfactant preferably distributes in aerated areas,102 alveolar recruitment is required prior to surfactant therapy.103 In PLV, deficiency of endogenous surfactant persists and respiratory insufficiency reappears after evaporation of the FC. In order to re-open atelectatic areas and substitute the inactivated surfactant, surfactant administration was used in animal studies both prior90,104–107 and after PLV,108 without clear beneficial results.101 In surfactant-depleted rats, administration of FC-in-surfactant emulsions (surfactant: Curosurf; FC: PFOB, perfluorooctane (PFO), or a blend of perfluorinated cyclic ethers (RM101)) led to a more homogenous distribution of gas (in the Curosurf group) and fluids (in the case of FC- or FC-in-Curosurf emulsion groups) and aeration of the lung than with surfactant alone (Fig. 4).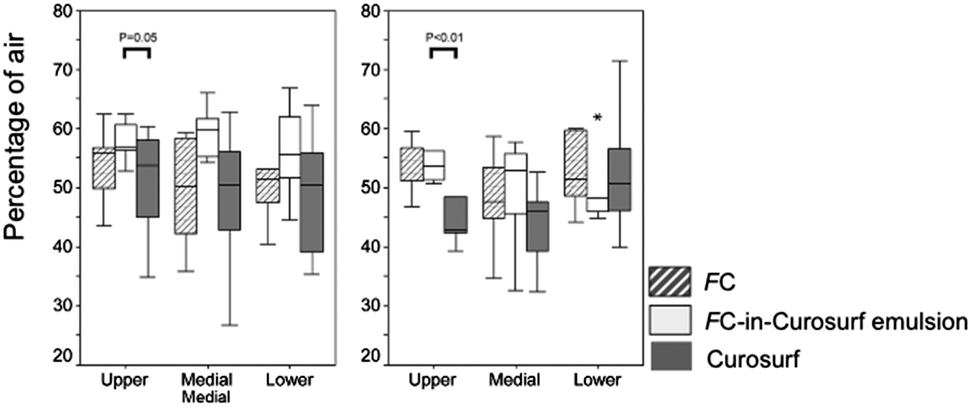 | ||
| Fig. 4 Percentage of gas-filled areas (in Curosurf group) or fluid-filled areas (in the FC or FC-in-Curosurf emulsion groups) in total parenchyma. Values are shown as median, range and outlayers (*) for the upper, middle and lower part of the left lung; from ref. 101. | ||
6.4. Fluorocarbon gases have a fluidising effect on DPPC monolayers, yet allow reaching low surface tensions, and prevent albumin adsorption
In vitro studies have shown that a series of fluorocarbon gases (gFCs), including PFOB, perfluorooctylethane, perfluorooctane, perfluorodecaline and bis(perfluorobutyl)ethene, are able to inhibit the liquid-expanded (LE)/liquid-condensed (LC) phase transition of DPPC Langmuir monolayers.109,110 The formation of semi-crystalline domains of an LC phase, which typically occurs in the LE/LC coexistence region upon compression of DPPC, is prevented when the atmosphere above the DPPC monolayer is saturated with a gFC (Fig. 5).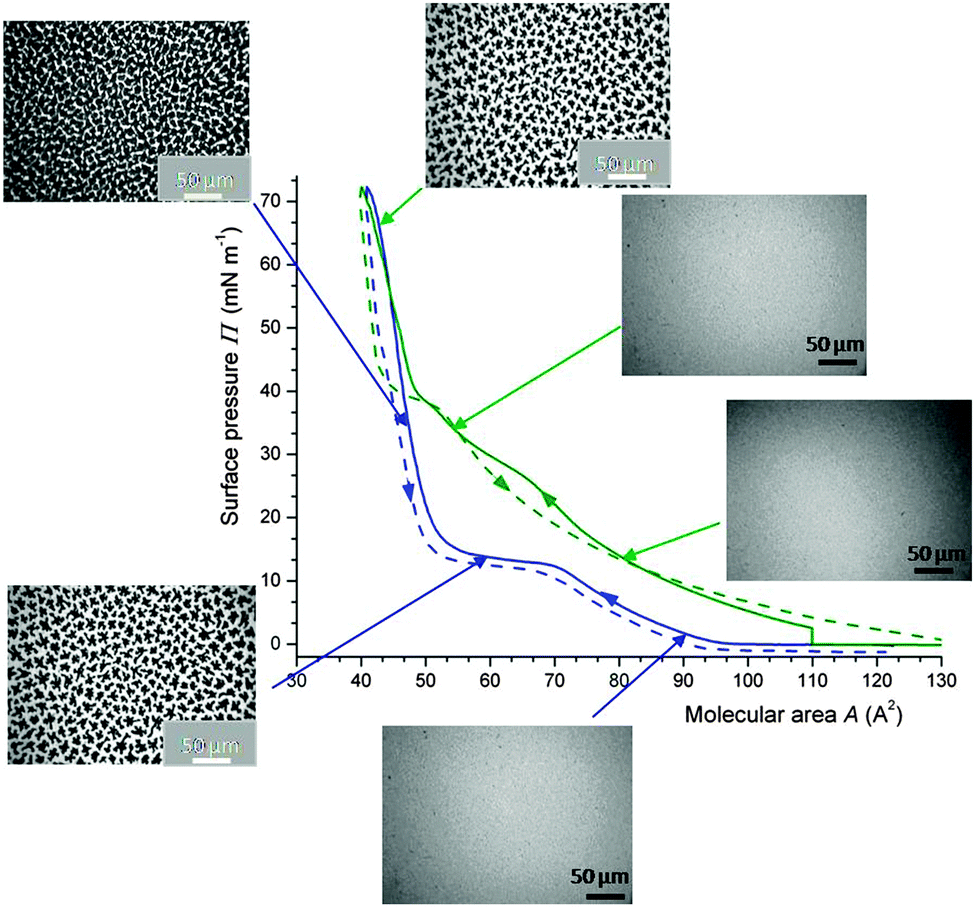 | ||
| Fig. 5 Fluidising effect of FC gases on DPPC monolayers as assessed by surface pressure Π/molecular area A isotherms and fluorescence optical microscopy (25 °C). Isotherms of DPPC (compression: solid lines; expansion: dashed lines) in an atmosphere of N2 (blue) or N2 saturated with PFOB (green). Insets: fluorescence images showing that the DPPC monolayer in contact with PFOB remains in the liquid expanded phase up to ∼38 mN m−1, while that in contact with N2 shows liquid-condensed phase domains at Π > ∼10 mN m−1. At maximum compression, PFOB is totally expelled from the DPPC monolayer and a high Π value (∼70 mN m−1) is reached. 15 μL of a DPPC solution in CHCl3 (1.0 × 10−3 mol L−1) were spread on the water surface. Adapted from ref. 109. | ||
When contacted with gFC, the DPPC monolayer remains in the LE phase for surface pressures lower than ∼40 mN m−1, as assessed by isotherms and fluorescence microscopy. The images were obtained using the fluorescent dye NBDC6-HPC, which is preferentially soluble in the disordered regions of the monolayer.111 Interestingly, the FC is totally expelled from the monolayer at maximal compression (i.e. for molecular area ∼40 Å2) and high surface pressure values (∼70 mN m−1) are reached. gFCs also induced the dissolution of pre-existing LC phase domains, thus facilitating re-spreading of DPPC molecules on the water surface, as shown by fluorescence microscopy and grazing incidence X-ray diffraction (Fig. 6).110 A unique situation is created in which gFCs have a highly effective fluidizing effect on the DPPC monolayer, without preventing achievement of low surface tension at maximal compression (Table 1).
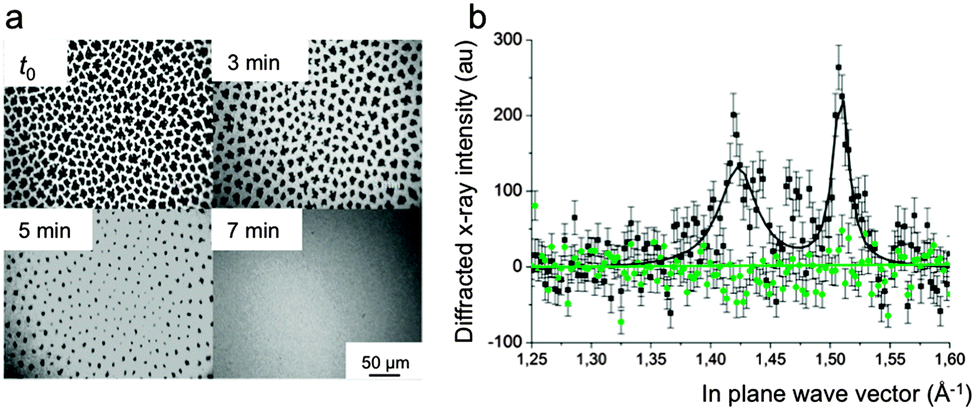 | ||
| Fig. 6 (a) Fluorescence micrographs of a DPPC monolayer compressed at 13 mN m−1 under N2. At time t0, the atmosphere of N2 above the monolayer was saturated with gPFOB. The LC domains progressively disappear over time. After 7 min, the monolayer is totally fluidized. (b) Intensity of the diffracted grazing X-rays as a function of the in-plane vector qxy. When compressed at 20 mN m−1, the DPPC monolayer shows the two Bragg peaks characteristic of the tilted LC phase of DPPC at 1.42 and 1.51 A−1 (black line). When the He atmosphere is saturated with gPFOB (green line), the peaks disappear rapidly, establishing the disappearance of the crystalline LC domains, allowing rapid respreading of the DPPC molecules. Adapted from ref. 110. | ||
Recently, an equation of state for insoluble monolayers was applied to describe the isotherms of phospholipids measured in the presence of a fluorocarbon in the gas phase.112 The observed co-adsorption mechanism of DPPC and FC molecules proposed in ref. 109 and 110 manifests itself by remarkable differences in cohesion surface pressure. Due to the interaction of the adsorbed FC molecules with DPPC, the mutual interaction energy between DPPC molecules is reduced, leading to a very effective fluidization of the monolayer.
Another study established that gPFOB could counteract the effects of albumin on DPPC Langmuir monolayers.113 When the FC gas is absent, albumin penetrates in the DPPC monolayer during compression, leading to the characteristic pattern shown in Fig. 7a (∼30 mN m−1) and to liquid condensed phases upon further compression. On the other hand, for pressure values higher than ∼30 mN m−1, gPFOB fluidises the monolayer (Fig. 7c), which means that it displaces the albumin from the interface. When expanded under air, the DPPC monolayer remains in a semi-crystalline LC phase, even at large molecular areas (80 Å2, Fig. 7b), whilst it is in the LE phase when gPFOB is present (Fig. 7d). The fact that the pressure is much lower under gPFOB than under air upon expansion (for areas larger than ∼50 Å2) supports the view that much less albumin is present in the monolayer. Altogether the incorporation of the PFOB molecules into the DPPC monolayer during expansion overcomes the incorporation of BSA, and prevents the latter from penetrating into the DPPC monolayer (Table 1).
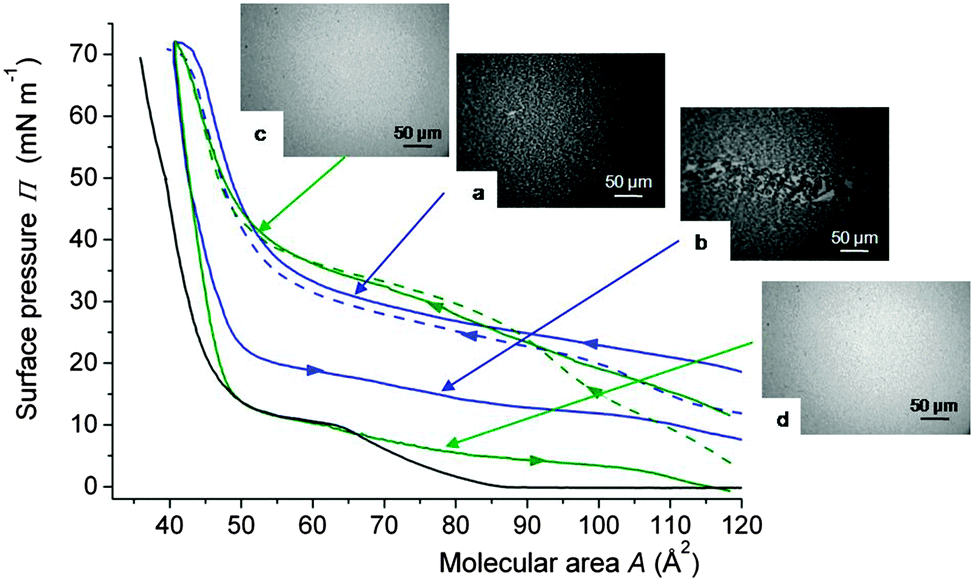 | ||
| Fig. 7 Compression isotherms of a Langmuir monolayer of DPPC spread on a sub-phase containing albumin (0.25 mg L−1) and compressed under air (blue) or gPFOB-saturated air (green) at 25 °C. The isotherm of DPPC, compressed under air, is shown as a reference (black). The first compression—expansion cycles are represented by solid lines and the second compressions by dashed lines. Fluorescence micrographs of the DPPC monolayer under air (a) upon compression at 30 mN m−1, and (b) upon expansion at 15 mN m−1; and under gPFOB saturated air (c) upon compression at 40 mN m−1, and (d) upon expansion at 5 mN m−1. DPPC: 15 μL of a 1.0 × 10−3 mol L−1 CHCl3 solution. Adapted from ref. 113. | ||
6.5. Combining the effects of fluorocarbon gases with an oscillatory regime
Applying sinusoidal oscillations to a film adsorbed on the surface of a millimetric bubble of air saturated with a fluorocarbon gas was recently found to constitute a mechanical means of reversing the outcome of the DPPC/albumin competitive adsorption.114,115 All the results presented in this section have been obtained using bubble profile analysis tensiometry (Tracker tensiometer, Teclis, Longessaigne, France).First, prolonged oscillations were applied to a DPPC monolayer adsorbed on the surface of an air bubble (Fig. 8).116
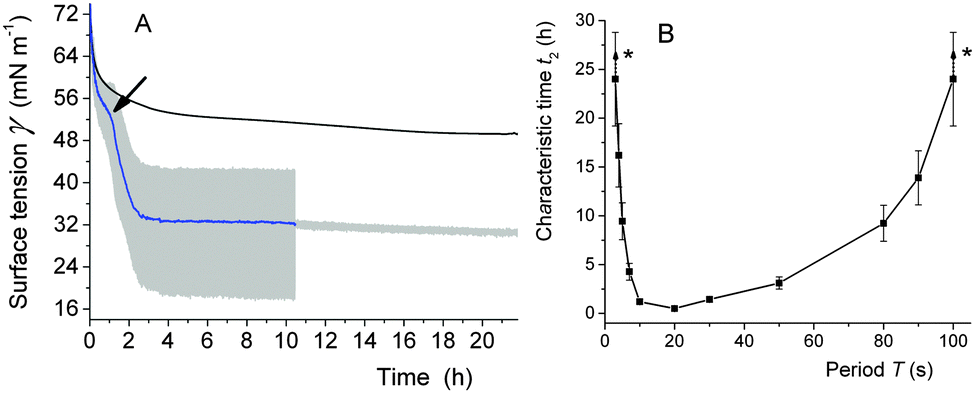 | ||
| Fig. 8 (A) Kinetics of adsorption of DPPC (10−3 mol L−1, provided as a dispersion of vesicles) at the surface of an air bubble at 37 °C. The bubble was static or submitted to oscillations (T 10 s, ΔA 15%). The oscillations were applied for ∼10 h, while γ was subsequently monitored for another 12 h. The arrow points to an increase in the adsorption rate. The grey area represents the fluctuations in surface tension associated with the oscillations. (B) Characteristic time t2 of the second regime of the DPPC adsorption at 37 °C, as a function of the oscillation period T (ΔA 15%). *These t2 values are higher than 24 h and could therefore not be determined precisely. From ref. 116. | ||
The adsorption of the phospholipid was drastically accelerated, a transition occurred between the dilute LE phase and the condensed LC phase and the surface tension γ was drastically reduced, reaching low values (30 mN m−1) (Fig. 8A). When the oscillations were stopped, γ remains low, meaning that the system has reached its equilibrium value 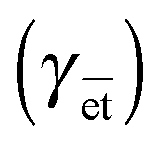 . Similar values have been measured for DPPC at equilibrium using different techniques.117 By contrast, when no oscillations were applied (static regime), the phospholipids adsorb very slowly at the air/water interface; the γ value remained very high even after 24 h. Maximum γ lowering effect was reached for a period range of 3–50 s (Fig. 8B). Interestingly, this range matches with the period range of respiration (15–20 cycles per min).
. Similar values have been measured for DPPC at equilibrium using different techniques.117 By contrast, when no oscillations were applied (static regime), the phospholipids adsorb very slowly at the air/water interface; the γ value remained very high even after 24 h. Maximum γ lowering effect was reached for a period range of 3–50 s (Fig. 8B). Interestingly, this range matches with the period range of respiration (15–20 cycles per min).
Another study investigated the influence of prolonged sinusoidal oscillations on the competitive adsorption of DPPC and albumin at the surface of an air bubble.114 It was found that the oscillations totally reversed the outcome of the competitive adsorption. After ∼5 h, albumin was progressively expelled and replaced by DPPC. Total replacement of albumin by DPPC was obtained in ∼10 h (Fig. 9A).
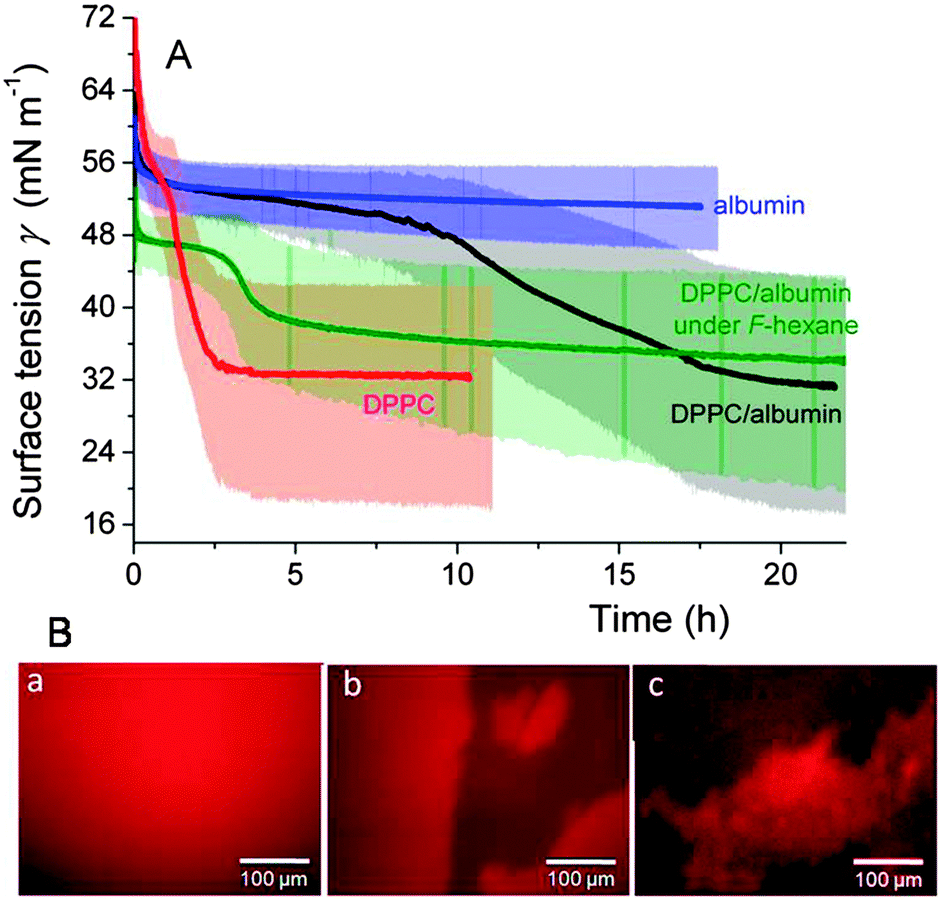 | ||
| Fig. 9 (A) Kinetics of adsorption (37 °C) at the surface of an air bubble having a shell of DPPC (10−3 mol L−1), albumin (7.5 × 10−7 mol L−1), or a DPPC/albumin (1 × 10−3:7.5 × 10−7 mol L−1) combination. Also shown is the absorption kinetics of DPPC/albumin at the F-hexane-saturated air/water interface. The bubbles were submitted to oscillations (T 10 s, ΔA 15%) throughout the experiments. The lightly colored areas represent the fluctuations in surface tension associated with the oscillations. (B) Fluorescence micrographs of the sequential adsorption of albumin and DPPC (injected 1.5 h after albumin) at the air/buffer interface of a Langmuir trough in the presence or absence of F-hexane. (a) at t = 0, albumin (+2 mol% of albumin-Texas-Red) is injected in the sub-phase. DPPC is injected 1.5 h later. It takes ∼1.5 h (in the presence of F-hexane) and ∼6 h to replace albumin from ∼60% of the interface. Adapted from ref. 114 and 115. | ||
Next, it was demonstrated that introduction of F-hexane in the air phase of the bubble accelerated the displacement of albumin and its replacement by DPPC by roughly an order of magnitude (Fig. 9A).115
The replacement of albumin by DPPC was also visualized by fluorescence microscopy on Langmuir films (Fig. 9B). A possible mechanism was proposed in which F-hexane rapidly goes to the air/water interface. Once there, it is incorporated into the hydrophobic site of albumin and promotes its unfolding, thus facilitating its expulsion towards the aqueous phase.115 The unique aptitude of fluorocarbon gases to recruit molecules initially solubilised in an aqueous phase at the interface across a monolayer of phospholipids has recently been utilised to immobilise an hypoxia biomarker in a phospholipid monolayer.118
These results suggest that combinations of DPPC and a fluorocarbon gas might be useful for the treatment of lung conditions resulting from deterioration of the native lung surfactant function due to plasma proteins, such as in ARDS. Further investigation of the effects of FC gases on additional potential inhibitors is needed in order to assess the clinical relevance of this approach, as it was recently shown that albumin is probably not the most important agent responsible for the inhibition of the lung surfactant.119 The delivery system most adapted to fluorocarbons has to be selected and the mode of administration investigated. Microbubbles, which have been reported for delivering Survanta,120 are potential candidates as they are often stabilised by fluorocarbon gases.
6.6. Fluorinated surfactants in combination with DPPC provide mixed monolayers with improved resistance to inactivation
Fluorinated and partially fluorinated surfactant additives have been assessed for tailoring potential lung surfactant replacement preparation properties. Fluorinated surfactants are highly surface-active and can generate lower surface tensions than their hydrocarbon analogues (typically ∼15 mN m−1versus ∼30 mN m−1). Fluorinated surfactants can be miscible, partially miscible or entirely immiscible with hydrocarbon surfactants, in particular with DPPC, depending on their structure. When they are mixed with DPPC in molar fractions lower than ∼0.4, high surface pressures (>65 mN m−1) can be reached. Fluorinated surfactants are generally chemically stable over a wide range of aggressive conditions. Despite concerns related to possible toxicity (perfluorooctane sulfonic acid was found to interact with DPPC, and may thus interfere with the lung surfactant's normal physiological function,121,122) and propensity for bioaccumulation123,124 fluorinated surfactants are technologically useful. They could potentially be used for biomedical applications that usually require small quantities and for which exemptions are allowed.124 Provided their toxicity is fully assessed, fluorinated surfactants could contribute to tailoring lung surfactant formulation properties.The miscibility of per- or partially fluorinated surfactants, including alcohols, acids and single-chain derivatives of phosphocholine with DPPC has been investigated.125–131 Addition of the partially fluorinated (perfluorooctyl)pentanol (C8F17C5H10OH) to DPPC-Hel 13-5 peptide mixtures yielded significant enhancement of surfactant recovery when submitted to repeated compression–expansion cycles, and allowed reaching very low surface tension values upon film compression (Table 1).132 A mixed fluorocarbon–hydrocarbon ion pair compound (C8F17SO3− (C2H5)3N+ (CH2OCH2)10C16H33) was shown to reduce the inhibitory effect of fibrinogen by selectively interacting with DPPC and mimicking some of the surface properties of SP-C.133,134 In particular, it was found that fibrinogen could not penetrate in DPPC monolayers formed in the presence of this partially fluorinated compound.
7. Conclusions
The design of exogenous surfactant preparations efficient for the treatment of pulmonary conditions that involve inactivation of the lung surfactant (ARDS/ALI, MAS) is still a challenge. Analogues of the specific surfactant proteins and simple peptides have shown potential in the treatment of ARDS/ALI. Addition of polymers to existing surfactant substitute formulations represents a cost-efficient and versatile approach. Further work is needed however to test the new polymer-containing formulations in appropriate animal models, examine the pulmonary fluid balance and the gas transfer properties. Liquid fluorocarbons, which have shown efficacy in liquid ventilation procedures, could be utilised in the form of targeted emulsions to deliver exogenous surfactant in zones of the lung where it is most needed. Use of fluorocarbon gases, which have demonstrated anti-inflammatory properties and surface activity, and would be easy to administrate, certainly warrants preclinical investigations. The protecting effect of fluorocarbon gases against surfactant inhibitors other than albumin warrant further investigation. Fluorinated surfactants that have demonstrated resistance to inactivation by potent inhibitors show promise, as well as, possibly, combinations of fluorinated surfactants and fluorocarbon gases.Acknowledgements
We thank the French National Research Agency (ANR-14-CE35-0028-01) for funding, and Teclis Instruments (Longessaigne, France) for discussion and technical help.Notes and references
- L. B. Ware and M. A. Matthay, N. Engl. J. Med., 2000, 1334–1349 CrossRef CAS PubMed.
- H. W. Taeusch, J. Bernardino de la Serna, J. Perez-Gil, C. Alonso and J. A. Zasadzinski, Biophys. J., 2005, 89, 1769–1779 CrossRef CAS PubMed.
- A. Dushianthan, R. Cusack, V. Goss, A. D. Postle and M. P. W. Grocott, Crit. Care, 2012, 16, 238–248 CrossRef PubMed.
- R. Marshall, G. Bellingan and G. Laurent, Thorax, 1998, 53, 815–817 CrossRef CAS.
- G. R. Bernard, A. Artigas, K. L. Brigham, J. Carlet, K. Falke, L. Hudson, M. Lamy, J. R. Legall, A. Morris and R. Spragg, Am. J. Respir. Crit. Care Med., 1994, 149, 818–824 CrossRef CAS PubMed.
- B. D. Kamath, E. R. MacGuire, E. M. McClure, R. L. Goldenberg and A. H. Jobe, Pediatrics, 2011, 127, 1139–1146 CrossRef PubMed.
- Y. Y. Zuo, R. A. W. Veldhuizen, A. W. Neumann, N. O. Petersen and F. Possmayer, Biochim. Biophys. Acta, 2008, 1778, 1947–1977 CrossRef CAS PubMed.
- N. El-Gendy, A. Kaviratna, C. Berkland and P. Dhar, Ther. Delivery, 2013, 4, 951–980 CrossRef CAS PubMed.
- G. D. Rubenfeld, E. Caldwell, E. Peabody, J. Weaver, D. P. Martin, M. Neff, E. J. Stern and L. D. Hudson, N. Engl. J. Med., 2005, 353, 11685–11693 CrossRef PubMed.
- R. E. Hoekstra, J. C. Jackson, T. F. Myers, I. D. Franz, M. E. Stern, W. F. Powers, M. Maurer, J. R. Raye, S. T. Carrier, J. H. Gunkel and A. J. Gold, Pediatrics, 1991, 88, 10–18 CAS.
- H. W. Taeusch, Biol. Neonate, 2000, 77, 2–8 CrossRef CAS PubMed.
- J. G. Riess, Chem. Rev., 2001, 101, 2797–2920 CrossRef CAS PubMed.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, Chem. Rev., 2015, 115, 1106–1129 CrossRef CAS PubMed.
- R. H. Notter, Lung Surfactants: Basic Science and Clinical Applications, Marcel Dekker, New York, 2000 Search PubMed.
- J. Perez-Gil, Biochim. Biophys. Acta, 2008, 1778, 1676–1695 CrossRef CAS PubMed.
- S. Yu, P. G. Harding, N. Smith and F. Possmayer, Lipids, 1983, 18, 522–529 CrossRef CAS.
- A. D. Postle, E. L. Heeley and D. C. Wilton, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2001, 129, 65–73 CrossRef CAS.
- E. Parra and J. Perez-Gil, Chem. Phys. Lipids, 2015, 185, 153–175 CrossRef CAS PubMed.
- J. Goerke, Biochim. Biophys. Acta, 1998, 1408, 79–99 CrossRef CAS.
- F. Possmayer, Am. Rev. Respir. Dis., 1990, 142, 749–752 CrossRef CAS PubMed.
- J. R. Wright, Nat. Rev. Immunol., 2005, 5, 58–68 CrossRef CAS PubMed.
- H. Sano and Y. Kuroki, Mol. Immunol., 2005, 42, 279–287 CrossRef CAS PubMed.
- P. R. Lawson and K. B. Reid, Immunol. Rev., 2000, 173, 66–78 CAS.
- A. G. Serrano and J. Perez-Gil, Chem. Phys. Lipids, 2006, 141, 105–118 CrossRef CAS PubMed.
- J. Johansson and T. Curstedt, Eur. J. Biochem., 1997, 244, 675–676 CAS.
- N. Wüstneck, R. Wüstneck, J. Perez-Gil and U. Pison, Biophys. J., 2003, 84, 1940–1949 CrossRef.
- J. Johansson, Biochim. Biophys. Acta, 1998, 1408, 161–172 CrossRef CAS.
- J. A. Zasadzinski, J. Ding, H. E. Warriner, F. Bringezu and A. J. Waring, Curr. Opin. Colloid Interface Sci., 2001, 6, 506–513 CrossRef CAS.
- S. Schürch, R. Qanbar, H. Bachofen and F. Possmayer, Biol. Neonate, 1995, 67, 61–76 CrossRef PubMed.
- J. Kesecioglu, R. Beale, T. E. Stewart, G. P. Findlay, J. J. Rouby, L. Holzapfel, P. Bruins, E. J. Steenken, O. K. Jeppesen and B. Lachmann, Am. J. Respir. Crit. Care Med., 2009, 180, 989–994 CrossRef CAS PubMed.
- H. Meng, Y. Sun, J. Lu, S. Fu, Z. Meng, M. Scott and Q. Li, J. Cardiothorac. Vasc. Anesth., 2012, 26, 849–856 CrossRef CAS PubMed.
- R. G. Spragg, J. F. Lewis, W. Wurst, D. Häfner, R. P. Baughman, M. D. Wewers and J. J. Marsh, Am. J. Respir. Crit. Care Med., 2003, 167, 1562–1566 CrossRef PubMed.
- C. G. Cochrane, S. D. Revak, T. A. Merritt, G. P. Heldt, M. Hallman, M. D. Cunningham, D. Easa, A. Pramanik, D. K. Edwards and M. S. Alberts, Am. J. Respir. Crit. Care Med., 1996, 153, 404–410 CrossRef CAS PubMed.
- H. Nakahara, S. Lee, G. Sugihara, C.-H. Chang and O. Shibata, Langmuir, 2008, 3370–3379 CrossRef CAS PubMed.
- Y. Nakamura, K. Yukitake, H. Nakahara, S. Lee, O. Shibata and S. Lee, Biochim. Biophys. Acta, 2014, 1838, 2046–2052 CrossRef CAS PubMed.
- J. M. van Zyl, J. Smith and A. Hawtrey, Drug Des., Dev. Ther., 2013, 7, 139–148 CrossRef CAS PubMed.
- J. A. Zasadzinski, P. C. Stenger, I. Shieh and P. Dhar, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 801–828 CrossRef CAS PubMed.
- B. A. Holm, Z. Wang and R. H. Notter, Pediatr. Res., 1999, 46, 85–93 CrossRef CAS PubMed.
- A. Ishizaka, T. Matsuda, K. H. Albertine, H. Koh, S. Tasaka, N. Hasegawa, N. Kohno, T. Kotani, H. Morisaki, J. Takeda, M. Nakamura, X. H. Fang, T. R. Martin, M. A. Matthay and S. Hashimoto, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2004, 286, L1088–L1094 CrossRef CAS PubMed.
- M. Hallman, V. Glumoff and M. Ramet, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2001, 129, 287–294 CrossRef CAS.
- A. Günther, C. Ruppert, R. Schmidt, P. Markart, F. Grimminger, D. Walmrath and W. Seeger, Respir. Res., 2001, 2, 353–364 CrossRef.
- S. A. Tabak and R. H. Notter, J. Colloid Interface Sci., 1977, 59, 293–300 CrossRef CAS.
- B. A. Holm, R. H. Notter and J. N. Finkelstein, Chem. Phys. Lipids, 1985, 38, 287–298 CrossRef CAS.
- B. A. Holm, G. Enhorning and R. H. Notter, Chem. Phys. Lipids, 1988, 49, 49–55 CrossRef CAS.
- B. A. Holm, A. R. Venkitaraman, G. Enhorning and R. H. Notter, Chem. Phys. Lipids, 1990, 52, 243–250 CrossRef CAS.
- A. M. Cockshutt, J. Weitz and F. Possmayer, Biochemistry, 1990, 29, 8424–8429 CrossRef CAS.
- A. R. Venkitaraman, S. B. Hall, J. A. Whitsett and R. H. Notter, Chem. Phys. Lipids, 1990, 56, 185–194 CrossRef CAS.
- W. Seeger, C. Grube, A. Günther and R. Schmidt, Eur. Respir. J., 1993, 6, 971–977 CAS.
- Z. Wang and R. H. Notter, Am. J. Respir. Crit. Care Med., 1998, 158, 28–35 CrossRef CAS PubMed.
- W. Friedrich, G. Schmalisch, P. A. Stevens and R. R. Wauer, Eur. J. Med. Res., 2000, 19, 277–282 Search PubMed.
- H. E. Warriner, J. Ding, A. J. Waring and J. A. Zasadzinski, Biophys. J., 2002, 82, 835–842 CrossRef CAS.
- J. A. Zasadzinski, T. F. Alig, C. Alonso, J. Bernardino de la Serna, J. Perez-Gil and H. W. Taeusch, Biophys. J., 2005, 89, 1621–1629 CrossRef CAS PubMed.
- M. Martínez Sarrasague, A. Cimato, E. Rubin de Celis and G. Facorro, Respir. Physiol. Neurobiol., 2011, 175, 316–321 CrossRef PubMed.
- K. Nag, A. Hillier, K. Parsons and F. M. Garcia, Respir. Physiol. Neurobiol., 2007, 157, 411–424 CrossRef CAS PubMed.
- J. Li, R. Sanders, K. McAdam, C. Hales, B. Thompson, J. Gelfand and J. Burke, J. Trauma, 1989, 29, 1690–1697 CrossRef CAS PubMed.
- A. Saenz, A. Lopez-Sanchez, J. Mojica-Lazaro, L. Martinez-Caro, N. Nin, L. Bagatolli and C. Casals, FASEB J., 2010, 24, 3662–3673 CrossRef CAS PubMed.
- T. I. Yamada, M. B. L. Tabbor and A. H. Jobe, Am. Rev. Respir. Dis., 1990, 142, 754–757 CrossRef CAS PubMed.
- B. Sun, T. Curstedt, G. Lindgren, B. Franzen, A. A. Alaiya, A. Calkovska and B. Robertson, Eur. Respir. J., 1997, 10, 1967–1974 CrossRef CAS.
- K. W. Lu, J. Perez-Gil, M. Echaide and H. W. Taeusch, Biochim. Biophys. Acta, 2011, 1808, 2366–2373 CrossRef CAS PubMed.
- L. Gomez-Gil, D. Schürch, E. Goormaghtigh and J. Perez-Gil, Biophys. J., 2009, 97, 2736–2745 CrossRef CAS PubMed.
- Z. Wang, A. L. Schwan, L. L. Lairson, J. S. O'Donnell, G. F. Byrne, A. Foye, B. A. Holm and R. H. Notter, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2003, 285, L550–L559 CrossRef CAS PubMed.
- J. D. Amirkhanian, R. Bruni, A. J. Waring, C. Navar and H. W. Taeusch, Biochim. Biophys. Acta, 1993, 1168, 315–320 CrossRef CAS.
- F. J. Walther, J. M. Hernadez-Juviel, P. E. Mercado, L. M. Gordon and A. J. Waring, Biol. Neonate, 2002, 8, 181–187 CrossRef PubMed.
- A. Sato and M. Ikegami, PLoS One, 2012, 7, e39392 CAS.
- M. Seehase, J. J. P. Collins, E. Kuypers, R. K. Jellena, D. R. M. G. Ophelders, O. L. Ospina, J. Perez-Gil, F. Bianco, R. Garzia, R. Razzetti and B. W. Kramer, PLoS One, 2012, 7, e47631 CAS.
- H. W. Taeusch, K. W. Lu, J. Goerke and J. A. Clements, Am. J. Respir. Crit. Care Med., 1999, 164, 1531–1536 Search PubMed.
- T. Kobayashi, K. Ohta, K. Tashiro, K. Nishizuka, W.-M. Chen, S. Ohmura and D. Yamamoto, J. Appl. Physiol., 1999, 86, 1778–1784 CAS.
- K. Tashiro, X. Cui, T. Kobayashi, T. Curstedt and B. Robertson, Biol. Neonate, 2003, 83, 49–56 CrossRef CAS PubMed.
- K. W. Lu, H. W. Taeusch, B. Robertson, J. Goerke and J. A. Clements, Am. J. Respir. Crit. Care Med., 2000, 162, 623–628 CrossRef CAS PubMed.
- I. Minkov, K. Mircheva, N. Grozev, T. Ivanova and I. Panaiotov, Colloids Surf., B, 2013, 101, 135–142 CrossRef CAS PubMed.
- P. C. Stenger, S. G. Isbell, D. St. Hillaire and J. A. Zasadzinski, Langmuir, 2009, 25, 10045–10050 CrossRef CAS PubMed.
- G. A. Georgiev, C. Vassilieff, A. Jordanova, A. Tsanovad and Z. Lalcheva, Soft Matter, 2012, 8, 12072–12079 RSC.
- E. M. Scarpelli, Anat. Rec., 1998, 251, 491–527 CrossRef CAS.
- N. Kang, Z. Policova, G. Bankian, M. L. Hair, Y. Y. Zuo, A. W. Neumann and E. J. Acosta, Biochim. Biophys. Acta, 2008, 1778, 291–302 CrossRef CAS PubMed.
- E. J. Acosta, Z. Policova, S. Lee, A. Dang, M. L. Hair and A. W. Neumann, Biochim. Biophys. Acta, 2010, 1798, 489–497 CrossRef CAS PubMed.
- M. P. Krafft and J. G. Riess, Chem. Rev., 2009, 109, 1714–1792 CrossRef CAS PubMed.
- M. P. Krafft, Acc. Chem. Res., 2012, 45, 514–524 CrossRef CAS PubMed.
- M. P. Krafft and J. G. Riess, J. Polym. Sci., Polym. Chem. Ed., 2007, 45, 1185–1198 CrossRef CAS PubMed.
- E. S. Schutt, D. H. Klein, R. M. Mattrey and J. G. Riess, Angew. Chem., Int. Ed., 2003, 42, 3218–3235 CrossRef CAS PubMed.
- J. R. Lindner, Nat. Rev. Drug Discovery, 2004, 3, 527–532 CrossRef CAS PubMed.
- S. Sirsi and M. Borden, Bubble Sci., Eng., Technol., 2009, 1, 3–17 CrossRef CAS PubMed.
- E. Unger, T. Porter, J. Lindner and P. Grayburn, Adv. Drug Delivery Rev., 2014, 72, 110–126 CrossRef CAS PubMed.
- S. Schürch, Respir. Physiol., 1982, 48, 339–355 CrossRef.
- J. G. Riess and M. P. Krafft, Mater. Res. Soc. Bull., 1999, 24, 42–48 CrossRef CAS.
- M. Schmutz, B. Michels, P. Marie and M. P. Krafft, Langmuir, 2003, 19, 4889–4894 CrossRef CAS.
- P. N. Nguyen, T. T. Trinh Dang, G. Waton, T. Vandamme and M. P. Krafft, ChemPhysChem, 2011, 12, 2646–2652 CrossRef CAS PubMed.
- L. C. Clark and F. Gollan, Science, 1966, 152, 1755–1756 CAS.
- L. C. Clark, S. Kaplan, F. Becattini and G. Benzing, Fed. Proc., 1970, 29, 1764–1770 CAS.
- D. G. Ashbaugh, D. B. Bigelow, T. Petty and B. E. Levine, Lancet, 1967, 319–323 CrossRef CAS.
- M. R. Wolfson, N. E. Kechner, R. F. Roache, J. Dechadarevian, H. E. Friss, S. D. Rubenstein and T. H. Shaffer, J. Appl. Physiol., 1998, 84, 624–640 CAS.
- C. L. Leach, J. S. Greenspan, D. Rubenstein, T. H. Shaffer, M. R. Wolfson, J. C. Jackson, R. DeLemos and B. P. Fuhrman, N. Engl. J. Med., 1996, 335, 761–766 CrossRef CAS PubMed.
- M. Sukumar, M. Bommaraju, J. E. Fisher, F. C. Morin, M. C. Papo, B. P. Fuhrman, L. J. Hernan and C. L. Leach, J. Appl. Physiol., 1998, 84, 327–334 CAS.
- A. S. Tütuncü, N. S. Faithfull and B. Lachmann, Crit. Care Med., 1993, 21, 962–969 CrossRef PubMed.
- A. S. Tutuncu, K. Akpir, P. Mulder, W. Erdmann and B. Lachmann, Anesthesiology, 1993, 79, 1083–1093 CrossRef CAS PubMed.
- R. B. Hirschl, A. Parent, R. Tooley, M. McCracken, K. Johnson, T. H. Shaffer and M. R. Wolfson, Ann. Surg., 1995, 221, 79–88 CrossRef CAS PubMed.
- R. B. Hirschl, T. Pranikoff, C. Wise, M. C. Overbeck, P. Gauger, R. J. Schreiner, R. Dechert and R. H. Bartlett, J. Am. Med. Assoc., 1996, 275, 383–389 CrossRef CAS.
- P. G. Gauger, T. Pranikoff, R. J. Schreiner, F. W. Moler and R. B. Hirschl, Crit. Care Med., 1996, 24, 16–22 CrossRef CAS PubMed.
- M. A. Croce, T. C. Fabian, J. H. Patton, S. M. Melton, M. Moore and L. L. Trenthem, J. Trauma, 1998, 45, 273–282 CrossRef CAS PubMed.
- T. Koch, M. Ragaller, D. Haufe, A. Hofer, M. Grosser, D. M. Albrecht, M. Kotzsch and T. Luther, Anesthesiology, 2001, 94, 101–109 CrossRef CAS PubMed.
- J. U. Bleyl, M. Ragaller, U. Tschöh, M. Regner, M. Hübler, M. Kanzow, O. Vincent and M. Albrecht, Crit. Care Med., 2002, 30, 1340–1347 CrossRef CAS PubMed.
- W. Burkhardt, S. Kraft, M. Ochs, H. Proquitté, L. Mense and M. Rüdiger, PLoS One, 2012, 7, e47923 CAS.
- R. V. Dimel, M. Walch, H. P. Haagsman and G. Putz, Crit. Care Med., 2002, 30, 1083–1090 CrossRef PubMed.
- M. F. Krause, C. Jäckel, J. Haberstroh, J. Schulte-Möntig, J. U. Leititis and M. Orlowska-Volk, Pediatr. Res., 2001, 50, 34–43 CrossRef CAS PubMed.
- A. Hartog, D. Gommers, J. J. Haitsma and B. Lachmann, Br. J. Anaesth., 2000, 2000, 752–756 CrossRef PubMed.
- S. E. Chappell, M. Wolfson and T. Shaffer, Respir. Med., 2001, 95, 612–617 CrossRef CAS PubMed.
- S. Wolf, H. Lohbrunner, T. Busch, A. Sterner-Kock, M. Deja, A. Sarrafzadeh, U. Neumann and U. Kaisers, Br. J. Anaesth., 2001, 87, 593–601 CrossRef CAS PubMed.
- M.-J. Jeng, Y. R. Kou, C.-C. Sheu and B. Hwang, Crit. Care Med., 2003, 31, 1166–1174 CrossRef PubMed.
- J. D. Mrozek, K. M. Smith, S. C. Simonton, D. R. Bing, P. A. Meyers, J. E. Connett and M. C. Mammel, Crit. Care Med., 1999, 27, 1916–1922 CrossRef CAS PubMed.
- F. Gerber, M. P. Krafft, T. F. Vandamme, M. Goldmann and P. Fontaine, Angew. Chem., Int. Ed., 2005, 44, 2749–2752 CrossRef CAS PubMed.
- F. Gerber, M. P. Krafft, T. F. Vandamme, M. Goldmann and P. Fontaine, Biophys. J., 2006, 90, 3184–3192 CrossRef CAS PubMed.
- C. M. Knobler and R. C. Desai, Annu. Rev. Phys. Chem., 1992, 43, 207–236 CrossRef CAS.
- M. P. Krafft, V. B. Fainerman and R. Miller, Colloid Polym. Sci., 2015 DOI:10.1007/s00396-015-3622-8.
- F. Gerber, M. P. Krafft and T. F. Vandamme, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 490–494 CrossRef CAS PubMed.
- P. N. Nguyen, G. Waton, T. Vandamme and M. P. Krafft, Soft Matter, 2013, 9, 9972–9976 RSC.
- P. N. Nguyen, M. Veschgini, M. Tanaka, G. Waton, T. Vandamme and M. P. Krafft, Chem. Commun., 2014, 50, 11576–11579 RSC.
- P. N. Nguyen, G. Waton, T. Vandamme and M. P. Krafft, Angew. Chem., Int. Ed., 2013, 52, 6404–6408 CrossRef CAS PubMed.
- S. Lee, D. H. Kim and D. Needham, Langmuir, 2001, 17, 5544–5550 CrossRef CAS.
- G. Yang, M. O'Duill, V. Gouverneur and M. P. Krafft, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201502677R2.
- A. B. Kharge, Y. Wu and C. E. Perlman, J. Appl. Physiol., 2014, 117, 440–451 CrossRef PubMed.
- S. Sirsi, C. Pae, D. K. T. Oh, H. Blomback, A. Koubaa, B. Papahadjopoulos-Sternberg and M. Borden, Soft Matter, 2009, 5, 4835–4842 RSC.
- S. M. Vyas, J. Turánek, P. Knötigová, A. Kašná, V. Kvardová, V. Koganti, S. E. Rankin, B. Knutson and H.-J. Lehmler, New J. Chem., 2006, 30, 944–951 RSC.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Toxicol. Sci., 2007, 99, 366–394 CrossRef CAS PubMed.
- C. Lau, Reprod. Toxicol., 2012, 33, 405–409 CrossRef CAS PubMed.
- M. P. Krafft and J. G. Riess, Chemosphere, 2015, 129, 4–19 CrossRef CAS PubMed.
- H. Lehmler, M. Jay and P. Bummer, Langmuir, 2000, 16, 10161–10166 CrossRef CAS.
- T. Hiranita, S. Nakamura, M. Kawachi, H. M. Courrier, T. F. Vandamme, M. P. Krafft and O. Shibata, J. Colloid Interface Sci., 2003, 265, 83–92 CrossRef CAS.
- H. Nakahara, S. Nakamura, H. Kawasaki and O. Shibata, Colloids Surf., B, 2005, 41, 285–298 CrossRef CAS PubMed.
- H. J. Lehmler and P. M. Bummer, Colloids Surf., B, 2005, 44, 74–81 CrossRef CAS PubMed.
- S. Nakamura, H. Nakahara, M. P. Krafft and O. Shibata, Langmuir, 2007, 23, 12634–12644 CrossRef CAS PubMed.
- H. Nakahara, S. Lee, M. P. Krafft and O. Shibata, Langmuir, 2010, 26, 18256–18265 CrossRef CAS PubMed.
- A. Eftaiha, S. M. K. Brunet and M. F. Paige, Langmuir, 2012, 28, 15150–15159 CrossRef CAS PubMed.
- H. Nakahara, M. P. Krafft, A. Shibata and O. Shibata, Soft Matter, 2011, 7, 7325–7333 RSC.
- N. Aydogan, B. Uslu and H. Tanaci, J. Colloid Interface Sci., 2011, 360, 163–174 CrossRef CAS PubMed.
- G. U. Dilli, H. B. Uslu and N. Aydogan, Colloids Surf., B, 2014, 122, 566–575 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |