
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein dynamics: from rattling in a cage to structural relaxation
S.
Khodadadi
ab and
A. P.
Sokolov
*c
aFaculty of Applied Sciences, Delft University of Technology, Delft, The Netherlands
bDelft Projectmanagement B.V., Delft University of Technology, Delft, The Netherlands
cJoint Institute for Neutron Sciences, University of Tennessee, Knoxville, TN, USA. E-mail: sokolov@utk.edu
First published on 1st June 2015
Abstract
We present an overview of protein dynamics based mostly on results of neutron scattering, dielectric relaxation spectroscopy and molecular dynamics simulations. We identify several major classes of protein motions on the time scale from faster than picoseconds to several microseconds, and discuss the coupling of these processes to solvent dynamics. Our analysis suggests that the microsecond backbone relaxation process might be the main structural relaxation of the protein that defines its glass transition temperature, while faster processes present some localized secondary relaxations. Based on the overview, we formulate a general picture of protein dynamics and discuss the challenges in this field.
 S. Khodadadi | Sheila Khodadadi received her PhD in Polymer Science in 2009 from the University of Akron, USA. In 2010, she was awarded two years NRC Postdoctoral Research Associateship to conduct her research at NIST Center for Neutron Research, National Institute of Standards and Technology, USA. Currently, she is a researcher at Delft University of Technology in the Netherlands. Her main research interest is understanding protein–solvent dynamics and interactions in pharmaceutically relevant conditions. |
 A. P. Sokolov | Alexei Sokolov received an MS in Physics in 1981 from the Novosibirsk State University, and a PhD in Physics in 1986 from the Russian Academy of Sciences. He worked several years in Germany before joining faculty at the University of Akron, USA, in 1998. In 2009 he accepted Governor's Chair position at the University of Tennessee and Oak Ridge National Laboratory, where he leads the Soft Matter group. His current research interest focuses primarily on dynamics of soft materials, including topics of glass transition, polymer dynamics and dynamics of biological macromolecules, composite materials and materials for energy applications. |
I. Introduction
Biological macromolecules play a crucial role in all processes of life, from catalysis of biochemical reactions, to transport and to genetic codes. Proteins, RNAs and DNA work as small machines executing their functions through stochastic thermal fluctuations. It is well accepted that dynamics of these macromolecules are the key to their biochemical activity and function. Biological macromolecules in their normal state (e.g. under physiological conditions) do not possess a fixed structure. They fluctuate all the time between many conformational states.1–11 Currently we have reasonable knowledge of the structure of many proteins, and a clear understanding of the importance of protein dynamics and conformational fluctuations to their function. Yet our knowledge and understanding of the internal protein dynamics is very limited. The same is true for dynamics of RNA and DNA. Even a qualitative atomistic picture of dynamics of biological macromolecules is still absent. This is in contrast to the dynamics of other soft materials (e.g. synthetic polymers), where general classification of various relaxation processes exists and their characteristic behavior is known (although in many cases detailed microscopic mechanisms remain to be explained).12 There are several approaches where coarse-grained models are applied to describe longer time dynamics of biomacromolecules.13–17 They include traditional polymer models, such as worm-like and Rouse/Zimm models with internal friction,13,15 and colloidal models.16,17 However, as it has been discussed in several papers, these models either fail to describe all the data, or have to involve additional free parameters describing e.g. internal friction that is directly related to conformational fluctuations in biomacromolecules.11,17Many experimental and computational methods have been applied to studies of dynamics of biological macromolecules. Among them, NMR, molecular dynamics (MD) simulations and neutron scattering present the methods that analyze atomic motions directly. NMR is actively used in studies of structure and dynamics of proteins and results of these studies are discussed in several reviews.18–20 NMR presents very local information on motions of specific groups in protein structures. A very broad array of NMR techniques was able to analyze dynamics of specific groups and residues in the time range from tens of ps up to ms.21–26 Neutron scattering is a unique technique that provides analysis of dynamics not only in a broad time range, but also reveals the geometry of the underlying motions, can probe large scale (e.g. inter-domain) and collective dynamics.11,27–39 The high contrast between neutron scattering of hydrogen and deuterium atoms provides additional advantage for this technique in studies of biological molecules. For example, deuteration of solvent or a part of the molecule helps to drastically reduce their contribution to the neutron scattering spectra and separate the dynamics of the targeted part (containing H-atoms). Moreover, the energy of neutrons used in these experiments is in the meV range and they are not destructive for biological systems (in contrast to X-ray). Of course, MD simulations provide the most direct visualization of the atomic motions in proteins.6,9,10,40–43 The results, however, might depend strongly on force-fields and approximations used. The best approach in this case is a combination of MD-simulations with neutron scattering, because the measured experimental parameters can be directly calculated from MD simulated atomic trajectories. This also provides calibration and validation of simulations.
A significant drawback of neutron scattering is the limited frequency (time) range and low accuracy of the measured spectra. In contrast, broadband dielectric spectroscopy provides dynamic measurements in an extremely broad frequency range with very high accuracy. The measured spectra reflect reorientational motions of dipoles and translational motion of charges.44 This technique, however, does not provide microscopic information on the molecular motions involved. Combining dielectric spectroscopy with NMR, neutron scattering and MD simulations can be a very powerful approach to study details of molecular motions in a broad frequency and temperature range.41,45–50
In recent years significant progress has been achieved in single molecular studies of biomolecules. Using various fluorescence techniques, atomic force microscopy, optical and magnetic tweezers, coarse-grained dynamics and fluctuations of many biomacromolecules have been studied.13,15,51–55 These studies usually provide dynamics on a time scale longer than milliseconds and length scales of a few nm, missing atomistic details. Combination of the results obtained with these techniques with atomistic details learned from NMR, neutrons and MD-simulations might result in a significant breakthrough in our understanding of biomolecular dynamics. This topic, however, is out of scope of the current review.
Here we present an overview of studies of intra-molecular protein dynamics based mostly on neutron scattering, broadband dielectric spectroscopy and MD simulation results, although results from other techniques are also included. We focus on equilibrium fluctuations and conformational changes in the folded state on time scales from sub-picosecond to microseconds, and discuss the influence of temperature and hydration level on protein dynamics. We intentionally exclude the discussion of the folding/unfolding process, because it presents a separate important topic. In this review we aim to formulate a general atomistic picture of protein dynamics with classification of major relaxation processes that control conformational fluctuations. This classification is based on analogy with dynamics of soft materials, and aims at providing a framework to describe complex dynamics of biological macromolecules. We emphasize specificity of the relaxation processes in proteins that differentiate them from the classical dynamic processes of other soft materials.
II. Dynamics of soft materials
Soft Matter includes a wide range of materials from polymers and glass-forming liquids, to colloids and biological systems. The particular properties that differentiate soft materials from other materials include the existence of a large number of metastable states with comparable potential energies that are separated by relatively small energy barriers. So, there are constant fluctuations between different conformational states that result in a delicate balance between the entropic and enthalpic contributions to the free energy, and large thermodynamically equilibrium fluctuations, significant dynamic heterogeneity and strong sensitivity to small external forces and perturbations.12,56 These properties emphasize the distinct characteristics of the soft materials: permanent transitions between multiple meta-stable conformational states under relatively small perturbations or due to equilibrium fluctuations. So, the dynamics, i.e. motions of molecular (or other structural) units are the key to the main macroscopic properties of Soft Matter. This is directly applicable to biological macromolecules. They exhibit all the properties listed above and are very sensitive to slight variations in temperature, pH and other external perturbations.The dynamical processes that take place in soft materials are very complex and the microscopic mechanisms of many of them remain poorly understood. Cooperativity and dynamic heterogeneities are characteristic features of dynamics in soft materials.56–61Fig. 1 presents an overview of the general picture of the dynamic processes in soft materials with the example of polymer – polyisoprene (PI). One of them is fast dynamics that includes low-frequency collective vibrations, the so-called boson peak, and fast picosecond fluctuations.62,63 The former present excess vibrational modes relative to what is expected in a simple Debye model for acoustic modes.64,65 Debye-like behavior in density of vibrational states is usually observed in crystals, but all disordered systems show excess vibrational density of states. The nature of these excess vibrations remains a topic of active discussion, but most authors ascribe them to heterogeneity in elastic constants66,67 or to soft modes.68,69 The fast picosecond fluctuations are usually ascribed to rattling of a structural unit in a cage formed by neighboring units and is considered to be a precursor of structural relaxation.70,71 Its characteristic time scale has weak temperature variations reflecting very low energy barriers.72
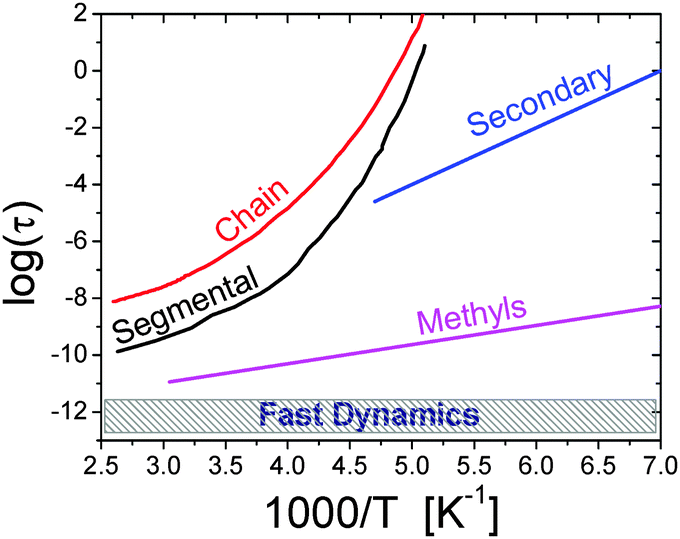 | ||
| Fig. 1 Traditional relaxation map of a soft material is illustrated with the example of the polymer polyisoprene.27 It includes (i) chain and (ii) segmental relaxation processes with strongly non-Arrhenius temperature variations; (iii) a secondary relaxation; (iv) methyl group rotation with Arrhenius temperature dependence; and (v) a fast picosecond relaxation with a rather weak temperature dependence of the characteristic relaxation time.27 The characteristic relaxation times are usually estimated from the maximum of the loss spectra (e.g. mechanical, dielectric, etc.), width of quasielastic neutron or light scattering spectra, or time decay of correlation functions of the corresponding relaxation process. | ||
Secondary relaxations are usually chemically specific and present mainly local conformational changes.12 The characteristic relaxation time has Arrhenius temperature dependence, τ = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(E/kBT), where τ is the relaxation time, Ea is the activation energy, kB is the Boltzman constant and T is temperature (Fig. 1). The term secondary relaxation includes a broad class of relaxation processes from some intra-molecular and side-group motions (e.g. methyl group rotation), to localized processes that are reminiscent of structural relaxation, the so-called Johari–Goldstein β-relaxation.12 Most of the secondary relaxations separate clearly from the main structural relaxation only at lower temperatures close to Tg, although some motions, such as methyl group rotation, remain faster than structural dynamics up to very high temperatures (Fig. 1).
exp(E/kBT), where τ is the relaxation time, Ea is the activation energy, kB is the Boltzman constant and T is temperature (Fig. 1). The term secondary relaxation includes a broad class of relaxation processes from some intra-molecular and side-group motions (e.g. methyl group rotation), to localized processes that are reminiscent of structural relaxation, the so-called Johari–Goldstein β-relaxation.12 Most of the secondary relaxations separate clearly from the main structural relaxation only at lower temperatures close to Tg, although some motions, such as methyl group rotation, remain faster than structural dynamics up to very high temperatures (Fig. 1).
The main structural relaxation (often called α-relaxation) is the main relaxation process that defines transition from a solid (jammed) to a liquid (flow) state. This relaxation process leads to the glass transition and is a collective motion that involves many structural units. It controls viscosity and diffusion in non-polymeric systems. It is called segmental relaxation in polymers where viscosity is defined by chain relaxation (see below). The main structural relaxation in most of the systems exhibits strongly non-Arrhenius temperature dependence that is traditionally described by the Vogel–Fulcher–Tammann (VFT) equation: τ = τ0exp[B/(T − T0)], where B and T0 are material dependent constants (Fig. 1). While the mechanism of this sharp slowing down of structural relaxation time with temperature decrease still remains a subject of active discussions,12,56,73,74 the role of dynamic cooperativity and heterogeneity becomes the center of attention in current research.56,57,75,76
The main structural relaxation is the longest relaxation process in non-polymeric systems. However, chain connectivity in polymers leads to an even slower relaxation process in macromolecules. Chain relaxation (Fig. 1) is the slowest relaxation process in polymers that defines their viscosity and macromolecular diffusion. Chain relaxation depends on the length of the chain (e.g. molecular weight) and is usually described by Rouse and reptation models.77
III. Dynamics of biomolecules
There is an analogy between dynamics of biological macromolecules (proteins, DNA and RNA) and other soft materials with many similarities, while there are also clear differences due to the globular nature of the proteins. Unlike in many other soft materials, a generally accepted picture for the protein dynamics even on a qualitative level is still missing. One of the traditional pictures for biomolecular dynamics is hierarchy of the energy landscape that defines motions in various tiers.2–5,78 There is a clear analogy between this approach and energy landscape models used for other soft materials.58 In this review, however, we will follow the same classification for dynamics of the soft materials as has been described in the previous section on example of a polymer. Studies discussed in this review cover a broad temperature range, far outside of the usual ambient temperature range. Although such a broad temperature range is not directly relevant to most of the biological functions and processes (except bio- and cryo-preservation), it is a traditional approach in physics that helps to disentangle various dynamic processes, analyze potential energy landscape, separate enthalpic and entropic contributions.III.1 Fast dynamics
The boson peak and fast picosecond relaxation have been observed in all proteins, DNA and RNA in the frequency range ∼100 GHz–1 THz.32,79–82 Neutron scattering spectra are usually presented as a dynamic structure factor S(Q,ν), which is the measured intensity normalized to the sample and scattering geometry; here Q – is the scattering wave vector and hν is the change in energy between the incoming and scattered neutrons (h is the Planck constant and ν is the frequency). The boson peak appears as an inelastic peak in S(Q,ν) at frequencies ∼1 THz, while fast relaxation appears as a quasielastic broadening at lower frequencies (Fig. 2). Analysis of neutron and light scattering spectra demonstrates that the fast dynamics depend strongly on hydration level (Fig. 2) and temperature.33,39,83,84 According to simulations,85 the boson peak vibrations present collective motions of amino acids that involve the whole protein and even water of hydration. Moreover, collective protein modes seem to propagate to the hydration water.86 It has been discovered recently that the boson peak in many proteins has a universal spectral shape that is similar to the spectral shape observed in glass-forming systems.87 The properties of the fast picosecond relaxation are similar to those of the fast relaxation known in all soft materials. It depends strongly on temperature and solvent, and can be ascribed to a “rattling” of amino acids in a cage formed by neighbor residues and solvent molecules.39 It can also be described as small-scale conformational fluctuations between different conformational states separated by a very low-energy barrier. The amplitude of these fluctuations is relatively small with mean-squared displacements (MSD) 〈r2〉 ∼ 0.15–0.4 Å2 under ambient conditions.37,39,88 It has been demonstrated that the fast protein dynamics is coupled strongly to the fast solvent dynamics.79,89–91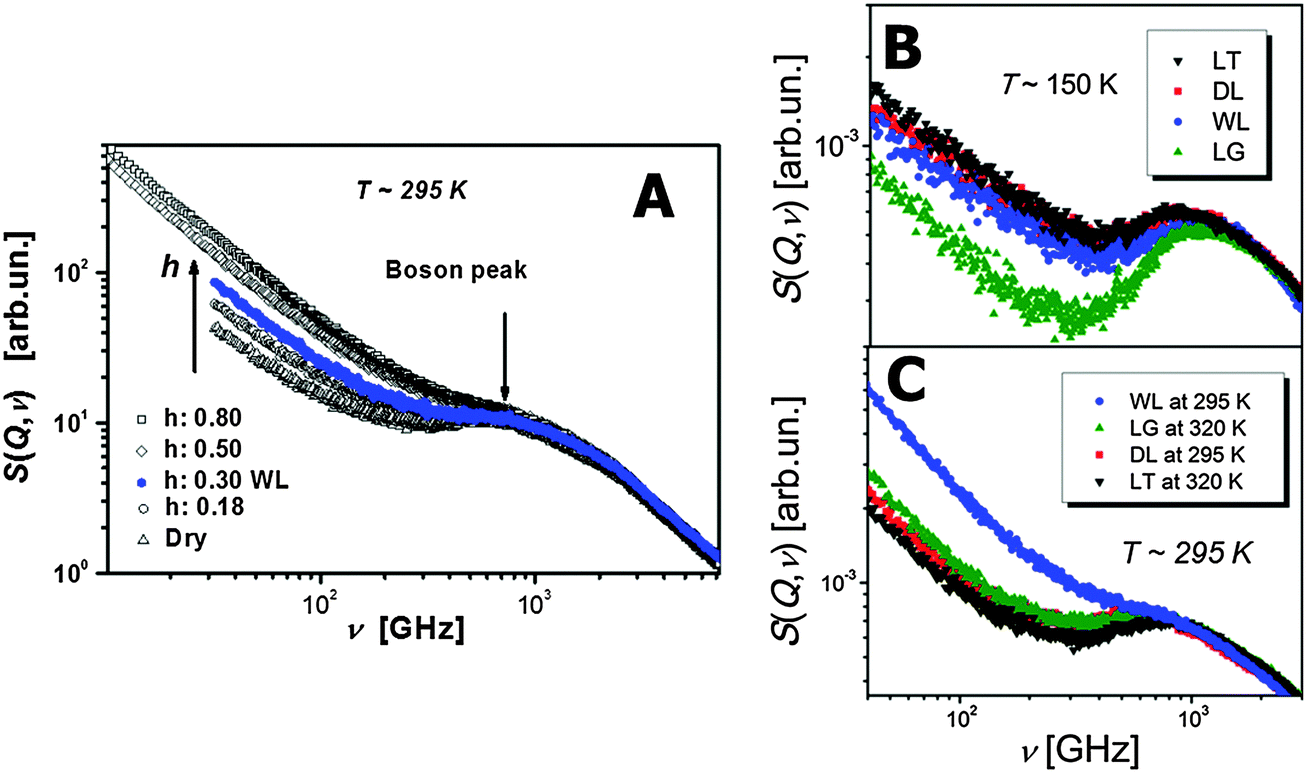 | ||
| Fig. 2 Dynamic structure factor S(Q,ν) of lysozyme at (A) different hydration levels39 (h refers to grams of water, D2O, per one gram of protein), and (B, C) different temperatures and solvents (DL – dry lysozyme; WL – hydrated lysozyme; LG – lysozyme in glycerol; LT – lysozyme in trehalose).39,90 (A) As the hydration level increases the Boson peak shifts to higher frequencies and the quasielastic contribution coming mainly from the fast relaxation process strongly increases. (B) At low T = 150 K the fast relaxation process is strongly suppressed in LG. While as the temperature is increased to ambient T ∼ 295 K. (C) WL has the most contribution from the fast relaxation process. This behavior of the protein's fast relaxation follows the behavior of the solvent.79,89 | ||
III.2 Methyl dynamics
Various side group motions (e.g. methyl group rotations, phenyl ring flips) exist in proteins. These relaxations usually have a broad distribution of relaxation times with Arrhenius-like temperature dependence. Methyl group dynamics deserve particular attention and has been actively studied using NMR, neutron scattering and simulations.18,19,39,42,84,92–95 All proteins contain a significant number of methyl groups. They provide proteins hydrophobicity, and they also play an important role in facilitating protein dynamics. Combined analysis of NMR and MD-simulations data suggests that methyl group dynamics can be used as an entropy meter for entire protein,96 because their NMR order parameter correlates well with conformational motions of side groups. Methyl group rotation has rather low energy barriers (E ∼ 10–20 kJ mol−1),39 and remains fast even at low hydrations and low temperatures.18,19,39,42,84,92–95 These observations suggest that methyl groups might play the role of internal plasticizers by facilitating protein dynamics even under extreme conditions of low hydration or temperature. The methyl groups show broad distribution of relaxation times that depends on the chemical structure of the residues and their position in the protein. It has been noticed in lysozyme and in myoglobin that the fastest methyl groups are placed around the active sites.19,39,42,94,97,98 Apparently there might be a particular role that nature assigns to methyl groups in proteins. It is possible that methyl groups and their position in proteins play the key role in providing conformational flexibility and additional entropy for active sites. It is important to notice that RNA has no methyl groups and only one nucleic acid of DNA has a single methyl group (Fig. 3C).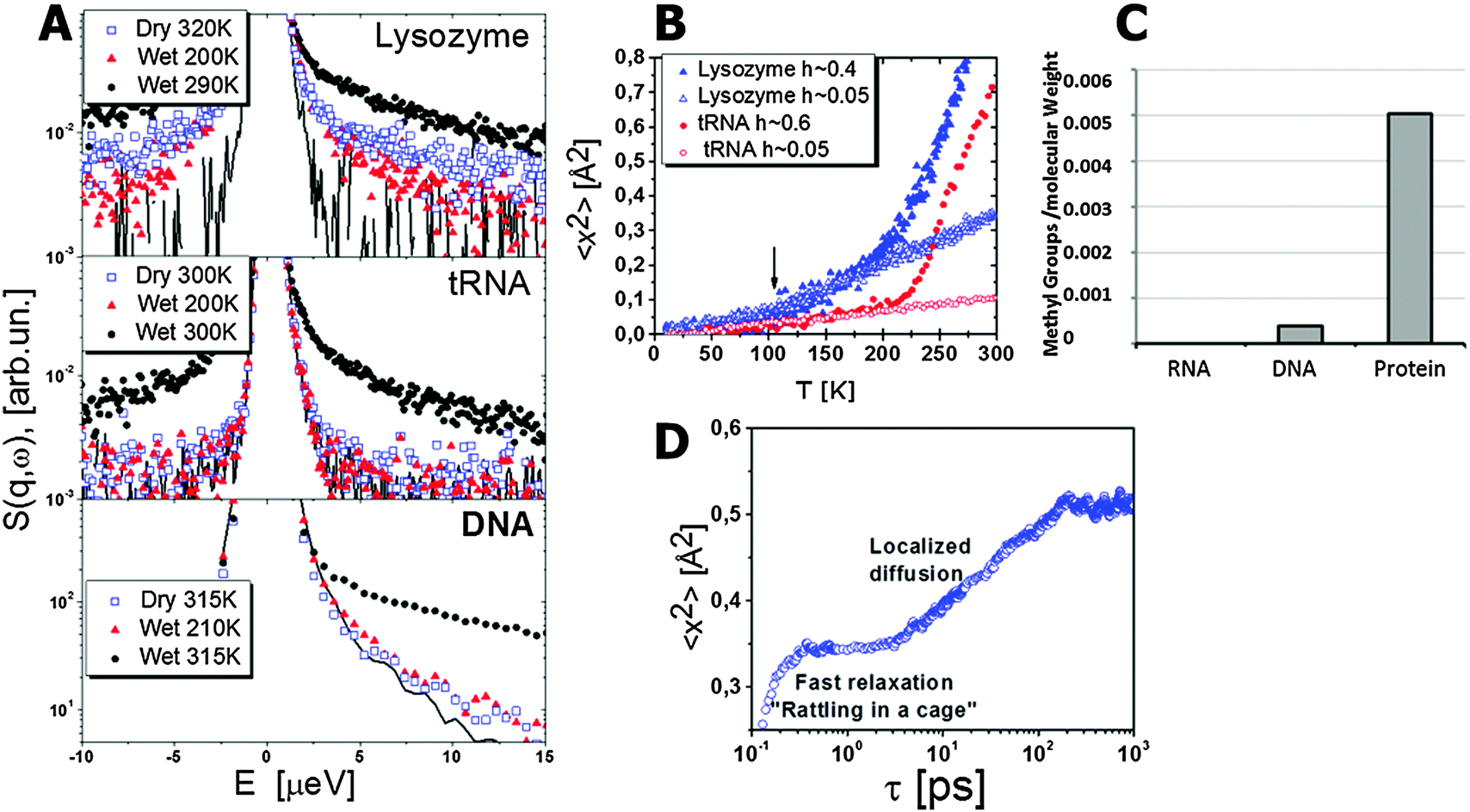 | ||
| Fig. 3 (A) Dynamic structure factor S(Q, E) of lysozyme, tRNA and DNA. Data for lysozyme and tRNA are obtained from ref. 92, for DNA from ref. 99. The relaxation process appears as a quasielastic scattering, i.e. extra broadening in comparison to the resolution function of the spectrometer (line). Lysozyme spectra exhibit relaxation contribution even in the dry state and at T = 200 K, while no quasielastic scattering is observed for dry tRNA and DNA, or for hydrated tRNA and DNA below 210 K. This broadening for lysozyme is related to the methyl group rotation. (B) Mean squared displacements demonstrate the onset of methyl group rotation at T ∼ 100 K for lysozyme as marked by the arrow. This onset is absent in MSD of tRNA. A sharp rise of MSD in hydrated biomacromolecules at T above ∼220 K is usually called the dynamic transition. (C) Methyl groups in different classes of biological macromolecules are presented in numbers of methyl groups per molecular weight.94 Proteins have a significant number of methyl groups in comparison to DNA (one out of four nucleic acids has a single methyl group) and RNA (no methyl in primary structure of RNA). (D) An example of MSD of hydrogen atoms in lysozyme involved in the localized diffusion motion as a function of time obtained from MD-simulations.6 It demonstrates fast dynamics and caging (plateau) at times shorter than ∼3 ps, followed by the localized diffusion with an almost logarithmic increase in MSD up to ∼200 ps. MSD saturates at longer times indicating localized motion. | ||
III.3 Sub-nanosecond process: coupled protein–solvent relaxation
Neutron scattering revealed an extremely stretched relaxation process in hydrated proteins with characteristic relaxation time τ ∼10–30 ps at room temperature (Fig. 4).6,31,35,39,48,84,100–102 Using hydrogen/deuterium contrast in neutron scattering, researchers were able to separate dynamics of hydration water and protein dynamics.102–105 The time scale of the protein process and its temperature dependence appear to be very similar to those of the hydration water (Fig. 5). The latter is ∼2–4 times slower than bulk water.102,103 However, analysis of the Q dependence revealed that hydration water shows diffusive translational motion, while this protein process is a localized motion, usually described as a “localized diffusion”.6,102 Analysis of MSD (Fig. 3D) demonstrates this point very clearly: MSD slowly increases due to this process, but saturates at time scales longer than ∼100–200 ps. Analysis of the atomic trajectories in MD-simulations6 revealed Brownian-like motions in a confined space, with radius of confinement ∼1–1.5 Å. This leads to the name of the process – ‘localized diffusion’.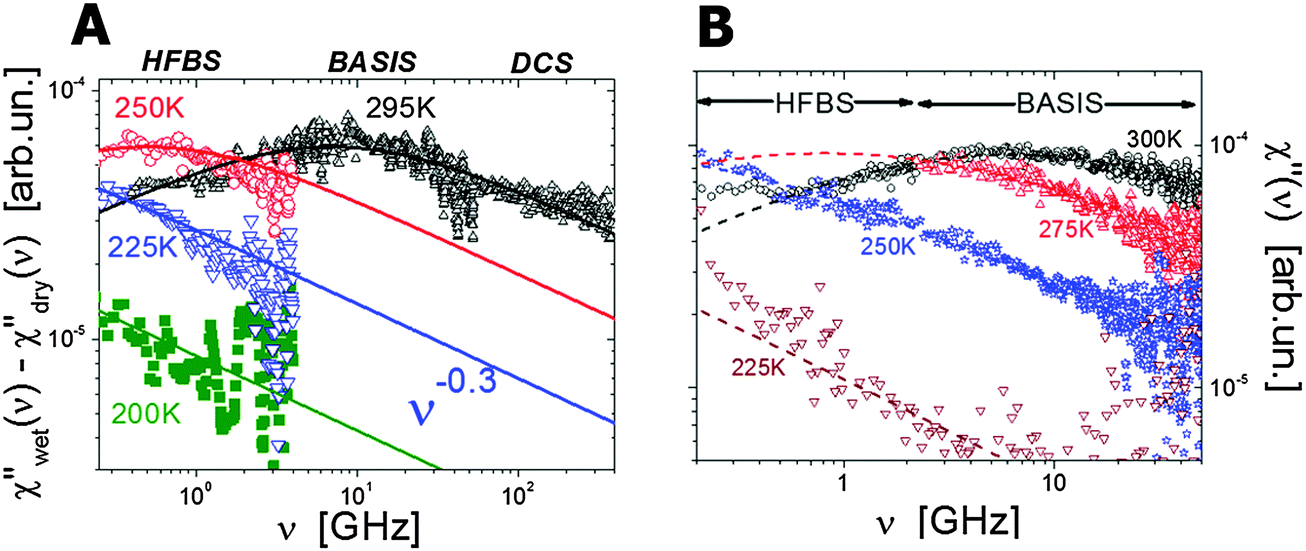 | ||
| Fig. 4 (A) Neutron scattering susceptibility spectra of the localized motion in hydrated lysozyme (h ∼ 0.35) at different temperature after correction for methyl group rotation.106 (B) Neutron scattering susceptibility of the localized motion in hydrated t-RNA (h ∼ 0.65) at different temperatures.122 Symbols are experimental data and lines are the fit to Cole–Cole distribution functions. Names at the top present different neutron scattering spectrometers used in these studies. | ||
 | ||
| Fig. 5 (A) Temperature dependence of characteristic relaxation times of hydrated lysozyme,48,122 myoglobin123 and tRNA,122 as estimated from broadband dielectric (closed symbols) and neutron scattering (open symbols) spectroscopies. In all cases the relaxation time was estimated from the susceptibility maximum (Fig. 4) or dielectric loss maximum, τ = (2πνmax)−1. (B) Temperature dependence of characteristic relaxation times estimated from quasielastic neutron scattering spectra of tRNA,122 lysozyme,48,122 and DNA124 hydrated with D2O; (C) characteristic relaxation times of hydration water of RNA,121 lysozyme,103 and DNA,120 estimated using quasielastic neutron scattering spectra. | ||
Since the pioneering work of Doster and co-workers,32 the protein relaxation spectra have also been analyzed in susceptibility presentation:
| χ′′(Q,ν) ∝ S(Q,ν)/n(ν,T) ≈ S(Q,ν) × hν/kT, | (1) |
(hν/kT) − 1]−1 is the Bose population factor. This presentation is similar to dielectric or mechanical loss spectra and has several advantages in comparison to the traditional dynamic structure factor. The susceptibility spectra of the protein process have symmetric spectral shape that is usually well described by a Cole–Cole distribution function, χ′′(Q,ν) ∝ 1/[1 + (iωτ)α], where ω is angular frequency, with the stretching parameter α ∼ 0.25–0.4 (Fig. 4).32,39,106 Such a strongly stretched process suggests significant dynamic heterogeneity in this motion. The appearance of this process and its relaxation time (defined from the frequency of the maximum τ = 1/ωmax) depends strongly on protein hydration,39,48 and solvent in general.91,107 This process exhibits slightly non-Arrhenius temperature dependence (Fig. 5A), and is associated with the so-called dynamic transition observed in hydrated protein as a sharp rise in MSD at T ∼ 200–230 K (Fig. 3B).31,37,92,106–111
Neutron scattering and simulations provide estimates of the localization length of this motion to be ∼1–3 Å.6,39,84,101 The spectral shape (symmetric stretching, Fig. 4) and localized nature of this process suggest that it can be ascribed to some kind of secondary relaxation in protein dynamics, although it seems to involve all (or most of) the residues.6,39–41,112 MD-simulations demonstrate that not only surface, but also core residues are involved in this motion. However, according to various simulations this process might involve mostly side group motions with much smaller involvement of the backbone.9,40,41 According to analysis of MD-simulations of lysozyme and Green Fluorescence Protein (GFP), this process can be described as an over-damped diffusive motion in a harmonic potential well.6,113 So, these motions are not jumps between different well-defined conformational states, but rather present a diffusion in a restricted space with a particular friction.
This process has also been observed in dielectric relaxation spectra and in this case is traditionally ascribed to hydration water dynamics.114–116 However, NMR, neutron scattering and simulations all reveal the existence of protein dynamics on the same time scale ∼10–50 ps.6,18,38,39,48,84,101,102,106,117 Coincidence of the characteristic time scales and similar temperature dependences of hydration water and protein dynamics (Fig. 5B and C) suggest strong coupling of those two processes. Apparently the translational motion of the hydration water molecules is strongly coupled to this localized motion of proteins. This has been clearly demonstrated from MD-simulations where the translational motion of water molecules has been artificially restricted.118 This restriction of water motions strongly suppressed the protein relaxation on the sub-nanosecond time scale.
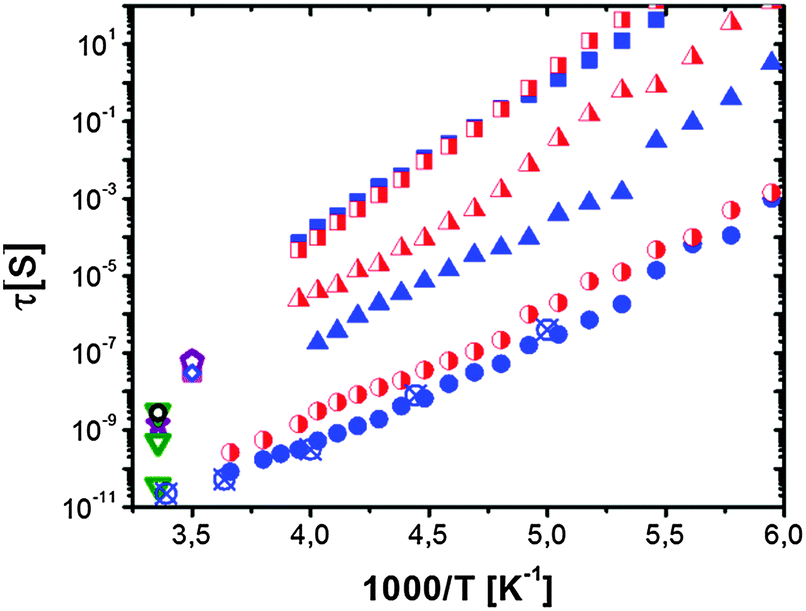
We emphasize that a similar relaxation process has also been observed in RNA and DNA dynamics (Fig. 4B), but in t-RNA it has a significantly larger localization length ∼7 Å.119 In the case of nucleic acids it exhibits stronger temperature dependence, with t-RNA showing slower dynamics than proteins like lysozyme and myoglobin, and DNA showing even slower dynamics than t-RNA (Fig. 5B). In all these cases it has been found that dynamics of hydration water shows the same difference (Fig. 5C).103,120,121 This result suggests that dynamics of biomolecules not only strongly coupled to hydration water dynamics, but also strongly influence the latter. There is no simple ‘slaving’ of biomolecular dynamics by its hydration water, rather there is a coupled motion where the chemical and 3-D structure of biomolecules play important roles.
III.4 Nanosecond dynamics: conformational jumps
Detailed analysis of MD-simulations suggested the existence of jump-like motions in proteins on the nanosecond time scale.6,9 Neutron scattering on protein solutions also detected relaxation processes on the time scale ∼10–30 ns.10,125,126 They present rather large atomic displacements ∼8 Å, and were ascribed to domain-like motions.10,28,29,125 The amplitude and time scale of this process changes upon ligand binding.21–23,25,125 A similar process has recently been reported for an intrinsically disordered protein Myelin basic protein.11 Apparently this nanosecond process does not necessarily present domain-like motion, but can be ascribed to more general conformational changes in proteins. In contrast to above-described “localized diffusion” process on the sub-nanosecond time scale, the nanosecond process presents jumps between well-defined conformational states.6,10,28,29,125 Protein relaxations in the ns time range has also been detected in many NMR studies.18,19 Recent detailed MD-simulation studies suggested broad distribution of energy barriers for conformational motions of side groups.127,128 They span from ∼10 kJ mol−1 up to ∼35 kJ mol−1 and higher.127 Methyl groups dominate the lower energy barriers motions, while aromatic side groups usually have higher energy barriers.127,128 There is also a heterogeneity in the behavior of hydrophobic and hydrophilic groups.128Dielectric spectroscopy revealed nanosecond relaxation in protein solutions and hydrated powders (Fig. 6).41,48,115,116,126,129–134 While this dielectric process is often ascribed to hydration water strongly bound to proteins,115,123,129,132,134,135 its large dielectric amplitude would require hundreds of tightly bound hydration water molecules per protein with dynamics of about ∼102–103 times slower than bulk water molecules.41 Such a behavior of hydration water contradicts current neutron scattering, NMR and simulations studies.6,10,18,19,28,29,38,41,101,125,136 Thus the nanosecond relaxation process should be attributed to the internal protein dynamics. The latter cause water continuum motions that might induce additional large dielectric response.
 | ||
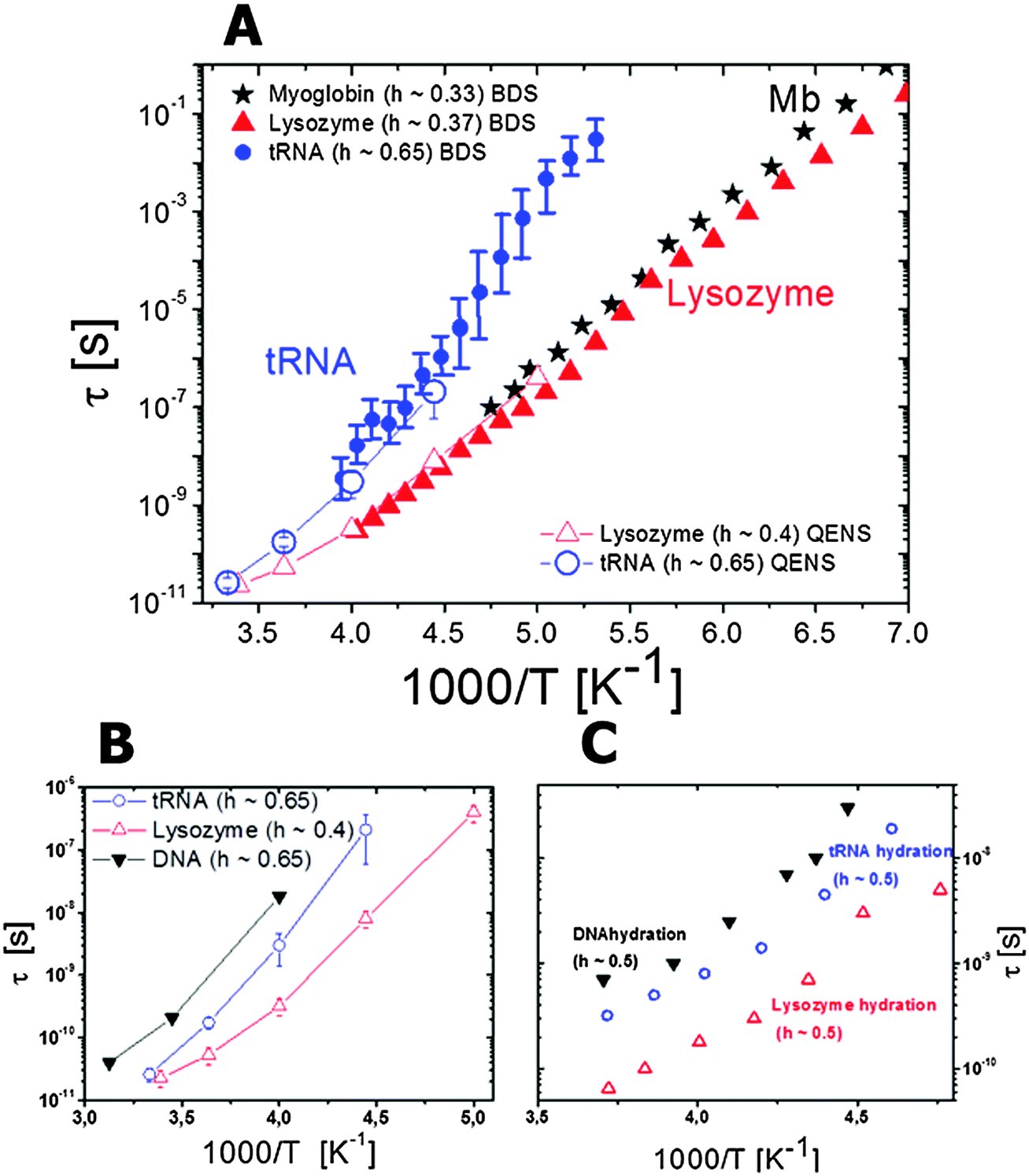
| Fig. 6 Temperature dependence of the relaxation times observed by dielectric spectroscopy for hydrated lysozyme (h ∼ 0.4; filled symbols) and for hydrated myoglobin (h ∼ 0.33; half-filled symbols) powders. Neutron scattering data for hydrated lysozyme (h ∼ 0.4) are shown as crossed circles (data taken from ref. 48 and 130). Circles are localized diffusion (sub-nanosecond coupled protein–solvent motion), triangles are domain motions (nanosecond process) and squares are segmental relaxation (microsecond dielectric process). For comparison the dielectric relaxation times in protein solutions at 298 K are also included: RNase A (open triangles116 and star137), lysozyme137 (open circle). Characteristic τ of the nanosecond relaxation estimated from neutron spin echo measurements are also shown: alcohol dehydrogenase28 (open diamond); PGK bound (open square) and unbound to the substrate (open pentagon) at 283 K (data from ref. 125). | ||
Temperature dependence of the nanosecond process has been analyzed mostly by dielectric relaxation spectroscopy.48,130,131,133 It shows slightly non-Arrhenius behavior similar to the sub-nanosecond relaxation process, and is ∼102–103 times slower than the latter (Fig. 6). This process in dielectric spectra slows down strongly with a decrease in the hydration level.48,130,131,133 The process is also strongly stretched and its characteristic relaxation time, according to dielectric spectroscopy, is comparable in different globular proteins, regardless of their size.137 This is consistent with a rather universal distribution of energy barriers for side group motions of different proteins suggested from MD-simulation analysis in ref. 127. This process definitely presents conformational jumps between well-defined states, and analysis of MD-simulation results suggests that the nanosecond process involves more side group motions than the motion of the backbone.9,41,128 It is most probably dominated by their conformational jumps with relatively large amplitude, and energy barriers controlled by the chemical structure, interactions and steric constraints. It involves the protein surface and core.41,128 According to neutron scattering, motions of secondary structures, e.g. domain-like motions in the case of multi-domain proteins, also appear in the ns time range. Its temperature dependence is most probably controlled by the behavior of the sub-nanosecond localized diffusion process that effectively defines a local friction for the nanosecond process. More detailed studies using MD-simulations and neutron scattering would be very helpful in unravelling microscopic details of the proteins nanosecond relaxation.
III.5 Microsecond dynamics: main structural relaxation?
Very recent dielectric spectroscopy studies of several hydrated protein powders revealed a relaxation process in the microsecond time scale.130,131,133 This process has an anomalously large dielectric amplitude, Δε ∼ 103–104.130,131,133 This is ∼10–100 times larger than the dielectric amplitude of relaxation in water, one of the most polar solvent. It is not possible to analyze this process in solution using dielectric spectroscopy because its characteristic time scale (∼1–20 μs) is much longer than the protein tumbling time (∼10–100 ns).116,129,132,134,137 This also creates a problem for studying this process using traditional NMR techniques. However there are NMR studies confirming the existence of backbone conformational dynamics at the microsecond timescale.138,139 Current neutron scattering spectroscopy is limited to hundreds of ns and cannot reach this time scale, and current MD-simulations are usually limited to hundreds of ns. Nevertheless, a few simulations that reach millisecond times indeed identified protein relaxations in microsecond time scales (Fig. 7).9,26 In the case of bovine pancreatic trypsin inhibitor (BPTI), it has been shown that rearrangement of the sulfide bridge accompanied by proximal loop motions appear in the microsecond time range.9 However for ubiquitin, it has been shown that the jumps in the microsecond time range are between multiple conformational states, not just between two favorable states, and present the main component of the backbone dynamics.26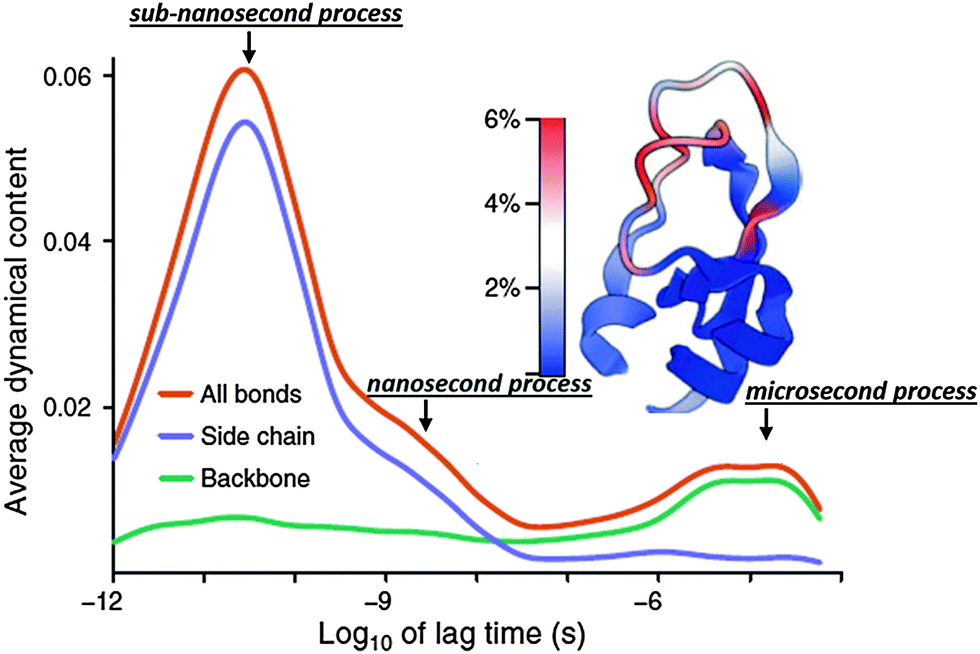 | ||
| Fig. 7 Dynamical content of the P2 internal correlation function and its decomposition into side chain and backbone contribution for bovine pancreatic trypsin inhibitor (BPTI) obtained from millisecond simulations.9 It demonstrates how dynamics of different protein parts spread over a wide range of time scale. Most important, it clearly indicates existence of three relaxation modes: (i) sub-nanosecond; (ii) nanosecond and (iii) microsecond relaxations. The latter is the only one dominated by the backbone motions. Adopted with permission from ref. 9. | ||
Thus analyses of MD simulations and NMR studies suggest that the microsecond process presents the main backbone motion, its rotation, and jumps between well-defined basins in potential energy landscape of the protein structures.9,26,138,139 However the general microscopic picture of this relaxation remains unknown. Its anomalously large dielectric amplitude provides some hints to its nature. Analysis of temperature dependence of the microsecond process in dielectric relaxation spectra suggests direct connection of this process to the glass transition in the hydrated protein system.130,131,133 It has strong dependence on the hydration level that agrees with calorimetric measurements of Tg in the same samples.140–142 In that case the microsecond relaxation presents the main structural relaxation of hydrated proteins. This connection makes it especially intriguing to understand the microscopic nature of this process. Moreover, this process appears on time scales usual for many biochemical processes and might be directly connected to the proteins function. Advanced MD-simulations, NMR and complementary scattering and spectroscopic studies might provide detailed microscopic information on the mechanism of the microsecond relaxation, and its dependence on protein structure, temperature and hydration.
III.6 Dynamics at longer times
Many single molecular studies revealed protein and nucleic acids dynamics on time scales much longer than microseconds.13–15,51–55 These dynamics are usually ascribed to chain-like motions using worm-like, or Rouse/Zimm models with internal friction. The latter is obviously controlled by the internal dynamics discussed above. However, the connection between atomistic details of dynamics on ps–μs time scales to these longer time dynamics remains to be studied, and is out of scope of the present review.IV. General picture of protein dynamics
Based on the presented overview of the experimental and computational results we propose a general picture of internal protein dynamics (Fig. 8) and analyze its similarity and differences with dynamics of other soft materials. First of all, there is clearly a broad spectrum of conformational fluctuations and transitions in all hydrated biomolecules. The internal dynamics under ambient conditions span over a wide time range from faster than picosecond to longer than microsecond, with atomic displacements ranging from smaller than 0.5 Å to ∼10 Å. As already emphasized by many researchers,9,143,144 protein samples contain a large amount of conformational states and resemble much closer a liquid than a solid state. Therefore the use of a protein crystallographic structure as a state of active protein is misleading.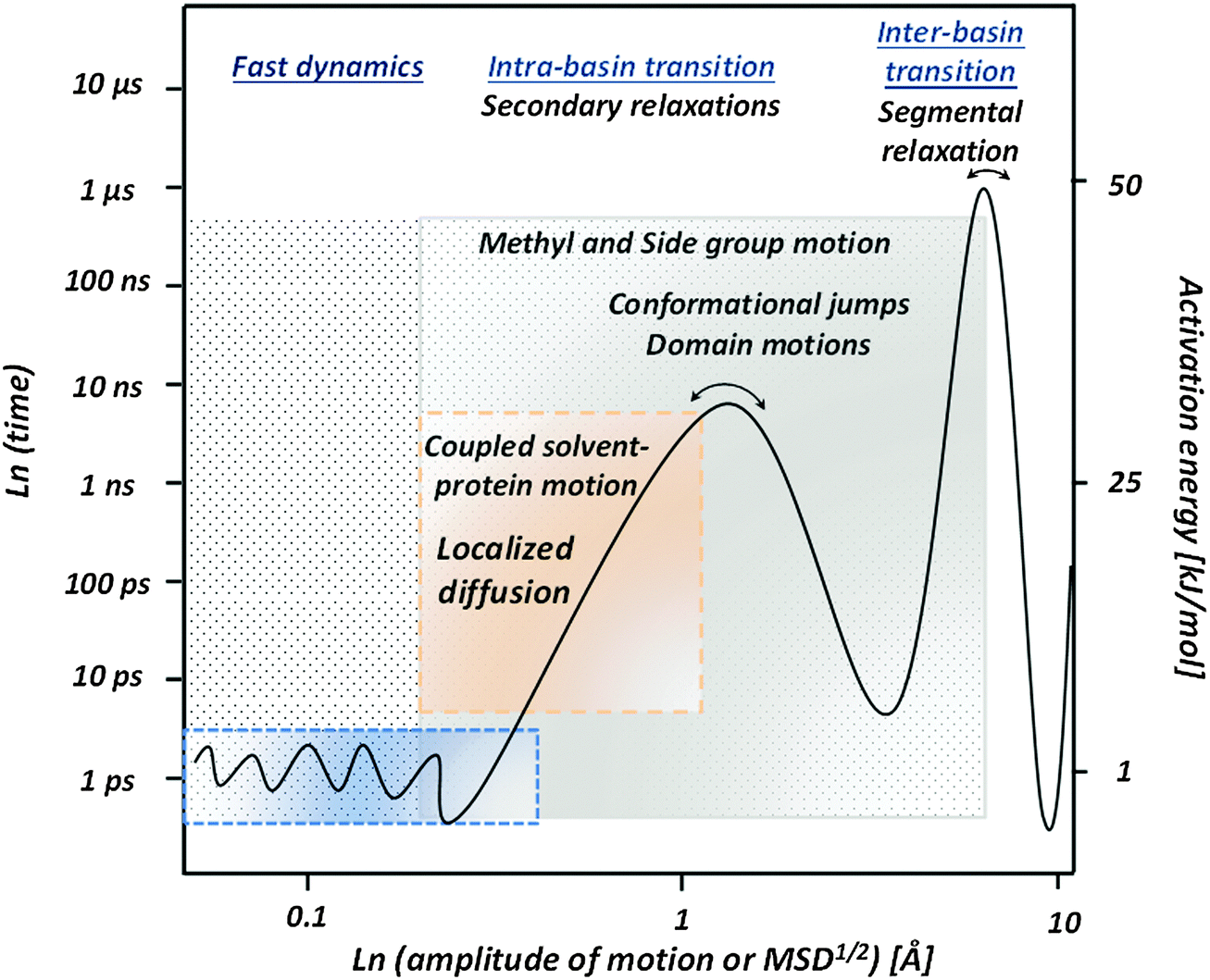 | ||
| Fig. 8 Proposed qualitative picture of hydrated protein dynamics at ambient temperature. Fast dynamics present small scale (∼0.1–0.5 Å) conformational fluctuations on a ps time scale. Intra-basin transitions present kinds of secondary relaxations. They include a coupled protein–solvent process on the sub-nanosecond time scale (localized diffusion) with an amplitude of 1–3 Å, methyl and other side group motions on time scales from ∼10 ps to many ns, domain motions and other conformational jumps with amplitude extending to several Å. Inter-basin transitions that can be considered as segmental relaxation in proteins appear on a microsecond time scale and should have an amplitude of several Å. The right axis shows the corresponding energy barriers that vary from a few kJ mol−1 for fast dynamics to more than 50 kJ mol−1 for segmental relaxation. | ||
On the fast time scale, ∼1 ps and faster, there are small conformational fluctuations reflecting ‘rattling in a cage’ with usual amplitude MSD ∼0.2–0.4 Å2. These motions have extremely low energy barriers of ∼2–5 kJ mol−1, and are characteristic for dynamics of all soft materials.69,71,72 The amplitude of this motion is strongly coupled to the fast dynamics in solvents79,89,90,145,146 (Fig. 2B), and its suppression was shown to be important for effective long-term biopreservation.90,145–147
Several relaxation processes are observed on the time scale ∼10–50 ps at ambient temperature (Fig. 8). They include methyl group rotations that apparently play an important role in facilitating protein dynamics and remain fast even at low hydration levels and lower temperatures.18,19,42,84,94,95,102,148 We speculate that the absence (or very low number) of methyl groups in nucleic acids (RNA and DNA) might be one of the reasons for much stronger slowing down of their dynamics with a decrease in temperature (Fig. 5). Methyl group dynamics in proteins is similar to their dynamics in synthetic polymers and can be considered as a usual secondary relaxation. According to NMR data, there are many other side group fluctuations and rotations on the same time scale.18–21,23,25,138
Translational and rotational dynamics of hydration water at ambient temperature also appears on the same time scale (Fig. 5 and 6). NMR, neutron scattering and MD-simulations all suggest that the hydration water is slowed down on average ∼2–4 times relative to the dynamics of bulk water.6,102,116,126 This slowing down is spatially heterogeneous and is stronger around protein polar groups.149–152 Moreover, most of the hydration water molecules exchange with the bulk water on a time scale much faster than one nanosecond.41 The characteristic activation energy barrier for hydration water motion is ∼15–18 kJ mol−1, but it increases with a decrease in temperature, leading to the VFT-like temperature dependence of the characteristic relaxation time (Fig. 5, 6 and 9). The VFT-like behavior is typical for Soft Matter and is usually ascribed to the cooperative nature of the relaxation process that involves several molecular units in a single relaxation event. We emphasize that dynamics of hydration water and its temperature dependence are different for proteins, t-RNA and DNA (Fig. 5C), reflecting the importance of the biomolecular structure in the behavior of hydration water.122
Proteins ‘localized diffusion’ also appears on the same time scale and presents conformational fluctuations of residues, side groups and backbone with an amplitude of ∼1–3 Å. Its time scale and temperature dependence (Fig. 5, 6, 8 and 9) suggest strong coupling of the localized diffusion to the dynamics of hydration water. Essentially this relaxation process presents a coupled hydration water – biomolecule relaxation, and its VFT-like behavior reflects the cooperative nature of this process. However, in contrast to translational motions of water, this process in proteins presents localized fluctuations (see, e.g.Fig. 3D). These conformational fluctuations involve the entire protein, its surface and core.41,112,153 The extremely strong stretching of its relaxation spectrum reflects significant dynamic heterogeneity apparently caused by the difference in the chemical structure and position of residues in proteins. This process depends strongly on the hydration level, and can be suppressed in dry proteins or in proteins placed in a glassy matrix.37–39,48,90,91,101,102,107,130 Its characteristic time scale and temperature dependence are dictated by the behavior of hydration water that together with internal rigidity/flexibility of the biomacromolecules control the friction of the sub-nanosecond process.
The sub-nanosecond coupled protein–solvent process is associated with the dynamic transition observed as a sharp increase in the MSD of hydrated proteins at T ∼ 220 K (Fig. 3B). Moreover, strong correlations of behavior of this process with biochemical or enzymatic activity of proteins31,39,108,110,154–157 suggest its importance for protein functions. Although this process is much faster than the characteristic time of biochemical reactions, it might present necessary precursors for slower protein dynamics, in particular, for motions directly involved in protein functions. The strength of this process reflects conformational flexibility of the protein on a ns time scale that might be critical for protein activity. In particular, the temperature dependence of the slower nanosecond process follows well the temperature dependence of the localized diffusion48,130 (Fig. 6 and 9).
The symmetrically stretched spectral shape and localized nature of this process are similar to the behavior of a secondary relaxation in other Soft Matter. However, its non-Arrhenius temperature dependence differs from temperature variations usually observed for secondary relaxation. Moreover, usual secondary relaxation would merge with structural relaxation at time scales ∼10−6–10−8 s, while the localized diffusion remains as a well separated process in biomacromolecules even at τ ∼ 10−11 s. The reason for the VFT-like behavior of this process might be its coupling to the structural relaxation of hydration water. This process is certainly not the main structural relaxation responsible for the glass transition of hydrated proteins at T ∼ 190–170 K. In fully hydrated lysozyme, this process reaches a relaxation time of ∼10−4 s at T ∼ 190–170 K,131,133,142 instead of the usual structural relaxation of ∼102–103 s at Tg. However, it is worth noticing that this process has an inflection point in its temperature dependence at T ∼ Tg.123,142,158–160 Thus we will classify this sub-nanosecond process as a specific secondary relaxation in biomacromolecules (Fig. 8). This assignment is supported by the dominating contribution of side group motions to this process (Fig. 7).
Larger scale conformational jumps with displacements on scale ∼1–10 Å appear on the nanosecond time scale6,8,10,11,28,125 (Fig. 6–9). The characteristic activation energy of this process should be ∼20–40 kJ mol−1 at ambient temperature, and its characteristic time scale shows VFT behavior that is similar to the VFT dependence of the sub-nanosecond process, but is slower than the latter by ∼102–103 times. It depends strongly on protein hydration, even stronger than the localized diffusion. Detailed analysis of neutron scattering suggests that in the case of proteins phosphoglycerate kinase125 and alcohol dehydrogenase28 the nanosecond relaxation is well described by domain motions. Detection of a similar process in intrinsically disordered proteins11,161 suggests that it can have a more general nature. It would be of great importance to understand whether this process can be described as rigid like motions and twist/bending of secondary structures, or more complicated motions have to be involved to describe the nanosecond process in various proteins. At the same time, NMR and MD-simulations studies reveal strong contribution of side group motions in this time range.9,18,19,40,128 They also present conformational jumps between different conformations of the side groups and show broad distribution of characteristic time scales, depending on the chemical structure and position of the residue in the protein. The nanosecond process is also analogous to secondary relaxation in other soft materials, and its temperature dependence most probably is controlled by the temperature dependence of the coupled solvent–protein process.
Recent dielectric relaxation studies on hydrated protein powders and MD-simulations extended to the millisecond time range both detected protein relaxations in the microsecond time range (Fig. 6–8). This process has an activation energy of the order of ∼50 kJ mol−1 at ambient temperature, exhibits slightly non-Arrhenius temperature dependence and has a strong dependence on hydration.130,131,133 Its possible relation to the glass transition temperature of hydrated proteins makes especially intriguing to unravel the microscopic nature of this process. It might be the main structural (segmental) relaxation of this system, analogous to structural relaxation in usual soft materials, and might present the motions directly involved in protein functions. This process can be considered as inter-basin jumps (Fig. 8). Unfortunately, very little is known currently about this process. Studies on this process are complicated due to several reasons: (i) its time scale is longer than the characteristic protein tumbling time in solutions; (ii) modern neutron scattering spectroscopy cannot reach this time; and (iii) only a few simulations have been performed at long enough time to equilibrate the system and detect the process. Dielectric spectroscopy reveals an anomalously large amplitude of this process,130,131,133 which remains a puzzle. At the same time, MD-simulations suggest that this process presents the main relaxation of the backbone.9,26 However even the length scale of this relaxation is not known, but obviously it should be larger than the length scale of the nanosecond process. It is also important that the characteristic energy barriers controlling this process are comparable to the barriers characteristic for peptide backbone rotation ∼50–100 kJ mol−1.162–164 Thus the microsecond process indeed can be segmental relaxation – the main structural relaxation in proteins.
The structural relaxation in folded proteins will have similarity and difference with segmental relaxation in synthetic polymers. The latter usually form random coils and on a long time scale segments can have a MSD comparable to the size of the coil, i.e. MSD ∼ Rg2. This is not possible for motions of residues in the protein as long as it stays folded. So, dynamics of residues will be always more localized/restricted than segmental dynamics in synthetic polymers. However, on the time scale of structural relaxation segments in synthetic polymers move only ∼1–2 Å, and this is possible even for residues in folded proteins. Backbone motions are the key for the segmental dynamics in polymers, and the microsecond process in proteins presents the backbone motions.9,26,138,139 Thus the proposed assignment of this process to structural relaxation of proteins seems justified. More studies are required to shed light on this probably most biologically relevant process. It is possible that this process reflects large scale changes in secondary structures and partial protein unfolding.
We want to emphasize the strong similarity of the proposed picture to the analysis of atomistic millisecond simulations of BPTI (Fig. 7): one can identify sub-nanosecond and nanosecond processes as large amplitude motions of side groups and the microsecond relaxation process dominated by the backbone motions. Based on this analysis we can propose the following hierarchy of protein dynamics (Fig. 8): at a time scale of about picosecond there are small conformational fluctuations with a characteristic length scale of ∼0.3–0.5 Å, and low energy barriers of ∼2–5 kJ mol−1. This is characteristic for the fast dynamics of all soft materials and presents some rattling in a cage formed by neighboring structural units. Methyl group dynamics appear on time scales of several ps and seem to facilitate protein dynamics even at very low temperatures or hydration levels. Methyl rotation has low energy barriers for rotation (∼10–20 kJ mol−1) and remains fast even at lower temperatures or low hydrations (Fig. 9). Dynamics of hydration water also appear on a time scale of ∼10–50 ps, but it has much stronger temperature dependence than methyl groups (Fig. 9). Dynamics of hydration water is coupled to protein's sub-nanosecond process, and the latter presents a kind of diffusion localized in a harmonic potential with the length scale limited to 1–3 Å (Fig. 8). The friction coefficient of this motion is defined by hydration water and internal protein friction (flexibility). As a result, the characteristic time scale of the localized diffusion depends strongly on the hydration level and follows temperature dependence of hydration water dynamics (Fig. 9). This motion might be a precursor for conformational jumps that occur on a nanosecond time scale and has an amplitude of ∼2–8 Å (Fig. 8). It has characteristic energy barriers ∼20–40 kJ mol−1 and presents a kind of intra-basin conformational jumps. Its temperature variations seem to be defined by the temperature dependence of the sub-nanosecond process (Fig. 9). But it depends on hydration even stronger than the latter.48,130,131 Both these processes, localized diffusion and nanosecond relaxation, involve mostly side group motions in the protein core and surface (Fig. 7). Thus they might be considered as secondary relaxations in biomolecules. Finally, the inter-basin relaxation appears at the microsecond time scale (Fig. 8). This microsecond process involves mostly the backbone rotation9,26 (Fig. 7) and apparently can be considered as segmental (structural) relaxation of the protein. It has a characteristic energy barrier >50 kJ mol−1 and depends strongly on hydration.130,131 This process might be the most directly related to protein functions, folding and other biologically relevant processes, including glass transition.
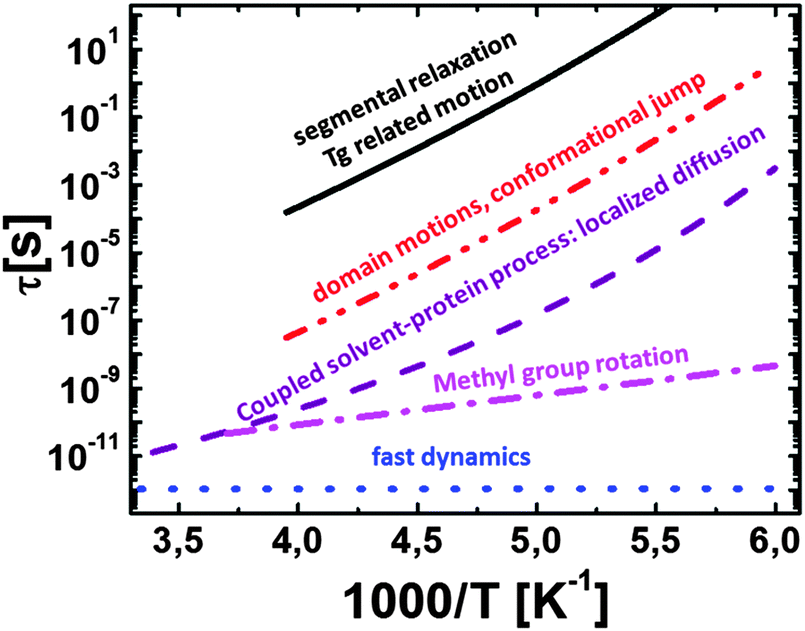 | ||
| Fig. 9 Schematic of temperature dependence of different internal motions of protein including: (i) main structural (segmental) relaxation reaching τ ∼ 100 s at the glass transition temperature Tg of hydrated proteins; (ii) domain motions, conformational jumps with nanosecond relaxation times at room temperature; (iii) coupled protein–solvent motions, presents “localized diffusion” of residues and side group motions with a relaxation time of tens of picosecond at room temperature; (iv) methyl group rotation with Arrhenius temperature dependence; and (v) fast picosecond relaxation with a rather weak temperature dependence of the characteristic relaxation time. Processes (ii)–(iv) are apparently kinds of secondary relaxation in proteins. | ||
In the final part, we would like to comment on the studies of protein dynamics at longer time and length scales, in many cases performed using single molecular experiments.13–17,51–55 These studies include different optical and pulling experiments with characteristic time scales usually longer than ms and displacements in the range 10–100 Å. Analysis of these experiments usually can be done using coarse-grained models where atomistic details of studied processes are hidden in friction terms and other phenomenological parameters. For example, authors of ref. 15 analyzed retraction of an extended DNA chain on the millisecond time scale using a classical worm-like model. This process reflects the chain-like motion of DNA and should be significantly slower than segmental (structural) relaxation. The relaxation processes discussed in this review at ps–μs time scales define the internal friction coefficient for such worm-like motions. The importance of internal conformational dynamics for coarse-grained models of proteins (e.g. colloidal-like approach, Zimm-like models) has also been emphasized in many papers.11,17 At present, these two worlds of protein studies, atomistically detailed analysis presented here and coarse-grained single molecular studies, do not overlap much. It would be extremely important to bridge these two approaches, and in this way broaden and deepen our understanding of the biomolecular dynamics.
V. Current challenges and future perspectives
Despite significant efforts towards studies on biomolecular dynamics, we are still missing a general atomistic picture of internal protein dynamics, classification of different relaxation processes and their connection to protein function and activity. There is no accepted theory or model of the protein dynamics even on a qualitative phenomenological level. In this review we attempt to formulate a general atomistic picture of biomolecular dynamics with clear separation of various relaxation processes. The presented picture (Fig. 8) is consistent with an earlier review by Henzler-Wildman and Kern,4 but provides more specific classification and more microscopic details based on neutron scattering, MD-simulations and dielectric spectroscopy studies.The presented review did not touch the important question of heterogeneity in protein dynamics caused by difference in rigidity of secondary structures, flexibility of loops and disordered regions of the protein structure. Based on hydrogen exchange rate studies, ideas of knots in protein structures with strongly suppressed dynamics have been proposed.165 MD simulations also revealed a smaller amplitude of motions in the protein core relative to the motions on the protein surface.41,112 The intrinsic dynamic heterogeneity might be a key to protein stability and unfolding, enzymatic activity and other protein functions. Unfortunately, this topic of protein research remains in its early stage (see e.g., ref. 128), and no systematic picture can be drawn at this time. Creating a general picture of protein dynamics and finding details of intrinsic heterogeneities is one of the challenges in future research on protein dynamics.
In this review we did not discuss the role of secondary and tertiary structure in protein dynamics, flexibility and stability. There is a large class of intrinsically disordered proteins that have no significant secondary structure in their natively unfolded state.166–168 Analysis of their dynamics demonstrated that they have higher flexibility and large amplitude of motions on a picosecond–nanosecond time scale than regular globular proteins.36,161,169,170 Whether intrinsically disordered proteins exhibit dynamics significantly different from the dynamics of globular proteins considered here remains to be studied.
We also did not discuss the role of solvents in biomolecular dynamics. This is another important topic that is directly related to the field of bio-preservation. Experimental and computation studies revealed a strong coupling of the fast dynamics and the sub-nanosecond process to the dynamics of solvents, such as water, glycerol and sugars.89–91,93,145–147,171 But detailed understanding of the role of the solvents in protein dynamics, activity and stability remains a great challenge that affects many life science and biotechnological applications. Developing a general concept of protein dynamics is one of the keys to address this challenge.
One of the important challenges is to reach longer time scales (milliseconds) with MD-simulations and with experimental techniques that provide microscopic information, such as neutron or X-ray scattering. While the former is feasible with developments of more powerful computers, faster algorithms and proper coarse-graining, the latter does not look promising in the near future. However, development of intense X-ray sources with short pulses opens a new possibility for analysis of kinetics of conformational changes in proteins.172 This might help in analysis of dynamics of biological macromolecules on time scales longer than microsecond. Also detailed dielectric and NMR studies combined with MD-simulations might provide the required information on protein dynamics on longer time scales, once the millisecond time scale becomes more accessible for atomistic MD. In addition, combining these atomistic studies with optical and single molecular studies might provide a breakthrough in understanding the connection between conformational dynamics and enzymatic activity of proteins.
The main challenge, in our view, remains the development of analytical models describing protein dynamics. They most probably will be based on traditional Langevin dynamics with frictions estimated from experimental data or atomistic MD-simulations. These models should provide a description of atomic motions, motions of secondary structures and loops, and even partial folding/unfolding of secondary structures due to thermodynamically equilibrium fluctuations. We hope that the presented review and the proposed general picture will help to develop such a model of the internal protein dynamics that will describe the sub-nanosecond localized diffusion, nanosecond conformational jumps and microsecond inter-basin transitions in a unified way.
Acknowledgements
We thank J. Smith and L. Hong for many helpful discussion. APS acknowledges NSF Polymer program for partial financial support under grant DMR-1408811.References
- A. J. Baldwin and L. E. Kay, Nature, 2012, 488, 165–166 CrossRef CAS PubMed.
- H. Frauenfelder, S. G. Sligar and P. G. Wolynes, Science, 1991, 254, 1598–1603 CAS.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129–5134 CrossRef CAS PubMed.
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- K. A. Henzler-Wildman, M. Lei, V. Thai, S. J. Kerns, M. Karplus and D. Kern, Nature, 2007, 450, 913–916 CrossRef CAS PubMed.
- L. Hong, N. Smolin, B. Lindner, A. P. Sokolov and J. C. Smith, Phys. Rev. Lett., 2011, 107, 148102 CrossRef.
- W. Köhler and J. Friedrich, J. Chem. Phys., 1989, 90, 1270–1273 CrossRef PubMed.
- O. F. Lange, N.-A. Lakomek, C. Farès, G. F. Schröder, S. Becker, K. F. A. Walter, J. Meiler, H. Grubmüller, C. Griesinger and B. L. De Groot, Science, 2008, 320, 1471–1475 CrossRef CAS PubMed.
- D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror, M. O. Eastwood, J. A. Bank, J. M. Jumper, J. K. Salmon, Y. Shan and W. Wriggers, Science, 2010, 330, 341–346 CrossRef CAS PubMed.
- N. Smolin, R. Biehl, G. R. Kneller, D. Richter and J. C. Smith, Biophys. J., 2012, 102, 1108–1117 CrossRef CAS PubMed.
- A. M. Stadler, L. Stingaciu, A. Radulescu, O. Holderer, M. Monkenbusch, R. Biehl and D. Richter, J. Am. Chem. Soc., 2014, 136, 6987–6994 CrossRef CAS PubMed.
- K. Ngai, Relaxation and Diffusion in Complex Systems, Springer, New York, 2011 Search PubMed.
- A. Soranno, B. Buchli, D. Nettels, R. R. Cheng, S. Müller-Späth, S. H. Pfeil, A. Hoffmann, E. A. Lipman, D. E. Makarov and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17800–17806 CrossRef CAS PubMed.
- R. R. Cheng, A. T. Hawk and D. E. Makarov, J. Chem. Phys., 2013, 138, 074112 CrossRef PubMed.
- O. Otto, S. Sturm, N. Laohakunakorn, U. F. Keyser and K. Kroy, Nat. Commun., 2013, 4, 1780 CrossRef PubMed.
- Y. Liu, L. Porcar, J. Chen, W.-R. Chen, P. Falus, A. Faraone, E. Fratini, K. Hong and P. Baglioni, J. Phys. Chem. B, 2011, 115, 7238–7247 CrossRef CAS PubMed.
- P. S. Sarangapani, S. D. Hudson, R. L. Jones, J. F. Douglas and J. A. Patha, Biophys. J., 2015, 108, 724–737 CrossRef CAS PubMed.
- M. Buck, J. Boyd, C. Redfield, D. A. MacKenzie, D. J. Jeenes, D. B. Archer and C. M. Dobson, Biochemistry, 1995, 34, 4041–4055 CrossRef CAS.
- A. Krushelnitsky and D. Reichert, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 1–25 CrossRef CAS PubMed.
- P. R. L. Markwick, T. Malliavin and M. Nilges, PLoS Comput. Biol., 2008, 4, e000168 Search PubMed.
- L. E. Kay, Nat. Struct. Biol., 1998, 5, 513–517 CrossRef CAS PubMed.
- L. E. kay, J. Magn. Reson., 2005, 173, 193–207 CrossRef CAS PubMed.
- A. Mittermaier and L. E. Kay, Science, 2006, 312, 224–228 CrossRef CAS PubMed.
- A. G. Palmer III, Curr. Opin. Struct. Biol., 1997, 7, 732–737 CrossRef.
- A. G. Palmer, Chem. Rev., 2004, 104, 3623–3640 CrossRef CAS PubMed.
- P. Maragakis, K. Lindorff-Larsen, M. P. Eastwood, R. O. Dror, J. L. Klepeis, I. T. Arkin, M. Ø. Jensen, H. Xu, N. Trbovic, R. A. Friesner, A. G. Palmer III and D. E. Shaw, J. Phys. Chem. B, 2008, 112, 6155–6158 CrossRef CAS PubMed.
- A. P. Sokolov and V. Garcia Sakai, in Dynamics of Soft Matter: Neutron Applications, ed. V. Garcia Sakai, C. Alba-Simionesco and S. H. Chen, Springer Science & Business Media, New York, 2012, ch. 1, pp. 1–23 Search PubMed.
- R. Biehl, B. Hoffmann, M. Monkenbusch, P. Falus, S. Préost, R. Merkel and D. Richter, Phys. Rev. Lett., 2008, 101, 138102 CrossRef.
- R. Biehl, M. Monkenbusch and D. Richter, Soft Matter, 2011, 7, 1299–1307 RSC.
- D. J. E. Callaway, B. Farago and Z. Bu, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 9891–9898 Search PubMed.
- W. Doster, S. Cusack and W. Petry, Nature, 1989, 337, 754–756 CrossRef CAS PubMed.
- W. Doster, S. Cusack and W. Petry, Phys. Rev. Lett., 1990, 65, 1080 CrossRef CAS.
- W. Doster, Neutron Scattering in Biology, Springer, 2006 Search PubMed.
- W. Doster, H. Nakagawa and M. S. Appavou, J. Chem. Phys., 2013, 139, 45105 CrossRef CAS PubMed.
- F. Gabel, D. Bicout, U. Lehnert, M. Tehei, M. Weik and G. Zaccai, Q. Rev. Biophys., 2002, 35, 327–367 CrossRef CAS.
- F.-X. Gallat, A. Laganowsky, K. Wood, F. Gabel, L. van Eijck, J. Wuttke, M. Moulin, M. Härtlein, D. Eisenberg, J.-P. Colletier, G. Zaccai and M. Weik, Biophys. J., 2012, 103, 129–136 CrossRef CAS PubMed.
- G. Zaccai, Science, 2000, 288, 1604–1607 CrossRef CAS.
- J. D. Nickels, S. Perticaroli, H. O'Neill, Q. Zhang, G. Ehlers and A. P. Sokolov, Biophys. J., 2013, 105, 2182–2187 CrossRef CAS PubMed.
- J. H. Roh, J. E. Curtis, S. Azzam, V. N. Novikov, I. Peral, Z. Chowdhuri, R. B. Gregory and A. P. Sokolov, Biophys. J., 2006, 91, 2573–2588 CrossRef CAS PubMed.
- D. C. Glass, M. Krishnan, D. R. Nutt and J. C. Smith, J. Chem. Theory Comput., 2010, 6, 1390–1400 CrossRef CAS.
- S. Khodadadi, J. E. Curtis and A. P. Sokolov, J. Phys. Chem. B, 2011, 115, 6222–6226 CrossRef CAS PubMed.
- M. Krishnan, V. Kurkal-Siebert and J. C. Smith, J. Phys. Chem. B, 2008, 112, 5522–5533 CrossRef CAS PubMed.
- J. C. Smith, Q. Rev. Biophys., 1991, 24, 227–291 CrossRef CAS.
- Broadband Dielectric Spectroscopy ed. F. Kremer and A. Schönhals, Springer, New York, 2003 Search PubMed.
- A. Arbe, J. Colmenero and D. Richter, in Broadband Dielectric Spectroscopy, ed. F. Kremer and A. Schönhals, Springer, New York, 2003, ch. 18, pp. 685–718 Search PubMed.
- A. Arbe, A.-C. Genix, S. Arrese-Igor, J. Colmenero and D. Richter, Macromolecules, 2010, 43, 3107–3119 CrossRef CAS.
- J. Colmenero, A. Arbe and A. Alegría, Phys. A, 1993, 201, 447–452 CrossRef CAS.
- S. Khodadadi, S. Pawlus and A. P. Sokolov, J. Phys. Chem. B, 2008, 112, 14273–14280 CrossRef CAS PubMed.
- A. Oleinikova, N. Smolin and I. Brovchenko, Biophys. J., 2007, 93, 2986–3000 CrossRef CAS PubMed.
- M. Tyagi, A. Alegría and J. Colmenero, J. Chem. Phys., 2005, 244909 CrossRef PubMed.
- X. Michalet, S. Weiss and M. Jäger, Chem. Rev., 2006, 106, 1785–1813 CrossRef CAS PubMed.
- A. A. Deniz, S. Mukhopadhyay and E. A. Lemke, J. R. Soc., Interface, 2008, 5, 15–45 CrossRef CAS PubMed.
- K. Bavishi and N. S. Hatzakis, Molecules, 2014, 19, 19407–19434 CrossRef PubMed.
- S. Mukhopadhyay, R. Krishnan, E. A. Lemke, S. Lindquist and A. A. Deniz, Proc. Natl. Acad. Sci. U. S. A., 2006, 104, 2649–2654 CrossRef PubMed.
- Y. Choi, I. S. Moody, P. C. Sims, S. R. Hunt, B. L. Corso, I. Perez, G. A. Weiss and P. G. Collins, Science, 2012, 335, 319–324 CrossRef CAS PubMed.
- L. Berthier, G. Biroli, J.-P. Bouchaud, L. Cipelletti and W. van Saarloos, Dynamical heterogeneities in glasses, colloids and granular materials, Oxford University Press, Oxford, 2011 Search PubMed.
- L. Berthier, G. Biroli, J.-P. Bouchaud, L. Cipelletti, D. El Masri, D. L'Hôte, F. Ladieu and M. Pierno, Science, 2005, 310, 1797–1800 CrossRef CAS PubMed.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS PubMed.
- X. H. Qiu and M. D. Ediger, J. Phys. Chem. B, 2003, 107, 459–464 CrossRef CAS.
- E. V. Russell and N. E. Israeloff, Nature, 2000, 408, 695–698 CrossRef CAS PubMed.
- E. R. Weeks, J. C. Crocker, A. C. Levitt, A. Schofield and D. A. Weitz, Science, 2000, 287, 627–631 CrossRef CAS.
- J. Jäckle, Amorphous Solids: Low-Temperature Properties, Springer, New York, 1981 Search PubMed.
- A. Sokolov, E. Rössler, A. Kisliuk and D. Quitmann, Phys. Rev. Lett., 1993, 71, 2062–2065 CrossRef CAS.
- U. Buchenau, A. Nücker and A. J. Dianoux, Phys. Rev. Lett., 1986, 53, 2316 CrossRef.
- V. K. Malinovsky, V. N. Novikov, P. P. Parshin, A. P. Sokolov and M. G. Zemlyanov, Europhys. Lett., 1990, 11, 43–47 CrossRef.
- W. Schirmacher, G. Ruocco and T. Scopigno, Phys. Rev. Lett., 2007, 98, 025501 CrossRef CAS.
- A. P. Sokolov, R. Calemczuk, B. Salce, A. Kisliuk, D. Quitmann and E. Duval, Phys. Rev. Lett., 1997, 78, 2405–2408 CrossRef CAS.
- V. L. Gurevich, D. A. Parshin, J. Pelous and H. R. Schober, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 16318–16331 CrossRef CAS.
- D. A. Parshin, H. R. Schober and V. L. Gurevich, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 064206 CrossRef.
- W. Gotze and L. Sjogren, Rep. Prog. Phys., 1992, 55, 241–376 CrossRef.
- W. Gotze and L. Sorgen, J. Phys.: Condens. Matter, 1989, 1, 4203–4222 CrossRef.
- N. V. Surovtsev, J. Wiedersich, V. N. Novikov, E. Rössler and A. P. Sokolov, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 14888–14891 CrossRef CAS.
- J. Dudowicz, K. F. Freed and J. F. Douglas, J. Chem. Phys., 2006, 124, 064901 CrossRef PubMed.
- S. Mirigian and K. S. Schweizer, J. Phys. Chem. Lett., 2013, 4, 3648–3653 CrossRef CAS.
- C. Dalle-Ferrie, C. Thibierge, C. Alba-Simionesco, L. Berthier, G. Biroli, J. P. Bouchaud, F. Ladieu, D. L'Hôte and G. Tarjus, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 041510 CrossRef.
- L. Hong, V. N. Novikov and A. P. Sokolov, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 061508 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- H. Frauenfelder and B. McMahon, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4795–4797 CrossRef CAS.
- G. Caliskan, A. Kisliuk, A. M. Tsai, C. L. Soles and A. P. Sokolov, J. Chem. Phys., 2003, 118, 4230–4236 CrossRef CAS PubMed.
- M. Kataoka, H. Kamikubo, J. Yunoki, F. Tokunaga, T. Kanaya, Y. Izumi and K. Shibata, J. Phys. Chem. Solids, 1999, 60, 1285–1289 CrossRef CAS.
- H. Leyser, W. Doster and M. Diehl, Phys. Rev. Lett., 1999, 82, 2987–2990 CrossRef CAS.
- A. Paciaroni, A. Orecchini, E. Cornicchi, M. Marconi, C. Petrillo, M. Haertlein, M. Moulin, H. Schober, M. Tarek and F. Sacchetti, Phys. Rev. Lett., 2008, 101, 148104 CrossRef CAS.
- S. Cusack and W. Doster, Biophys. J., 1990, 58, 243–251 CrossRef CAS.
- W. Doster and M. Settle, Biochim. Biophys. Acta, 2005, 1749, 173–186 CrossRef CAS PubMed.
- M. Tarek and D. Tobias, J. Chem. Phys., 2001, 115, 1607–1612 CrossRef CAS PubMed.
- V. C. Nibali, G. D'Angelo, A. Paciaroni, D. J. Tobias and M. Tarek, J. Phys. Chem. Lett., 2014, 5, 1181–1186 CrossRef.
- S. Perticaroli, J. D. Nickels, G. Ehlers and A. P. Sokolov, Biophys. J., 2014, 106, 2667–2674 CrossRef CAS PubMed.
- W. Doster, Eur. Biophys. J., 2008, 37, 591–602 CrossRef CAS PubMed.
- G. Caliskan, D. Mechtani, J. H. Roh, A. Kisliuk, A. P. Sokolov, S. Azzam, M. T. Cicerone, S. Lin-Gibson and I. Peral, J. Chem. Phys., 2004, 121, 1978–1983 CrossRef CAS PubMed.
- V. G. Sakai, S. Khodadadi, M. T. Cicerone, J. E. Curtis, A. P. Sokolov and J. H. Roh, Soft Matter, 2013, 9, 5336–5340 RSC.
- A. Paciaroni, S. Cinelli and G. Onori, Biophys. J., 2002, 83, 1157–1164 CrossRef CAS.
- G. Caliskan, R. M. Briber, D. Thirumalai, V. Garcia-Sakai, S. A. Woodson and A. P. Sokolov, J. Am. Chem. Soc., 2006, 128, 32–33 CrossRef CAS PubMed.
- E. Cornicchi, M. Marconi, G. Onori and A. Paciaroni, Biophys. J., 2006, 91, 289–297 CrossRef CAS PubMed.
- J. D. Nickels, J. E. Curtis, H. O'Neill and A. P. Sokolov, J. Biol. Phys., 2012, 38, 497–505 CrossRef CAS PubMed.
- K. Wood, D. J. Tobias, B. Kessler, F. Gabel, D. Oesterhelt, F. A. A. Mulder, G. Zaccai and M. Weik, J. Am. Chem. Soc., 2010, 132, 4990–4991 CrossRef CAS PubMed.
- V. Kasinath, K. A. Sharp and A. J. Wand, J. Am. Chem. Soc., 2013, 135, 15092–15100 CrossRef CAS PubMed.
- V. S. Bajaj, P. C. A. van der Wel and R. G. Griffin, J. Am. Chem. Soc., 2009, 131, 118–128 CrossRef CAS PubMed.
- K. B. Wong and V. Daggett, Biochemistry, 1998, 37, 11182–11192 CrossRef CAS PubMed.
- A. P. Sokolov, H. Grimm and A. Kisliuk, J. Biol. Phys., 2001, 27, 313–327 CrossRef CAS.
- J. Fitter, R. E. Lechner, G. Buldt and N. A. Dencher, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7600–7605 CrossRef CAS.
- J. D. Nickels, V. G. Sakai and A. P. Sokolov, J. Phys. Chem. B, 2013, 117, 11548–11555 CrossRef CAS PubMed.
- J. D. Nickels, H. O'Neill, L. Hong, M. Tyagi, G. Ehlers, K. Weiss, Q. Zhang, Z. Yi, E. Mamontov, J. C. Smith and A. P. Sokolov, Biophys. J., 2012, 103, 1566–1575 CrossRef CAS PubMed.
- S.-H. Chen, L. Liu, E. Fratini, P. Baglioni, A. Faraone and E. Mamontov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9012–9016 CrossRef CAS PubMed.
- K. Wood, M. Plazanet, F. Gabel, B. Kessler, D. Oesterhelt, D. J. Tobias, G. Zaccai and M. Weik, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18049–18054 CrossRef CAS PubMed.
- K. Wood, A. Frölich, A. Paciaroni, M. Moulin, M. Härtlein, G. Zaccai, D. J. Tobias and M. Weik, J. Am. Chem. Soc., 2008, 130, 4586–4587 CrossRef CAS PubMed.
- S. Khodadadi, S. Pawlus, J. H. Roh, V. Garcia Sakai, E. Mamontov and A. P. Sokolov, J. Chem. Phys., 2008, 128, 195106 CrossRef CAS PubMed.
- A. M. Tsai, D. A. Neumann and L. N. Bell, Biophys. J., 2000, 79, 2728–2732 CrossRef CAS.
- R. M. Daniel, J. L. Finney, V. Réat, R. Dunn, M. Ferrand and J. C. Smith, Biophys. J., 1999, 77, 2184–2190 CrossRef CAS.
- F. G. Parak, Rep. Prog. Phys., 2003, 66, 103–129 CrossRef CAS.
- B. F. Rasmussen, A. M. Stock, D. Ringe and G. A. Petsko, Nature, 1992, 357, 423–424 CrossRef CAS PubMed.
- A. P. Sokolov, H. Grimm and R. Kahn, J. Chem. Phys., 1999, 110, 7053–7057 CrossRef CAS PubMed.
- L. Hong, X. Cheng, D. C. Glass and J. C. Smith, Phys. Rev. Lett., 2012, 108, 238102 CrossRef.
- L. Hong, D. C. Glass, J. D. Nickels, S. Perticaroli, Z. Yi, M. Tyagi, H. O'Neill, Q. Zhang, A. P. Sokolov and J. C. Smith, Phys. Rev. Lett., 2013, 110, 028104 CrossRef.
- N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013–2045 CrossRef CAS PubMed.
- N. Miura, Y. Hayashi and S. Mashimo, Biopolymers, 1996, 39, 183–187 CrossRef CAS.
- A. Oleinikova, P. Sasisanker and H. Weingartner, J. Phys. Chem. B, 2004, 108, 8467–8474 CrossRef CAS.
- J. Perez, J.-M. Zanotti and D. Durand, Biophys. J., 1999, 77, 454–469 CrossRef CAS.
- M. Tarek and D. J. Tobias, Phys. Rev. Lett., 2002, 88, 138101 CrossRef CAS.
- J. H. Roh, R. M. Briber, A. Damjanovic, D. Thirumalai, S. A. Woodson and A. P. Sokolov, Biophys. J., 2009, 96, 2755–2762 CrossRef CAS PubMed.
- S.-H. Chen, L. Liu, X. Chu, Y. Zhang, E. Fratini, P. Baglioni, A. Faraone and E. Mamontov, J. Chem. Phys., 2006, 125, 171103 CrossRef PubMed.
- X.-Q. Chu, E. Fratini, P. Baglioni, A. Faraone and S.-H. Chen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 011908 CrossRef.
- S. Khodadadi, J. H. Roh, A. Kisliuk, E. Mamontov, M. Tyagi, S. A. Woodson, R. M. Briber and A. P. Sokolov, Biophys. J., 2010, 98, 1321–1326 CrossRef CAS PubMed.
- J. Swenson, H. Jansson and R. Bergman, Phys. Rev. Lett., 2006, 96, 247802 CrossRef.
- A. P. Sokolov and R. B. Gregory, in Neutron Scattering in Biology: Techniques and Applications, ed. J. Fitter, T. Gutberlet and J. Katsaras, Springer Science & Business Media, New York, 2006, ch. 21, pp. 485–502 Search PubMed.
- R. Inoue, R. Biehl, T. Rosenkranz, J. Fitter, M. Monkenbusch, A. Radulescu, B. B. Farago and D. Richter, Biophys. J., 2010, 99, 2309–2317 CrossRef CAS PubMed.
- F. Sterpone, G. Stirnemann and D. Laage, J. Am. Chem. Soc., 2012, 134, 4116–4119 CrossRef CAS PubMed.
- R. Rajeshwar, J. C. Smith and M. Krishnan, J. Am. Chem. Soc., 2014, 136, 8590–8605 CrossRef PubMed.
- Y. Miao, Z. Yi, D. C. Glass, L. Hong, M. Tyagi, J. Baudry, N. Jain and J. C. Smith, J. Am. Chem. Soc., 2012, 134, 19576–19579 CrossRef CAS PubMed.
- C. Cametti, S. Marchetti, C. M. C. Gambi and G. Onori, J. Phys. Chem. B, 2011, 115, 7144–7153 CrossRef CAS PubMed.
- M. Nakanishi and A. P. Sokolov, J. Non-Cryst. Solids, 2015, 407, 478–485 CrossRef CAS PubMed.
- A. K. A. Panagopoulou, N. Shinyashiki and P. Pissis, J. Phys. Chem. B, 2012, 116, 4593–4602 CrossRef CAS PubMed.
- R. Pethig, Annu. Rev. Phys. Chem., 1992, 43, 177–205 CrossRef CAS PubMed.
- N. Shinyashiki, W. Yamamoto, A. Yokoyama, T. Yoshinari, S. Yagihara, R. Kita, K. L. Ngai and S. Capaccioli, J. Phys. Chem. B, 2009, 113, 14448–14456 CrossRef CAS PubMed.
- M. Wolf, R. Gulich, P. Lunkenheimer and A. Loidl, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 723–730 CrossRef CAS PubMed.
- H. Jansson, R. Bergman and J. Swenson, J. Phys. Chem. B, 2005, 109, 24134–24141 CrossRef CAS PubMed.
- J. Lal, P. Fouquet, M. Maccarini and L. Makowski, J. Mol. Biol., 2010, 397, 423–435 CrossRef CAS PubMed.
- N. Miura, N. Asaka, N. Shinyashiki and S. Mashimo, Biopolymers, 1994, 34, 357–364 CrossRef CAS PubMed.
- J. D. Haller and P. Schanda, J. Biomol. NMR, 2013, 57, 263–280 CrossRef CAS PubMed.
- F. Massi, M. J. Grey and A. G. Palmer III, Protein Sci., 2005, 14, 735–742 CrossRef CAS PubMed.
- F. Demmel, W. Doster, W. Petry and A. Schulte, Eur. Biophys. J., 1997, 26, 327–335 CrossRef CAS.
- T. M. Y. Miyazaki and H. Suga, J. Phys. Chem. B, 2000, 104, 8044–8052 CrossRef.
- S. Khodadadi, A. Malkovskiy, A. Kisliuk and A. P. Sokolov, Biochim. Biophys. Acta, 2010, 1804, 15–19 CrossRef CAS PubMed.
- R. B. B. Lindorff-Larsen, M. A. DePristo, C. M. Dobson and M. Vendruscolo, Nature, 2005, 433, 128–132 CrossRef PubMed.
- D. V. Y. Zhou and M. Karplus, J. Mol. Biol., 1999, 285, 1371–1375 CrossRef PubMed.
- T. E. Dirama, G. A. Carri and A. P. Sokolov, J. Chem. Phys., 2005, 122, 244910 CrossRef PubMed.
- T. E. Dirama, J. E. Curtis, G. A. Carri and A. P. Sokolov, J. Chem. Phys., 2006, 124, 034901 CrossRef PubMed.
- M. T. Cicerone and C. L. Soles, Biophys. J., 2004, 86, 3836–3845 CrossRef CAS PubMed.
- J. H. Roh, V. N. Novikov, R. B. Gregory, J. E. Curtis, Z. Chowdhuri and A. P. Sokolov, Phys. Rev. Lett., 2005, 95, 038101 CrossRef CAS.
- J. T. King, E. J. Arthur, C. L. Brooks and K. J. Kubarych, J. Phys. Chem. B, 2012, 116, 5604–5611 CrossRef CAS PubMed.
- O. Rahaman, S. Melchionna, D. Laage and F. Sterpone, Phys. Chem. Chem. Phys., 2013, 15, 3570–3576 RSC.
- D. Russo, G. Hura and T. Head-Gordon, Biophys. J., 2004, 60, 1852–1862 CrossRef.
- W. Qiu, Y.-T. Kao, L. Zhang, Y. Yang, L. Wang, W. E. Stites, D. Zhong and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13979–13984 CrossRef CAS PubMed.
- K. Wood, F.-X. Gallat, R. Otten, A. J. V. Heel, M. Lethier, L. v. Eijck, M. Moulin, M. Haertlein, M. Weik and F. A. A. Mulder, Angew. Chem., Int. Ed., 2013, 52, 665–668 CrossRef CAS PubMed.
- T. Becker, J. A. Hayward, J. L. Finney, R. M. Daniel and J. C. Smith, Biophys. J., 2004, 87, 1436–1444 CrossRef CAS PubMed.
- W. Doster and M. Settles, Presented in part at the hydration processes in biology: theoretical and experimental approaches, 1998 Search PubMed.
- T. Kleinert, W. Doster, H. Leyser, W. Petry, V. Schwartz and M. Settles, Biochemistry, 1998, 37, 717–733 CrossRef CAS PubMed.
- H. Lichtenegger, W. Doster, T. Kleinert, A. Birk, B. Sepiol and G. Vogl, Biophys. J., 1999, 76, 414–422 CrossRef CAS.
- S. Cerveny, Á. Alegría and J. Colmenero, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 031803 CrossRef.
- J. Swenson, H. Jansson, J. Hedström and R. Bergman, J. Phys.: Condens. Matter, 2007, 19, 205109 CrossRef.
- K. L. Ngai, S. Capaccioli, S. Ancherbak and N. Shinyashiki, Philos. Mag., 2011, 91, 1809–1835 CrossRef CAS PubMed.
- G. Parigi, N. Rezaei-Ghaleh, S. B. Andrea Giachetti, C. Fernandez, M. Blackledge, C. Griesinger, M. Zweckstetter and C. Luchinat, J. Am. Chem. Soc., 2014, 136, 16201–16209 CrossRef CAS PubMed.
- G. Némethy and M. P. Printz, Macromolecules, 1972, 5, 755–758 CrossRef.
- G. Scherer, M. L. Kramer, M. Schutkowski, U. Reimer and G. Fischer, J. Am. Chem. Soc., 1998, 120, 5568–5574 CrossRef CAS.
- J. F. Yan, F. A. Momany, R. Hoffmann and H. A. Scheraga, J. Phys. Chem., 1970, 74, 420–433 CrossRef CAS.
- R. B. G. R. Lumry, Biopolymers, 1985, 24, 301–326 CrossRef PubMed.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins: Struct., Funct., Bioinf., 2000, 41, 415–427 CrossRef CAS.
- V. N. Uversky, Eur. J. Biochem., 2002, 269, 2–12 CrossRef CAS.
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS PubMed.
- A. M. Gaspar, M.-S. Appavou, S. Busch, T. Unruh and W. Doster, Eur. Biophys. J., 2008, 37, 573–582 CrossRef CAS PubMed.
- S. Perticaroli, J. D. Nickels, G. Ehlers, E. Mamontov and A. P. Sokolov, J. Phys. Chem. B, 2014, 118, 7317–7326 CrossRef CAS PubMed.
- T. E. Dirama, G. A. Carri and A. P. Sokolov, J. Chem. Phys., 2005, 122, 114505 CrossRef PubMed.
- M. Schmidt and D. K. Saldin, Struct. Dyn., 2014, 1, 024701 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |