
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation of poly(acrylic acid)-containing elastin-mimetic copolymers†
Bradford A.
Paik
a,
Marco A.
Blanco‡
b,
Xinqiao
Jia
ac,
Christopher J.
Roberts
*b and
Kristi L.
Kiick
*ac
aDepartment of Materials Science and Engineering, Delaware Biotechnology Institute, University of Delaware, Newark, DE 19716, USA. E-mail: kiick@udel.edu; Fax: +1 302-831-4545; Tel: +1 302-831-0201
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19176, USA. E-mail: cjr@udel.edu; Fax: +1 302-831-1048; Tel: +1 302-831-0838
cBiomedical Engineering Program, University of Delaware, Newark, DE 19716, USA
First published on 6th January 2015
Abstract
Polymer–peptide conjugates were produced via the copper-catalyzed azide–alkyne cycloaddition of poly(tert-butyl acrylate) (PtBA) and elastin-like peptides. An azide-functionalized polymer was produced via atom transfer radical polymerization (ATRP) followed by conversion of bromine end groups to azide groups. Subsequent reaction of the polymer with a bis-alkyne-functionalized, elastin-like peptide proceeded with high efficiency, yielding di- and tri-block conjugates, which after deprotection, yielded poly(acrylic acid) (PAA)-based diblock and triblock copolymers. These conjugates were solubilized in dimethyl formamide, and addition of phosphate buffered saline (PBS) induced aggregation. The presence of polydisperse spherical aggregates was confirmed by dynamic light scattering and transmission electron microscopy. Additionally, a coarse-grained molecular model was designed to reasonably capture inter- and intramolecular interactions for the conjugates and its precursors. This model was used to assess the effect of the different interacting molecular forces on the conformational thermodynamic stability of the copolymers. Our results indicated that the PAA's ability to hydrogen-bond with both itself and the peptide is the main interaction for stabilizing the diblocks and triblocks and driving their self-assembly, while interactions between peptides are suggested to play only a minor role on the conformational and thermodynamic stability of the conjugates.
Introduction
Peptide–polymer hybrid materials have the ability to combine advantageous chemical and physical properties, while overcoming the shortcomings of the individual component materials.1,2 Many previously reported hybrid materials have demonstrated enhanced biological functionality, or control of assembly over multiple length scales.1,3–6 This controlled assembly has resulted in versatile structures that have found application in numerous biological and non-biological applications.7–14 Most commonly these structures are driven by hydrophobic interactions or triggering of specific secondary structure in the peptide domains.6,9,15,16 Of interest among these conjugates are materials with assembly that can be controlled via temperature,17 pH,18 enzyme activity,19,20 and ion/cofactors.21 Triggered assembly by external stimuli allows for a greater level of control over the resulting structure of the hybrid conjugates.With the aim of producing conjugates that may capture the properties of select structural, multiblock proteins (e.g., elastin) and show triggered assembly, we have previously reported the synthesis of a hybrid multiblock copolymer comprising poly(acrylic acid) (PAA) and a short elastin-like peptide (ELP).22 PAA was chosen because of its pH responsiveness, chemical functionality, and common use in other block copolymer systems.23–26 The ELP was included in order to introduce a temperature-sensitive domain to confer assembly via triggering of a lower critical solution temperature (LCST)-like transition, allowing for thermoresponsiveness within.27–32 The resulting PAA–ELP multiblock copolymer was shown to form nanoparticles, although the assembly of these nanoparticles showed only pH responsiveness, and did not exhibit an LCST transition.22 We speculated that hydrogen bonding, along with some contributions from hydrophobic interactions, was the driving force for this nanoparticle formation.
In order to confirm this hypothesis and identify alternative opportunities to control the assembly of these chemically reactive multiblock polymers, a simplified PAA–ELP conjugate was designed, thus permitting study of the assembly pathway with coarse-grained modeling as a means to elucidate the molecular interactions between the PAA and ELP blocks. Coarse-grained molecular simulations provide a means to test the relative contributions from different molecular-scale interactions that mediate the assembly of the PAA and ELP components, as well as providing a visualization tool for considering the net effects on the free energy landscape(s) for assembly.
Here we report the synthesis and characterization of PAA–ELP di- and triblock copolymers, their aggregation into nanoparticles, and the study of the aggregation pathway via coarse-grained modeling. PAA–ELP conjugates were synthesized via CuAAC click chemistry; upon induced aggregation, the aggregates were characterized via dynamic light scattering (DLS) and transmission electron microscopy (TEM). A coarse-grained model analogous to those used previously was employed so as to maintain the ability to distinguish between interactions involving the polypeptide backbone and side chains, while avoiding the computational limitations of employing all-atom models for a macromolecular system.33,34 This combination of experimental and computational work sheds light on the use of PAA in peptide–polymer conjugates, and suggests physicochemical interactions that should be considered when potentially employing this chemically reactive block in the design of peptide–polymer conjugates.
Experimental
Materials
All reagents were used without further purification unless otherwise specified. Dimethyl 2,6-dibromoheptanedioate, ethyl 2-bromopropionate (EBP), copper(I) acetate, copper(I) bromide, N,N,N′N′,N′′-pentamethyldiethylenetriamine (PMDETA), tert-butyl acrylate, alumina inhibitor removal column, and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). tert-Butyl acrylate was purified by passing through an alumina inhibitor removal column. Sodium azide, acetic anhydride, N,N-dimethylformamide (DMF), tetrahydrofuran (THF), methanol, toluene, and dichloromethane (DCM) were purchased from Fisher Scientific (Pittsburgh, PA, USA). Fmoc-L-valine–OH, Fmoc-L-isoleucine–OH, Fmoc-L-glycine–OH, and Fmoc-L-proline–OH were purchased from Protein Technologies, Inc. (Tucson, AZ, USA), and Fmoc-L-propargylglycine–OH was purchased from AnaSpec, Inc. (Fremont, CA). Water was deionized and filtered through a Barnstead NANOpure Diamond water purification system (Thermo Scientific, Dubuque, IA, USA).Synthesis of triblock copolymers of poly(tert-butyl acrylate) and (VPGVG)2 [VG2–PtBA–VG2]
Poly(tert-butyl acrylate) diazide (N3–PtBA–N3, Mn = 2800 g mol−1, and X(VPGVG)2X (VG2) (where X = propargyl glycine, V = valine, P = proline, G = glycine) were synthesized and characterized in accordance with our previous work.22 Solutions of polymer N3–PtBA–N3 (0.028 mmol), peptide VG2 (0.07 mmol), and copper(I) acetate (1 equiv. to alkyne) were made in separate vials of 400 μL anhydrous-DMF; these solutions and copper wire were added to a nitrogen-purged, 50 mL round bottom flask, and a condenser was attached. The solution was stirred at 80 °C under nitrogen for 24 hours, and then precipitated into cold water. The solid was vacuum filtered, washed with water, and then dried under vacuum giving a brown solid in 56% yield. 1H NMR (DMSO-d6) δ (ppm): 7.10–8.48 (22H, varied, NHCO and triazole C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.18 (m, 2H, triazole-CH(NHCOCH3)CO), 3.95–4.48 (m, 18H, NH–CH–CO), 3.56 (m, 10H, CH2CH-triazole and proline NHCH2CH2CH2), 2.15 (m, 28H, PtBA CH2CH and proline NHCH2CH2CH2), 1.71–2.0 (m, 31H, PtBA CH2CH, valine CH(CH3)2, and proline NHCH2CH2CH2), 1.44 (s, 188H, PtBA CH2CH and C(CH3)3), 0.85 (m, 48H, valine CH(CH3)2).
CH), 5.18 (m, 2H, triazole-CH(NHCOCH3)CO), 3.95–4.48 (m, 18H, NH–CH–CO), 3.56 (m, 10H, CH2CH-triazole and proline NHCH2CH2CH2), 2.15 (m, 28H, PtBA CH2CH and proline NHCH2CH2CH2), 1.71–2.0 (m, 31H, PtBA CH2CH, valine CH(CH3)2, and proline NHCH2CH2CH2), 1.44 (s, 188H, PtBA CH2CH and C(CH3)3), 0.85 (m, 48H, valine CH(CH3)2).
Synthesis of diblock copolymers of poly(tert-butyl acrylate) and (VPGVG)2 [PtBA–VG2]
Poly(tert-butyl acrylate) mono-azide (PtBA–N3, Mn = 2800 g mol−1) was synthesized following a previously established procedure.35 Solutions of PtBA–N3 (0.028 mmol), VG2 (0.029 mmol), copper(I) acetate (1 equiv. to alkyne) were made in separate vials of in 400 μL anhydrous-DMF; these solutions and copper wire, were added to a nitrogen-purged, 50 mL round bottom flask and a condenser was attached. The solution was stirred at 80 °C, under nitrogen for 24 hours, and then precipitated into cold water. The solid was vacuum filtered, washed with water, and then dried under vacuum giving a brown solid in 60% yield. 1H NMR (DMSO-d6) δ (ppm): 7.10–8.48 (11H, varied, NHCO and triazole CCH), 5.18 (m, 1H, triazole-CH(NHCOCH3)CO), 3.95–4.48 (m, 9H, NH–CH–CO), 3.56 (m, 6H, CH2CH-triazole and proline NHCH2CH2CH2), 2.15 (m, 18H, PtBA CH2CH and proline NHCH2CH2CH2), 1.71–2.0 (m, 19H, PtBA CH2CH, valine CH(CH3)2, and proline NHCH2CH2CH2), 1.44 (s, 188H, PtBA CH2CH and C(CH3)3), 0.85 (m, 24H, valine CH(CH3)2).
Synthesis of PAA–VG2 triblock (VG2–PAA–VG2) and diblock (PAA–VG2) copolymers
VG2–PtBA–VG2 and PtBA–VG2 were added to a 80/20 TFA–DCM mixture (10 mL) and stirred for 24 hours at room temperature. The solution was diluted with DMF, added to regenerated cellulose dialysis membrane with a molecular weight cut-off (MWCO) of 1000 (Spectra/Por, Spectrum Laboratories, Inc., Rancho Dominguez, CA) and dialyzed against DMF, at room temperature, for two days (exchanged 3 times per day) to remove TFA and residual low molecular weight material. The solution (triblock and diblock at 0.6 mg mL−1 concentration in DMF) was then placed into a round bottom flask and refrigerated in order to prevent aggregation of conjugates. Aliquots of the cleaved di- and triblock polymer were also dialyzed against water under the same conditions, lyophilized, and characterized via1H NMR spectroscopy to verify the composition of the conjugates. Triblock: 1H NMR (DMSO-δ6) δ (ppm): 12.5–13.0 (s, COOH), 7.10–8.50 (m, 22H, NHCO and triazole CCH), 5.31 (m, 2H, triazole-CH(NHCOCH3)CO), 3.95–4.48 (m, 18H, NH–CH–CO), 3.56 (m, 10H, CH2CH-triazole and proline NHCH2CH2CH2), 2.20 (m, 32H, PAA CH2CH and proline NHCH2CH2CH2), 1.25–1.97 (m, 40H, PAA CH2CH, valine CH(CH3)2, and proline NHCH2CH2CH2), 0.85 (m, 48H, valine CH(CH3)2). Diblock: 1H NMR (DMSO-δ6) δ (ppm): 12.5–13.0 (s, COOH), 7.10–8.50 (m, 11H, NHCO and triazole CCH), 5.31 (m, 1H, triazole-CH(NHCOCH3)CO), 3.70–4.55 (m, 12H, NH–CH–CO), 3.56 (m, 6H, CH2CH-triazole and proline NHCH2CH2CH2), 2.20 (m, 26H, PAA CH2CH and proline NHCH2CH2CH2), 1.25–1.97 (m, 30H, PAA CH2CH, valine CH(CH3)2, and proline NHCH2CH2CH2), 0.85 (m, 24H, valine CH(CH3)2).
Aggregation of conjugates
A 1 mL solution of the diblock or triblock copolymers in DMF was added to a glass vial with a magnetic stir bar following a previously established procedure.36 While stirring, filtered PBS (pH 7.4) was then added to the solution at 1 mL h−1via a syringe pump (NE-1000, New Era Pump Systems, Inc., Farmingdale, NY), and aliquots were collected at PBS-to-DMF ratios of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 75:25, and 87.5:12.5. Each aliquot was characterized by DLS, with three measurements being recorded at 20 minute intervals. The final ratio of 87.5:12.5 was chosen for further study as the two other ratios yielded aggregates with sizes that would vary with time. The 87.5:12.5 sample was stirred overnight and was then measured by DLS and TEM. DLS data were processed by a moment-generated function of the measured correlation function, aggregate size was measured from the computed diffusion coefficients using the Stokes–Einstein equation. Aggregate sizes observed via TEM analysis were manually measured via ImageJ. Over 100 nanoparticles were manually analysed allowing for individual aggregates to be measured despite being in contact with other spherical aggregates. ImageJ identified aggregates by converting images to monochrome, and setting a threshold (values via ImageJ, 0 and 119) to distinguish aggregates from the background.
:50, 75:25, and 87.5:12.5. Each aliquot was characterized by DLS, with three measurements being recorded at 20 minute intervals. The final ratio of 87.5:12.5 was chosen for further study as the two other ratios yielded aggregates with sizes that would vary with time. The 87.5:12.5 sample was stirred overnight and was then measured by DLS and TEM. DLS data were processed by a moment-generated function of the measured correlation function, aggregate size was measured from the computed diffusion coefficients using the Stokes–Einstein equation. Aggregate sizes observed via TEM analysis were manually measured via ImageJ. Over 100 nanoparticles were manually analysed allowing for individual aggregates to be measured despite being in contact with other spherical aggregates. ImageJ identified aggregates by converting images to monochrome, and setting a threshold (values via ImageJ, 0 and 119) to distinguish aggregates from the background.
Coarse-grained modelling
To study the thermodynamic and conformational stability of the peptide–polymer conjugates, an implicit-solvent coarse-grained (CG) molecular model was used in which amino acids and monomer units are represented by 4 or 3 beads (Scheme 1). The CG model developed by Bereau and Deserno was used in the case of peptide fragments. In that model, these beads correspond to the amide group (N), central carbon (Cα), carbonyl group (C′), and a side chain (Cβ).37 The first three beads represent the peptide backbone and provide information regarding the flexibility and secondary structure propensity of the peptide chain. The fourth bead corresponds to the side chain for all non-glycine amino acids and is responsible for the hydrophobicity and specificity of each residue for favorable or unfavorable interactions between side chains. Interactions between these beads consider both local and non-local forces and only account for those interactions that have the largest effect on the structural stability of polypeptides.38 These interactions include dipole–dipole interactions, hydrogen bonding, hydrophobic attractions, steric repulsions, as well as different 4-body interactions, and were previously parameterized against NMR and crystallographic data in order to reproduce the different secondary and tertiary structures that can be adopted by polypeptides.37 The present work required a coarse-grained model in which the solvent was treated explicitly, so as to make the computations tractable for the large polypeptide and polymer conformational space that was sampled. As a result, one cannot correctly include explicit ions because explicit water is not present, and only screened electrostatics can be considered. For the experimental conditions used here, the ionic strength was so high that the screening length was too small for electrostatic interactions make appreciable contributions in the simulations.39 | ||
| Scheme 1 Schematic representation considered in the present coarse-grained model for the local geometry of (a) the protein chain and (b) the polymer chain. The solid beads highlighted indicate those used to represent a single amino acid (a) or monomer unit (b). The beads for neighboring units are represented by dashed lines.37 | ||
Similarly, for polymer chains, each monomer unit is represented by 3 beads (Scheme 1b). Although this representation is not intended to provide an accurate description of the geometry of PAA, it allows the same level of CG modeling and comparison between the effects of hydrogen-bond formation between peptide and non-peptide segments, while also capturing the greater freedom in torsional-angle space of the polymer versus that of the peptides. It is anticipated that this structural description of the polymer chain will sufficiently capture the qualitative impact of intermolecular interactions, as long as the balance of interactions within the molecule (i.e., interactions involving monomers and amino acid units) are reasonably captured.
Additionally, to ensure that the physicochemical properties of the polymer block were preserved in the CG model, the length of the simulated PAA chains was selected to yield a chain in which the average radius of gyration matched that of the experimental polymer (see Fig. S2†). Therefore, the net segment lengths were somewhat shorter than those in the experiment, but the relative ratios were similar. It is also a requirement that the force field for the interactions involving monomer units is on the same energy scale for that of the peptides. That is, the internal forces of a monomer unit of the polymer, as well as the non-local interactions of a monomer unit with another monomer unit or with an amino acid must be of a similar strength in order to avoid any bias of the CG model towards a given structure. Thus, interactions involving polymer chains are limited here to bond and angle forces, steric repulsions, and hydrogen bonding, and are represented by the same expressions used to model amino acids. However, unlike the interactions for polypeptides, 4-body interactions (e.g., torsional angles that enforce the planarity of the peptide bonds polypeptides) and hydrophobic interactions are not considered in the case of polymer chains in order to incorporate the high flexibility from the sp3 hybridization of the carbons as well as the high water-affinity of acrylic acid.
The force field for the present CG model is given by:
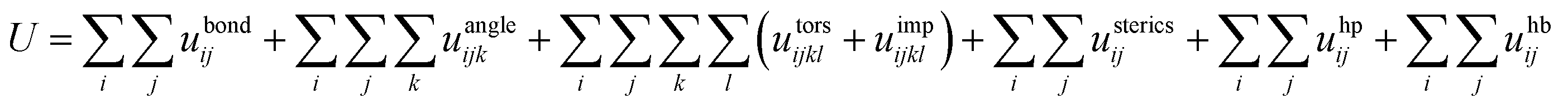 | (1) |
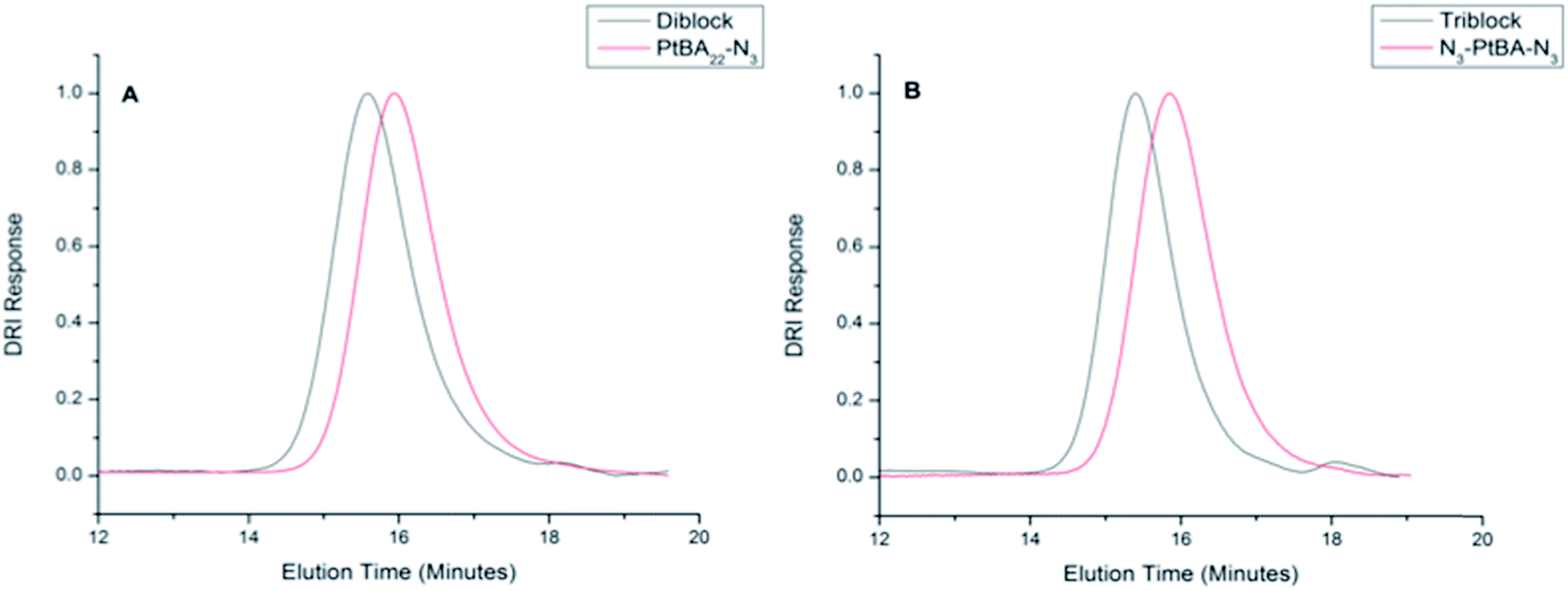 | ||
| Fig. 1 Gel permeation chromatography of polymers. (A) GPC traces of the azide, monofunctional PtBA22 and the PtBA22–VG2 diblock; (B) GPC traces of the azide, bifunctional PtBA22 precursor and the VG2–PtBA22–VG2 triblock. Monodisperse polystyrenes were used as standards. | ||
Computational methods
The thermodynamic and conformational stability of the selected peptides, PAA, and conjugates were assessed by performing Replica Exchange Molecular Dynamics (REMD) simulations on an ensemble at fixed volume and number of molecules.40 This method was coupled to a Nosé–Hoover chain thermostat to generate the correct canonical distribution for each replica in the simulation.41 REMD is a suitable method to determine accurate ensemble properties, as it helps to prevent simulations from becoming trapped in local basins on the free-energy landscape at low temperatures. Furthermore, combining REMD with weighted histogram analysis methods (WHAM) allows reconstruction of the density of states of the system (i.e., the population of each of the configurations that a given molecule can adopt) and calculation of thermodynamic observables over a continuous range of temperatures, such as free-energy and heat-capacity.42,43 During the REMD simulations, all lengths were measured in units of![[script L]](https://www.rsc.org/images/entities/char_e144.gif) = 1 Å, and energies were related to thermal energy
= 1 Å, and energies were related to thermal energy ![[scr E, script letter E]](https://www.rsc.org/images/entities/char_e140.gif) = kBTr, where kB is the Boltzmann constant and Tr = 300 K is a reference temperature. Masses were denoted in units of
= kBTr, where kB is the Boltzmann constant and Tr = 300 K is a reference temperature. Masses were denoted in units of ![[scr M, script letter M]](https://www.rsc.org/images/entities/char_e145.gif) ≈ 27.5 Da ≈ 4.6 × 10−26 kg, which yields the average weight of an amino acid (∼110 Da). This choice of units leads to a characteristic time for simulation time steps of
≈ 27.5 Da ≈ 4.6 × 10−26 kg, which yields the average weight of an amino acid (∼110 Da). This choice of units leads to a characteristic time for simulation time steps of 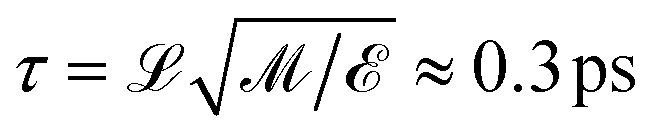 . Results are reported in units of kcal mol−1 for energy and free energy values, based on a reference value of kBT corresponding to RT ≈ 0.6 kcal mole−1 (ref. T = 300 K).
. Results are reported in units of kcal mol−1 for energy and free energy values, based on a reference value of kBT corresponding to RT ≈ 0.6 kcal mole−1 (ref. T = 300 K).
Polymer–peptide conjugates were modeled as polypeptide–polymer–polypeptide triblocks, but simulating only half the size of molecules that were experimentally synthesized. That is, each polypeptide block corresponds to the sequence VPGVG, while the polymer block represents a PAA molecule with an average radius of gyration of half of the experimental value (see Fig. S2 in ESI†). These modifications were employed in order to reduce computational cost and permit exploration of a larger number of macromolecular systems. Additionally, all REMD simulations presented here were performed for no more than three molecules in the simulation box. When considering more than one molecule, simulations were carried out into a cubic box of length L with periodic boundary conditions. The box length depended on the number of simulated molecules and was set to simulate a concentration of 5 mM. REMD simulations were carried out at solvent conditions corresponding to PBS (i.e., neutral pH and high salt concentration). A set of replicas in REMD were distributed between 120 K and 500 K, with the total number of replicas adjusted for a given set of conditions to assure acceptance ratios for the replica swaps between 30% and 40%. An integration time step δt = 0.005τ (≈ 2 fs) was used for all the REMD simulations, and swaps between replicas were attempted every 1τ for REMD steps. The initial configuration of each replica (given by the position of all of the beads in the molecule) was chosen randomly. Each simulation employed two short warming-up periods for thermal equilibration of 1 × 105τ each, using standard molecular dynamics simulations and REMD, respectively.40 Thereafter, a sampling period of 1 × 106τ was performed using REMD, where the configurations and energy values of each replica were stored every 1τ for further structural and thermodynamic analysis. Unless otherwise stated, simulations focused on the triblocks, so as to include the intramolecular competition between peptide–peptide and peptide–polymer interactions. Preliminary results from diblock simulations did not provide additional insights beyond those reported below.
Characterization
The successful synthesis of di- and triblock copolymers was confirmed by 1H NMR, gel permeation chromatography (GPC), and attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR). 1H NMR spectra were recorded on a Bruker AV600 (600 MHz) at ambient temperatures in DMSO-d6 or CDCl3 under standard quantitative conditions. All spectra were calibrated to the solvent peak and analysed with MNova software. Electrospray ionization mass spectroscopy (ESI-MS) was conducted on a Thermo Finnegan LCQ Advantage mass spectrometer with Surveyor MS pump. For these analyses, samples were dissolved in methanol at a concentration of 0.1 mg mL−1 and were filtered through a 0.22 μm PVDF filter. GPC was conducted using two Waters Styragel (HR1 and HR4) columns and a Waters 2695 autosampler pump (Waters Corporation, Milford, MA, USA) with THF as the mobile phase. All samples were analysed at a flow rate of 1.0 mL per minute, with detection via a Waters 2996 photodiode array and a Waters 2414 refractive index (RI) detector in series. Waters Empower software was used to construct calibration curves of narrow molecular weight polystyrene standards (Polyscience) for data analysis. ATR-FTIR was conducted using a Thermo Nicolet Nexus 670 (Thermo Scientific, Waltham, MA, USA) spectrometer with a DuraSamplIR II ATR accessory (Smiths Detection, Danbury, CT, USA). Samples were added as solids onto the silicon ATR crystal and gently pressed down during data acquisition (128 scans at 4 cm−1 resolution from 650–4000 cm−1). A background of the silicon crystal in air was subtracted from the sample spectra, and Omnic software was used to process the data.The aggregation of the di- and triblock copolymers was via DLS and TEM. DLS measurements were performed on a Malvern Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK) at a scattering angle of 173° using a square polystyrene cuvette with a pathlength of 12 mm. The data were processed by a moment-generated function of the measured correlation function, and aggregate size was measured from the computed diffusion coefficients using the Stokes–Einstein equation. Each reported measurement was conducted from the average of at least three runs from multiple different assessments of a on a given sample. TEM was conducted on a Tecnai 12 (FEI Company, Hillsboro, OR, USA) microscope using an accelerating voltage of 120 kV. TEM samples were prepared by adding a 5 μL drop of solution onto a 300 mesh ultrathin carbon-coated copper TEM grid (Electron Microscopy Sciences, Hatfield, PA). The sample was blotted with filter paper, and allowed to dry within a hood for three hours. No staining was required because of the copper present from the CuAAC conjugation.44
Results and discussion
Synthesis of block copolymers of PAA and VG2
Previously, we have reported the synthesis and association of [PAA–VG2]n peptide–polymer hybrid multiblock copolymers.22 In addition to the PAA segments, that multiblock copolymer contains peptides derived from the hydrophobic domains of tropoelastin, with an aim of producing a pH- and thermo-responsive material. Our previous study indicated that association was not mediated by changes in temperature, but rather was suggested to occur through hydrogen-bonding and hydrophobic interactions. In an effort to produce computationally tractable systems aimed at understanding our previous results, we have synthesized diblock and triblock copolymers, VG2–PAA–VG2 and PAA–VG2 (Scheme 2) to enable study of the aggregation pathway.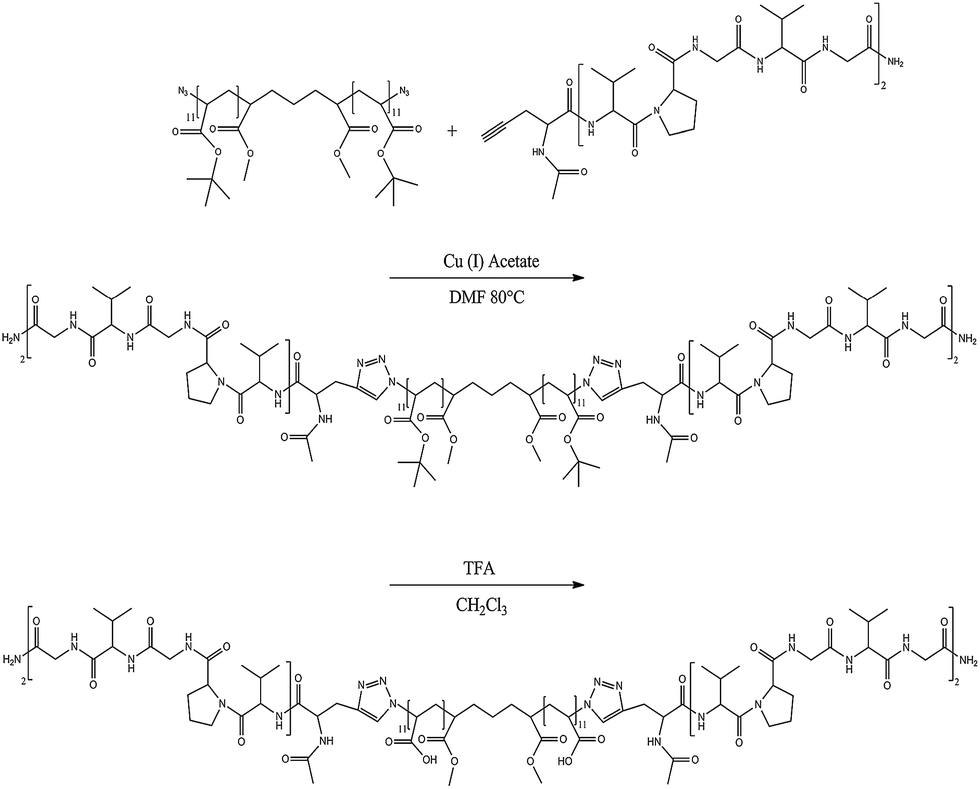 | ||
| Scheme 2 Synthetic protocols for CuAAC synthesis of the VG2–PAA–VG2 triblock copolymer. The coupling reaction for the diblock was conducted under the same conditions as those for the triblock copolymer, except for a different monomer molar ratio. | ||
The monofunctional, alkyne-functionalized peptide, VG2, and azide-functionalized N3–PtBA22–N3 were synthesized and purified according to our previous work.22 The monofunctional PtBA precursor (N3–PtBA22), of the same molecular weight, was synthesized via ATRP and characterized according to a previously reported method.35 The monofunctional polymer was functionalized via treatment with sodium azide in the same manner as that employed for the bifunctional polymer. The molecular weight of both polymers was calculated by comparing the ratio of the –CHC(O)O(CH3)3 proton (4.1 ppm) adjacent to the bromide or azide end groups to the –CHC(O)O(CH3)3 protons of the repeat unit (2.2 ppm). End group analysis yielded molecular weights of 2800 g mol−1 for all polymer species. As shown in Table 1, molecular weights determined by GPC are in good agreement with those calculated by NMR.
| Species | M n (NMR) (g mol−1) | M n (GPC) (g mol−1) | M W (GPC) (g mol−1) | PDI |
|---|---|---|---|---|
| Br–PtBA22 | 2800 | 2710 | 3200 | 1.17 |
| N3–PtBA22 | 2800 | 2520 | 2860 | 1.13 |
| Br–PtBA22–Br | 2800 | 2460 | 2770 | 1.12 |
| N3–PtBA22–N3 | 2800 | 2490 | 2810 | 1.12 |
| PAA22–VG2 | — | 3050 | 3580 | 1.17 |
| VG2–PAA22–VG2 | — | 3240 | 3750 | 1.14 |
The VG2 peptide and azide-functionalized PtBA22 were coupled to form di- and triblock copolymers via CuAAC in the presence of copper(I) acetate as shown in Scheme 2 (the synthesis of VG2–PAA–VG2 is shown). In the 1H NMR spectra (Fig. S1 and S2†), polymer and peptide peaks are both present and the integration of the peaks correspond to the molar ratios of the components in the conjugate (approximately 2 to 1, and 1 to 1 for the triblock and diblock, respectively). Furthermore, the coupling of the ELP to the polymer was confirmed by the presence of the triazole-CH(NHCOCH3)CO at 5.2 ppm for both conjugates, and the reduction of the intensity of the azide band (due to triazole formation) as measured via ATR-FTIR (data not shown). The molecular weights of the di- and triblock copolymers were characterized via GPC, as shown in Fig. 1 and Table 1. The chromatograms show a distinct shift in elution time to a higher molecular weight compared to the parent PtBA. The molecular weights of the diblock and triblock copolymers determined by GPC are lower than the expected molecular weights (approximately 4000 g mol−1 and 5000 g mol−1 for di- and triblock, respectively), which is likely due to the difference in hydrodynamic volume of the polystyrene calibration standards in comparison to the synthesized conjugates. Fig. 1 and Table 1 also show the low polydispersity of the diblock and triblock copolymers, indicating an efficient conjugation and uniform product. This robust conjugation efficiency is in agreement with the high efficiencies reported for conjugation reactions mediated by CuAAC.6,44–49
The tert-butyl groups of the polymer were hydrolyzed by treatment with TFA to yield the desired PAA-based di- and triblock copolymers. This cleavage was confirmed via1H NMR, (Fig. S1 and S2†), with the disappearance of the peak associated with the tert-butyl protons at 1.44 ppm, and the appearance of a broad peak associated with –COOH at 12.3–12.5 ppm. The peak remaining at 1.44 ppm is attributed to the methyl groups from the initiator in the PAA backbone.22 The fact that the diblock and triblock conjugates were not degraded is also indicated by retention of the triazole-CH(NHCOCH3)CO proton at 5.2 ppm, with an integrated area of one and two (relative to the peptide peaks) for the diblock and triblock, respectively. The observed lack of degradation is in agreement with our previous studies, and other reports of CuAAC click chemistry with peptides on resin.22,50
Aggregation of peptide–polymer conjugates
A solution titration method was used to study the association of the diblock and triblock copolymers. Solutions of the respective copolymers in DMF were prepared at a concentration of 0.6 mg mL−1. PBS (10 mM, pH 7.4) was then titrated into the solution until a 87.5:12.5 ratio of PBS to DMF was achieved. The 87.5:12.5 ratio was chosen as aggregates, under these conditions, did not vary in size with time, as compared to aggregates formed at the 50:50 and 75:25 ratios (data not shown). After stirring for 24 hours, the aggregates present in this solution composition were measured by DLS (Fig. 2). The data in Fig. 2 show the distribution of hydrodynamic diameters, P(Dh), of aggregates derived from solutions of the di- and triblock copolymers, and of those of a physical mixture of the component blocks, yielding average sizes of 320 nm, 360 nm, and 200 nm respectively. The aggregates formed from solutions of either the di- or triblock copolymers were found to be polydisperse, with calculated PDIs of 0.71 and 0.34 for the diblock and triblock copolymers, respectively. The physical mixture was not found to be polydisperse, with a PDI of 0.11.
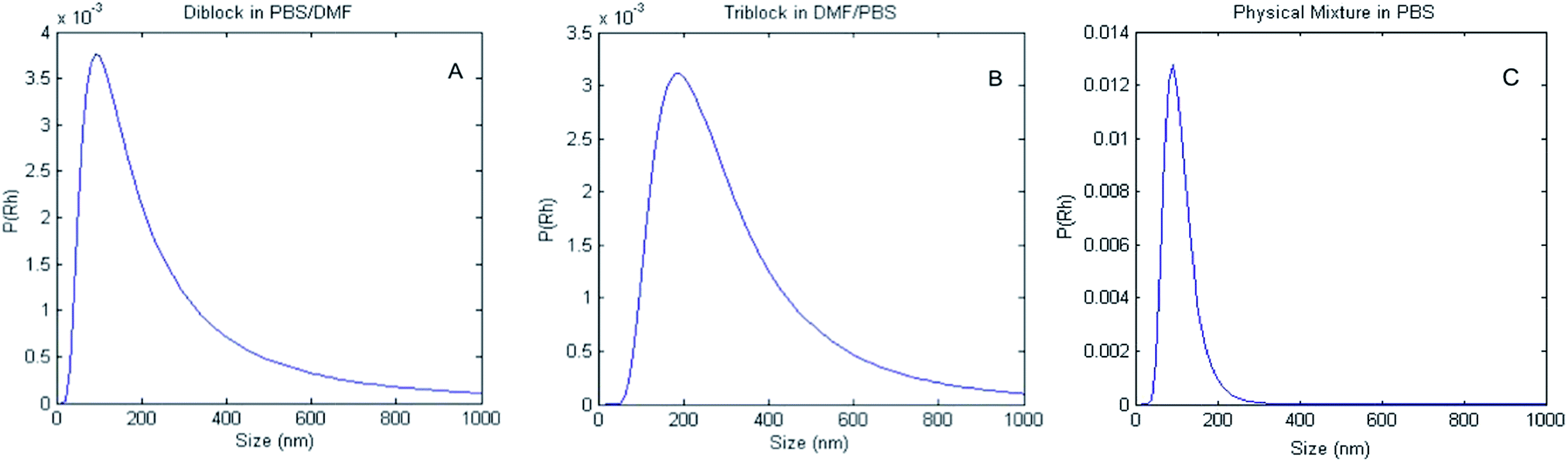 | ||
| Fig. 2 DLS was used to measure the average Rh value of PAA–VG2. (A) Diblock, (B) triblock and (C) physical mixture of PAA and VG2 in DMF–PBS solution. P(Rh) is the probability of finding an aggregate with a hydrodynamic radius (Rh), if one were to randomly select from the distribution of aggregates. | ||
This polydispersity in aggregate size, and the morphology of the aggregates was further studied by TEM (Fig. 3). Images were obtained directly from films cast from solution at room temperature; no staining was required because of the residual copper present from the CuAAC. The aggregates were found to be spherical in shape, and there was no perceivable difference in the morphology of the diblock and triblock. The observed spherical aggregates had diameters (calculated from analysis via ImageJ of over 100 nanoparticles in TEM images) of 165 ± 74 nm and 249 ± 70 nm for the diblock and triblock copolymers, respectively. The average diameter of these aggregates is smaller than those measured by DLS (320 nm and 360 nm for the diblock and triblock, respectively), which is likely due to the swelling of particles in solution, as compared to the dried state on a TEM grid.51,52 The average size measured by DLS and those of TEM cannot be directly compared because of the large polydispersity reported by the DLS, and the large standard deviation measured by TEM,52 but both do confirm the variation in average size present in both materials. Although one of the advantages of using CuAAC is the lack of a need for additional staining,44 any residual copper present in the samples here (from the ATRP synthesis and the CuAAC conjugation) was insufficient to permit any inner architecture of the aggregates to be visualized. Nevertheless, distinct micelle or vesicle formation was not expected from these materials, as there is not a distinct hydrophobic domain in the conjugates of the PAA22 and the VG2 peptide.
 | ||
| Fig. 3 TEM images of PAA–VG2 (A) and VG2–PAA–VG2 (B) assembled in DMF–PBS solution (87.5:12.5). Samples were not stained; the presence of copper from the synthesis of polymer–peptide conjugates provides necessary contrast for imaging. Scale bar is of 0.5 μm. | ||
Simulations of conjugates and parent materials
As a first step to study how the different interacting forces affect the structure of the hybrid conjugates, the effect of polymer block length on the conformational stability of the triblock conjugates was investigated via single-molecule REMD simulations, in order to computationally probe variations in the polymer's ability to hydrogen bond. The longest block length studied computationally is representative of the 22-repeat polymer made experimentally. Simulations were carried out for hybrid conjugates with polymer blocks of 5, 10, and 15 monomer units. Particularly, the effects of the polymer block were assessed via the heat-capacity profile (i.e., Cvversus T) as it is sensitive to conformational changes in the system, such as transitions from folded to unfolded structures or dissociation of molecular clusters. The ESI† provides details on the calculation of these profiles. Fig. 4 illustrates the heat capacity as a function of temperature for triblock copolymers with polymer blocks containing different number of monomers (N). For comparison, the heat capacity profiles for two polypeptides and a single PAA with 15 monomer units (N = 15) are also shown in the figure. The results illustrated in Fig. 4, as well as all those obtained from REMD simulations and shown below, correspond to simulations conducted under conditions that simulate those in PBS.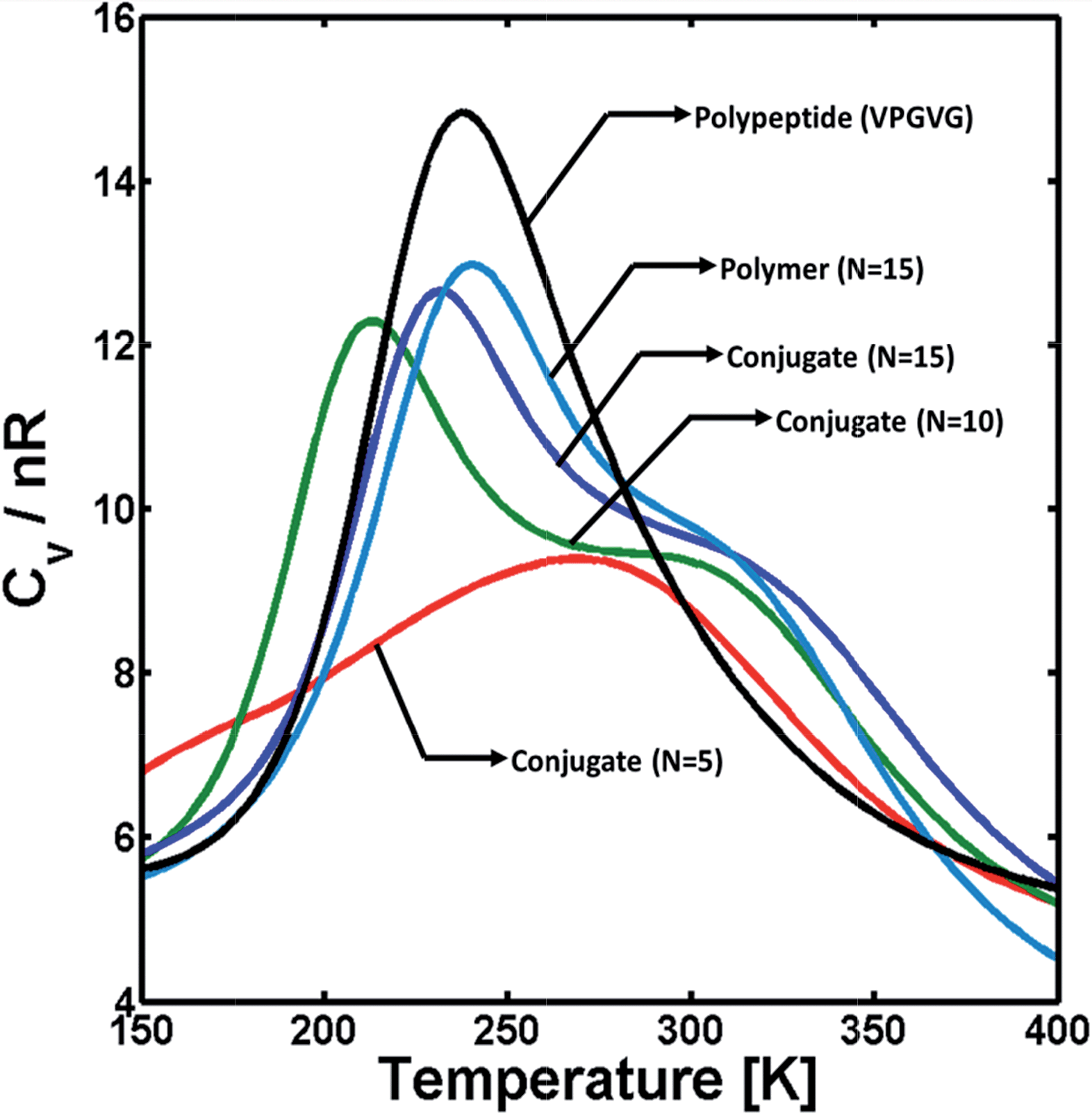 | ||
| Fig. 4 Heat capacity (Cv) as a function of temperature for conjugates with PAA blocks of different number N of monomeric units: N = 5 (red); N = 10 (green); and N = 15 (blue). Cv profiles for the dimerization of VPGVG (black) and the structural stability of PAA with N = 15 (teal), are also shown. Cv is normalized by the ideal gas constant R and the total number both amino acids and monomeric units of PAA (i.e., n = 10 + N). | ||
Qualitatively, thermodynamic transitions are identified in the heat-capacity profiles from a non-monotonic behavior of the Cvvs. T (i.e., the presence of one or more peaks or shoulders). Cv is a measure of the magnitude of fluctuations in intra- and intermolecular interactions relative to the thermal energy (see ESI†). Thus, a maximum in Cv is commonly interpreted as the “breaking” of multiple non-covalent molecular contacts that are important for the co-existence of more than one stable configuration in the system. Additionally, qualitative features of this maximum in Cv (e.g., the height and breadth of the peak, as well as the temperature at which this peak occurs) also provide information regarding the stability of the species involved in the transition as they are related to the change in internal energy during the transition (ΔU).53,54 Typically, short and/or broad Cv peaks are associated with poorly cooperative, unstructured species as they correspond to small ΔU, and vice versa. Therefore, the results in Fig. 4 illustrate that the transition between a folded, condensed state (low temperatures) and an unfolded, coil-like state (high temperatures) for the hybrid conjugates is dominated by the presence of the polymer block, but is less cooperative than the dissociation of VPGVG peptides when they are not linked by a PAA chain (black curve in the figure). For small polymer linker length within a triblock, there is a weakly cooperative thermodynamic transition in that it results in a small and broad Cv peak (red curve in the figure) indicating a poorly stable condensed or folded state. Conversely, as the length of the PAA block increases, there is a narrower and large Cv peak at ∼200 K which is consistent with a more well defined, cooperative unfolding transition, but it is convoluted with a second, weak transition (i.e., a shoulder at ∼280 K). Additionally, the Cv profile of the triblock molecule with large PAA chains presents a similar behavior to that of the polymer alone (teal curve in the figure), which suggests the PAA block plays a key role on the stability of the conjugates. Direct comparison to the experimental systems was not possible, as the experimental system is highly aggregated. Therefore, it does not show any discernable features in differential scanning calorimetry (data not shown), which is a common problem when dealing with net irreversible aggregates of proteins.
The effect of the different interactions or forces on the structural stability of the conjugate molecules was evaluated for the set of simulations illustrated above. Particularly, this effect is evident when looking at the free-energy of different configurations that hybrid conjugates can adopt at different temperatures as a function of the main interacting forces. The free-energy is calculated from the populations of the different stable configurations by combining REMD and WHAM (see ESI†). Fig. 5 shows free-energy landscapes as contour plots of the free-energy as a function of the hydrogen-bond energy for polymer–polymer (y-axis) and peptide–peptide (x-axis) interactions at different temperatures, and for the cases of conjugates with 5-monomer (top) and 15-monomer (bottom) PAA blocks. The color scale is shown next to each panel and is such that all the different configurations that lead to the same value of free-energy are represented with the same color. These landscapes allow identification of which interactions are more relevant for stabilizing the molecule (i.e., yielding the lowest free-energy) as the conjugates thermally unfold (across different temperatures) and for different polymer-chain lengths.
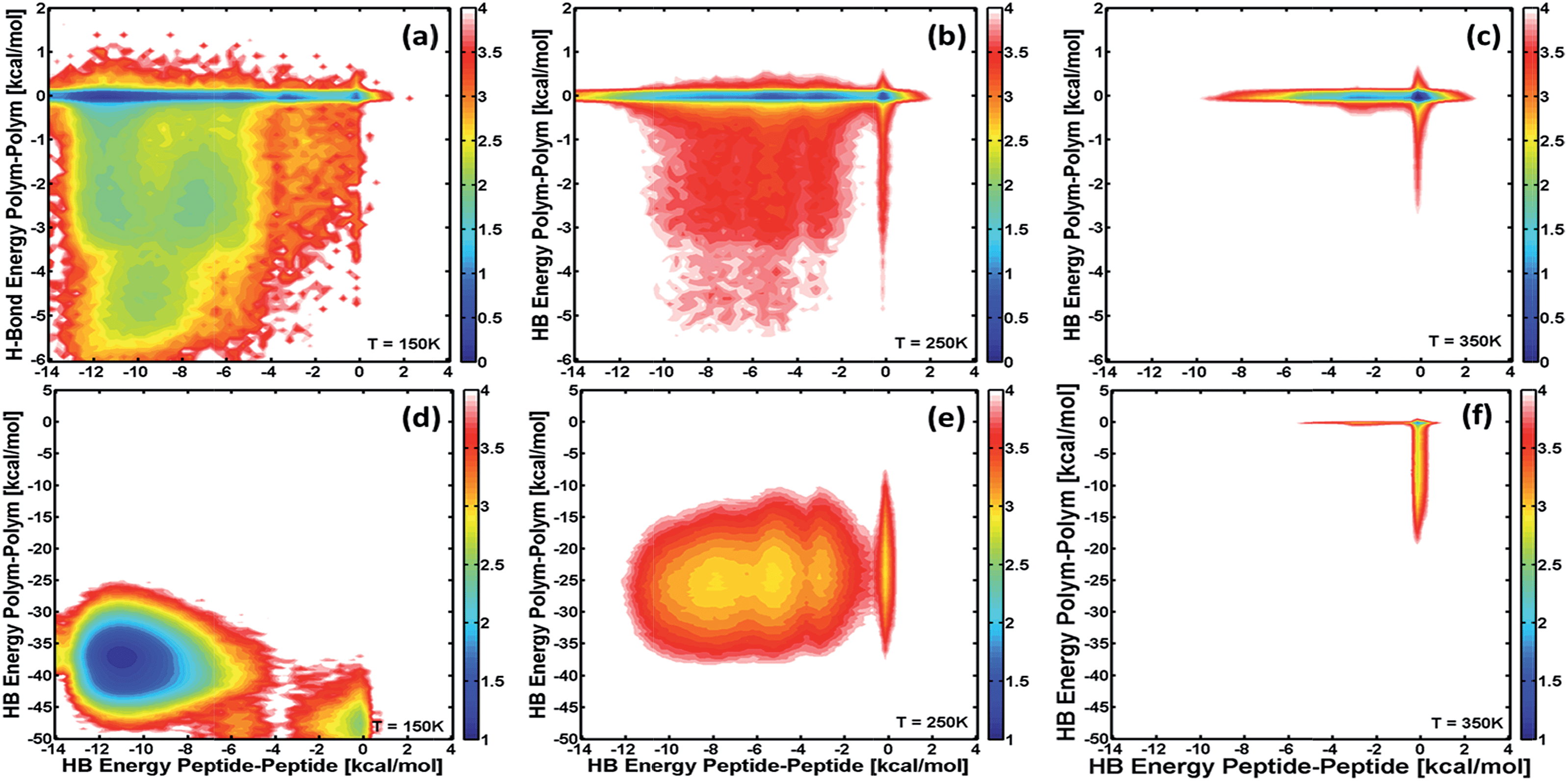 | ||
| Fig. 5 Free-energy as a function of the H-bond energy of the interactions between PAA monomers and the H-bond energy of the interaction between polypeptide blocks at T = 150 (left), 250 (middle), and 350 K (right). Free-energies are compared for two different sizes of PAA block: 5 monomeric units (top); and 15 monomeric units (bottom). Color code indicates the value of the Helmholtz free-energy in units of kcal mol−1, and is shown next to each panel. | ||
The results in Fig. 5a–c show that for a triblock conjugate with a small polymer block (5 monomers), interactions between peptides modulate the structural and thermodynamic stability of the conjugate. At low temperature, where the molecule is “folded”, the minimum free-energy or most stable structure (blue region in Fig. 5a) corresponds to those configurations where polymer–polymer interactions are negligible. Nevertheless, structures involving hydrogen-bonding between PAA monomers are significantly populated as it is observed by the broadness of the free-energy landscape in Fig. 5a. These latter configurations, however, are less stable and first “unfold” as temperature increases near the folding temperature (Fig. 5b). Thus, it is the “breaking” of the peptide–peptide interactions what determines the thermodynamic transition at ∼275 K (cf.Fig. 4 and 5b and c). In contrast, for the triblock with a larger polymer chain (15-monomer units; panels d–f, corresponding to the PAA block length of the molecules synthesized), the hydrogen-bonding between monomeric units of PAA are the main force (almost 5 times larger than those between peptides) in the folded structure. The thermodynamic transition for this conjugate at ∼200 K observed in the heat-capacity profile (cf.Fig. 4) is the result of the breaking of intra-polymer interactions. Nevertheless, the most stable configurations for this molecule involves the proximity of the peptide blocks as they are able to form hydrogen bonds, and thus they also affect the structure of the triblock. Comparison of the most stable, folded configurations for this conjugate with that of PAA alone (data not shown) shows that both molecules form a compact globular structure, but adding the peptide blocks to PAA creates a driving force for that compact state to have the ends of the PAA block to be close to one another and allow favorable peptide–peptide hydrogen-bond and hydrophobic interactions (see below).
The effect of the interactions between peptide blocks on the structural changes of the conjugates was analyzed further. The thermal unfolding of a series of point-mutations of the VPGVG sequence was simulated in order to modulate the hydrophobicity of the peptide block. The simulated triblock considered a fixed PAA block of 15 monomers, while the peptide blocks were substituted by either VPGIG or VPGEG so as to increase or decrease hydrophobic interactions, respectively. Morphological changes of the stable structure of the conjugates were assessed via the distribution of configurations as a function of the radius of gyration Rg (Fig. S3†). Although intramolecular interactions in the PAA block constitute the main force to stabilize the triblocks, the results from these simulations illustrate that hydrophobic interactions between peptide blocks play a major role on the morphology of the molecule. In general, these simulations indicate that the folded state of the triblock is formed by two configurations with Rg values of ∼0.6 and ∼0.8 nm, where the population of each of these configurations depends on hydrophobicity of the peptide block. Thus, by increasing hydrophobic interactions (i.e., mutating Val for Ile), configurations at Rg = 0.8 nm are suppressed, while decreasing hydrophobicity favors those configurations. Nevertheless, only the populations of these configurations are affected by the point-mutations, while their morphology remains unchanged (i.e., the Rg values are unaffected). This confirms that overall morphology of the triblocks is dictated by the PAA block, and it is such that allows close contact between the peptide blocks. These results demonstrate that the one might modulate the structure by altering peptide–peptide interactions, but these interactions must be balanced with the interactions involving PAA. As the length of PAA significantly exceeds that of the peptide blocks, it is perhaps not surprising that (on a per molecule basis) the overall energetics and morphology are strongly influenced by the PAA block. This is in agreement with experimental studies showing that the PAA blocks in amphiphilic block copolymers can tune the morphology of assemblies via volume fraction, PDI, and through control over the electrostatic repulsion of PAA chains.55–57
Finally, unlinked blocks were simulated (two peptide segments and one 15-monomer polymer) to understand how the different interaction forces between specific domains may affect the self-assembly propensity of the conjugated triblock. While this technique is not equivalent to modelling a block copolymer system with multiple molecules, it does provide valuable information about the preferential interactions of the blocks (or components, since the peptide and polymer segments are no longer covalently bonded to each other), and thus provides insight into the self-assembled system. Fig. 6 depicts the Helmholtz free-energy landscapes for the association of the different segments as a function of intermolecular hydrogen-bond interactions between two polypeptides, compared to those for a polypeptide and a polymer at different temperatures. These results are similar to the conformational stability of the single molecule. At low temperatures, those configurations with the lowest free-energy are dominated by peptide–polymer interactions with negligible peptide–peptide interactions. Despite this, there is a non-negligible population of configurations that involve peptide–peptide interaction, in agreement with the results above for the single-molecule examples. As temperature increases, the configurations involving strong interaction between the peptide and polymer remain stable, and there is a decrease in the free-energy of the configurations with hydrogen-bonding between peptides. These results further support that the self-assembled triblock molecules are likely stabilized to a large part by interactions with the polymer block (polymer–polymer or peptide–polymer), and to a lesser extent interactions between the peptide chains. Experimentally, it has been shown that short VPGVG peptides do not undergo a thermal transition,58 and indeed, an inverse transition temperature was not observed for the molecules in the previous multiblock study.22 Simulating bulk phase behavior – LCST or otherwise – requires large numbers of molecules in the simulation box; therefore, the present simulations are not appropriate for addressing questions regarding phase behavior of these systems.
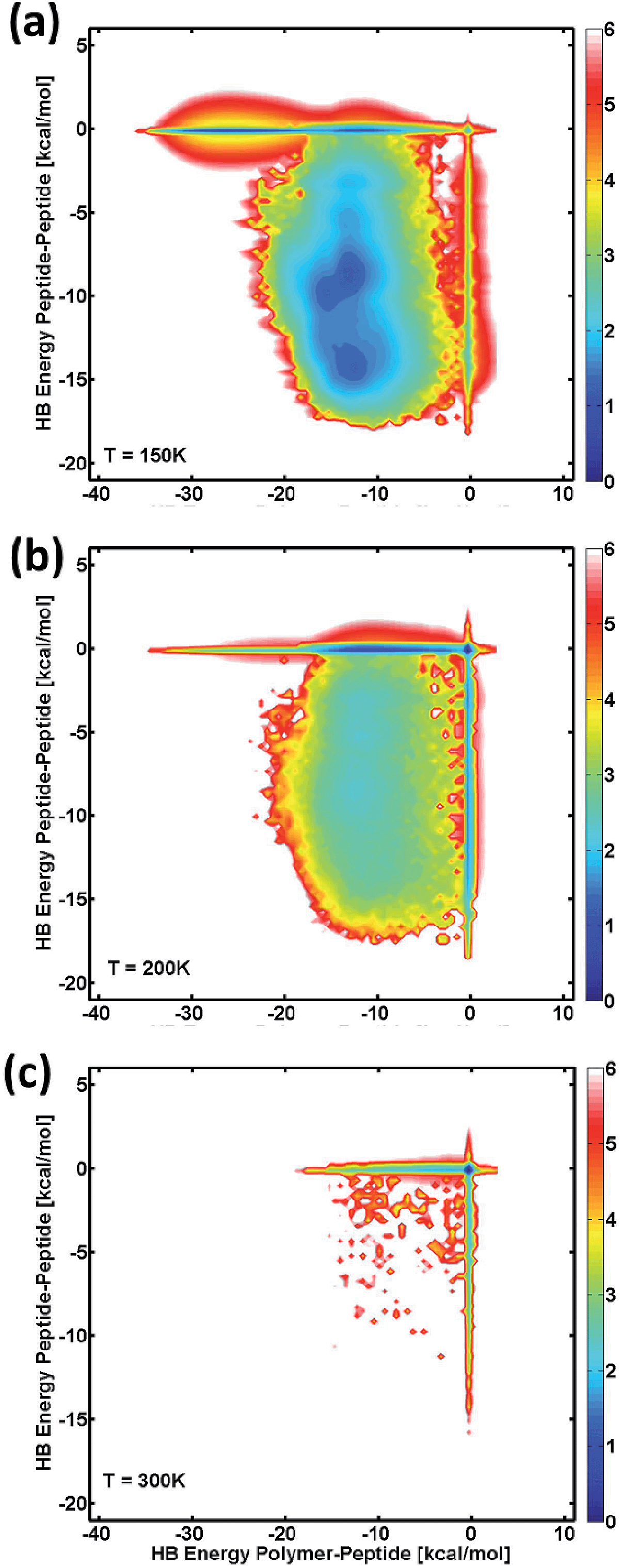 | ||
| Fig. 6 Free-energy landscapes as a function of the hydrogen-bond energies of the interactions between VPGVG molecules and the interaction between VPGVG and PAA at T = 150 (a), 200 (b), and 300 K (c). Simulated PAA molecule corresponds to a polymer chain with 15 monomer units. Color code indicates the value of the Helmholtz free-energy in units of kcal mol−1, and is shown next to each panel. | ||
The free-energy landscapes in Fig. 6 at higher temperatures show a similar behavior to those obtained for the analysis of the conformational stability of the single molecule (cf.Fig. 5). At low temperatures, the free-energy landscapes in Fig. 6a show that configurations involving both peptide–peptide and peptide–polymer interactions are relatively stable, and thus they present comparable low free-energies. However, those states where peptide–peptide hydrogen-bonding is negligible show the lowest free-energy (cf.Fig. 6a, deeper blue color along zero HB energy for peptide–peptide interactions). This suggests that peptide–polymer interactions may play a major role in stabilizing the self-assembly of the peptide–PAA system. As temperature increases (panels b and c), configurations involving strong interactions between the peptide and PAA remain stable, while there is an increase in the free-energy of those configurations with strong hydrogen-bonding between peptides. These results agree with those obtained for the single conjugated molecule in that the self-assembled triblock molecules are stabilized to a greater extent by interactions with the polymer block (polymer–polymer or peptide–polymer), than interactions between the peptide chains. Furthermore, the results from the coarse-grained model agree with previous experimental results for the coacervation of hydrophobic ELPs, showing that increased hydrophobicity of the peptide system can tune the association of the molecules through peptide–peptide hydrogen-bond interactions (Fig. S3†). Overall, the results here show that the contributions of the peptide–peptide interactions to the conformational stability of the molecules are non-negligible but are secondary to those of the PAA block (Fig. 5 and 6).
Traditionally, elastin-like polypeptides are used for their ability to undergo reversible aggregation due to hydrophobic dehydration, exhibiting a lower critical solution temperature.59 Less commonly studied are short ELPs (20 residues and below), as it has been shown that only very hydrophobic short ELPs (such as VPGFG) have an observable inverse phase transition.58 Despite this, others have incorporated single-repeat ELPs onto the side chains of polymers, and have observed inverse phase transition behavior.60–62 Ayres et al. observed aggregation via the formation of networks, as a result of the side-chain attachment of the ELP to the PMMA polymer.61 This provided an increased, hydrophobic environment enabling the phase transition. In the same vein, others have exploited the conjugation of hydrophobic groups to ELPs to induce the phase transition, and have found success in making spherical aggregates and fibers.63,64 Although this amphiphilicity is commonly used to direct the assembly of discrete architectures, through the inclusion of carefully chosen hydrophobic domains into conjugates,6,15,16 the conjugation of PEG to a (VPGVG)4 peptide did not yield diblock conjugates capable of aggregation.63 This indicates that short ELPs, like those reported here, are perhaps not sufficiently hydrophobic, and do not exhibit amphiphilic behavior when hydrophilic domains, such as PAA, are conjugated to them.
To overcome low hydrophobicity and drive assembly, hydrophobic moieties were added to a peptide terminus to facilitate the aggregation of conjugates.65 An ABA triblock with a central PEG block and flanking pentavaline blocks showed a decrease in aggregate size when the capping group was switched from an acetyl to Fmoc group. The authors also found that aggregates formed by peptides with acetyl caps yielded solutions with a high polydispersity, which is consistent with the findings in this study. Hwang et al. further studied the effects of hydrophobicity within Fmoc-capped penta-valine and PEG di- and triblock copolymers and found that the volume fraction of hydrophobic groups had a minimal impact on aggregation size while still resulting in polydisperse aggregates in solutions of both di- and triblock copolymers.52 The results of these previous studies are also in agreement with our study, which shows only a slight difference in aggregate size between the di- and triblock copolymers, both of which exhibit high polydispersity. It is possible that the increased hydrophobicity due to acetyl capping of the peptide does contribute to the formation of aggregates, as encapsulation of pyrene in PAA–VG2 multiblocks was possible.22 However in general, the short ELPs in the PAA-based conjugates are unlikely to contribute significantly to aggregation via hydrophobic interactions (see below), as is confirmed in both our experimental and computational analyses.
The use of PAA in bioconjugates has been of wide interest because of its chemical functionality and pH responsiveness, making it a useful material in biomedical applications.25 PAA has enabled electrostatic association and covalent conjugation of biomolecules through its carboxylic acid functional group, in brush, micelle and fiber structures.25,26,66,67 It is also commonly used in amphiphilic systems to form various architectures,68–71 and these architectures can be varied based on the PAA's ability to swell in solution due to pH-responsiveness.72 The ability of PAA to form hydrogen bonds is also of interest, and interpolymer complexes (IPC) of PAA and poly(ethylene oxide) (PEO) have previously been formed,73,74 and PAA–hydroxypropyl cellulose systems have been used to form nanoparticles through hydrogen bonding.23,75 Although most studies with IPCs employ higher molecular weight polymers than those employed in our studies, it has been shown that PAA with a molecular weight of 1.9 × 103 g mol−1 (which is similar to the 2.8 × 103 g mol−1 molecular weight used in the conjugates here) was able to form complexes.76 This is also in agreement with our findings that a physical mixture of PAA and VG2 forms aggregates. Relevant to our findings is that complex formation for conjugates in which PEO is grafted to PAA was shown to be more thermodynamically favorable than that for physical mixtures, as a result of the conjugation.77 Therefore, conjugation of the peptide directly to the PAA could increase the propensity of the conjugate to associate via hydrogen bonds.
Conjugation of peptides to PAA is less commonly studied. Self-assembly of PAA conjugated poly(valine) was explored, and showed assembly via hydrophobic interaction, but was shown that PAA and poly(valine) were able to interact via hydrogen-bonds.78,79 Our results are in agreement with these studies, as our computational assessment of the interactions of the conjugated PAA–VG2 triblock and of the individual blocks alone show that the most favorable interactions are a result of hydrogen bonding between polymer–polymer and polymer–peptide segments (Fig. 5 and 6). The data also show that the thermal stability of the folded state of the triblock is similar to that of the polymer alone, further supporting the idea that the conjugate is stabilized by polymer–polymer interactions, with smaller contributions from polymer–peptide interactions (Fig. 4). These computational results are in excellent agreement with our experimental data, showing that a physical mixture of PAA and VG2 also formed aggregates, and suggesting that peptide–peptide interactions are minimal with aggregation promoted primarily by the ability of PAA to form hydrogen bonds. Although PAA has proven useful as a domain in certain amphiphilic systems and in mediating assembly of IPCs, in bioconjugate applications with peptide domains, it is apparent that PAA can interact with itself and the peptide through hydrogen-bonds, introducing unwanted aggregation of polymer and peptide domains. Therefore alternative polymers may be of interest in producing functional materials with related polymer and peptide domains.
Conclusions
ELP–PAA di- and triblock copolymers were successfully synthesized via ATRP and CuAAC in order to study the potential affects of PAA's hydrogen bonding ability on aggregation, and to shed light on the assembly of our previous multiblock system. The conjugates were assembled through a slow-titration method, and yielded polydisperse solutions of spherical aggregates. In conjunction with coarse-grained modeling, we found that the assembly of these materials is dictated by the contributions due to intra- and intermolecular interactions of the polymer block, with minor contributions from peptide–peptide interactions. These results coupled with other examples within the literature show that while PAA may be utilized in amphiphilic systems, it may not be an appropriate choice for peptide–polymer hybrid applications in which assembly via the interactions of the peptide domain are of interest, due to the propensity of PAA to form hydrogen bonds with itself and peptides.Acknowledgements
This work was supported in part by funding from the National Science Foundation (CHE 1213728 to KLK (and XJ and CJR)), and the National Institutes of Health (NIH RO1 EB006006 to CJR and KLK). NMR and DLS characterization were supported by funding by the National Institutes of Health (NIH S10 RR026962-01 to Dr Joseph Fox and NIH NIDCD RO1 DC008965ZS2 to XJ). The authors thank Frank Kriss and Dr Jen Sloppy of the Keck Electron Microscopy Facility for help with TEM characterization and data interpretation. The contents of this manuscript do not necessarily reflect the views of the sponsors.References
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- G. Fuks, R. Mayap Talom and F. Gauffre, Chem. Soc. Rev., 2011, 40, 2475–2493 RSC.
- O. D. Krishna and K. L. Kiick, Biopolymers, 2010, 94, 32–48 CrossRef CAS PubMed.
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- I. W. Hamley, Biomacromolecules, 2014, 15, 1543–1559 CrossRef CAS PubMed.
- V. A. Sethuraman and Y. H. Bae, J. Controlled Release, 2007, 118, 216–224 CrossRef CAS PubMed.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2009, 94, 1–18 CrossRef PubMed.
- W. J. Kim, J. W. Yockman, M. Lee, J. H. Jeong, Y.-H. Kim and S. W. Kim, J. Controlled Release, 2005, 106, 224–234 CrossRef CAS PubMed.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS PubMed.
- H. G. Börner, Prog. Polym. Sci., 2009, 34, 811–851 CrossRef PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- R. Chandrawati, P. D. Odermatt, S.-F. Chong, A. D. Price, B. Städler and F. Caruso, Nano Lett., 2011, 11, 4958–4963 CrossRef CAS PubMed.
- J. Y. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–657 CrossRef CAS PubMed.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2013, 26, 507–518 CrossRef.
- E. Sahin and K. L. Kiick, Biomacromolecules, 2009, 10, 2740–2749 CrossRef CAS PubMed.
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- V. Castelletto, J. E. McKendrick, I. W. Hamley, U. Olsson and C. Cenker, Langmuir, 2010, 26, 11624–11627 CrossRef CAS PubMed.
- H. Kühnle and H. G. Börner, Angew. Chem., Int. Ed. Engl., 2009, 48, 6431–6434 CrossRef PubMed.
- R. I. Kühnle, D. Gebauer and H. G. Börner, Soft Matter, 2011, 7, 9616–9619 RSC.
- S. E. Grieshaber, B. A. Paik, S. Bai, K. L. Kiick and X. Jia, Soft Matter, 2013, 9, 1589–1599 RSC.
- Y. Chen, D. Ding, Z. Mao, Y. He, Y. Hu, W. Wu and X. Jiang, Biomacromolecules, 2008, 9, 2609–2614 CrossRef CAS PubMed.
- Y. Deng, S. Zhang, G. Lu and X. Huang, Polym. Chem., 2013, 4, 1289–1299 RSC.
- H. Jiang and F.-J. Xu, Chem. Soc. Rev., 2013, 42, 3394–3426 RSC.
- Z. Qu, K. Chen, H. Gu and H. Xu, Bioconjugate Chem., 2014, 25, 370–378 CrossRef CAS PubMed.
- E. Gil and S. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS PubMed.
- J. L. Osborne, R. Farmer and K. A. Woodhouse, Acta Biomater., 2008, 4, 49–57 CrossRef CAS PubMed.
- R. E. Sallach, W. Cui, J. Wen, A. Martinez, V. P. Conticello and E. L. Chaikof, Biomaterials, 2009, 30, 409–422 CrossRef CAS PubMed.
- O. S. Rabotyagova, P. Cebe and D. L. Kaplan, Biomacromolecules, 2011, 12, 269–289 CrossRef CAS PubMed.
- P. Shi, S. Aluri, Y.-A. Lin, M. Shah, M. Edman, J. Dhandhukia, H. Cui and J. A. MacKay, J. Controlled Release, 2013, 171, 330–338 CrossRef CAS PubMed.
- D. H. T. Le, R. Hanamura, D.-H. Pham, M. Kato, D. A. Tirrell, T. Okubo and A. Sugawara-Narutaki, Biomacromolecules, 2013, 14, 1028–1034 CrossRef CAS PubMed.
- F. Ding, J. M. Borreguero, S. V. Buldyrey, H. E. Stanley and N. V. Dokholyan, Proteins, 2003, 228, 220–228 CrossRef PubMed.
- M. Cheon, I. Chang and C. K. Hall, Proteins, 2010, 78, 2950–2960 CrossRef CAS PubMed.
- L. Xiao, C. Liu, J. Zhu, D. J. Pochan and X. Jia, Soft Matter, 2010, 5293–5297 RSC.
- J. Zhu, S. Zhang, F. Zhang, K. L. Wooley and D. J. Pochan, Adv. Funct. Mater., 2013, 23, 1767–1773 CrossRef CAS.
- T. Bereau and M. Deserno, J. Chem. Phys., 2009, 130, 235106–235121 CrossRef PubMed.
- A. Ben-Naim, J. Chem. Phys., 2006, 125, 24901–24911 CrossRef PubMed.
- M. A. Blanco, T. Perevozchikova, V. Martorana, M. Manno and C. J. Roberts, J. Phys. Chem. B, 2014, 118, 5817–5831 CrossRef CAS PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 2000, 329, 261–270 CrossRef CAS.
- M. Ikeguchi, J. Comput. Chem., 2004, 25, 529–541 CrossRef CAS PubMed.
- A. M. Ferrenberg, Phys. Rev. Lett., 1989, 6, 1195–1198 CrossRef.
- J. D. Chodera, W. C. Swope, J. W. Pitera, C. Seok and K. A. Dill, J. Chem. Theory Comput., 2007, 3, 26–41 CrossRef CAS.
- S. Dehn, V. Castelletto, I. W. Hamley and S. Perrier, Biomacromolecules, 2012, 13, 2739–2747 CrossRef CAS PubMed.
- S. Dehn, R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Rev., 2011, 51, 214–234 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed. Engl., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- M. Malke, H. Barqawi and W. H. Binder, ACS Macro Lett., 2014, 3, 393–397 CrossRef CAS.
- L. He and P. Theato, Eur. Polym. J., 2013, 49, 2986–2997 CrossRef CAS PubMed.
- S. Serim, S. V Mayer and S. H. L. Verhelst, Org. Biomol. Chem., 2013, 11, 5714–5721 CAS.
- I. Cho, J.-B. Kim and H.-J. Jung, Polymer, 2003, 44, 5497–5500 CrossRef CAS.
- E. E. Hwang, T. R. Wilson-hill, J. I. W. O. N. Ahn, A. P. Platt, K. E. Rutledge and S. L. Goh, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 871–878 CrossRef CAS.
- A. Bakk and R. Metzler, Chem. Phys. Lett., 2004, 398, 190–193 CrossRef CAS PubMed.
- Y. Zhou, C. K. Hall and M. Karplus, Protein Sci., 1999, 8, 1064–1074 CrossRef CAS PubMed.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- O. Terreau, C. Bartels and A. Eisenberg, Langmuir, 2004, 20, 637–645 CrossRef CAS.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS.
- H. Nuhn and H.-A. Klok, Biomacromolecules, 2008, 9, 2755–2763 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. a. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- L. Ayres, K. Koch, P. H. H. M. Adams and J. C. M. Van Hest, Macromolecules, 2005, 38, 1699–1704 CrossRef CAS.
- F. Fernadez-Trillo, A. Dureaulty, J. P. M. Bayley, J. C. M. Van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef.
- M. Pechar, J. Brus, L. Kostka, C. Konák, M. Urbanová and M. Slouf, Macromol. Biosci., 2007, 7, 56–69 CrossRef CAS PubMed.
- S. Aluri, M. K. Pastuszka, A. S. Moses and J. A. Mackay, Biomacromolecules, 2012, 13, 2645–2654 CrossRef CAS PubMed.
- S. L. Goh, A. P. Platt, K. E. Rutledge and I. Lee, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5381–5389 CrossRef CAS.
- S. P. Cullen, X. Liu, I. C. Mandel, F. J. Himpsel and P. Gopalan, Langmuir, 2008, 24, 913–920 CrossRef CAS PubMed.
- B. Xiang, G. Sun, K. S. Lam and K. Xiao, J. Biomed. Mater. Res., Part A, 2010, 95, 245–255 CrossRef PubMed.
- C. Chassenieux, B. Charleux and C. Burguie, Polymer, 2003, 44, 509–518 CrossRef.
- Z. Lei and S. Bi, J. Biotechnol., 2007, 128, 112–119 CrossRef CAS PubMed.
- A. C. Greene, J. Zhu, D. J. Pochan, X. Jia and K. L. Kiick, Macromolecules, 2011, 44, 1942–1951 CrossRef CAS PubMed.
- Y. Xing, C. Wang, P. Han, Z. Wang and X. Zhang, Langmuir, 2012, 28, 6032–6036 CrossRef CAS PubMed.
- J. Dai, Z. Bao, L. Sun, S. U. Hong, G. L. Baker and M. L. Bruening, Langmuir, 2006, 22, 4274–4281 CrossRef CAS PubMed.
- F. E. Bailey, R. D. Lukdberg and R. W. Callard, J. Polym. Sci., Part A: Polym. Chem., 1964, 2, 845–851 CAS.
- T. Ikawa, K. Abe, K. Honda and E. Tsuchida, J. Polym. Sci., Part A: Polym. Chem., 1975, 13, 1505–1514 CrossRef CAS.
- Y. Chen, X. Zheng, H. Qian, Z. Mao, D. Ding and X. Jiang, ACS Appl. Mater. Interfaces, 2010, 2, 3532–3538 CAS.
- H. T. Oyama, W. T. Tang and C. W. Frank, Macromolecules, 1987, 20, 474–480 CrossRef CAS.
- J. Hao, G. Yuan, W. He, H. Cheng, C. C. Han and C. Wu, Macromolecules, 2010, 43, 2002–2008 CrossRef CAS.
- A. Sinaga, T. A. Hatton and K. C. Tam, Biomacromolecules, 2007, 8, 2801–2808 CrossRef CAS PubMed.
- A. Sinaga, T. A. Hatton, K. C. Tam, R. V July, V. Re, M. Recei and V. August, Macromolecules, 2007, 40, 9064–9073 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sm02525c |
| ‡ Current affiliation: National Institute of Standards and Technology, 100 Bureau Drive, Stop 1070, Gaithersburg, MD 20899-1070, USA. |
| This journal is © The Royal Society of Chemistry 2015 |