
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-ranged and soft interactions between charged colloidal particles induced by multivalent coions
F. Javier
Montes Ruiz-Cabello
a,
Mohsen
Moazzami-Gudarzi
a,
Magdalena
Elzbieciak-Wodka†
a,
Plinio
Maroni
a,
Christophe
Labbez
b,
Michal
Borkovec
a and
Gregor
Trefalt
*a
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, Quai Ernest-Ansermet 30, 1205 Geneva, Switzerland. E-mail: gregor.trefalt@unige.ch
bLaboratoire Interdisciplinaire Carnot de Bourgogne, UMR 6303 CNRS, Université de Bourgogne, FR-21078, France
First published on 2nd January 2015
Abstract
Forces between charged particles in aqueous solutions containing multivalent coions and monovalent counterions are studied by the colloidal probe technique. Here, the multivalent ions have the same charge as the particles, which must be contrasted to the frequently studied case where multivalent ions have the opposite sign as the substrate. In the present case, the forces remain repulsive and are dominated by the interactions of the double layers. The valence of the multivalent coion is found to have a profound influence on the shape of the force curve. While for monovalent coions the force profile is exponential down to separations of a few nanometers, the interaction is much softer and longer-ranged in the presence of multivalent coions. The force profiles in the presence of multivalent coions and in the mixtures of monovalent and multivalent coions can be accurately described by Poisson–Boltzmann theory. These results are accurate for different surfaces and even in the case of highly charged particles. This behavior can be explained by the fact that the force profile follows the near-field limit to much larger distances for multivalent coions than for monovalent ones. This limit corresponds to the conditions with no salt, where the coions are expelled between the two surfaces.
Introduction
Electric double layer forces between solid substrates across aqueous solutions are currently a topic of intense research.1–8 Such forces are becoming routinely accessible by various methods, including the surface force apparatus, the colloidal probe technique, or total internal reflection microscopy.1–6,9–16 Double layer forces originate from overlapping diffuse layers forming near charged water–solid interfaces. Their range can be substantial at low salt levels, but decreases with increasing salt level due to progressive screening. The systems investigated mostly included simple monovalent electrolytes and surfaces of mica, latex, or various oxides (e.g., silica, alumina, titania).5,9–20 These studies reveal that at lower salt concentrations the force profiles in these systems are dominated by double layer forces, and they can be well described by the mean-field Poisson–Boltzmann (PB) theory or at lower charge densities by the linearized Debye–Hückel (DH) theory.2,21,22 To obtain accurate description of the force profiles at shorter distances, classical boundary conditions of constant charge (CC) and constant potential (CP) might be inaccurate, and a more detailed description of charge regulation effects may be necessary.13,23–25 The easiest way to incorporate such regulation effects is the constant regulation (CR) approximation. This approximation assumes a constant capacitance of the inner layer, and introduces only one additional parameter.23,26,27 The PB theory fails at smaller distances or higher salt levels, where one must consider additional interactions, especially van der Waals or hydration forces.11,14,15Forces between charged surfaces in solutions containing multivalent ions came into focus more recently.1,8,28–36 The existing studies support the picture that the PB theory provides a reasonably accurate description of the forces, at least at distances larger than several nanometers and in sufficiently dilute solutions. They further confirm that multivalent counterions interact strongly with charged surfaces.29,32,33,37 In this commonly studied case, multivalent ions are oppositely charged than the surface, and therefore they adsorb strongly. Thereby, they reduce the surface charge density, eventually to the point that the surface undergoes a charge reversal. Multivalent ions may also induce additional attractive forces, which can make the total force stronger than the van der Waals force.30–33 These additional attractions are likely related to ion correlation effects.35,38–40 These effects are not treated in the classical mean-field PB theory, but they could also be responsible for adsorption of multivalent ions.29,41,42
On the other hand, multivalent coions adsorb onto surfaces only weakly or not at all.37 In this case, the charge of the multivalent ions has the same sign as the charge of the surface. The strong electrostatic repulsion leads to a depletion of these ions from the surface, and to a modification of the structure of the diffuse layer.43,44 This situation has not been much investigated experimentally so far.7,8,37
The question whether the decay length of the double layer forces might deviate from the Debye length was equally raised.7,8,45,46 While PB theory predicts that double layer forces decay with the Debye length at large distances, theoretical studies of charge renormalization effects suggest that this decay length might be different, especially in asymmetric electrolytes.45,46 The currently available experimental results indicate that the measured screening lengths agree with the Debye length within experimental error, especially when complexation in solution is being considered.5,7,8,32,33 One should realize, however, that the theoretically predicted deviations from the Debye length are relatively small, and probably within experimental error.
Force measurements involving multivalent ions almost exclusively involved multivalent counterions, meaning that the ions are oppositely charged as the surface. Multivalent coions that are equally charged as the surface were hardly addressed. We are aware only of a limited number of studies, where these conditions were realized,7,8,37 but these studies did not analyze the respective force profiles in any detail. This situation prompted us to reinvestigate these conditions more carefully. We show that multivalent coions may induce unusually soft and long-ranged double layer forces, which can be accurately described by PB theory.
Poisson–Boltzmann (PB) theory
Electric double layer forces between colloidal particles can be analyzed by the classical PB theory.21,23,47,48 This approach treats all ions as point charges in a dielectric continuum. Their interactions are described in a mean-field fashion, whereby ion–ion correlations are being neglected. When the particles are sufficiently large, the situation can be simplified by calculating the net pressure Π(h) between two identical planar surfaces versus the surface separation h. Free energy per unit area can be then obtained by integrating the pressure profile, and the force between two particles follows from the Derjaguin approximation, which involves the effective radius.21 In the case of two spherical particles of the same size, the effective radius is half of the particle radius.The net pressure can be calculated by solving the PB equation, which defines the electric potential ψ(x) as a function of the position x, whose origin is taken at the mid-plane of the two surfaces located at x = ±h/2. When the solution contains different ions of number concentrations ci and valence zi, the PB equation reads23,47
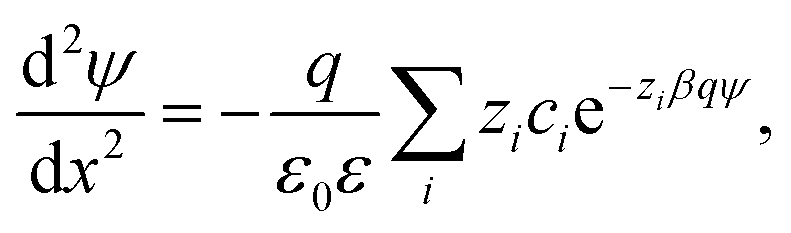 | (1) |
We assume that the solution contains a mixture of strong monovalent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and multivalent (1:z or z:1) salts of known concentrations from which the respective ionic concentrations ci can be evaluated. The net pressure is the difference between the pressure in the slit and the bulk pressure, and in the symmetric situation the former is obtained from the electric potential at the midplane ψM = ψ(0).21,47 The midplane potential is calculated from the PB equation numerically subject to the boundary condition dψ/dx = 0 at x = 0. To gain better insight into the different regimes, we further investigate two asymptotic solutions, namely the far-field and the near-field. The far-field regime reflects large separations and is equivalent to DH theory, where the solution composition enters only through the Debye length. The near-field regime describes small separations, where only the counterions neutralizing the surface charge contribute. This regime is equivalent to conditions without added salt.
:1) and multivalent (1:z or z:1) salts of known concentrations from which the respective ionic concentrations ci can be evaluated. The net pressure is the difference between the pressure in the slit and the bulk pressure, and in the symmetric situation the former is obtained from the electric potential at the midplane ψM = ψ(0).21,47 The midplane potential is calculated from the PB equation numerically subject to the boundary condition dψ/dx = 0 at x = 0. To gain better insight into the different regimes, we further investigate two asymptotic solutions, namely the far-field and the near-field. The far-field regime reflects large separations and is equivalent to DH theory, where the solution composition enters only through the Debye length. The near-field regime describes small separations, where only the counterions neutralizing the surface charge contribute. This regime is equivalent to conditions without added salt.
Far-field regime
At large separations, the electric potentials are small, and thus the PB equation can be linearized, leading to the DH equation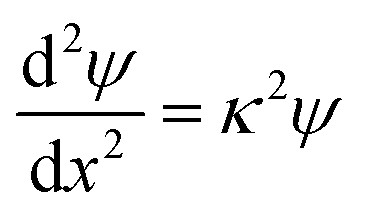 | (2) |
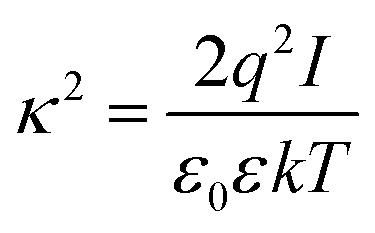 | (3) |
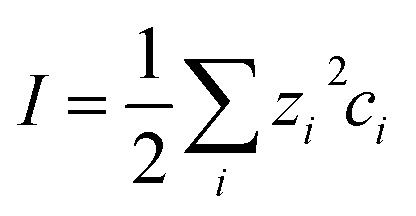 | (4) |
| Π = 2ε0εκ2ψeff2e−κh | (5) |
This relationship reflects the PB situation at larger separations, but ψeff depends on the surface potential in a non-linear fashion with the limiting behavior
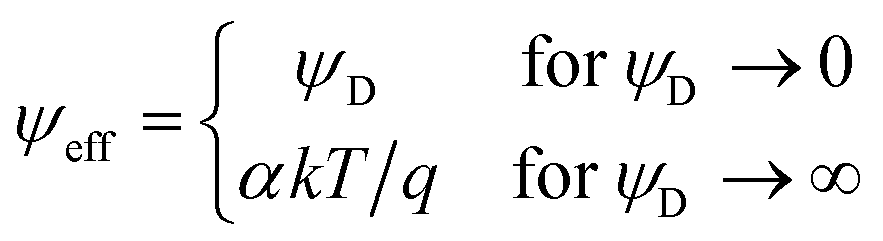 | (6) |
The above relationship applies when the potential is positive, otherwise the appropriate negative signs must be introduced. For pure electrolytes, the reduced values for the saturation potential α are known,43,44 and they are summarized for multivalent coions in Table 1. The characteristic feature is that these values strongly increase with valence. Note that the far-field regime is independent of the boundary conditions.
:1 electrolytes containing multivalent coions
| z | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| α | 4 | 6 | 8.707 | 12.314 | 17.337 |
| ψ eff (mV) | 102.7 | 154.0 | 223.5 | 316.1 | 445.0 |
Near-field regime
When the distance between the surfaces is small, the coions are expelled, and their charge is fully neutralized by the remaining counterions. This situation corresponds to no added salt, and the PB equation simplifies to42–44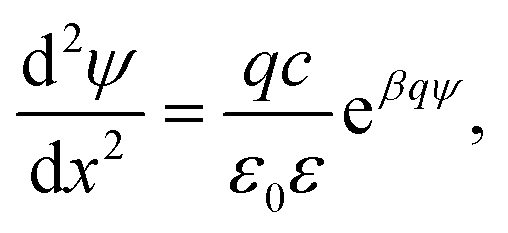 | (7) |
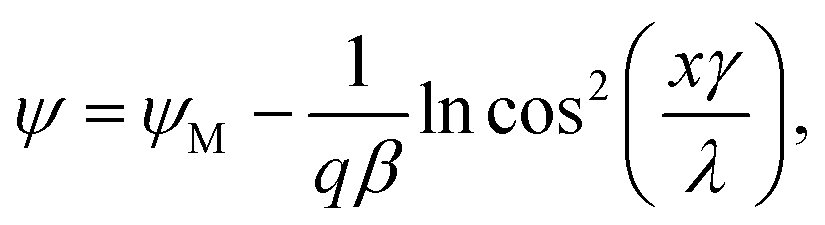 | (8) |
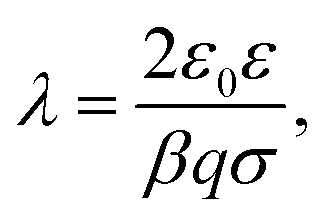 | (9) |
| Π = kTceβqψM | (10) |
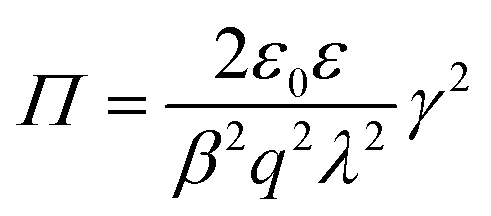 | (11) |
By invoking the potential profile given by eqn (8) and using the constant regulation boundary conditions one finds that the pressure is determined by
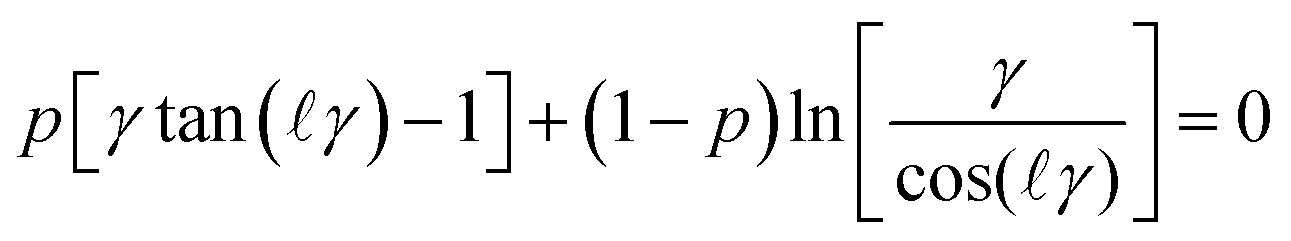 | (12) |
![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) = h/(2λ) and p is the regulation parameter. When one analyses eqn (12) for large separation distances, the pressure profile again becomes independent of the boundary conditions, and reads
= h/(2λ) and p is the regulation parameter. When one analyses eqn (12) for large separation distances, the pressure profile again becomes independent of the boundary conditions, and reads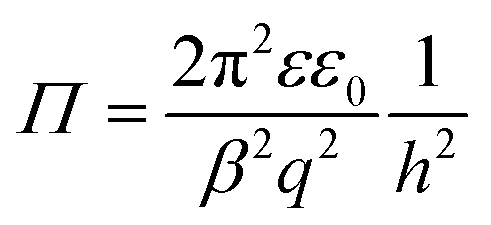 | (13) |
Experimental
Materials
Two different types of polystyrene latex particles were used, namely sulfate-terminated latex (SL) and amidine-terminated latex (AL), and they were obtained from Invitrogen. Further experiments were carried out with silica particles obtained from Bangs Laboratories Inc., USA. Table 2 summarizes the mean diameters and polydispersities as determined by the manufacturer.| Particles | Abbreviation | Diameter (μm) | Polydispersity (%) | RMS roughness (nm) | pH | Charge density (mC m−2) | Regulation parameter p |
|---|---|---|---|---|---|---|---|
| Sulfate latex | SL | 3.0 | 4.1 | 0.8 | 5.6 | −9.3 | 0.64 |
| Amidine latex | AL | 0.95 | 3.6 | 0.5 | 4.0 | +7.5 | 0.31 |
| Silica | SiO2 | 5.2 | 10 | 1.4 | 10.0 | −4.4 | 0.66 |
Experiments were carried out at 22 ± 2 °C in solutions of KCl, K2SO4, LaCl3, K3Fe(CN)6, and K4Fe(CN)6 in Milli-Q water. For the SL particles pH 5.6 was used, while for AL particles the solutions were adjusted to pH 4.0 with HCl, and for the silica particles with KOH to pH 10.0. The specific pH values were chosen in order to increase the magnitude of the surface charge density, while keeping the ionic strength low. Some experiments were also performed in solutions of aliphatic polyamine, namely of 3,6,9,12-tetraazatetradecane-1,14-diamine (N6) with chemical formula H2N–(CH2–CH2–NH)4–CH2–CH2–NH2, obtained from Sigma-Aldrich (St. Louis, USA). At pH 4.0, La3+ does not hydrolyze, and N6 has the +4 cation as the predominant species.51 The experiments were carried out at ionic strengths between 1 and 3.1 mM. The ionic strength was chosen as low as possible, such that the low salt regime is well developed, while high enough, such that ions originating from the self-dissociation of water or carbonate dissolution remain negligible.
Direct force measurements
Forces between particles were measured with a closed-loop AFM (MFP-3D, Asylum Research) mounted on an inverted optical microscope (Olympus IX70).13,52 The particles were attached on tip-less cantilevers (MikroMasch, HQ CSC37, without Al coating) and to the substrate. A pair of particles was centered laterally through optical fringes viewed in the optical microscope with an accuracy of about 100 nm. For one pair of particles, we typically recorded about 100 vertical approach-retraction cycles with an approach-retraction velocity of 300 nm s−1, a sampling rate of 5 kHz, and a cycle frequency of 0.5 Hz. The contact point was obtained from the constant compliance region. The cantilever deflection recorded in the approach part was transformed into force profiles by means of the spring constant of the cantilever. The latter constant was measured by the method developed by Sader et al.,53 which relies on the frequency response of the cantilever and its geometrical dimensions. Typical values of the spring constants were 0.1–0.3 N m−1. These values agreed within about 10% with those determined through the thermal fluctuations of the cantilever.54 The forces were subsequently down-sampled to 150 Hz and averaged over the different approach curves, leading to a force resolution of about 1 pN. All measurements were repeated with at least three pairs of different particles, and they were well reproducible. The relative error between the resulting double layer potentials, which was determined by fitting the force curves for every pair of particles under the same conditions, was below 15%.Latex particles were mounted in solution within the AFM fluid cell. A glass plate fitting the fluid cell was used as the substrate. The glass plate and cantilevers were cleaned overnight in a piranha solution, which consists of H2SO4 98% and H2O2 30% mixed in ratio 3:1. They were further rinsed with water, dried, and cleaned in an air-plasma for 20 min. Silanization of the plate and cantilevers was carried out overnight in an evacuated container aside two drops of 20 μL of 3-ethoxydimethylsilylpropylamine and 100 μL of (3-glycidoxypropyl)dimethylethoxysilane. The silanized plate was then introduced into the AFM fluid cell, and the cantilever into the cantilever holder. Before the experiments, the stock latex particle suspension was purified by dialysis against Milli-Q (Millipore) until the conductivity of the dialysate reached the value of the pure water. The particle suspensions were prepared in the respective electrolyte solutions at a particle concentration of 80 mg L−1, and injected into the fluid cell. The particles were allowed to settle for a few hours, and the cell was then rinsed with the pure electrolyte solution. By pressing the particle against the substrate with the cantilever, one could firmly attach a particle to the cantilever. For the latex particles, the constant compliance region could be identified after the jump-in in the load range of 5–10 nN with an absolute accuracy of about 0.3 nm.
The silica particles were attached by sintering in the dry state. Cantilevers were cleaned in air-plasma for 5 minutes. Few silica particles were placed on a glass slide and tiny drops of about 5 μL of glue (Araldite 2000+) were deposited in their proximity. The cantilever was mounted in the AFM, brought in contact with the glue with the translation stage, and used to pick up the particle. The cantilever was removed from the AFM, and placed into an oven at 1150 °C for 3 h. The same particles were attached to the glass slide sealing the AFM fluid cell as follows. The slide was cleaned with piranha solution for 2 hours, then rinsed with water, and dried under a stream of nitrogen. Subsequently, particles were spread onto a square quartz slide of 19 mm (Edmund Optics), and then sintered at 1150 °C for 3 hours. The reverse side of the slide with the sintered particles was glued (Pattex 100% Repair Gel) onto a glass slide sealing the AFM cell. After sintering, the substrate and the cantilevers were again cleaned in an air-plasma. This procedure leads to a firm attachment of the particles to the substrate and to the cantilever, while completely removing traces of the glue and of any organic impurities. For the silica particles, the contact point was identified at a load of about 5 nN. The precision of the contact position is inferior to the latex particles, but typically below 1 nm.
Particle roughness
The root mean square (RMS) roughness of the particles was measured by AFM imaging. The latex particles were deposited for about 1 hour on a piranha-cleaned and silanized glass slide with dimensions about 1 cm × 1 cm. These slides were silanized overnight in an evacuated glass container aside a 50 μL drop of 3-ethoxydimethylsilylpropylamine (for SL) or (3-glycidoxypropyl)dimethylethoxysilane (for AL). After the deposition of the particles, the substrate was rinsed in 10 mM KCl solution. The images were recorded in liquid with a Cypher AFM instrument (Asylum Research, Santa Barbara, USA) with BioLever Mini cantilevers (BL-AC40TS, Olympus, Japan). They had a nominal tip radius of <9 nm and a resonance frequency of around 30 kHz in water. The scan rate was 2.0 Hz, the scan size 0.5 μm × 0.5 μm, and the free oscillation amplitude (FOA) 20 nm. The set-point was fixed at around 70% of the FOA.The roughness of the silica particles was determined as follows. The particles sintered onto a quartz slide, which was previously cleaned with piranha solution, rinsed with water, and finally treated in air plasma for 20 min. The particles were imaged in air with the MFP-3D in amplitude modulation mode. Silicon cantilevers (OMCL-AC240TS, Olympus) with a nominal tip radius <10 nm and a resonance frequency of about 70 kHz were used. Images were acquired with a scan rate of 2 μm s−1, an FOA of about 40 nm, and a set point around 80% of the FOA. The roughness was determined for an area of 1 μm × 1 μm. The RMS roughness of the particles is summarized in Table 2. These values are <1 nm for the latex particles, and 1.4 nm for the silica particles. All particles used can be thus considered to be smooth for the relevant surface separations considered.
Results and discussion
We present direct force measurements between pairs of similar colloidal particles in aqueous solutions containing multivalent coions. These multivalent ions have the same sign of the charge as the surface, and they do not adsorb on the surface. With respect to the monovalent situation, however, one obtains much softer and long-ranged force profiles. Several types of particles and different coions are compared with PB theory, which captures the observed profiles very well.Pressure profiles
Let us first discuss pressure profiles between two charged plates calculated with PB theory. These profiles are shown in Fig. 1, and they reveal the relevant features more clearly than the force profiles discussed below. At larger distances, the profiles decay exponentially as one expects from the far-field linearized DH solution. This decay is evident from the linear dependence in the semi-logarithmic representation. As the separation decreases, the coions are progressively being expelled between the plates. When their concentration becomes negligible, the pressure profile follows the near-field solution of the PB equation. This solution assumes that the charge of the plates is fully neutralized by counterions, which corresponds to the situation of no added salt.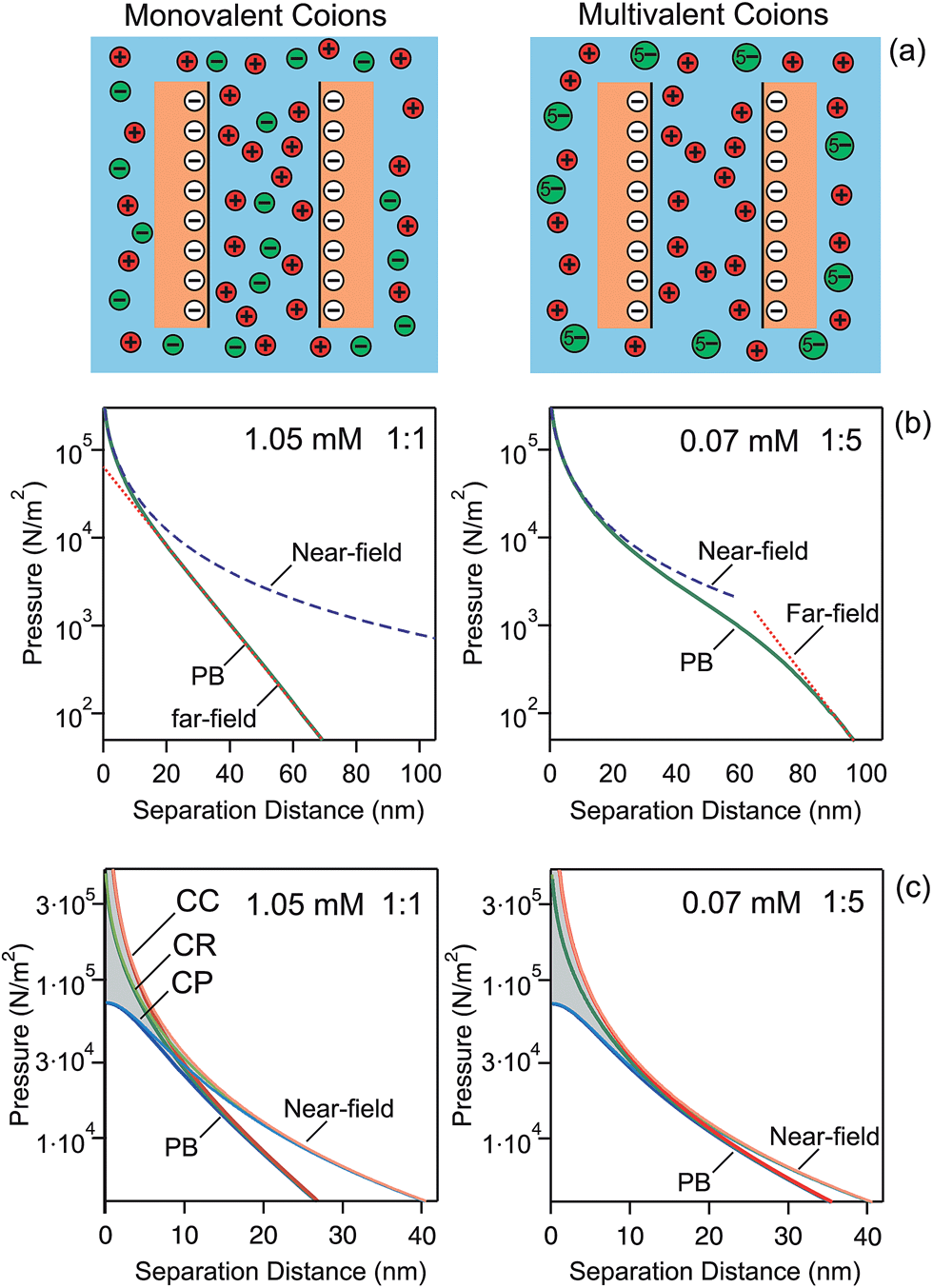 | ||
| Fig. 1 (a) Schematic presentation of expulsion of the multivalent coions between the charged walls. (b) Net pressure between charged plates in the presence of 1.05 mM 1:1 and 0.05 mM 1:5 electrolyte calculated with the PB theory. The near and far-field asymptotes are denoted by dashed lines. (c) The effect of the CC, CR, and CP boundary conditions for the same surfaces. Near-field solution is also shown. A surface charge density of −10 mC m−2 and regulation parameter p = 0.5 are used throughout. Ionic strength is 1.05 mM. | ||
Fig. 1b shows the calculated force profiles for plates of a surface change density of −10 mC m−2 in a solution of an ionic strength of 1.05 mM. This surface charge density corresponds to the diffuse layer potentials −87 mV and −114 mV in 1:1 and 1:5 electrolytes, respectively. The left panel shows the familiar situation of monovalent 1:1 salt. The pressure profile is dominated by the exponential far-field DH profile down to separations of a few nanometers. The effective potential is −69 mV, which is about half of the saturation value. The near-field profile sets in at small separation distances only. For a pentavalent 1:5 electrolyte, the profile is much softer and longer-ranged, and features a sigmoidal shape. At larger distances the pressure is again exponential, but this dependence only sets in at relatively large distances, around 80 nm here. The effective potential is now −293 mV, which is still substantially below the saturation value. At smaller distances, the pressure follows the near-field regime, but this regime now sets in at much larger distances than for the monovalent ones, about 30 nm in this case. This wider region of validity is related to the larger magnitude of the effective potential. This larger value results from the fact that the multivalent coions get expelled from the slit at larger distances than the monovalent ones. This difference is a consequence of the higher charge of the multivalent ions, which leads to their higher electrostatic energy when they are situated between the plates.
The details of the pressure profiles at smaller distances are shown in Fig. 1c. At small separations, the effect of boundary conditions is substantial. The CC conditions, characterized by the regulation parameter p = 1, result in the strongest repulsion by the double layer forces. Regulation effects are reflected by smaller regulation parameters and they lead to the decrease of the strength of the repulsion, as illustrated with p = 0.5 and p = 0 (CP conditions). At larger distances, the effect of boundary conditions disappears, and the near-field solution converges to eqn (13). The near-field solution reproduces the full PB calculations at small distances. However, its region of validity is much wider for the multivalent coions than for the monovalent ones.
Characteristic length scales in the far-field and near-field regimes are the Debye length κ−1 and Gouy–Chapman length λ. In this example, we approximately have κ−1 = 9.5 nm and λ = 3.6 nm. The Debye length reflects the range of the exponential decay at larger distances, while the Gouy–Chapman length measures the thickness of the layer containing counterions only.
Sulfate latex particles
Let us first discuss the effect of multivalent coions on experimentally measured force profiles between negatively charged SL particles of 3 μm in diameter. Fig. 2 compares forces in 1.0 mM KCl solutions and 0.1 mM K4Fe(CN)6 solutions. These strong electrolytes fully dissociate into Fe(CN)64− or Cl−, which are the coions, K+ being the counterion. The ionic strength of both solutions is the same, namely 1.0 mM. The force profile in the monovalent electrolyte shown in Fig. 2a is exponential down to a few nanometers, as was the case for the pressure. The profile in the presence of multivalent coions shown in Fig. 2b is much softer, and features a sigmoidal shape. In this case, the exponential dependence sets in only around 60 nm. In both cases, one observes a jump-in close to contact, which occurs due to additional van der Waals attraction and hydrophobic forces.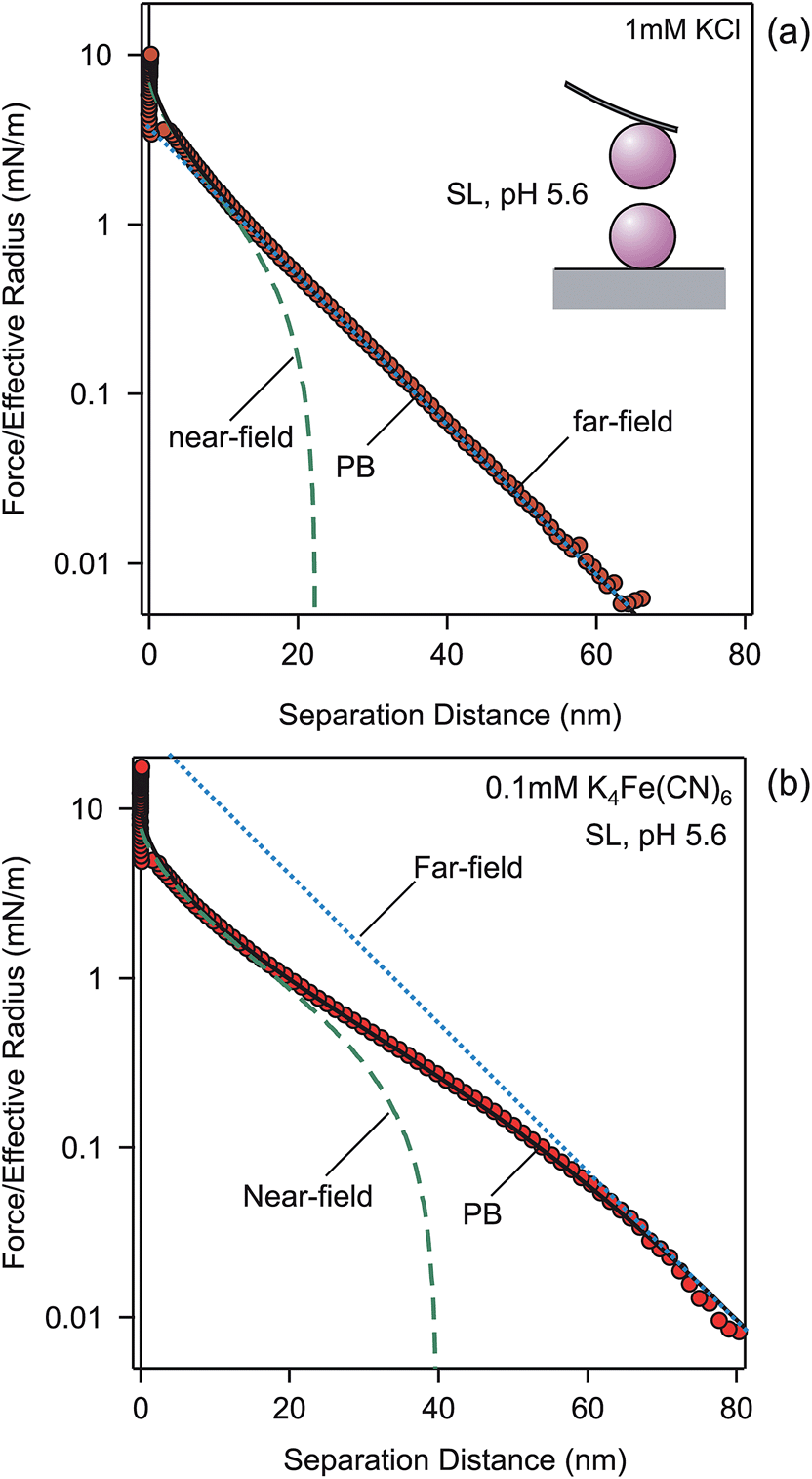 | ||
| Fig. 2 Forces between SL particles in (a) 1 mM KCl and (b) 0.1 mM K4Fe(CN)6 at pH 5.6. Points designate the experiments, while solid lines are PB calculations with a surface charge density of −9.3 mC m−2 and regulation parameter p = 0.64. The far-field and near-field asymptotes are presented with dashed lines. | ||
The measured force profiles can be described with PB theory perfectly well. By fitting the force profile measured in the KCl solution, we extract the surface charge density σ = −9.3 ± 0.1 mC m−2 and a regulation parameter p = 0.64 ± 0.02 (see also Table 2). The calculation uses the fixed analytical KCl concentration of 1.0 mM. When this concentration would be adjusted, the fitted concentration will remain within 5% of this value. We now keep the same surface parameters, and predict the force profile in 0.10 mM K4Fe(CN)6 solution with the PB theory. Again, the analytical concentration is being fixed. The calculation reproduces the sigmoidal shape of the measured force curve extremely well. The fact that the force profiles are consistent with the same surface charge density for monovalent and multivalent coions suggests that the multivalent ions adsorb onto the surface very weakly, or not at all. The lack of adsorption is obviously related to the strong electrostatic repulsion between multivalent coions and the surface.
The reason for the different appearance of the force curves is related to the fact that multivalent coions are being expelled from the gap between the surfaces at much larger separations than the monovalent ones. Therefore, the near-field solution provides a good description of the force profile to larger distances, albeit not as large as was the case for the pressure profile. The sigmoidal dependence is reinforced by the larger magnitude of the effective potential entering the far-field profile for the multivalent salts. For KCl, the effective potential is ψeff = −67 mV, while for K4Fe(CN)6 one has ψeff = −176 mV. The magnitude of these values is still substantially below the saturation values given in Table 1. At the same time, the force profile must converge into the near-field profile at small distances. While for KCl, the far-field asymptote lies below the near-field profile, for K4Fe(CN)6 the far-field asymptote lies above it, and thus the force curve makes the sigmoidal transition converge into the near-field profile.
Further force measurements with other solution compositions confirm that PB theory describes the force profiles quantitatively. Fig. 3 shows measurements in K4Fe(CN)6 solutions of varying concentrations. Thereby, the charge density and regulation properties of the surface were kept at the previous values (Table 2) and the solution concentrations were fixed to the known analytical concentrations of the solutions. These calculations contain no adjustable parameters, and provide very satisfactory results.
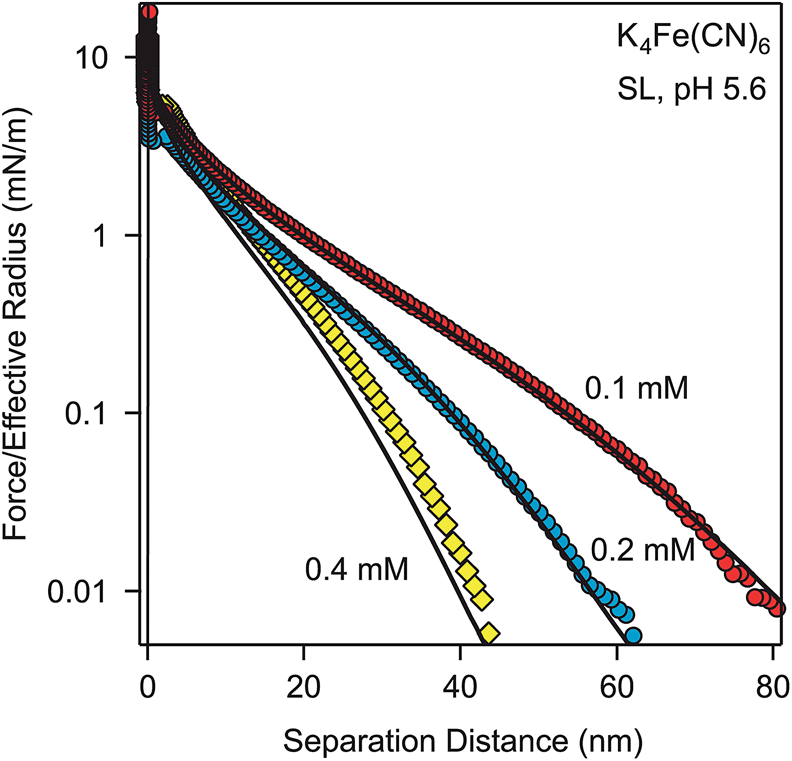 | ||
| Fig. 3 Forces between SL particles in different concentrations of K4Fe(CN)6 at pH 5.6. Points designate the experiments, while solid lines are predictions with the PB theory with a surface charge density of −9.3 mC m−2 and regulation parameter p = 0.64. | ||
Fig. 4a shows another test of the PB theory, as these experiments were carried out in mixtures of KCl and K4Fe(CN)6 solutions at a constant ionic strength of 1.0 mM. The calculations rely on the solution of the PB equation for mixed electrolytes, and they predict the observed force profiles very well. Again, no adjustable parameters were used.
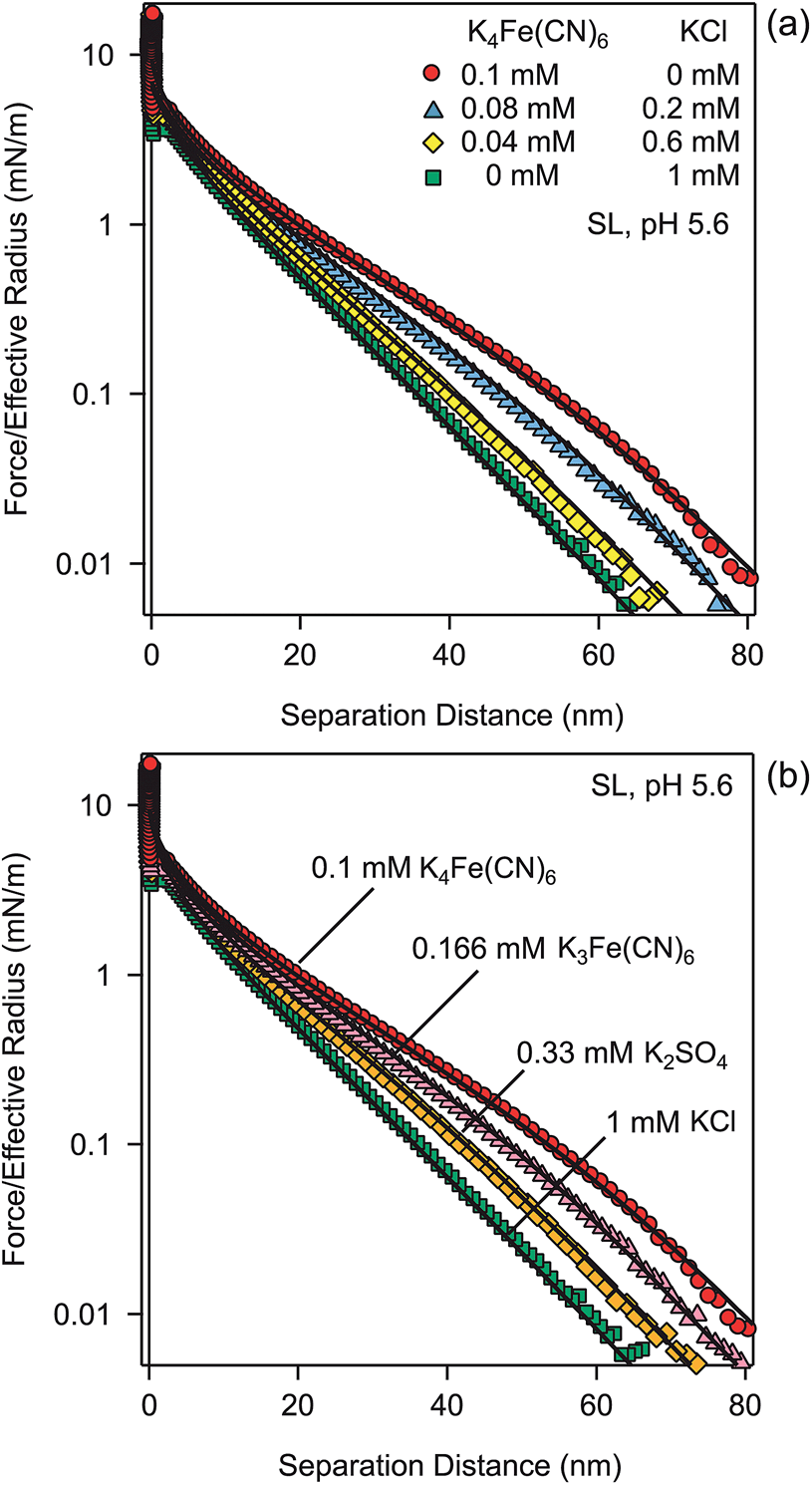 | ||
| Fig. 4 (a) Forces between SL particles in a mixture of KCl and K4Fe(CN)6 at pH 5.6 and fixed ionic strength of 1 mM. (b) Forces in KCl, K2SO4, K3Fe(CN)6, and K4Fe(CN)6 solutions at pH 5.6 and constant ionic strength of 1 mM. Points designate the experiments, while solid lines are predictions of the PB theory with a surface charge density of −9.3 mC m−2 and regulation parameter p = 0.64. | ||
Fig. 4b summarizes the effect of the valence of different coions. These experiments were carried out in solutions of KCl, K2SO4, K3Fe(CN)6, and K4Fe(CN)6, thus covering valences of coions of −1, −2, −3, and −4. The ionic strength was fixed to 1.0 mM. The corresponding predictions of the PB theory, which contain no adjustable parameters, are again in excellent agreement with experiment.
In all cases, a fixed value of surface charge density satisfactorily describes the forces, which implies that the ions involved do not adsorb onto the particles. This behavior is characteristic for multivalent coions, and is in contrast to multivalent counterions, which readily adsorb onto the surfaces and modify the surface charge or even induce charge reversal.29,32,33 However, the assumption of a fixed surface charge density leads to minor discrepancies, for example, for the highest concentration shown in Fig. 3. These discrepancies are probably related to small variations of the surface charge density and the surface regulation properties. These variations could be caused by weak adsorption of the ions involved onto the surface.
All calculations used the analytical concentrations. While the analytical concentrations are accurate within <1%, some modification might result from chemical decomposition of Fe(CN)64− due to oxidation. We find that the analytical concentrations are consistent with the measured force profiles to an accuracy of about <10%. This agreement is comparable to previous studies that attempted to determine the salt concentrations from measured force profiles.5,7,8,32,33
Amidine latex particles
A similar set of experiments was carried out with positively charged AL particles of a diameter of 0.95 μm. For these particles, the cations are the coions, and for this reason we investigated solutions containing La3+ and K+. The forces in mixtures of KCl and LaCl3 solutions at a fixed ionic strength of 3.1 mM are shown in Fig. 5a. One observes the same features as in the previously discussed case with the SL particles. In the monovalent salt, the decay is exponential of larger part of the accessible distance range, typically down to a few nanometers. For multivalent coions, the exponential decay sets in at larger distances, at about 30 nm. Again, the characteristic soft and sigmoidal force profile is observed. For the AL particles, one observes a more pronounced jump-in close to contact, which probably occurs since the hydrophobic forces are stronger for AL than for SL .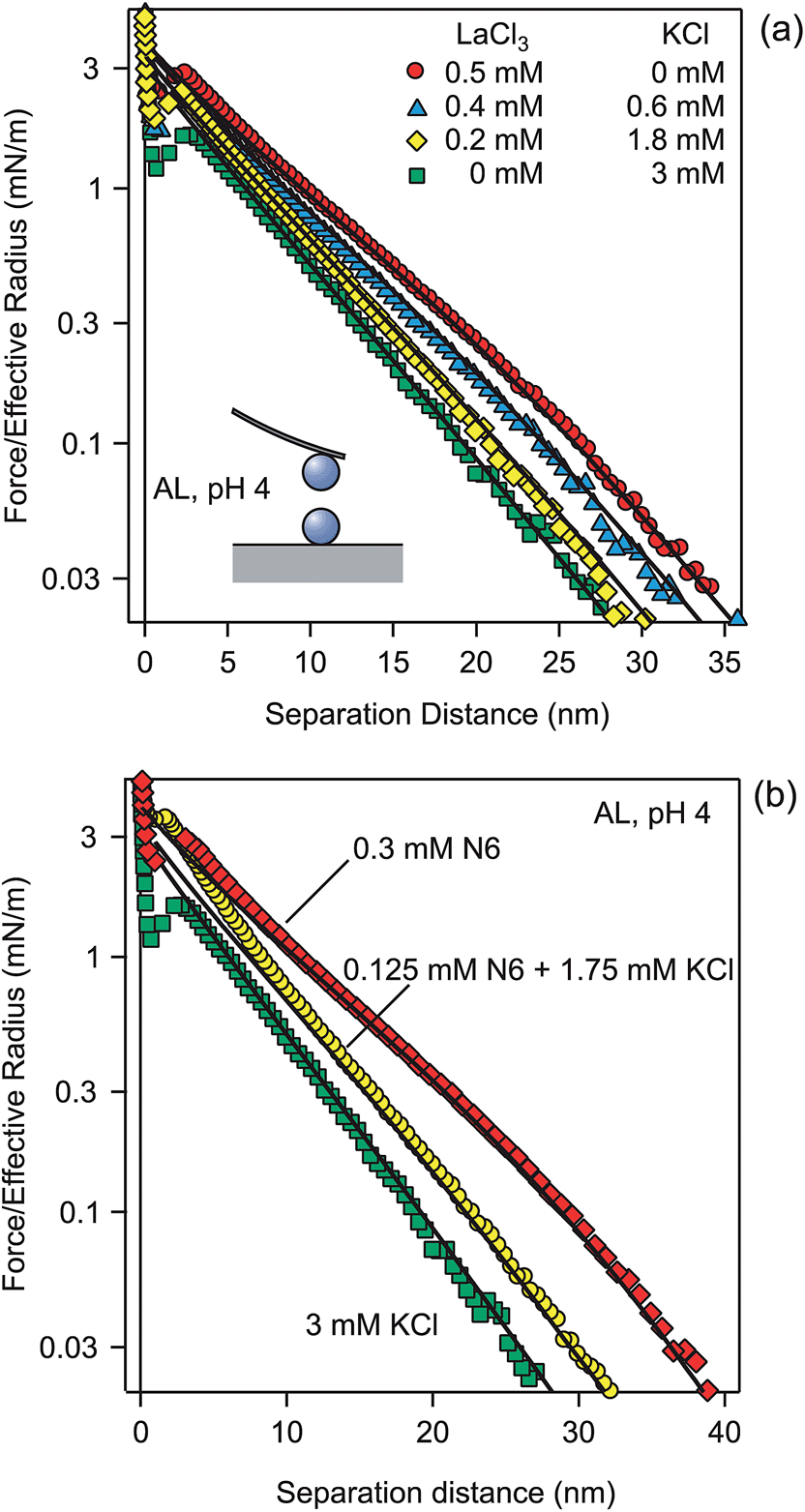 | ||
| Fig. 5 (a) Forces between AL particles in a mixture of KCl and LaCl3 at pH 4 and a fixed ionic strength of 3.1 mM. (b) Forces in the presence of N6 polyamine and its mixture with KCl. Points designate the experiments, while solid lines are predictions with the PB theory with a surface charge density of +7.5 mC m−2 and regulation parameter p = 0.31. | ||
The surface properties of the particles were again determined by fitting the force profile in KCl solutions, whereby the solution concentrations were fixed to the analytical values. The respective surface charge density was σ = +7.5 ± 0.1 mC m−2 and the regulation parameter p = 0.31 ± 0.02 (Table 2). With these values, one can correctly predict the force profiles in all mixtures. These calculations contain again no adjustable parameters, since the analytical concentrations and the surface properties are kept fixed (Table 2). We have also fitted the electrolyte concentrations, and we found that they agreed within 10% with the analytical ones. A small discrepancy could also be related to the formation of LaCl2+ complexes in solution.33,55 The reason for the softer repulsion is again related to the more effective exclusion of the multivalent coions from the gap and to the larger effective potentials for the multivalent coions (Table 1). The near-field profile again remains a good approximation at much larger distances for multivalent coions than for monovalent ones.
Fig. 5b shows a similar effect with highly charged organic aliphatic polyamine. N6 is a linear amine, which does not ionize fully, and the prevalent species has a valence of +4. The forces in the presence of N6 and its mixture with KCl are predicted with PB theory only by invoking the respective concentrations. Again the ionic strength is fixed to 3.1 mM. No parameter adjustment was made, as the same surface properties as before were used. Good agreement with the observed force profiles was found. The minor discrepancies could be related to weak adsorption onto the surface or due to the contribution of other valences, which occur in small concentrations due to the multi-step dissociation equilibria.
Silica particles
To confirm that the described effects are generic for various types of materials, forces between negatively charged silica particles of 5.2 μm in diameter were measured in solutions of K4Fe(CN)6, KCl, and their mixtures at an ionic strength of 2.1 mM (Fig. 6). As for the previously discussed systems, one observes softer profiles in the presence of multivalent coions. The main difference to the previous force profiles is that forces remain repulsive down to contact and that one cannot observe a jump-in at short distances. This feature is probably related to repulsive short-ranged hydration forces acting between silica surfaces.17,19,20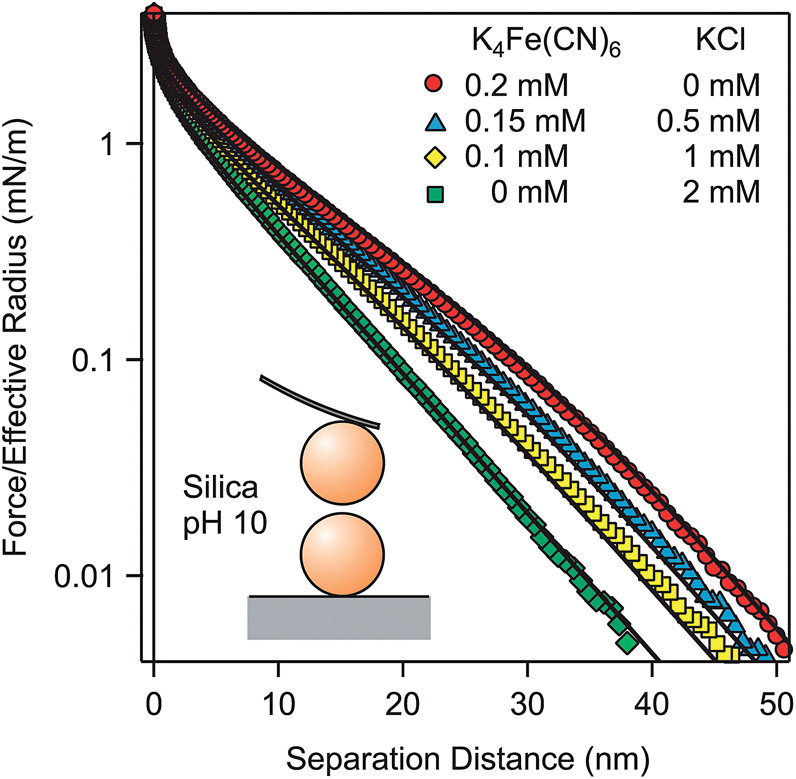 | ||
| Fig. 6 Forces between silica particles in a mixture of KCl and K4Fe(CN)6 at pH 10 and a fixed ionic strength of 2.1 mM. Points designate the experiments, while solid lines are predictions of PB theory with a surface charge density of −4.4 mC m−2 and regulation parameter p = 0.66. | ||
Surface properties of silica were determined by fitting the force profile in KCl solution with PB theory. Thereby, the analytical salt concentration was used as before. From this fit, we obtain the surface charge density σ = −4.4 ± 0.1 mC m−2 and a regulation parameter p = 0.66 ± 0.02 (Table 2). These parameters are then used to predict the force profiles in K4Fe(CN)6 solutions and their mixtures with KCl. Since the appropriate salt concentrations were kept fixed, no adjustable parameters enter the calculation. The PB theory again describes the measured force profiles very well. The characteristic features observed for the force profiles in the presence of multivalent coions do not depend on the nature of the particles. The obtained surface charge density is well comparable to the published values measured for silica surfaces at pH ∼ 10 at low salt concentrations.17–20,56 These values were determined by force measurements and they are in the range from −8 to −4 mC m−2.
Conclusions
Direct force measurements were carried out with different types of negatively and positively charged colloidal particles in aqueous electrolyte solutions containing multivalent coions. In all situations studied, the multivalent ions have the same sign of charge as the particles. While these ions hardly adsorb onto such surfaces, they strongly modify the structure of the diffuse layer. As a consequence, one observes unusually softer and longer-ranged force profiles than for monovalent electrolytes. These profiles have a sigmoidal appearance in the popular semi-logarithmic representation and they can be described with PB theory for asymmetric electrolytes very well. These characteristic features reflect the fact that multivalent coions get expelled from the gap between the surfaces at larger distances than for monovalent ones. At short distances, the force profile converges to the near-field solution of the PB equation, which reflects the situation where the surface charge is neutralized by counterions only, as is the case without added salt. In the presence of multivalent coions, the near-field profile represents a good approximation up to larger separation distances than for monovalent coions. This fact is also reflected by the effective potentials entering the far-field DH theory, which are higher for the multivalent coions than for the monovalent ones. These findings demonstrate that the PB theory can accurately describe forces in the presence of multivalent coions even when surfaces are highly charged.The measured force profiles reflect the analytical electrolyte concentrations very well. While the observed discrepancies are typically <10%, these deviations could also be related to complexation in solution or to a chemical decomposition of the ions. The present work confirms the findings of previous studies,7,8 which concluded that salt concentrations determined from direct force measurements are in agreement with the analytical ones. Systematic deviations between experimentally observed decay lengths in the far-field regime and the Debye lengths cannot be confirmed even in the presence of multivalent ions. While these deviations might exist, they are beyond the resolution of the surface force instruments currently in use.
The present work contributes to the current discussion on the role of multivalent ions in electrostatic interactions between charged surfaces.8,29–34,36 Thereby, a major difference between multivalent counterions and multivalent coions must be stressed. Multivalent counterions strongly adsorb onto charged surfaces, and they may eventually lead to overcharging. Multivalent coions do not adsorb onto such surfaces, or eventually only weakly. Therefore, they do not affect the surface charge, but they strongly modify the structure of the electric double layer and the resulting interaction forces. Therefore, addition of multivalent coions provides a new means of tuning repulsive double layer forces. This possibility might be of relevance in colloidal self-assembly, where delicate balance between interactions must be achieved in order to obtain the regular particle arrangements.57,58 In this respect, a more detailed understanding how multivalent ions influence forces between dissimilar surfaces would be desirable as well.
Acknowledgements
This research was supported by the Swiss National Science Foundation, University of Geneva, COST Action CM1101, and the Swiss Secretariat of Education and Research. We acknowledge helpful discussions with Jordi Faraudo and Peter Kralchevsky.References
- R. M. Pashley and J. N. Israelachvili, J. Colloid Interface Sci., 1984, 97, 446–455 CrossRef CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1992 Search PubMed.
- T. Baimpos, B. R. Shrestha, S. Raman and M. Valtiner, Langmuir, 2014, 30, 4322–4332 CrossRef CAS PubMed.
- H. J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS PubMed.
- A. Tulpar, V. Subramanian and W. Ducker, Langmuir, 2001, 17, 8451–8454 CrossRef CAS.
- M. Borkovec, I. Szilagyi, I. Popa, M. Finessi, P. Sinha, P. Maroni and G. Papastavrou, Adv. Colloid Interface Sci., 2012, 179–182, 85–98 CrossRef CAS PubMed.
- M. Nayeri, Z. Abbas and J. Bergenholtz, Colloids Surf., A, 2013, 429, 74–81 CrossRef CAS PubMed.
- M. M. Kohonen, M. E. Karaman and R. M. Pashley, Langmuir, 2000, 16, 5749–5753 CrossRef CAS.
- R. M. Pashley, J. Colloid Interface Sci., 1981, 83, 531–546 CrossRef CAS.
- G. Toikka, R. A. Hayes and J. Ralston, Langmuir, 1996, 12, 3783–3788 CrossRef CAS.
- M. Elzbieciak-Wodka, M. Popescu, F. J. Montes Ruiz-Cabello, G. Trefalt, P. Maroni and M. Borkovec, J. Chem. Phys., 2014, 140, 104906 CrossRef PubMed.
- F. J. Montes Ruiz-Cabello, P. Maroni and M. Borkovec, J. Chem. Phys., 2013, 138, 234705 CrossRef PubMed.
- I. Popa, P. Sinha, M. Finessi, P. Maroni, G. Papastavrou and M. Borkovec, Phys. Rev. Lett., 2010, 104, 228301 CrossRef.
- R. M. Espinosa-Marzal, T. Drobek, T. Balmer and M. P. Heuberger, Phys. Chem. Chem. Phys., 2012, 14, 6085–6093 RSC.
- M. A. Bevan and D. C. Prieve, Langmuir, 1999, 15, 7925–7936 CrossRef CAS.
- M. Dishon, O. Zohar and U. Sivan, Langmuir, 2009, 25, 2831–2836 CrossRef CAS PubMed.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Langmuir, 1992, 8, 1831–1836 CrossRef CAS.
- K. Hu and A. J. Bard, Langmuir, 1997, 13, 5114–5119 CrossRef CAS.
- M. Giesbers, J. M. Kleijn and M. A. Cohen Stuart, J. Colloid Interface Sci., 2002, 248, 88–95 CrossRef CAS PubMed.
- J. Morag, M. Dishon and U. Sivan, Langmuir, 2013, 29, 6317–6322 CrossRef CAS PubMed.
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal Dispersions, Cambridge University Press, Cambridge, 1989 Search PubMed.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle Deposition and Aggregation: Measurement, Modeling, and Simulation, Butterworth-Heinemann Ltd., Oxford, 1995 Search PubMed.
- R. Pericet-Camara, G. Papastavrou, S. H. Behrens and M. Borkovec, J. Phys. Chem. B, 2004, 108, 19467–19475 CrossRef CAS.
- B. V. Zhmud, A. Meurk and L. Bergstrom, J. Colloid Interface Sci., 1998, 207, 332–343 CrossRef CAS PubMed.
- S. Rentsch, H. Siegenthaler and G. Papastavrou, Langmuir, 2007, 23, 9083–9091 CrossRef CAS PubMed.
- S. L. Carnie and D. Y. C. Chan, J. Colloid Interface Sci., 1993, 161, 260–264 CrossRef CAS.
- S. H. Behrens and M. Borkovec, J. Phys. Chem. B, 1999, 103, 2918–2928 CrossRef CAS.
- R. M. Pashley, J. Colloid Interface Sci., 1984, 102, 23–35 CrossRef CAS.
- K. Besteman, M. A. G. Zevenbergen, H. A. Heering and S. G. Lemay, Phys. Rev. Lett., 2004, 93, 170802 CrossRef CAS.
- O. Zohar, I. Leizerson and U. Sivan, Phys. Rev. Lett., 2006, 96, 177802 CrossRef.
- M. Dishon, O. Zohar and U. Sivan, Langmuir, 2011, 27, 12977–12984 CrossRef CAS PubMed.
- P. Sinha, I. Szilagyi, F. J. Montes Ruiz-Cabello, P. Maroni and M. Borkovec, J. Phys. Chem. Lett., 2013, 4, 648–652 CrossRef CAS.
- F. J. Montes Ruiz-Cabello, G. Trefalt, Z. Csendes, P. Sinha, T. Oncsik, I. Szilagyi, P. Maroni and M. Borkovec, J. Phys. Chem. B, 2013, 117, 11853–11862 CrossRef PubMed.
- D. Ebeling, D. van den Ende and F. Mugele, Nanotechnology, 2011, 22, 305706 CrossRef PubMed.
- R. Kjellander, S. Marcelja, R. M. Pashley and J. P. Quirk, J. Chem. Phys., 1990, 92, 4399–4407 CrossRef CAS PubMed.
- C. Gutsche, U. F. Keyser, K. Kegler and F. Kremer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 031403 CrossRef CAS.
- F. J. Montes Ruiz-Cabello, G. Trefalt, P. Maroni and M. Borkovec, Langmuir, 2014, 30, 4551–4555 CrossRef CAS PubMed.
- J. Forsman, J. Phys. Chem. B, 2004, 108, 9236–9245 CrossRef CAS.
- M. M. Hatlo and L. Lue, Soft Matter, 2009, 5, 125–133 RSC.
- K. Bohinc, J. M. A. Grime and L. Lue, Soft Matter, 2012, 8, 5679–5686 RSC.
- M. Kanduc, A. Naji, J. Forsman and R. Podgornik, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 011502 CrossRef.
- B. I. Shklovskii, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1999, 60, 5802–5811 CrossRef CAS.
- G. Tellez, Philos. Trans. R. Soc., A, 2011, 369, 322–334 CrossRef CAS PubMed.
- M. Han and X. J. Xing, J. Stat. Phys., 2013, 151, 1121–1139 CrossRef.
- R. Kjellander and D. J. Mitchell, J. Chem. Phys., 1994, 101, 603–626 CrossRef CAS PubMed.
- A. McBride, M. Kohonen and P. Attard, J. Chem. Phys., 1998, 109, 2423–2428 CrossRef CAS PubMed.
- G. Trefalt, I. Szilagyi and M. Borkovec, J. Colloid Interface Sci., 2013, 406, 111–120 CrossRef CAS PubMed.
- S. H. Behrens and M. Borkovec, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1999, 60, 7040–7048 CrossRef CAS.
- M. Borkovec and S. H. Behrens, J. Phys. Chem. B, 2008, 112, 10795–10799 CrossRef CAS PubMed.
- A. C. Cowley, N. L. Fuller, R. P. Rand and V. A. Parsegian, Biochemistry, 1978, 17, 3163–3168 CrossRef CAS.
- A. E. Martell, R. M. Smith and R. J. Motekaitis, Critically Selected Stability Constants of Metal Complexes: Version 8.0., National Insitutue of Standards and Technology, Gainesburg, 2004 Search PubMed.
- I. Popa, G. Gillies, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2009, 113, 8458–8461 CrossRef CAS PubMed.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS PubMed.
- C. F. Baes and R. E. Mesmer, The Hydrolysis of Cations, Krieger Publishing, Malabar 1976 Search PubMed.
- V. Kuznetsov and G. Papastavrou, J. Phys. Chem. C, 2014, 118, 2673–2685 CAS.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381–384 CrossRef CAS PubMed.
- F. Li, D. P. Josephson and A. Stein, Angew. Chem., Int. Ed., 2011, 50, 360–388 CrossRef CAS PubMed.
Footnote |
| † Present address: Jerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Science, 30-239 Krakow, Poland. |
| This journal is © The Royal Society of Chemistry 2015 |