
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic aminopyrrolic receptors have apoptosis inducing activity†
Seong-Hyun
Park
a,
Yoon Pyo
Choi
a,
Jinhong
Park
b,
Andrew
Share
c,
Oscar
Francesconi
d,
Cristina
Nativi
d,
Wan
Namkung
b,
Jonathan L.
Sessler
c,
Stefano
Roelens
e and
Injae
Shin
*a
aCenter for Biofunctional Molecules, Department of Chemistry, Yonsei University, 03722 Seoul, Korea. E-mail: injae@yonsei.ac.kr
bCollege of Pharmacy, Yonsei Institute of Pharmaceutical Sciences, Yonsei University, 21983 Incheon, Korea
cDepartment of Chemistry, The University of Texas at Austin, 78712-1224 Austin, Texas, USA
dDepartment of Chemistry and INSTM, University of Florence, Polo Scientifico e Tecnologico, 50019 Sesto Fiorentino, Firenze, Italy
eIstituto di Metodologie Chimiche (IMC), Consiglio Nazionale delle Ricerche (CNR), Department of Chemistry and INSTM, University of Florence, Polo Scientifico e Tecnologico, 50019 Sesto Fiorentino, Firenze, Italy
First published on 2nd October 2015
Abstract
We report two synthetic aminopyrrolic compounds that induce apoptotic cell death. These compounds have been previously shown to act as receptors for mannosides. The extent of receptor-induced cell death is greater in cells expressing a high level of high-mannose oligosaccharides than in cells producing lower levels of high-mannose glycans. The ability of synthetic receptors to induce cell death is attenuated in the presence of external mannosides. The present results provide support for the suggestion that the observed cell death reflects an ability of the receptors to bind mannose displayed on the cell surface. Signaling pathway studies indicate that the synthetic receptors of the present study promote JNK activation, induce Bax translocation to the mitochondria, and cause cytochrome c release from the mitochondria into the cytosol, thus promoting caspase-dependent apoptosis. Such effects are also observed in cells treated with mannose-binding ConA. The present results thus serve to highlight what may be an attractive new approach to triggering apoptosis via modes of action that differ from those normally used to promote apoptosis.
Introduction
Apoptosis or programmed cell death is an essential biological event occurring in multicellular organisms.1,2 Normally, unwanted or damaged cells in multicellular organisms are removed by this biological event. Even though apoptosis plays a number of key roles in normal physiological processes, abnormal apoptosis results in a range of human diseases.3–5 For instance, retarded apoptosis leads to the progression of various cancers by interfering with tissue homeostasis and thus extending cancer cell survival. Because apoptosis is associated with both normal physiology and diverse human diseases, apoptosis-regulating agents have been extensively exploited for basic biological studies and therapeutic applications. In particular, extensive effort has been devoted to the discovery of new apoptosis-inducing small molecules that may have potential as antitumor pharmaceuticals.6–9It is now well appreciated that plant lectins with glycan binding properties are able to induce apoptosis in various cancer cells, although the molecular mechanism underlying apoptotic cell death remains to be clarified.10,11 Changes in cell surface glycosylation is one of the general features of malignant transformation and tumor progression.12 In particular, high-mannose type oligosaccharides are expressed on the surface of various cancer cells; however, they are not commonly found on the surface of normal cells.13 Because mannose is prevalent as the terminal sugar of glycans in cancer cells, terminal oligomannosides are attractive therapeutic targets. In this context it is noteworthy that mannose-binding lectins, including Concanavalin A (ConA), Sophora flavescens lectin (SFL) and Polygonatum cyrtonema lectin (PCL), display cytotoxicity in various cancer cells as a result of stimulating apoptosis, presumably by interacting with extracellular and maybe also intracellular mannosides.14 However, owing to the natural cytotoxicity and the short serum half-life of plant lectins, small molecule-based glycan binding receptors15 that target glycans displayed in cancer cells might represent an attractive new approach to generating apoptosis-inducing anticancer agents. At present, the only small molecules that recognize mannosides and are known to induce apoptosis in cancer cells are pradimicin, a naturally occurring antibiotic, and its derivatives.16 Cancer cell death induced by these substances is closely correlated to the expression of high-mannose glycans on the cell surface. This lends support to the notion that small molecules that target mannosides could emerge as new anticancer agents. Here, we report progress towards this goal.
Recently, some of us reported a new set of synthetic aminopyrrolic receptors that bind mannosides with selectivity over other monosaccharides.17 Some of the compounds in question were found to possess antibiotic activity, an effect rationalized in terms of an ability to bind to cell-surface glycans thereby interfering with key cellular processes.17a Recognizing the potential link between antibiotic and anticancer activity that might exist for synthetic, mannoside-recognizing receptors, we decided to conduct an investigation of the effect of these wholly synthetic receptors on cell viability. As detailed below, we have found evidence supporting the conclusion that the synthetic receptors promote cell death via caspase-dependent apoptosis. As in the case of pradimicin, this activity is presumed to reflect an ability to recognize the cell-surface glycans.
Results and discussion
Synthetic receptors have cell death activity
Compounds 1–7 used in the present study (Fig. 1) were prepared according to previously described procedures.17 This series of compounds was chosen so as to span a range of supramolecular recognition space. Specifically, it includes synthetic receptors whose octyl mannoside binding affinities are low (1, 5), moderate (2–4) and high (6, 7) (Table S1†).17 Synthetic aminopyrrolic receptors were found to bind to highly mannosylated glycoproteins, as inferred from surface plasmon resonance (SPR) studies.18 On the basis of these observations, we hypothesized that the synthetic mannose receptors might have cell death activity as observed in lectin treated cells.14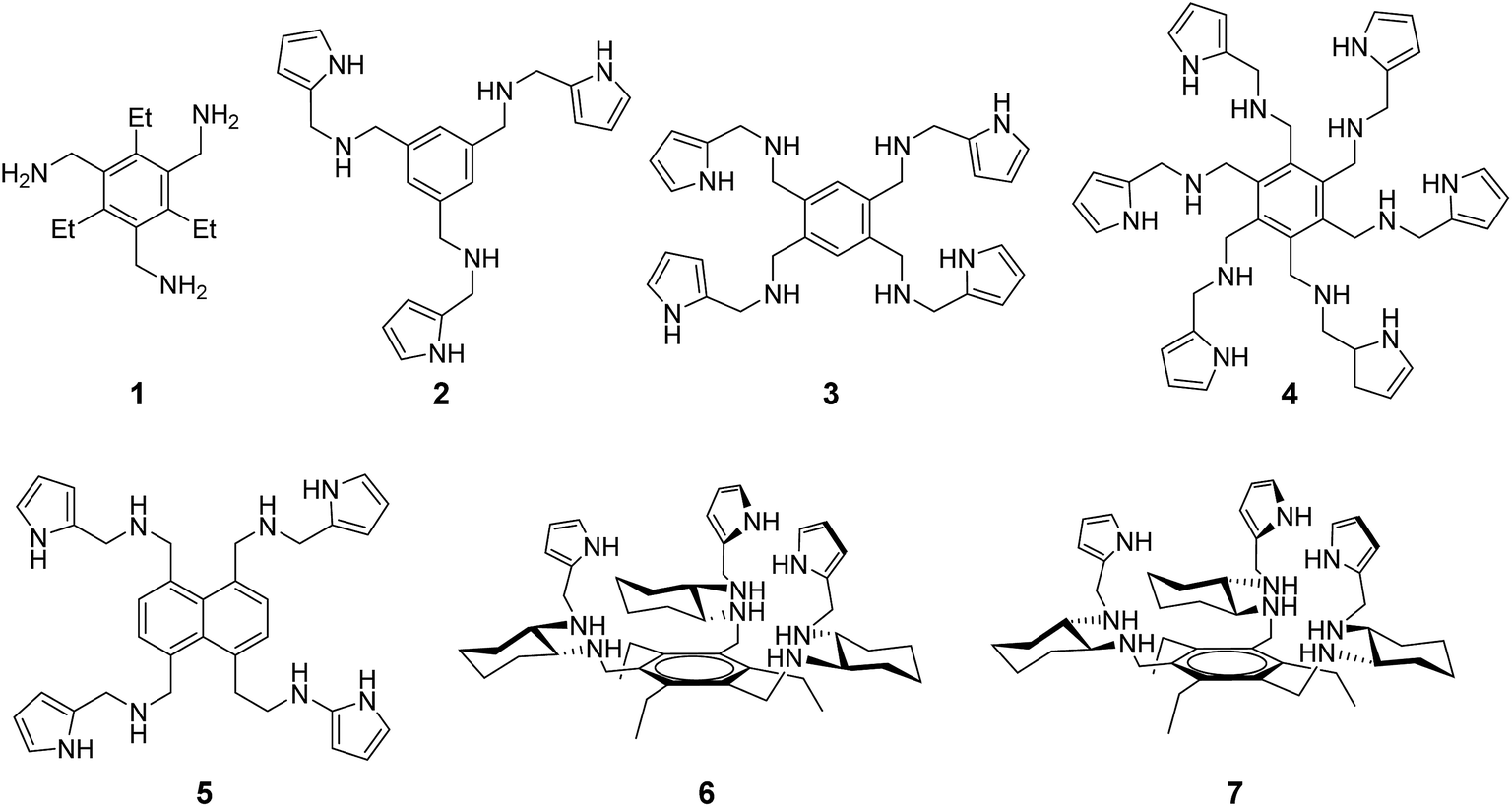 | ||
| Fig. 1 Structures 1–7. | ||
Prior to testing this proposal, the expression level of high-mannose glycans on the cell surface was measured by using mannose-binding ConA. Several cells, including HeLa (human cervical cancer cells), PLC/PRF/5 (human hepatocellular carcinoma cells), A549 (human lung adenocarcinoma epithelial cells) and KG-1 cells (human myeloid leukemia cells), were treated with biotin–conjugated ConA for 1 h in the absence and presence of a competitor, mannan. Flow cytometry analysis of cells stained with Cy3-streptavidin reveals that KG-1 cells exhibit a weak fluorescence signal while other types of cells show a strong fluorescence emission, which is markedly attenuated when mannan is present (Fig. 2). This finding indicates that the tested cells other than KG-1 cells express a high level of high-mannose oligosaccharides.
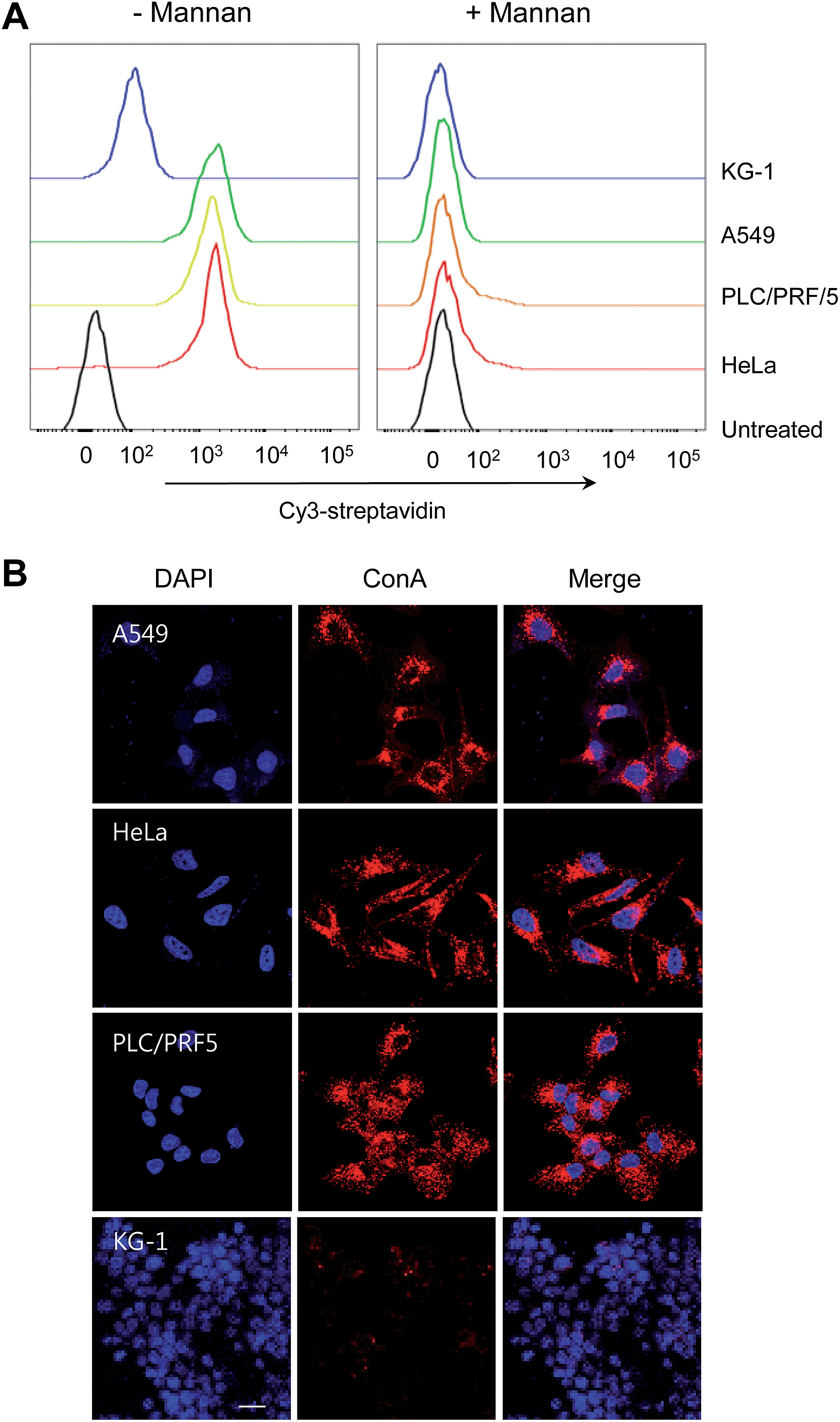 | ||
| Fig. 2 Expression levels of high-mannose glycans on the cell surface. (A) Flow cytometry of cells treated with 10 μM of biotin–ConA in the absence or presence of a competitor mannan for 1 h and then stained with Cy3-streptavidin. (B) Confocal microscopy images of cells treated with 10 μM of biotin–ConA for 1 h and then stained with Cy3-streptavidin (scale bar = 20 μM). DAPI was used to stain the nucleus. | ||
The effect of compounds 1–7 on cell viability was then examined by incubating HeLa and PLC/PRF/5 cells, both of which express a high level of high-mannose glycans, making them of particular interest in the context of the present study. Cell viabilities were measured using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays. Compounds 6 and 7 were found to be highly cytotoxic. In contrast, compounds 1–5 displayed a little or no cytotoxic activity at the tested concentrations (Fig. S1†).
Compounds 6 and 7 bind to mannosides considerably more strongly than 1–5 (Table S1†). We thus sought to test whether the cytotoxic activity induced by 6 and 7 is dependent on the expression level of high-mannose glycans on the cell surface. Toward this end, the cytotoxicity of 6 and 7 was measured in cells expressing high (HeLa, PLC/PRF/5 and A549 cells) and low (KG-1 cells) levels of high-mannose glycans. As a negative control, cells were also incubated with compound 1 with a very low mannose-binding affinity. After incubating the cells with 6 and 7, along with a control 1, for 18 h, the IC50 values were determined. They were found to be respective 1.3 μM and 2.7 μM for 6 and 7 in cells expressing high levels of high-mannose glycans (Fig. S2†). However, 6 and 7 displayed lower cytotoxicity in the case of KG-1 cells (Fig. S2†). In contrast, cell death activity induced by a control 1 was not detected.
We then examined the effect of a mannose competitor on the synthetic receptor-induced cell death. For this study, HeLa and PLC/PRF/5 cells were exposed to 6 and 7 in the presence of methyl α-mannoside as a competitor at concentrations where cytotoxicity was not observed (Fig. S3†). The number of viable cells was found to increase in a competitor concentration-dependent manner (Fig. S4†). It is worthwhile mentioning that because a monomeric mannoside is an inefficient competitor in water to block association of the synthetic receptors with cell-surface oligomannosides, a relatively high concentration of a competitor is needed to protect against cell death induced by 6 and 7. To further examine whether cytotoxicity induced by 6 and 7 is attributable to their binding properties toward cell surface mannosides, we conducted the competition experiments of compounds against mannose-binding ConA. When HeLa cells pretreated with 6 or 7 were incubated with ConA, the ability of ConA to bind cells was attenuated significantly in the presence of either compound (Fig. S5†). Collectively, the results provide support for the proposal that the cell death elicited by 6 and 7, if not all, is caused by their ability to bind to the mannose glycans displayed on the cell surface.
Compounds 6 and 7 are pyrrole-containing receptors that bear some chemical similarity to ion transporters, such as calixpyrroles and prodigiosin analogues,19 that exhibit cell death activity by mediating through-membrane anion transport. Thus, we considered it possible that such ion transport effects could be playing a role in promoting cell death. Accordingly, we investigated whether 6 and 7 were capable of inducing changes in intracellular concentrations of several ions in mammalian cells. Also studied was compound 1, a simple triamine that was not expected to be an effective ion transporter at the physiological pH. To evaluate changes in intracellular chloride concentrations, FRT (Fischer rat thyroid epithelial) cells were incubated with each compound for 2 h. These cells express a mutant yellow fluorescent protein (YFP-F46L/H148Q/I152L) whose fluorescence is quenched sensitively by chloride ions in the cytosol.19a The intracellular chloride concentrations were unaltered in cells treated with 1, 6 or 7, as inferred from the observation that fluorescence of the mutant YFP in treated cells was not changed (Fig. S6A†). To test if 1, 6, or 7 caused changes in intracellular concentrations of the sodium, potassium and calcium ions, cells, which were pretreated for 1–1.5 h with fluorescent probes for the sodium (SBFI-AM), potassium (PBFI-AM) or calcium ions (Fluo-4 NW), were exposed to each compound for 2 h. No appreciable difference in the fluorescence intensity was observed in any of these experiments (Fig. S6B–S6D†), leading us to infer that treatment with 1, 6, or 7 fails to induce a change in the intracellular concentrations of the sodium, potassium, and calcium ions. These findings indicate that the cell death induced by 6 and 7 does not result from receptor-mediated changes in the intracellular ion concentrations.
Synthetic receptors induce apoptotic cell death
Several previous studies have served to demonstrate that plant lectins induce cancer cell death through regulation of apoptotic pathways.14 To examine whether the cytotoxic synthetic receptors of this study could also induce apoptosis, HeLa and PLC/PRF/5 cells were incubated with either 6 or 7, as well as with the relatively non-cytotoxic control 1, and then treated with a mixture of fluorescein–annexin V and propidium iodide (PI). The results of flow cytometry analysis reveal that 6 and 7 induce apoptosis, as inferred from positive annexin V and PI staining (Fig. 3A and S7A†).6b,c In addition, analysis of time-dependent flow cytometry of cells treated with a compound shows that annexin V staining precedes PI staining, indicating that cells undergo apoptosis (Fig. S8†). The extent of cell shrinkage was measured as a further marker of apoptosis. Analysis of cell size by flow cytometry reveals that cells treated with 6 and 7 are characterized by a large degree of cell shrinkage (Fig. S9†). This result also serves to exclude cell death via necrosis as a dominant mechanism because necrosis generally leads to cell swelling. The loss of mitochondrial membrane potential, which is a hallmark of apoptosis, was also investigated by using a JC-1 probe that is sensitive to membrane potential.19a The intensity of the dye-derived red fluorescence in cells treated with 6 or 7 for 18 h decreases significantly, as would be expected for apoptotic cell death (Fig. 3B and S7B†). An increase in DNA fragmentation, another hallmark of apoptosis, was also observed in cells treated with 6 or 7 for 18 h (Fig. 3C and S7C†). In marked contrast to 6 and 7, no evidence of apoptosis induction was seen in the case of the control compound 1.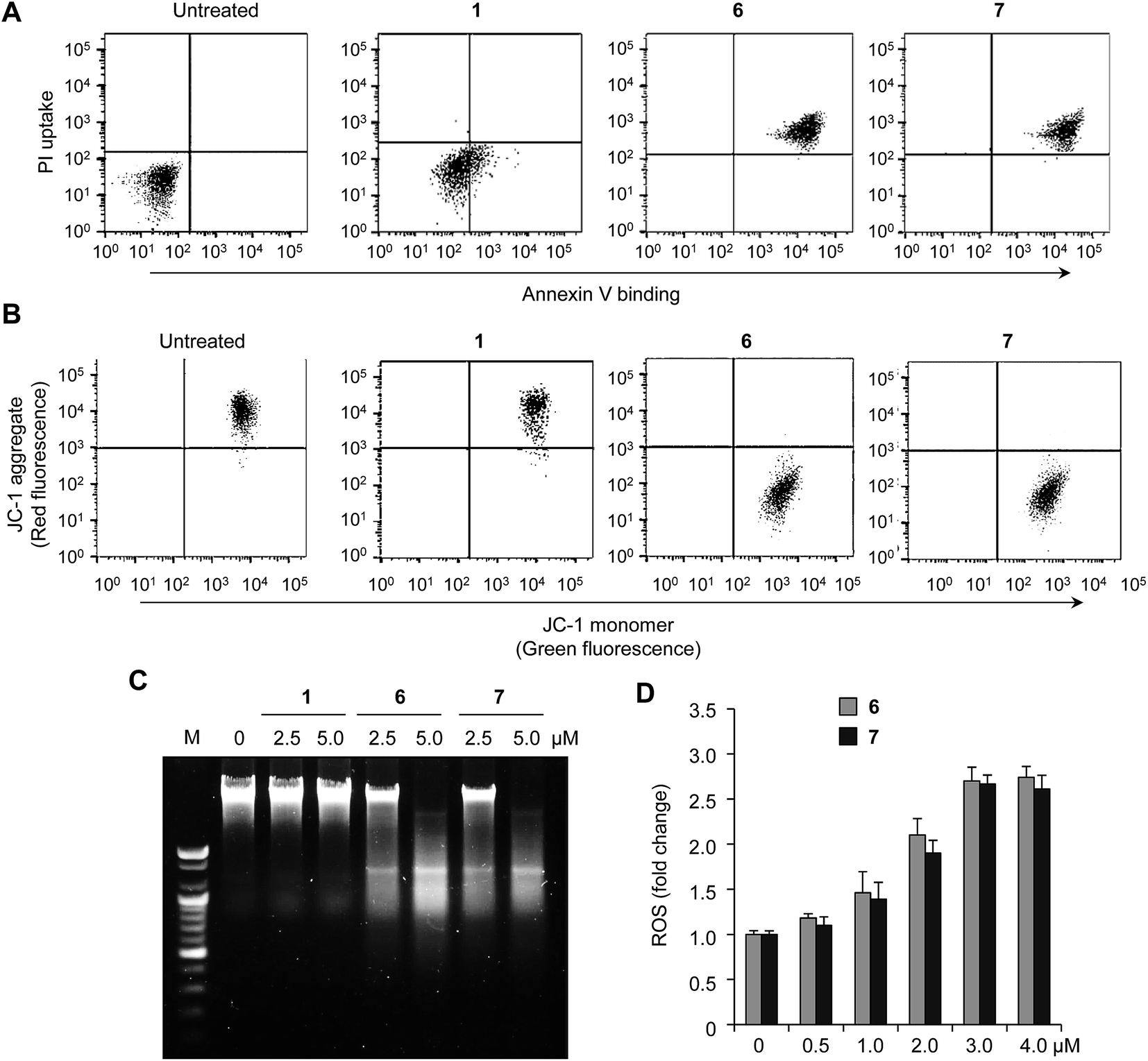 | ||
| Fig. 3 Synthetic receptors induce apoptosis. (A) Flow cytometry of HeLa cells treated with 5 μM of each of the indicated compounds for 18 h and then stained with a mixture of fluorescein–annexin V and PI (annexin V binding versus PI uptake). (B) Flow cytometry of HeLa cells treated with 5 μM of each of the indicated compounds for 18 h and stained with JC-1. Shown is a dot plot of red fluorescence (FL2, JC-1 aggregate) versus green fluorescence (FL1, JC-1 monomer). (C) HeLa cells were treated with each of the indicated compounds for 18 h. The DNA fragments were then visualized by staining with RedSafe™ Nucleic Acid Staining Solution. (D) HeLa cells were treated with various concentrations of each of the indicated compounds for 8 h and then stained with 10 μM PF1 for 1 h so as to monitor the ROS production (mean ± s.d., n = 3). | ||
It is widely appreciated that reactive oxygen species (ROS) may serve as an intracellular signal of the apoptotic cascade.20 To test if 6 and 7 enhance ROS generation in cells, HeLa and PLC/PRF/5 cells were treated with 6 or 7 at various concentrations for 8 h. The levels of intracellular ROS were then measured by treating the cells with 10 μM of a boronate-based fluorescent H2O2 probe, PF1, for 1 h.21 An increase in the fluorescence intensity was observed in cells treated with either 6 or 7 (Fig. 3D and S7D†). To determine whether ROS production is a cause or consequence of apoptosis, we investigated the temporal relationship between ROS production and apoptosis. As shown in Fig. S10,† ROS is produced in cells after the short time (1–2 h) incubation with a compound but apoptosis takes place after ca. 6 h treatment, indicating that ROS production is likely to be a cause of apoptosis.
HeLa cells treated with 6 for 6 h were also subjected to gene expression profiling analysis of mRNAs using DNA chips. It was found that after treatment with 6, positive regulators of apoptosis were upregulated in cells. In contrast, negative regulators of apoptosis were down-regulated (Table S2†). Taken together, the results provide evidence to support that the synthetic receptors induce apoptotic cell death.
Synthetic receptors induce caspase activation
Previous studies have led to the suggestion that plant lectins induce apoptotic cell death via a caspase-dependent pathway.22 We thus sought to examine whether caspase activation was involved in the receptor-mediated apoptosis noted above. With such a consideration in mind, HeLa and PLC/PRF/5 cells were incubated with 6 and 7, as well as with 1, for 18 h. The proteolytic activities associated with caspases were then measured using a colorimetric peptide substrate for caspases, Ac-DEVD-pNA (pNA, p-nitroaniline). It was found that caspase activities increased in the case of cells treated with 6 or 7, but not in cells treated with 1 (Fig. 4A and S11A†). When Ac-DEAD-CHO, a known inhibitor of caspases, was added to the lysates of the cells treated with 6 or 7, no evidence of caspase activity was seen.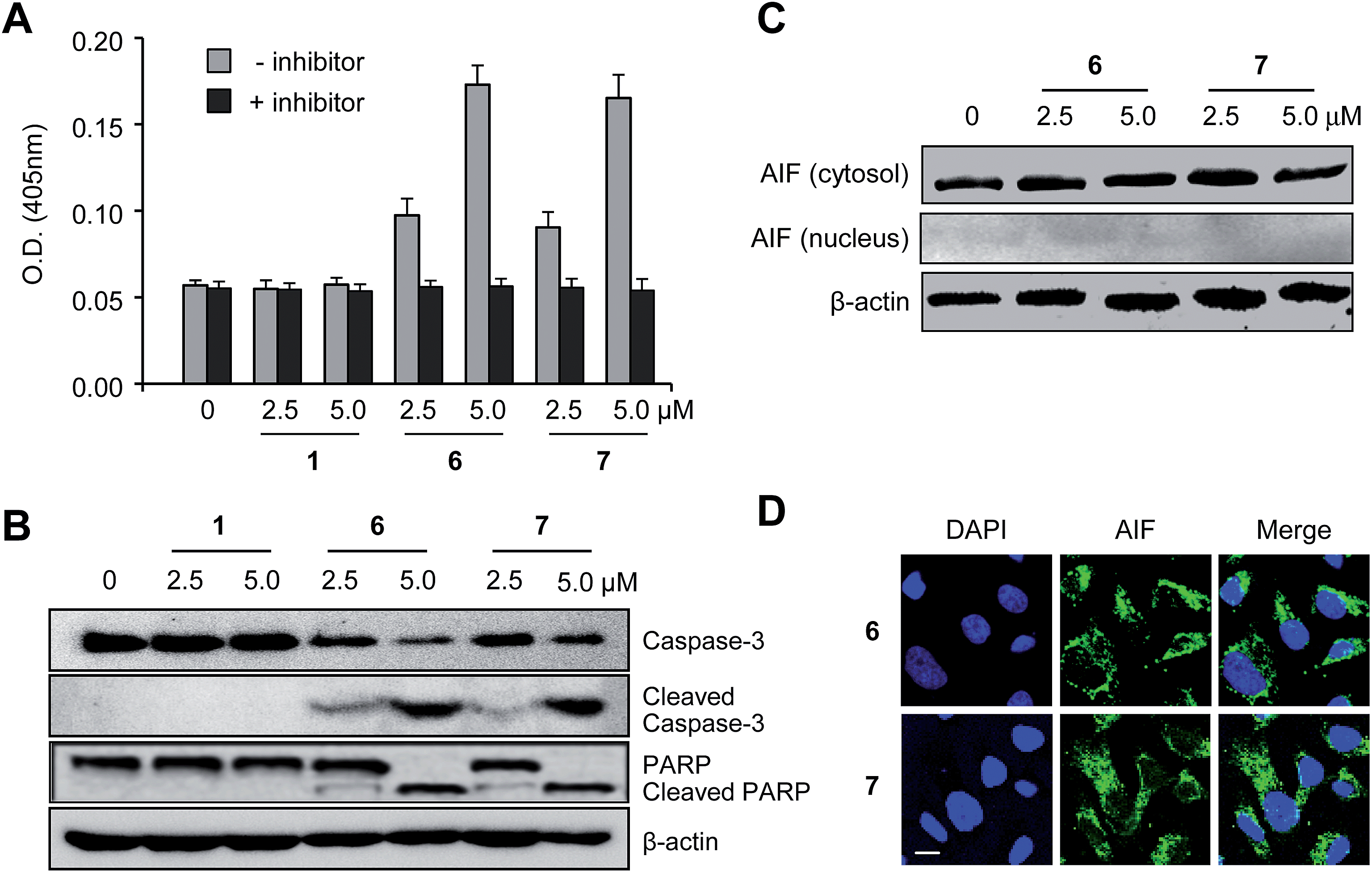 | ||
| Fig. 4 Synthetic receptors induce caspase activation but do not activate the AIF-associated caspase-independent apoptotic pathway. (A) The caspase activity of lysates of HeLa cells treated with each of the indicated compounds for 18 h were measured using acetyl-DEVD-pNA in the absence (grey) or presence (black) of 200 μM Ac-DEVD-CHO (mean ± s.d., n = 3). (B) HeLa cells were treated with each of the indicated compounds for 18 h. The indicated proteins were then immunoblotted using the appropriate corresponding antibodies. (C) HeLa cells were treated with each of the indicated compounds for 18 h. Western blotting was performed using an anti-AIF antibody. (D) HeLa cells were treated with 5.0 μM of each of the indicated compounds for 18 h. Immunocytochemistry was then performed using an anti-AIF antibody. The nuclei were stained with DAPI (scale bar: 10 μm). | ||
Next, we tested if the pan-caspase inhibitor ZVAD-FMK (benzyloxycarbonyl-Val-Ala-Asp(OMe) fluoromethylketone), which is a cell-permeable, irreversible caspase inhibitor with broad specificity,23 protected cells against the effect of 6 or 7. HeLa and PLC/PRF/5 cells, pretreated with ZVAD-FMK (0, 10 and 20 μM) for 3 h, were incubated with either 6 or 7 (0–4 μM) for 18 h. Under these conditions the cells were greatly protected from apoptotic cell death induced by compounds (Fig. 5).
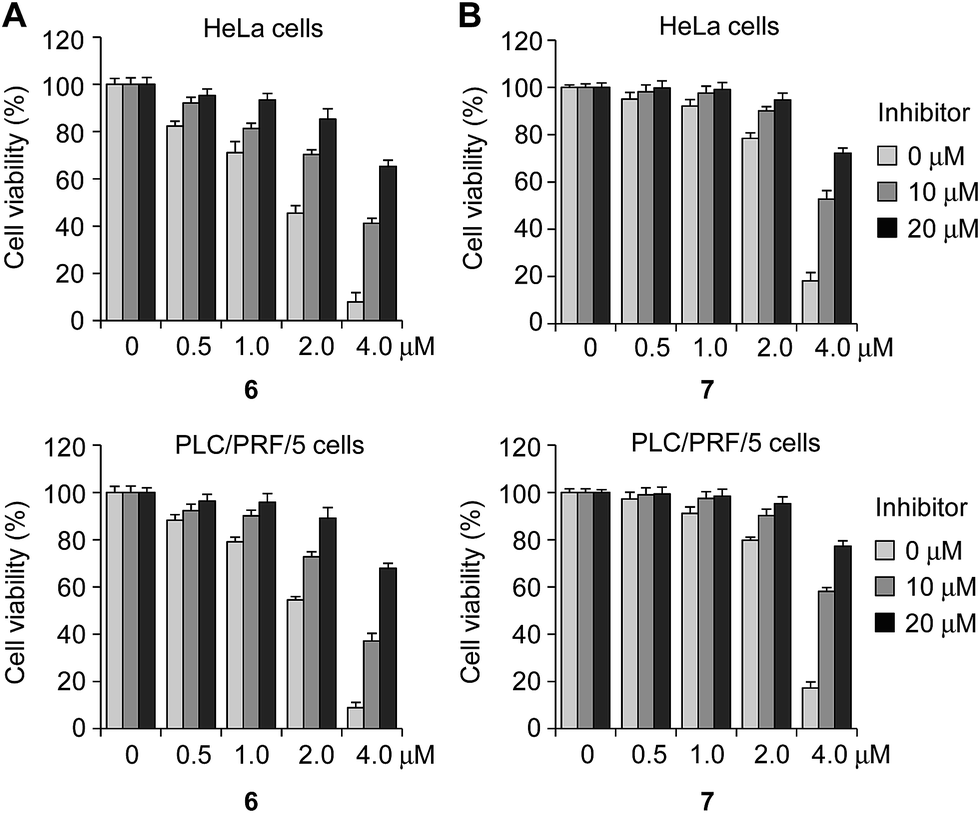 | ||
| Fig. 5 HeLa and PLC/PRF/5 cells were pre-incubated with the caspase inhibitor ZVAD-FMK for 3 h, and then treated with various concentrations of (A) 6 and (B) 7 for 18 h. The effect of this caspase inhibitor on cell survival was determined using an MTT assay (mean ± s.d., n = 3). | ||
During caspase-dependent apoptosis, cytochrome c, which is located in the space between the inner and outer mitochondrial membranes, is released from the mitochondria into the cytosol where it binds to Apaf-1 to form the apoptosome.24 The cytochrome c/Apaf-1 complex activates caspase-9, which subsequently induces activation of caspase-3 through the cleavage of procaspase-3. The effect of the synthetic receptors on caspase-3 activation in cells was thus tested in order to further provide evidence for the involvement of caspase activation in the receptor-mediated apoptosis. HeLa and PLC/PRF/5 cells were incubated with compounds 6 and 7, as well as the control 1, for 18 h. The results of western blot analysis reveal that procaspase-3 is proteolytically cleaved to produce caspase-3 in cells treated with 6 and 7 but not those treated with 1 (Fig. 4B and S11B†). This study also served to reveal that an endogenous caspase substrate, poly(ADP-ribose)polymerase (PARP), was cleaved in cells treated with 6 and 7 but not those treated with 1.
Apoptosis is also known to take place through caspase-independent pathways.25 Thus, we determined whether the synthetic receptors 6 and 7 promoted caspase-independent apoptosis which is mediated by apoptosis inducing factor (AIF). It has been shown that AIF is translocated into the nucleus during caspase-independent apoptosis.26 Therefore, comparisons of the AIF levels in the cytosolic and nuclear fractions may be used to determine whether or not apoptosis takes place in a caspase-independent fashion. With such considerations in mind, HeLa cells were incubated with 6 and 7 for 18 h and then subjected to immunostaining analyses using an AIF antibody. The results of western blot and immunocytochemical analyses indicate that treatment with 6 and 7 does not promote AIF translocation into the nucleus (Fig. 4C and D). Taken together, the findings leads us to conclude that synthetic receptors 6 and 7 induce apoptotic cell death by activating caspases but do not activate the AIF-associated caspase-independent apoptotic pathway.
Finally, experiments designed to probe the molecular mechanisms underlying synthetic receptor-mediated apoptosis were carried out. It has been suggested that mannose binding lectins induce caspase-dependent apoptosis by activating the c-Jun N-terminal Kinase (JNK) signaling pathway after they bind to cell-surface glycans.22 When JNK is phosphorylated and activated, the proapoptotic protein Bax is translocated from the cytosol to the mitochondria. Translocation of Bax into the mitochondria subsequently promotes cytochrome c release from the mitochondria into the cytosol to induce caspase-dependent apoptosis.24 In light of these observations, the effect of the synthetic receptors 6 and 7 on the JNK pathway was tested by initially measuring the level of phosphorylated JNK. HeLa and PLC/PRF-5 cells were separately treated with 6 and 7, as well as with a positive control ConA, for 18 h. The levels of phosphorylated JNK were then measured. The results of western blot analysis reveal that treatment with these two synthetic receptors leads to production of phosphorylated JNK, a phenomenon which is also observed in ConA treated cells (Fig. 6). It was also found that cytosolic Bax was translocated into the mitochondria of cells treated with 6 and 7, as well as in cells treated with ConA. Moreover, cytochrome c was released from the mitochondria to the cytosol in the treated cells. Taken in concert, these signaling pathway studies are consistent with the notion that the two synthetic receptors 6 and 7 induce caspase-dependent apoptosis via JNK activation, a phenomenon which is also observed in cells treated with ConA. Because synthetic receptors are known to bind to highly mannosylated glycoproteins18 and compete with ConA for cell surface mannose binding, it is likely that synthetic receptor-mediated cellular events occur as the result of recognizing cell-surface glycans.
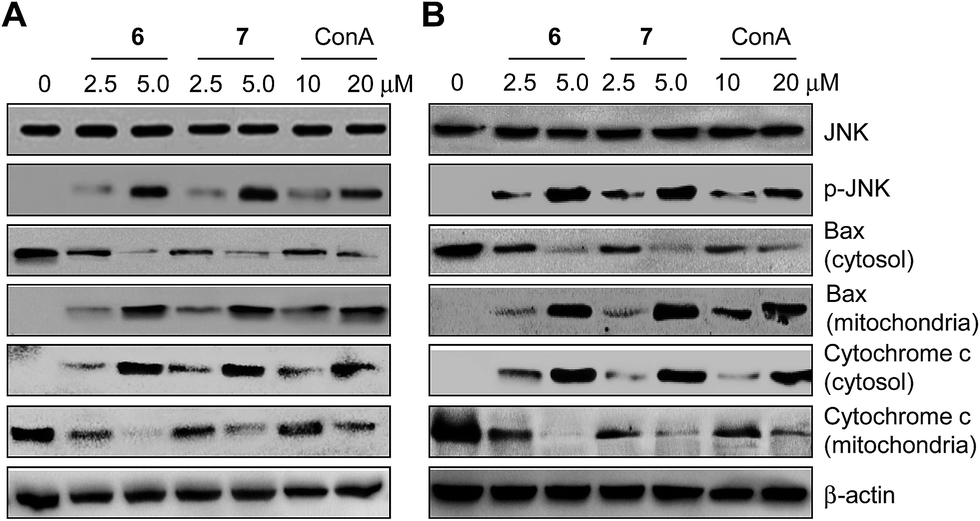 | ||
| Fig. 6 Synthetic receptors activate the JNK signaling pathway. (A) HeLa and (B) PLC/PRF-5 cells were treated with 6 and 7 as well as a control ConA for 18 h. The indicated proteins were then immunoblotted using appropriate corresponding antibodies. | ||
Conclusion
In summary, synthetic aminopyrrolic receptors 6 and 7, which are capable of binding to mannose, induce apoptotic cell death. In contrast, compounds 1–5, which display very weak mannose-binding affinities, do not promote cell death. The receptor-associated cellular response is observed to be dependent on the expression of high-mannose glycans on the cell surface. We have also shown that the two synthetic receptors promote JNK activation, enhance Bax translocation into the mitochondria, cause cytochrome c release from the mitochondria into the cytosol, and induce caspase activation, phenomena which are seen in cells exposed to a mannose-binding lectin. On the other hand, the synthetic receptors do not affect the AIF-associated caspase-independent pathway. Taken together, the results presented here provide support for the notion that appropriately designed receptors can be used to induce cell death through caspase-dependent apoptosis. The present study also leads us to suggest that synthetic glycan-recognizing receptors, such as 6 and 7, could emerge as useful chemical probes that can be used to trigger apoptosis via modes of action that differ from the approaches generally employed to promote programmed cell death.Experimental section
Cell culture
HeLa (human cervical cancer cells), PLC/PRF/5 (human hepatocellular carcinoma cells), A549 (human lung adenocarcinoma epithelial cells) and KG-1 cells (human myeloid leukemia cells) were cultured in RPMI 1640 (Invitrogen) or DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 50 units per mL penicillin and 50 units per mL streptomycin. Calu-3 (airway adenocarcinoma cells) cells were cultured in a mixture of DMEM and Coon's modified F12 medium supplemented with 10% FBS, 50 units per mL penicillin and 50 units per mL streptomycin. FRT (Fischer rat thyroid epithelial) cells were cultured in Coon's modified F12 medium supplemented with 10% FBS, 50 units per mL penicillin and 50 units per mL streptomycin. FRT cells were stably transfected with the plasmid containing a mutant YFP (YFP-F46L/H148Q/I152L) which is quenched sensitively by chloride ions in the cytosol. Cells were maintained at 37 °C under a humidified atmosphere containing 5% CO2.Measurement of expression levels of high-mannose glycans on the cell surface
HeLa, A549, PLC/PRF/5 and KG-1 cells were treated with 10 μM of biotin–ConA in culture media for 1 h. After washing, cells were treated with Cy3-streptavidin. Flow cytometry was performed using a BD FACSVerse™ instrument (BD Biosciences) and a FlowJo™ software (BD Biosciences). For confocal microscopy analysis, the nucleus of cells was stained with DAPI and cell images were then obtained using confocal fluorescence microscopy (Zeiss, Germany).Measurement of cell death
Cells in culture media were incubated with various concentrations of each of the compounds subject to study. MTT assays were performed according to standard procedures. The absorbance at 570 nm was measured using an Infinite® 200 PRO multimode microplate reader.Flow cytometry
HeLa and PLC/PRF/5 cells were treated with 5 μM of each of the compounds subject to study in culture media for 18 h. Untreated cells were used as a negative control. After washing with PBS twice, the cells were incubated with 0.5 mL of trypsin–EDTA (0.05% trypsin, 0.02% EDTA, Sigma-Aldrich) for 5–10 min at 37 °C and collected. Cells were re-suspended in binding buffer (500 μL, 10 mM HEPES/NaOH, pH 7.5 containing 1.4 M NaCl and 2.5 mM CaCl2) and treated with a mixture of fluorescein–annexin V (final concentration = 0.5 μg mL−1) and propidium iodide (final concentration = 2 μg mL−1) for 10 min at room temperature.For measurements of cell size, HeLa and PLC/PRF/5 cells were treated with 5 μM of each compound subject to study for 18 h. After washing with PBS twice, cell size was measured with a flow cytometer by exciting with a 488 nm argon laser and determining their distribution by means of a forward scatter versus side scatter dot plot. Light scattered in the forward direction is proportional to cell size, and light scattered at a 90° angle (side scatter) is proportional to cell density.
For JC-1 staining, HeLa and PLC/PRF/5 cells were treated with 5 μM of each compound subject to study for 18 h. After washing with PBS twice, the cells were incubated with JC-1 (final concentration = 2.5 μg mL−1, Anaspec) in PBS for 15 min. The red fluorescence signal was measured using an excitation wavelength of 550 nm and an emission wavelength of 600 nm. The green fluorescence was measure using an excitation wavelength of 485 nm and monitoring the emission at 535 nm. Flow cytometry was performed using a BD FACSVerse™ instrument using the FlowJo™ software.
DNA fragmentation assays
DNA fragmentation was analyzed via electrophoresis on an agarose gel. HeLa and PLC/PRF/5 cells were incubated with 2.5 and 5.0 μM of each compound subject to study for 18 h. The cells were lysed in a buffer containing 10 mM Tris, 1 mM EDTA, and 0.2% Triton X-100, pH 8.0. Samples were incubated in 100 μg mL−1 RNase A (30 min, 37 °C) and 100 μg mL−1 proteinase K (10 min, 56 °C). The DNA was precipitated by addition of 0.5 M NaCl–isopropyl alcohol and washed with 70% ethanol. Samples were loaded on a 1.5% agarose gel and subject to electrophoresis at 100 V for 0.5 h in TBE (Tris/borate/EDTA) buffer (0.5×). The DNA ladder was stained with RedSafe™ Nucleic Acid Staining Solution (Intron, Korea) and analyzed by using a G:BOX Chemi Fluorescent & Chemiluminescent Imaging System (Syngene).Isolation of cytosolic and mitochondrial fractions
Mitochondrial and cytosolic fractions of cells were prepared using a mitochondrial/cytosol fractionation kit (Biovision). Cells were harvested by centrifugation at 600 × g for 5 min and washed twice with cold PBS buffer. The cells were re-suspended in 250 μL of an extraction buffer, which contained the protease inhibitor mixture and dithiothreitol (Biovision). After incubation on ice for 30 min, the cells were homogenized on ice and centrifuged at 700 × g for 10 min at 4 °C, and the supernatant was collected. The collected supernatant was centrifuged again at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 30 min at 4 °C. The resulting supernatant was harvested and used as cytosolic fractions. The pellets were re-suspended and used as mitochondrial fractions.
000 × g for 30 min at 4 °C. The resulting supernatant was harvested and used as cytosolic fractions. The pellets were re-suspended and used as mitochondrial fractions.
Western blot analysis
Proteins were separated by 6–15% SDS-PAGE. Rabbit caspase-3 (H-277) polyclonal (1:1000, Santa Cruz Biotechnology), rabbit cleaved caspase-3 (Asp175) polyclonal (1:1000, Cell Signaling Technology), rabbit PARP polyclonal (1:1000, Cell Signaling Technology), rabbit AIF polyclonal (1:1000, Santa Cruz Biotechnology), rabbit JNK polyclonal (1:1000, Cell Signaling Technology), rabbit phospho-JNK polyclonal (1:1000, Cell Signaling Technology), rabbit Bax polyclonal (1:1000, Santa Cruz Biotechnology), mouse cytochrome c monoclonal (1:1000) (Biovision), and mouse β-actin (1:1000, Santa Cruz Biotechnology) antibodies were used as primary antibodies. Horse peroxidase–conjugated goat anti-rabbit IgG (1:2000, Santa Cruz Biotechnology) and goat anti-mouse IgG (1:2000, Santa Cruz Biotechnology) were used as the secondary antibodies. The blots were developed using a West-ZOL® plus Western Blot Detection System (Intron Biotechnology Inc., South Korea). The western blot signal was analyzed by means of a G:BOX Chemi Fluorescent & Chemiluminescent Imaging System.
Caspase activity assay
HeLa and PLC/PRF/5 cells were incubated with 2.5 and 5.0 μM of each compound subject to study for 18 h. The cells were lysed in a buffer containing 50 mM HEPES, pH 7.4, 5 mM CHAPS, 5 mM DTT. Cell lysates were placed into 96-well plate and then assay buffer containing 20 mM HEPES, pH 7.4, 0.01% CHAPS, 5 mM DTT, 2 mM EDTA was added to each well. The caspase inhibitor Ac-DEVD-CHO (20 μM) was added to the wells. Caspase activity was determined by adding acetyl-DEVD-pNA (200 μM) (Sigma-Aldrich) to each well. The enzyme-catalyzed release of pNA was monitored at 405 nm using an Infinite® 200 PRO multimode microplate reader.Determination of ROS generation
HeLa and PLC/PRF/5 cells were treated with each compound subject to study for 8 h. The treated cells were incubated with 10 μM PF1 for 1 h. After washing with PBS, ROS production within the cells was then measured using an Infinite® 200 PRO multimode microplate reader (λex = 485 nm, λem = 535 nm).Acknowledgements
This work was supported by the National Creative Research Initiative (2010-0018272 to I. S.) program in Korea. The work in Austin was supported by the U.S. Department of Energy Office of Basic Energy Sciences (grant DOE: DE-FG02-01ER15186 to J. L. S.).Notes and references
- D. D. Newmeyer and S. Ferguson-Miller, Cell, 2003, 112, 481–490 CrossRef CAS.
- A. Ashkenazi and V. M. Dixit, Science, 1998, 281, 1305–1308 CrossRef CAS.
- C. B. Thompson, Science, 1995, 267, 1456–1462 CrossRef CAS.
- M. P. Mattson, Nat. Rev. Mol. Cell Biol., 2000, 1, 120–129 CrossRef CAS PubMed.
- J. C. Reed, Nat. Rev. Drug Discovery, 2002, 1, 111–121 CrossRef CAS PubMed.
- (a) H. J. Cho, H. Y. Gee, K.-H. Baek, S.-K. Ko, J.-M. Park, H. Lee, N.-D. Kim, M. G. Lee and I. Shin, J. Am. Chem. Soc., 2011, 133, 20267–20276 CrossRef CAS PubMed; (b) D. R. Williams, S.-K. Ko, S. Park, M.-R. Lee and I. Shin, Angew. Chem., Int. Ed., 2008, 47, 7466–7469 CrossRef CAS PubMed; (c) S.-K. Ko, J. Kim, D. C. Na, S. Park, S.-H. Park, J. Y. Hyun, K.-H. Baek, N. D. Kim, N.-K. Kim, Y. N. Park, K. Song and I. Shin, Chem. Biol., 2015, 22, 391–403 CrossRef CAS PubMed; (d) K.-H. Baek, J. Park and I. Shin, Chem. Soc. Rev., 2012, 41, 3245–3263 RSC.
- A. Kamal, S. Faazil and M. S. Malik, Expert Opin. Ther. Pat., 2014, 24, 339–354 CrossRef CAS PubMed.
- M. Hassan, H. Watari, A. AbuAlmaaty, Y. Ohba and N. Sakuragi, BioMed Res. Int., 2014, 2014, 150845 Search PubMed.
- A. Masood, A. S. Azmi and R. M. Mohammad, Cancers, 2011, 3, 1527–1549 CrossRef CAS PubMed.
- L. L. Fu, C. C. Zhou, S. Yao, J. Y. Yu, B. Liu and J. K. Bao, Int. J. Biochem. Cell Biol., 2011, 43, 1442–1449 CrossRef CAS PubMed.
- E. G. de Mejía and V. I. Prisecaru, Crit. Rev. Food Sci. Nutr., 2005, 45, 425–445 CrossRef PubMed.
- (a) A. Kobata and J. Amano, Immunol. Cell Biol., 2005, 83, 429–439 CrossRef CAS PubMed; (b) D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed.
- (a) T. G. Johns, I. Mellman, G. A. Cartwright, G. Ritter, L. J. Old, A. W. Burgess and A. M. Scott, FASEB J., 2005, 19, 780–782 CAS; (b) M. L. de Leoz, L. J. Young, H. J. An, S. R. Kronewitter, J. Kim, S. Miyamoto, A. D. Borowsky, H. K. Chew and C. B. Lebrilla, Mol. Cell. Proteomics, 2011, 10, M110.0027171 CrossRef PubMed; (c) S. C. Tao, Y. Li, J. Zhou, J. Qian, R. L. Schnaar, Y. Zhang, I. J. Goldstein, H. Zhu and J. P. Schneck, Glycobiology, 2008, 18, 761–769 CrossRef CAS PubMed.
- (a) Z. Shi, N. An, S. Zhao, X. Li, J. K. Bao and B. S. Yue, Cell Proliferation, 2013, 46, 86–96 CrossRef CAS PubMed; (b) Z. Shi, J. Chen, C. Y. Li, N. An, Z. J. Wang, S. L. Yang, K. F. Huang and J. K. Bao, Acta Pharmacol. Sin., 2014, 35, 248–256 CrossRef CAS PubMed; (c) Z. Liu, B. Liu, Z. T. Zhang, T. T. Zhou, H. J. Bian, M. W. Min, Y. H. Liu, J. Chen and J. K. Bao, Phytomedicine, 2008, 15, 867–875 CrossRef CAS PubMed; (d) A. R. M. Ruhul Amin, M. L. Oo, T. Senga, N. Suzuki, G. S. Feng and M. Hamaguchi, Cancer Res., 2003, 63, 6334–6339 CAS; (e) S. Y. Wang, Q. J. Yu, J. K. Bao and B. Liu, Biochem. Biophys. Res. Commun., 2011, 406, 497–500 CrossRef CAS PubMed.
- (a) Y. Ferrand, M. P. Crump and A. P. Davis, Science, 2007, 318, 619–622 CrossRef CAS PubMed; (b) Y. Ferrand, E. Klein, N. P. Barwell, M. P. Crump, J. Jiménez-Barbero, C. Vicent, G. J. Boons, S. Ingale and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 1775–1779 CrossRef CAS PubMed.
- (a) T. Oki, Y. Yamazaki, T. Furumai and Y. Igarashi, Biosci., Biotechnol., Biochem., 1997, 61, 1408–1410 CrossRef CAS; (b) T. Oki, Y. Yamazaki, N. Nomura, T. Furumai and Y. Igarashi, J. Antibiot., 1999, 52, 449–454 CrossRef CAS; (c) Y. Nakagawa, Y. Masuda, K. Yamada, T. Doi, K. Takegoshi, Y. Igarashi and Y. Ito, Angew. Chem., Int. Ed., 2011, 50, 6084–6088 CrossRef CAS PubMed; (d) Y. Nakagawa, T. Doi, Y. Masuda, K. Takegoshi, Y. Igarashi and Y. Ito, J. Am. Chem. Soc., 2011, 133, 17485–17493 CrossRef CAS PubMed.
- (a) C. Nativi, O. Francesconi, G. Gabrielli, I. de Simone, B. Turchetti, T. Mello, L. Di Cesare Mannelli, C. Ghelardini, P. Buzzini and S. Roelens, Chem.–Eur. J., 2012, 18, 5064–5072 CrossRef CAS PubMed; (b) A. Ardá, F. J. Cañada, C. Nativi, O. Francesconi, G. Gabrielli, A. Ienco, J. Jiménez-Barbero and S. Roelens, Chem.–Eur. J., 2011, 17, 4821–4829 CrossRef PubMed.
- O. Francesconi, C. Nativi, G. Gabrielli, I. de Simone, S. Noppen, J. Balzarini, S. Liekens and S. Roelens, Chem.–Eur. J., 2015, 21, 10089–10093 CrossRef CAS PubMed.
- (a) S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. V. Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed; (b) J. L. Sessler, L. R. Eller, W.-S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989–5992 CrossRef CAS PubMed; (c) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed; (d) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed; (e) M. S. Melvin, J. T. Tomlinson, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 2000, 122, 6333–6334 CrossRef CAS.
- M. L. Circu and T. Y. Aw, Free Radical Biol. Med., 2010, 48, 749–762 CrossRef CAS PubMed.
- (a) S. Park, G.-H. Kim, S.-H. Park, J. Pai, D. Rathwell, J.-Y. Park, Y.-S. Kang and I. Shin, J. Am. Chem. Soc., 2015, 137, 5961–5968 CrossRef CAS PubMed; (b) M. C. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS PubMed.
- Q. L. Jiang, S. Zhang, M. Tian, S. Y. Zhang, T. Xie, D. Y. Chen, Y. J. Chen, J. He, J. Liu, L. Ouyang and X. Jiang, Cell Prolif., 2015, 48, 17–28 CrossRef CAS PubMed.
- D. W. Nicholson, A. Ali, N. A. Thornberry, J. P. Vaillancourt, C. K. Ding, M. Gallant, Y. Gareau, P. R. Griffin, M. Labelle, Y. A. Lazebnik, N. A. Munday, S. M. Raju, M. E. Smulson, T. T. Yamin, V. L. Yu and D. K. Miller, Nature, 1995, 376, 37–43 CrossRef CAS PubMed.
- P. Li, D. Nijhawan, I. Budihardjo, S. M. Srinivasula, M. Ahmad, E. S. Alnemri and X. Wang, Cell, 1997, 91, 479–489 CrossRef CAS.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- N. Joza, S. A. Susin, E. Daugas, W. L. Stanford, S. K. Cho, C. Y. Li, T. Sasaki, A. J. Elia, H. Y. Cheng, L. Ravagnan, K. F. Ferri, N. Zamzami, A. Wakeham, R. Hakem, H. Yoshida, Y. Y. Kong, T. W. Mak, J. C. Zúñiga-Pflücker, G. Kroemer and J. M. Penninger, Nature, 2001, 410, 549–554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03200h |
| This journal is © The Royal Society of Chemistry 2015 |