
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA NIR dye with high-performance n-type semiconducting properties†
Jiajun
Xie
,
Ke
Shi
,
Kang
Cai
,
Di
Zhang
,
Jie-Yu
Wang
,
Jian
Pei
* and
Dahui
Zhao
*
Beijing National Laboratory for Molecular Sciences, Centre of Soft Matter Science and Engineering, Key Lab of Polymer Chemistry & Physics of Ministry of Education, College of Chemistry, Peking University, China. E-mail: dhzhao@pku.edu.cn; jianpei@pku.edu.cn
First published on 6th October 2015
Abstract
A novel hetero-polycyclic aromatic compound manifesting strong near-infrared (NIR) absorption as well as high-performance n-type semiconducting properties is developed. With an exceptionally low LUMO level at −4.7 eV, this NIR dye (λmax ≈ 1100 nm, ε ≈ 105 mol−1 L cm−1) exhibits adequate stability under ambient conditions, with electron mobility up to 0.96 cm2 V−1 s−1 measured in solution-processed organic field-effect transistors. A special metal-free C–C coupling serves as a pivotal step in constructing the polycyclic π-framework of this low-bandgap chromophore, by fusing an electron-deficient naphthalenediimide moiety with an electron-donating naphthalenediamine. Such a rare combination of extraordinary optical and semiconductive attributes is quite valuable for organic small molecules, and promising for unique applications in the opto-electronic field.
Introduction
Organic molecules with pronounced near-infrared (NIR) absorptions are valuable optical, opto-electronic, and bio-applicable materials.1–3 However, in spite of all the efforts made to acquire the low-bandgap feature, up to now only a limited number of air-stable closed-shell, neutral organic small molecules have been developed possessing optimal NIR optical activity. Moreover, their absorptions mostly fall in the range of 700–900 nm. Very few neutral organic small molecules possess a desirable extinction ability beyond 1000 nm.4,5 The challenges in designing potent NIR dyes lie in the fact that, in order to attain a low-energy bandgap with a significant extinction coefficient, fine tuning the ground and excited electronic states is required to achieve not only suitable energy levels but also an adequate transition dipole moment. Particular care should also be taken to avoid a detrimental effect on the chemo-stability, which could result from over-boosting the HOMO or over-depressing the LUMO.A common strategy to attain a low-energy bandgap in an organic structure relies on incorporating electron donator (D) and acceptor (A) moieties into the same molecule, preferably linked by a π-conjugated spacer to extend the effective conjugation length.6 However, in many cases separately installed D and A subunits impart a very limited extinction coefficient to the low-energy excited state. Also, the charge transfer nature may render the optical behavior highly sensitive to environmental polarity variation.7
Another effective approach to developing low-bandgap chromophores utilizes large polycyclic π-systems.8 A major advantage of polycyclic aromatic compounds is their readily tuned HOMO/LUMO energy levels. Through chemical modifications, D/A substituents can also be incorporated. Moreover, large π-systems with delocalized frontier orbitals also promote enhanced light-absorbing ability. Previously, we developed a number of N-hetero-polycyclic dicarboximide molecules manifesting absorptions around 800–900 nm.9 The syntheses of these molecules harnessed 2,3,6,7-tetrabromo-1,4,5,8- tetracarboxydiimide (4Br-NDI) as an important synthon.10 The highly electron-deficient NDI allows exploitation of facile nucleophilic substitution reactions to functionalize and expand the polycyclic π-skeleton. Moreover, the strongly electron-pulling dicarboximide groups help confer an adequately low LUMO to the designed products, favorable for inducing NIR optics. In these previous designs, benzene-1,2-diamine and 1,2,4,5-tetramine were applied to react with 4Br-NDIvia their N-nucleophilic sites. Thus, polycyclic products were formed featuring both electron-accepting NDI and electron-rich dihydrophenazine moieties, bringing about the NIR-absorbing attributes.
In the current work, we integrate naphthalene-1,5-diamine with two equivalents of 4Br-NDI to construct a polycyclic NIR chromophore, via a tandem reaction entailing a nucleophilic aromatic substitution (SNAr) followed by a unique metal-free C–C coupling. A large N-hetero-polycyclic tetraimide molecule 1 (Scheme 1) exhibiting absorptions at ca. 1000 nm was thus obtained. Upon further functionalizing with 2-(dimercaptomethylene)malononitrile, molecule 2 was achieved, impressively manifesting major absorption around 1100 nm (ε > 105 mol−1 L cm−1). Besides the very narrow optical bandgap, compound 2 also possessed an exceptionally low LUMO level at −4.72 eV. Such a low LUMO level greatly favoured the electron-transporting ability. A remarkable electron mobility up to 0.96 cm2 V−1 s−1 was determined for 2 in a solution-processed thin-film transistor. The notable stability of 2 was underscored by comparison to molecule 3 with a similarly low LUMO level. Such a rare combination of high stability, strong NIR absorption, and optimal n-type semiconducting performance endows molecule 2 with great potential for opto-electronic applications.
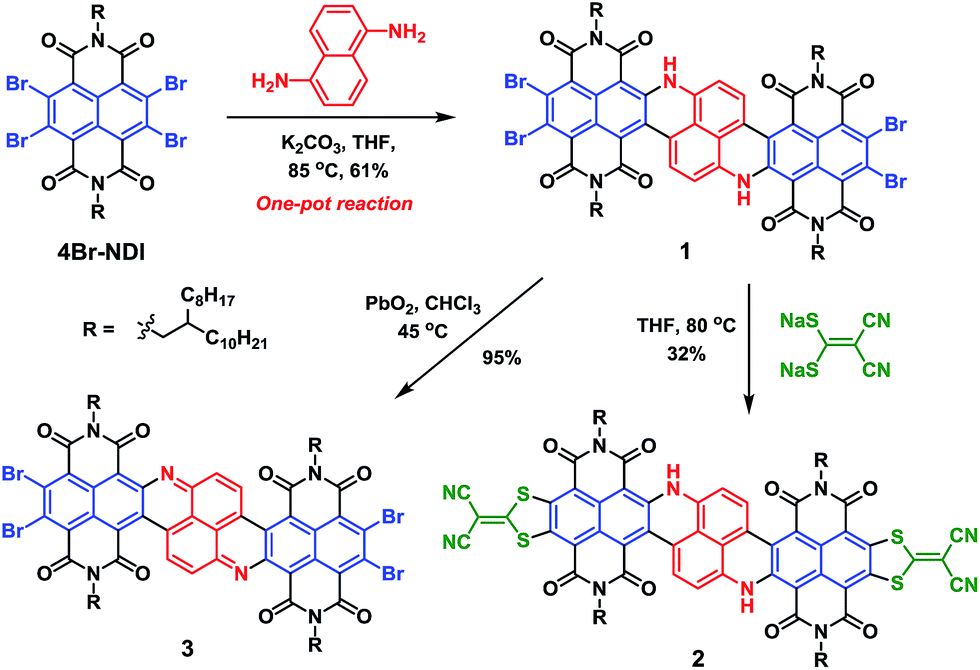 | ||
| Scheme 1 Synthetic route. | ||
Results and discussion
By subjecting naphthalene-1,5-diamine to excess 4Br-NDI, we were initially expecting a double SNAr product 1a′ (Scheme S1†), which was then planned to be transformed to 1 under separate Heck-coupling conditions. However, after working up the reaction of naphthalenediamine and 4Br-NDI in the presence of K2CO3, the 1H NMR spectrum indicated that two aromatic protons were missing from the expected 1a′. The mass spectroscopy also revealed that, while the generated molecule incorporated two NDI units, it contained only four bromine atoms. After analyzing the complete characterization results, we concluded that molecule 1 was produced in one pot from naphthalene-1,5-diamine and 4Br-NDI in the absence of the Pd catalyst (Scheme 1).The mechanism of this tandem process was then examined in more detail. Molecule 4 was obtainable in decent yield at a shortened reaction time and lowered temperature, while 4a was not isolated. Since 4Br-NDI was known to undergo substitutions with various nucleophiles,10 SNAr between 4Br-NDI and naphthalenediamine reasonably happened (Scheme 2). Since debromination was not detected with 4a or 1a, the following intramolecular C–C coupling was unlikely to involve redox processes.11 Moreover, it was found that molecule 1 was formed much faster in tetrahydrofuran than toluene, with nearly identical yields, which also suggested a polar mechanism. We thus proposed that the intramolecular C–C coupling was a SNAr process for the bromo-NDI moiety, with C(sp2)H as the nucleophile (Scheme 2). It was suspected that the strongly electron-donating NH was a critical activator in this electrophilic aromatic substitution of naphthalenediamine, by conferring high nucleophilicity to its para-position. To prove this hypothesis, we then carried out a reaction between 4Br-NDI and 1,3-phenylenediamine. As expected, molecule 5 was generated, substantiating the notion that NH activated its para-CH and realized a 5-membered ring annulation (Schemes 2 and S2†).
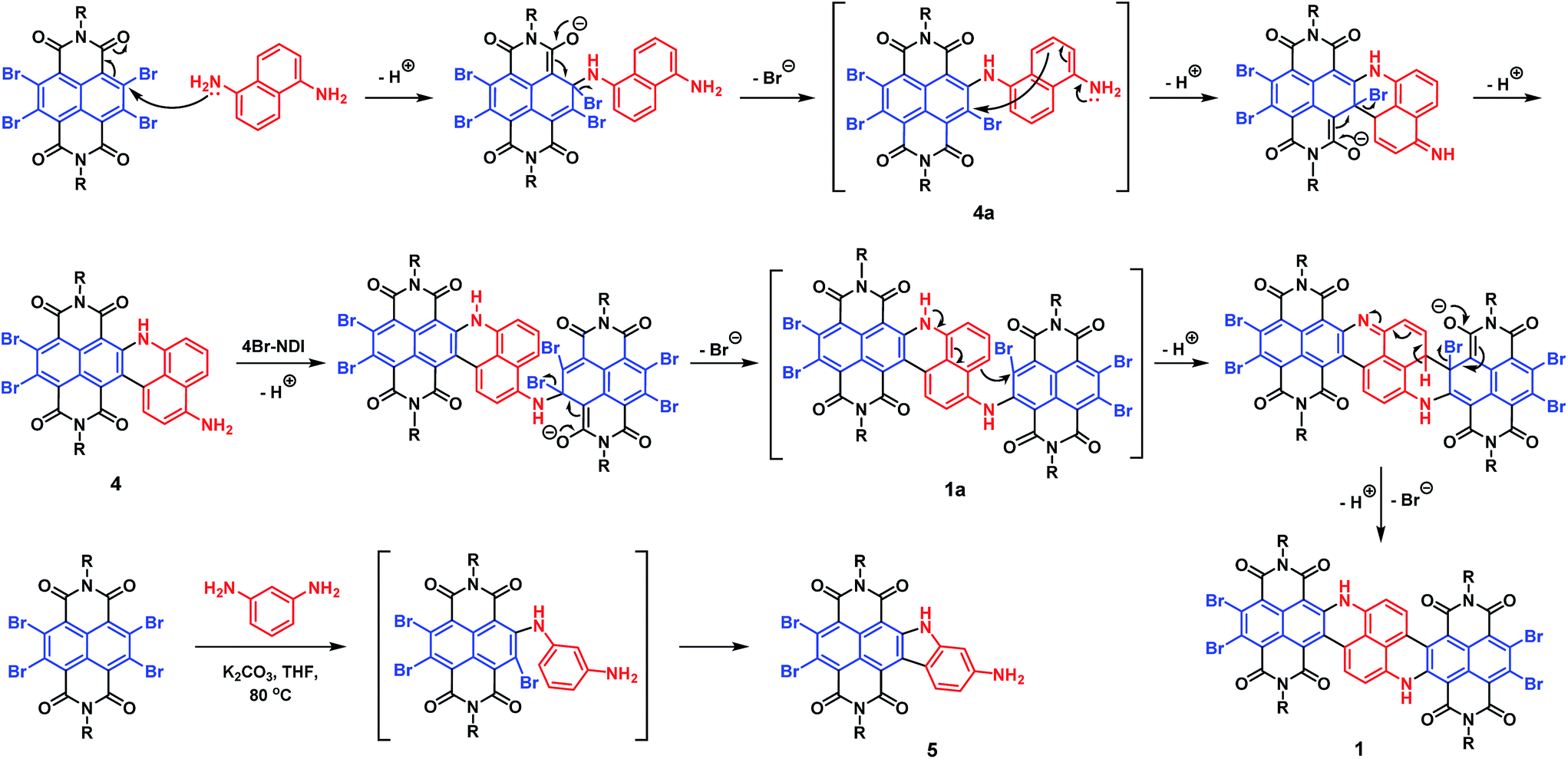 | ||
| Scheme 2 Proposed reaction pathways to 1 and 5. | ||
The UV/vis/NIR absorption spectrum of 1 showed a broad absorption band in the range of 600–1200 nm (Fig. 1a), and the maximum (λmax) emerged at ca. 1000 nm, with a large molar extinction coefficient (ε) of 1.2 × 105 L mol−1 cm−1. The vibronic structures were clearly observable, consistent with the highly rigid polycyclic skeleton of the chromophore.5,12,13
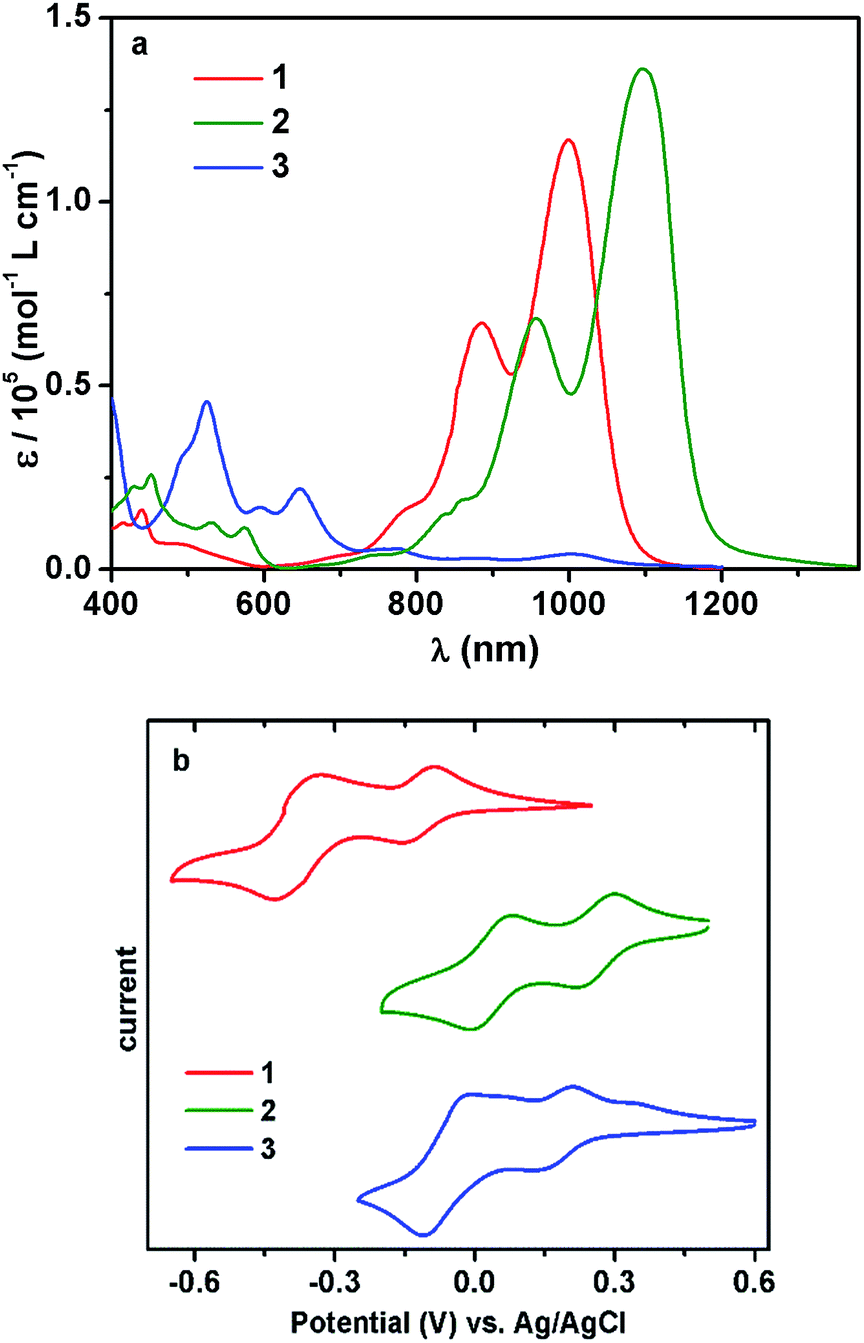 | ||
| Fig. 1 (a) UV-vis-NIR absorption spectra (at 1.0 × 10−5 M in CHCl3); (b) cyclic voltammograms of 1–3 recorded in CHCl3. | ||
Impressed by the remarkable NIR optical properties of 1, we were intrigued to investigate whether the bandgap could be further narrowed through chemical modifications. After various attempts, molecule 2 was successfully prepared by subjecting compound 1 to sodium 1,1-dicyanoethylene-2,2-dithiolate. Such a modification was anticipated to further lower the bandgap since the resultant 2 possessed a further expanded polycyclic π-system with more intense D–A characteristics. Desirable results were observed when the absorption spectrum of 2 was collected. While the band shape remained quite similar to that of 1, the overall spectrum was shifted to a longer wavelength by about 100 nm, giving rise to λmax at ca. 1100 nm with ε > 1.3 × 105 L mol−1 cm−1 (Fig. 1a). Such strong absorptions around 1100 nm are observed for the first time with closed-shell polycyclic dicarboximide dye molecules.
The absorption spectra of 1 and 2 at varied concentrations suggested that these molecules were weakly aggregating in chloroform (Fig. S1 and S2†). Both 1 and 2 were weakly luminescent, exhibiting small Stokes shifts of 412 and 257 cm−1, respectively (Fig. S4 and S5†).
Subsequent electrochemical study unveiled that the much narrowed bandgap of 2 was mainly attributable to its substantially lowered LUMO level. As shown by the cyclic voltammograms (CV), both 1 and 2 displayed two reduction and two oxidation waves (Fig. 1b and S8†). All these redox processes were reversible. A particularly low LUMO at −4.72 eV was displayed by 2, in comparison to the LUMO at −4.35 eV for 1 (Table 1). On the other hand, the HOMO energy levels of the two molecules were separated by 0.03 eV, which explained the much narrower bandgap of 2. It is noteworthy that, in spite of such a low-lying LUMO and narrow bandgap, compound 2 was fairly stable under ambient conditions. No detectable changes were observed in the absorption or NMR spectra after storing under ambient conditions for months.
| λ abs [nm] | ε [mol−1 L cm−1] | λ em [nm] | LUMOd [eV] | HOMO [eV] | LUMO–HOMOd [eV] | E g [eV] | |
|---|---|---|---|---|---|---|---|
| a Absorption maxima of the S0 → S1 transition in CHCl3 solution. b Molar extinction coefficient at λabs. c Emission maxima in CHCl3. d Data from CV. e Optical bandgap from the absorption onset. f Calculated from the optical bandgap and LUMO in CV. | |||||||
| 1 | 1000 | 1.2 × 105 | 1043 | −4.35 | −5.27d | 0.92 | 1.09 |
| 2 | 1101 | 1.4 × 105 | 1133 | −4.72 | −5.30d | 0.58 | 0.99 |
| 3 | 1004 | 4.7 × 103 | — | −4.66 | −5.80f | — | 1.14 |
The remarkable stability of 2 was further underlined by comparison to molecule 3 with a similarly low LUMO. Compound 1 could be oxidized to 3 in nearly quantitative yield using PbO2 (Scheme 1). This redox process was well reversible, and the reduction of 3 back to 1 was realized, also quantitatively, with 1,4-phenylenediamine. The absorption spectrum showed that molecule 3 also possessed a low-energy S0 → S1 band in the NIR regime, exhibiting a local maximum at about 1000 nm, but this low-energy transition displayed minimal extinction ability (Fig. 1a). The overall absorption maximum at a much higher energy emerged around 525 nm. Not surprisingly, CV revealed that the dehydrogenated molecule 3 displayed a considerably lowered LUMO at −4.66 eV compared to 1, actually slightly higher than that of 2 (Table 1). However, unlike compound 2, inferior chemo-stability was observed with 3. Both absorption and NMR spectra indicated that molecule 3 was partially reduced to 1 after storing under ambient conditions for only a few days. The similar LUMO level but superior chemo-stability of 2 compared to 3, by virtue of the dihydro-structure, further stressed the precious values of the former.
Time-dependent density functional theory (TD-DFT) calculation results confirmed the experimental observations by showing that the S0 → S1 transition in compound 3, corresponding to electron excitation from HOMO to LUMO, possessed a much smaller oscillator strength compared to its higher energy transitions (Fig. S14†). DFT and TD-DFT calculations were also performed for 1 and 2. Remarkably, the HOMO and LUMO of 1 and 2 were extensively delocalized over the entire polycyclic π-framework (Fig. 2). Consistent with the experimentally observed strong NIR absorbing abilities of 1 and 2, TD-DFT calculations also verified that both molecules manifested very large transition dipole moments and significant oscillator strengths with their low-energy S0 → S1 (HOMO to LUMO) transitions.
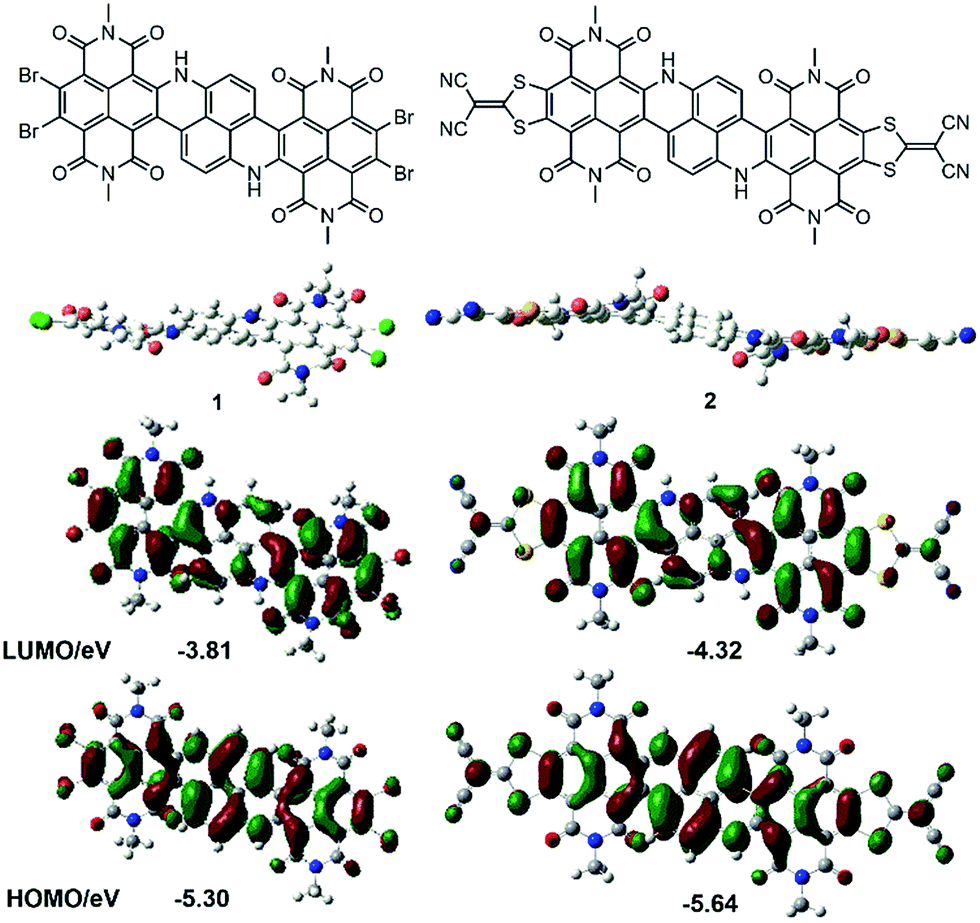 | ||
| Fig. 2 DFT calculated geometry (side view) and HOMO/LUMO (top view) of 1 and 2 (alkyl side groups are replaced by methyl in the calculations). | ||
The low-lying LUMO level and delocalized frontier orbitals14 of 2 prompted us to examine its electron-transporting semiconducting capability.15 To this end, organic field-effect transistors (OFET) with top-gate/bottom-contact configuration were fabricated. Using the solution processing technique, the active layer was deposited by spin-casting solutions of 2 in trichloroethylene (10 mg mL−1) on patterned Au(source–drain)/SiO2/Si substrates. After thermal annealing the semiconducting materials, a poly(perflurobutenylvinylether) (CYTOP) solution was spin-coated on top of it as the dielectric layer, followed by thermally evaporating a layer of aluminum as the gate electrode. All devices were fabricated in a glove box but tested under ambient conditions (RH = 50−60%). As expected, molecule 2 exhibited typical n-type transport characteristics. Quite impressively, optimal electron mobility (μe) up to 0.96 cm2 V−1 s−1 (Fig. 3) and an average μe of 0.93 cm2 V−1 s−1 were determined. In comparison, an electron mobility of merely 0.007 cm2 V−1 s−1 (Fig. S11†) was measured for 1 under similar conditions, while compound 3 was not completely stable (partially reduced) during the device fabrication.
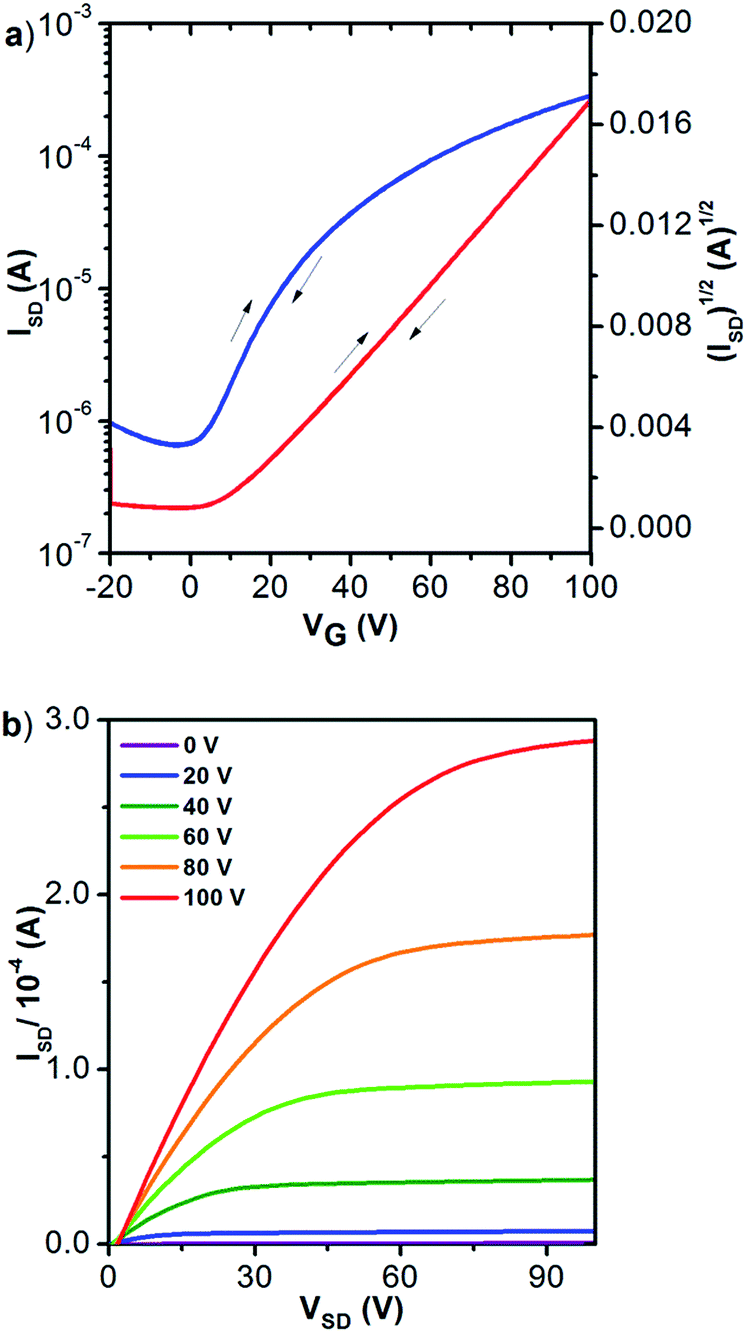 | ||
| Fig. 3 (a) Transfer (VDS = 100 V) and (b) output profiles of 2 (annealed at 220 °C) in OFET (μe = 0.96 cm2 V−1 s−1). | ||
Notably, the transfer and output characteristics of 2 showed negligible hysteresis, which has rarely been observed for n-type organic materials and could be attributable to the low LUMO of the molecule. Besides, no contact resistance was observed in the output curves, suggesting good contact between molecule 2 and the gold electrode.
In the device characterizations, it was noticed that the electron mobility of 2 was highly sensitive to the annealing temperature (Fig. S10†). TGA and DSC characterizations confirmed adequate thermal stability of molecule 2 (Fig. S9†), so a relatively wide range of annealing temperatures was examined. When the semiconductor was annealed at 100 °C, only a moderate electron mobility of 0.12 cm2 V−1 s−1 was obtained. Whereas, if the annealing temperature was elevated to 150 °C, the electron mobility was significantly improved to 0.73 cm2 V−1 s−1. More desirable performances were achieved between 180 and 250 °C, with the mobility fluctuating in the range of 0.8–1.0 cm2 V−1 s−1 (Fig. S10†). Moreover, at the optimized annealing temperature, very low VTh values of −5 to 0 V were observed. Subsequently, AFM and X-ray diffraction studies elucidated that the variation in the device performance was clearly correlated to the crystallinity of the semiconducting layer. Highly crystalline morphology and best device performance were both obtained after annealing at about 220 °C (Fig. 4 and 5). Such high electron mobility, as well as the low VTh value, is believed to benefit from the low-lying LUMO and suitable frontier orbital distribution of 2. The S⋯S interactions may have helped to induce a favorable molecular packing motif.
 | ||
| Fig. 4 AFM height images of thin films of 2 upon thermal annealing at (a) 100 °C, (b) 180 °C and (c) 220 °C. | ||
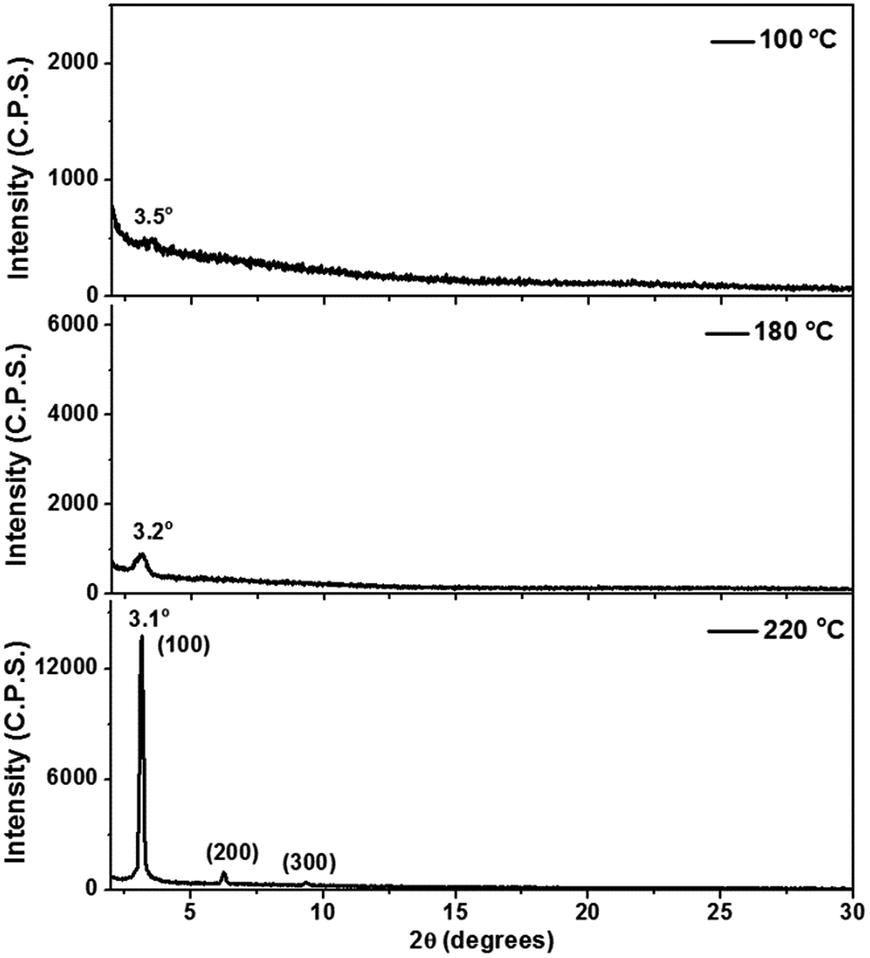 | ||
| Fig. 5 XRD profiles of thin films of 2 after thermal annealing at varied temperatures. | ||
Conclusions
In conclusion, a neutral organic small molecule 2 exhibiting strong NIR absorption around 1100 nm (ε ≈ 105 mol−1 L cm−1) and an electron mobility up to 0.96 cm2 V−1 s−1 is developed. The synthesis of the molecule is accomplished via a tandem process, involving a unique metal-free C–C coupling reaction between an electrophilic aryl bromide and an electron-rich aryl amine with dual nucleophilic sites of NH and CH groups. Having an exceptionally low-lying LUMO at −4.72 eV, the advantageously high chemo-stability of 2 is highlighted by comparison to molecule 3, which has a slightly higher LUMO than 2 but undergoes auto-reduction under ambient conditions. Such a combination of low-energy NIR-absorption, n-type semiconducting ability and optimal chemo-stability is rarely available for organic molecules. To the best of our knowledge, this is the first example of an organic small molecule that manifests both high performance n-type semiconducting properties and strong NIR absorption at wavelengths exceeding 1 μm. Such distinctive properties qualify the molecule for special applications as a transparent organic opto-electronic material.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation (No. 21174004, 21222403 and 51473003).Notes and references
- (a) S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS PubMed; (b) G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111 CrossRef CAS; (c) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. S. Challey and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed.
- (a) U. Mayerhöffer, K. Deing, K. Gruβ, H. Braunschweig, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2009, 48, 8776 CrossRef PubMed; (b) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC; (c) L. L. Li and E. W. G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC; (d) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119 CrossRef CAS PubMed.
- (a) Y. Wang, D. Gao, P. Zhang, P. Gong, C. Chen, G. Gao and L. Cai, Chem. Commun., 2014, 50, 811 RSC; (b) A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059 CrossRef CAS PubMed; (c) M. Li, X. Wu, T. Wang, Y. Li, W. Zhu and T. D. James, Chem. Commun., 2014, 50, 1751 RSC; (d) X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579 CrossRef CAS PubMed.
- (a) G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Wang, Chem. Mater., 2008, 20, 6208 CrossRef CAS; (b) G. Qian and Z. Wang, Can. J. Chem., 2010, 88, 192 CrossRef CAS; (c) G. Qian and Z. Wang, Chem.–Asian J., 2010, 5, 1006 CrossRef CAS PubMed.
- (a) C. Kohl, S. Becker and K. Müllen, Chem. Commun., 2002, 2778 RSC; (b) V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prudhomme, Chem. Mater., 2012, 24, 812 CrossRef CAS PubMed.
- (a) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS; (b) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC; (c) Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Chem.–Eur. J., 2014, 20, 7309 CrossRef CAS PubMed.
- Z. Guo, Z. Jin, J. Wang and J. Pei, Chem. Commun., 2014, 50, 6088 RSC.
- (a) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed; (b) W. Yue, J. Gao, W. Jiang, S. Motta, F. Negri and Z. Wang, J. Am. Chem. Soc., 2011, 133, 18054 CrossRef CAS PubMed; (c) L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938 RSC.
- (a) K. Cai, Q. Yan and D. Zhao, Chem. Sci., 2012, 3, 3175 RSC; (b) K. Cai, J. Xie and D. Zhao, J. Am. Chem. Soc., 2014, 136, 28 CrossRef CAS PubMed; (c) K. Cai, J. Xie, X. Yang and D. Zhao, Org. Lett., 2014, 16, 1852 CrossRef CAS PubMed.
- (a) C. Roger and F. Würthner, J. Org. Chem., 2007, 72, 8070 CrossRef PubMed; (b) J. Misek, A. Jentzsch, S. Sakurai, D. Emery, J. Mareda and S. Matile, Angew. Chem., Int. Ed., 2010, 49, 7680 CrossRef CAS PubMed; (c) C. Li, C. Xiao, Y. Li and Z. Wang, Org. Lett., 2013, 15, 682 CrossRef CAS PubMed; (d) S. Suraru, C. Burschka and F. Würthner, J. Org. Chem., 2014, 79, 128 CrossRef CAS PubMed; (e) S. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428 CrossRef CAS PubMed.
- (a) M. J. Lin, B. Fimmel, K. Radacki and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 10847 CrossRef CAS PubMed; (b) X. Fang, M. D. Guo, L. J. Weng, Y. Chen and M. J. Lin, Dyes Pigm., 2015, 113, 251 CrossRef CAS.
- S. Suraru and F. Würthner, J. Org. Chem., 2013, 78, 5227 CrossRef CAS PubMed.
- C. Tönshoff and H. F. Bettinger, Angew. Chem., Int. Ed., 2010, 49, 4125 CrossRef PubMed.
- Y. Yamaguchi, K. Ogawa, K. Nakayama, Y. Ohba and H. Katagiril, J. Am. Chem. Soc., 2013, 135, 19095 CrossRef CAS PubMed.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhu-khovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, characterization data, and DFT calculations. See DOI: 10.1039/c5sc03045e |
| This journal is © The Royal Society of Chemistry 2016 |