
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of cerium oxidation state through metal complex secondary structures†
Jessica R.
Levin
,
Walter L.
Dorfner
,
Patrick J.
Carroll
and
Eric J.
Schelter
*
Roy and Diana T. Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th St., Philadelphia, Pennsylvania 19104, USA. E-mail: schelter@sas.upenn.edu; Tel: +1 215-898-8633
First published on 11th August 2015
Abstract
A series of alkali metal cerium diphenylhydrazido complexes, Mx(py)y[Ce(PhNNPh)4], M = Li, Na, and K, x = 4 (Li and Na) or 5 (K), and y = 4 (Li), 8 (Na), or 7 (K), were synthesized to probe how a secondary coordination sphere would modulate electronic structures at a cerium cation. The resulting electronic structures of the heterobimetallic cerium diphenylhydrazido complexes were found to be strongly dependent on the identity of the alkali metal cations. When M = Li+ or Na+, the cerium(III) starting material was oxidized with concomitant reduction of 1,2-diphenylhydrazine to aniline. Reduction of 1,2-diphenylhydrazine was not observed when M = K+, and the complex remained in the cerium(III) oxidation state. Oxidation of the cerium(III) diphenylhydrazido complex to the Ce(IV) diphenylhydrazido one was achieved through a simple cation exchange reaction of the alkali metals. UV-Vis spectroscopy, FTIR spectroscopy, electrochemistry, magnetic susceptibility, and DFT studies were used to probe the oxidation state and the electronic changes that occurred at the metal centre.
Introduction
Cerium is unique among the lanthanides because of its accessible +4 oxidation state (E°(CeIV/III) = 1.40 V vs. Fc/Fc+).1–3 Considering its standard reduction potential, Ce(IV) complexes are best known as one-electron oxidants in inorganic and organic syntheses, as well as in materials chemistry.4–16 For example, ceric ammonium nitrate (CAN), has been used in water oxidation, oxidation of alcohols, oxidative carbon–carbon coupling reactions, and in oxidative deprotection of ketones and acetals.4,10 In materials chemistry cerium has been used in both oxidative and reductive contexts. Cerium(IV) dioxide (ceria) and related materials are applied in catalytic redox cycling devices, such as in fuel cells,15,17–21 active supports in 3-way catalytic converters,22–24 for the water gas shift reaction,25–28 and in heterogeneous catalysis for organic reactions and fuel production.11,12,29–32We have studied the electrochemical behaviour of a variety of cerium complexes,33,34 and found that, despite the isolated nature of the cerium 4f1 electron, electron donating ligands shift the Ce(IV/III) redox potential to reducing values, e.g. E1/2(CeIV/III[2-(tBuNO)py]4) = −1.95 V vs. Fc/Fc+, where 2-(tBuNO)py is N-tert-butyl-N-2-pyridylnitroxide.33,35 To further expand cerium redox chemistry we recently have focused on understanding the thermodynamic and kinetic factors that underlie cerium redox reactions. We demonstrated that in the cerium heterobimetallic frameworks, [M3(THF)n][Ce(BINOLate)3] M = Li, Na, K, Cs and BINOL = (S)-1,1′-bi-2-naphthol, the secondary coordination sphere, namely the identity of M+, impacted the rates and product outcomes of electron transfer (ET) reactions.36,37 Given these observations, we were compelled to investigate cerium heterobimetallic complexes with redox active ligands to express and modulate cerium–ligand intramolecular redox chemistry.
Herein, we report that the choice of alkali metal cation in the complexes Mx(py)y[Ce(PhNNPh)4], M = Li, Na, and K, x = 4 (Li and Na) or 5 (K), and y = 4 (Li), 8 (Na), or 7 (K), resulted in variable electronic structures. Our results showed the smaller, harder alkali metal cations Li+ and Na+ stabilized the tetravalent cerium cation whereas the softer K+ formed a cerium(III) complex. To the best of our knowledge, these results are the first examples of the use of secondary coordination sphere effects to modulate the oxidation state of a lanthanide cation.
Results and discussion
Synthesis and structural characterization of 1–3
Zdilla and coworkers recently reported a Li+ heterobimetallic diphenylhydrazido complex that effectively stabilized high valent Mn(IV) cations despite the reducing character of the ligand and the oxidizing character of Mn(IV) (see Scheme 1).38–40 Intrigued by these results and the relative scarcity of electrochemical properties reported for anionic nitrogen donors at cerium,41 we hypothesized that 1,2-diphenyl hydrazido ligands would similarly form cerium complexes with secondary structures governed by alkali metal cations.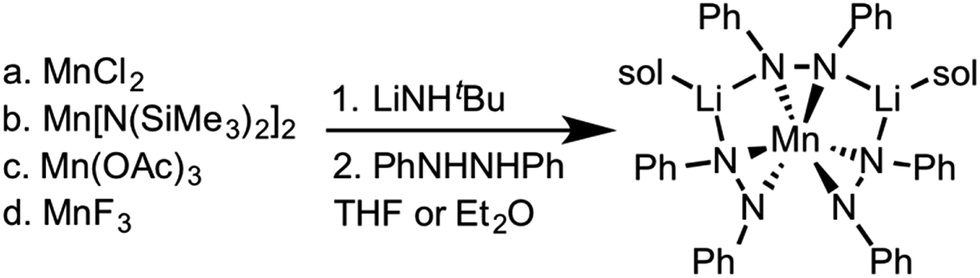 | ||
| Scheme 1 Synthesis of the lithium manganese diphenylhydrazido complex.40 | ||
Dark purple Li4(py)4[Ce(PhNNPh)4] (1) and Na4(py)8[Ce(PhNNPh)4] (2) were synthesized by layered reactions of Ce[N(SiMe3)2]3 with 4 equiv. 1,2-diphenylhydrazine and 4 equiv. MN(SiMe3)2, M = Li or Na, in a mixture of Et2O and pyridine. The yields for 1 and 2 were 75% and 63% respectively (Scheme 2). Because of the poor solubility of K5(py)7[Ce(PhNNPh)4] (3) in Et2O–pyridine mixtures, 3 was prepared in neat pyridine by reaction of Ce[N(SiMe3)2]3 with 4 equiv. 1,2-diphenylhydrazine and 5 equiv. KN(SiMe3)2. Complex 3 was isolated as dark brown needles in 65% yield following crystallization from a concentrated pyridine solution of the reaction mixture layered with hexanes.
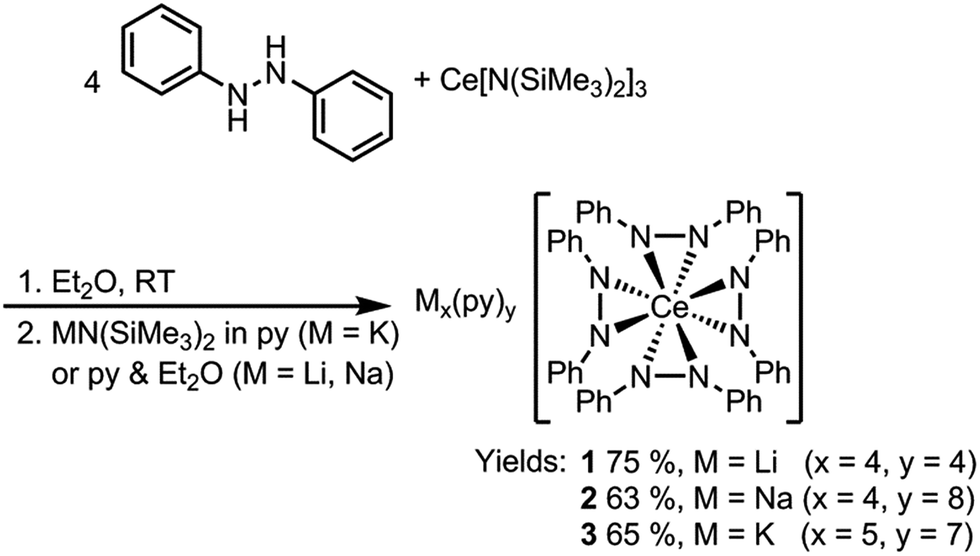 | ||
| Scheme 2 Syntheses of complexes 1, 2, and 3. | ||
X-ray crystal structures revealed that 1 and 2 both formed formally Ce(IV) complexes by charge balance, with four dianionic diphenylhydrazido ligands and four alkali metal cations per cerium cation in the formula unit (Fig. 1). Within the structures, the alkali metal cations bridged neighbouring 1,2-diphenylhydrazido units. Surprisingly, 3 formed an extended coordination polymer in which the potassium ions interacted both intramolecularly through bridging neighbouring hydrazido ligands and intermolecularly through K–arene interactions within the ligands at K(4) and K(5) (Fig. 1). The most notable difference in the structure of 3, however, was the presence of an additional K+ cation per formula unit, indicating that 3 was a formally Ce(III) complex.
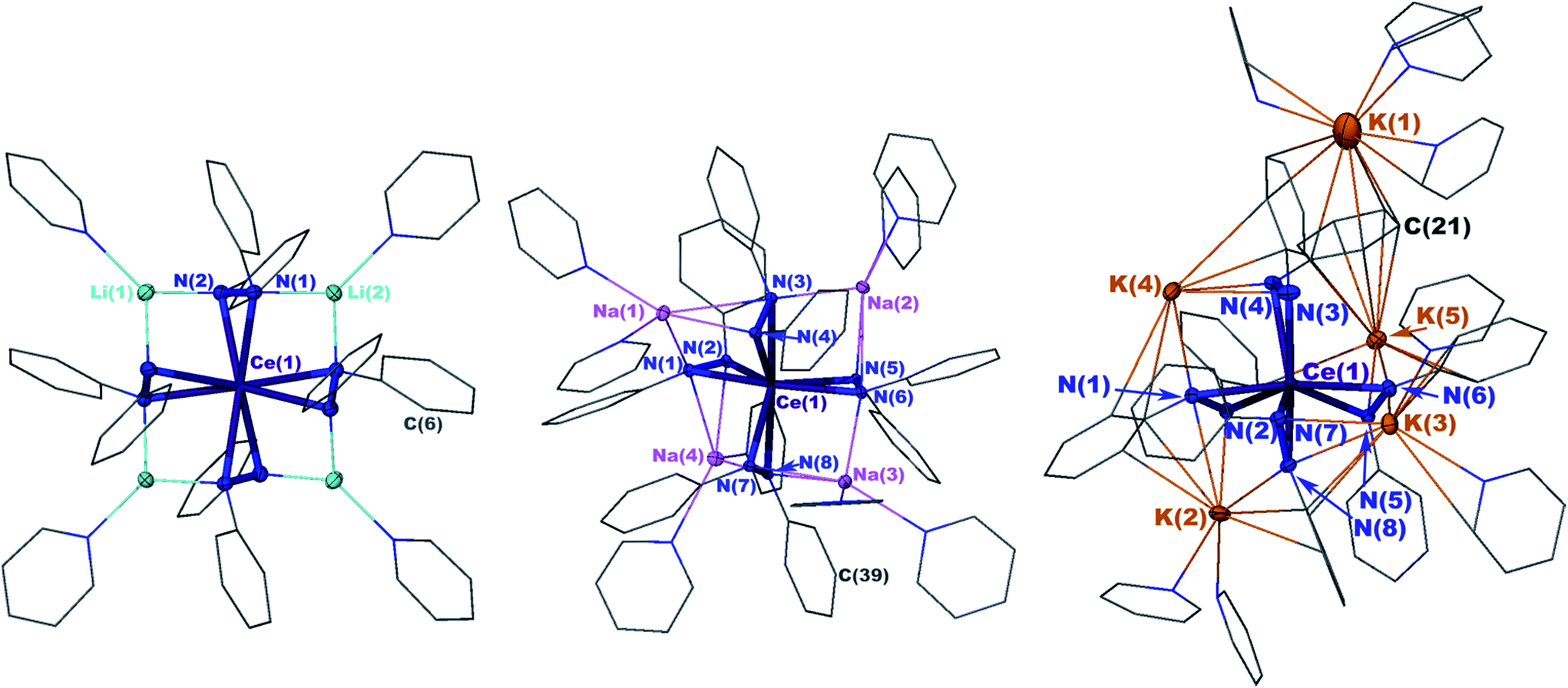 | ||
| Fig. 1 30% probability thermal ellipsoid plots of Li4(py)4[Ce(PhNNPh)4] (1) (left), Na4(py)8[Ce(PhNNPh)4] (2) (middle), and K5(py)7[Ce(PhNNPh)4] (3) (right) with the phenyl and pyridine rings shown in wire frame. Hydrogen atoms were omitted for clarity. Selected bond distances for 1 (Å): Ce(1)–N(1) 2.4408(13), Ce(1)–N(2) 2.4199(13), N(1)–N(2) 1.451(2), Li(2)–N(1) 2.018(3), Li(2)–N(1′) 2.018(3), Li(1)–N(2) 1.995(3). Selected bond distances for 2 (Å): Ce(1)–N(1) 2.390(3), Ce(1)–N(2) 2.373(2), N(1)–N(2) 1.462(3), Na(1)–N(1) 2.853(3), Na(1)–N(2) 2.630(3), Na(1)–N(3) 2.535(3). Selected bond distances for 3 (Å): Ce(1)–N(1) 2.564(3), Ce(1)–N(2) 2.480(4), N(1)–N(2) 1.465(5), K(2)–N(1) 3.044(4), K(2)–N(2) 2.877(4). | ||
The N–N bond lengths in complexes 1, 2, and 3 were consistent with single bonds ranging from 1.451(2)–1.466(3) Å (Table 1).39,42–46 The Ce–N distances for 1 ranged from 2.4199(13)–2.4408(14) Å while those for 2 were slightly shorter at 2.373(2)–2.398(2) (Table 1). The shortened Ce–N distances for 2 compared to 1 were consistent with the stronger Lewis acidity of Li+ cations in 1versus Na+ cations in 2. The Li+ cations reduced the relative charge density at the nitrogen atoms for binding with the cerium cation, compared to the Na+ cation in 2. This effect was reversed in 3, however, with Ce–N bonds ranging from 2.449(3)–2.636(4), in support of bonding to the larger cerium(III) cation in that complex. This set of bond distances also indicated that a change in secondary coordination sphere caused a change in cerium electronic structure.
| Complex | Ce(1)–N(x) (exp, Å) | Ce(1)–N(x) (calc, Å)a | N–N (exp, Å) | N–N (calc, Å)a | τ 4 (exp)b | τ 4 (calc)a,b |
|---|---|---|---|---|---|---|
| a Pyridine was replaced with OMe2 in the calculated structures, resulting in the following calculated complexes: Li4(OMe2)4[Ce(PhNNPh)4], Na4(OMe2)4[Ce(PhNNPh)4], and K4(OMe2)4[Ce(PhNNPh)4]−. b τ 4 values were calculated using the angles formed from the centroids of the N–N bonds and the cerium cations. | ||||||
| 1 | 2.4408(14) | 2.464 | 1.451(2) | 1.441 | 0.110 | 0.000 |
| 2.4199(13) | ||||||
| 2 | 2.390(3) | 2.439 | 1.462(3) | 1.441 | 0.663 | 0.498 |
| 2.373(2) | 2.443 | 1.461(3) | ||||
| 2.380(3) | 1.457(3) | |||||
| 2.381(2) | 1.466(3) | |||||
| 2.398(2) | ||||||
| 2.374(2) | ||||||
| 2.397(2) | ||||||
| 2.394(2) | ||||||
| 3 | 2.564(3) | 2.582 | 1.465(5) | 1.448 | 0.773 | 0.709 |
| 2.480(4) | 2.488 | 1.449(5) | ||||
| 2.415(4) | 1.459(5) | |||||
| 2.636(4) | 1.456(5) | |||||
| 2.499(3) | ||||||
| 2.482(4) | ||||||
| 2.449(3) | ||||||
| 2.494(3) | ||||||
To better understand the structural differences between complexes 1–3, we established the geometrical changes to the Ce primary coordination sphere using shape parameters for eight coordinate complexes (Table S1, see the ESI†).47–49 The shape parameters showed that each structure could be described by distinct eight-coordinate geometries with complexes 1 and 2 resembling dodecahedra (D2d), and complex 3 resembling a square antiprism (D4d). However, complexes 1–3 were heavily distorted from the idealized structures described by shape parameters, which made it difficult to express the three-dimensional structural differences between each of the complexes. Because of the distortions, the parameter τ4, which typically indicated the degree of planarity in four-coordinate structures, was found to be a more convenient metric to describe the system (Fig. 1 and Table 1).50 The four centroids between the N–N bonds of the ligands were used to calculate τ4 parameters for each complex, where a τ4 value of 0 indicated a planar distribution of the centroids and a value of 1 indicated a tetrahedral distribution of the centroids.50 The τ4 values revealed that as the ionic radius of the alkali metal cation increased, the geometry of the structure changed from pseudo-planar (1) τ4 = 0.110 to pseudo-tetrahedral (3) τ4 = 0.773 (Table 1 and Fig. 1).
We also examined the reaction mixtures of 1 and 2 by NMR spectroscopy for the reduction product from the spontaneous oxidation of Ce(III) to Ce(IV) (Scheme 3). Reductive cleavage of 1,2-diphenylhydrazine (E1/2 = −1.7 V versus Fc/Fc+ in DMSO)51 by metal complexes, including f-block metals, has been established to yield aniline, metal anilides, and metal diphenylamides.38–40,52–62 We postulated the balanced equation in Scheme 3 was at work in the synthesis of complexes 1 and 2, where the electron transfer occurred from a Ce(III) cation to 0.5 equiv. 1,2-diphenylhydrazine to form aniline.40 Indeed, aniline was detected by 1H NMR spectroscopy of the reaction mixture of complex 1 (see the ESI, Fig. S1–S4†). As expected, the reaction mixture for complex 3 did not show any evidence of aniline formation.
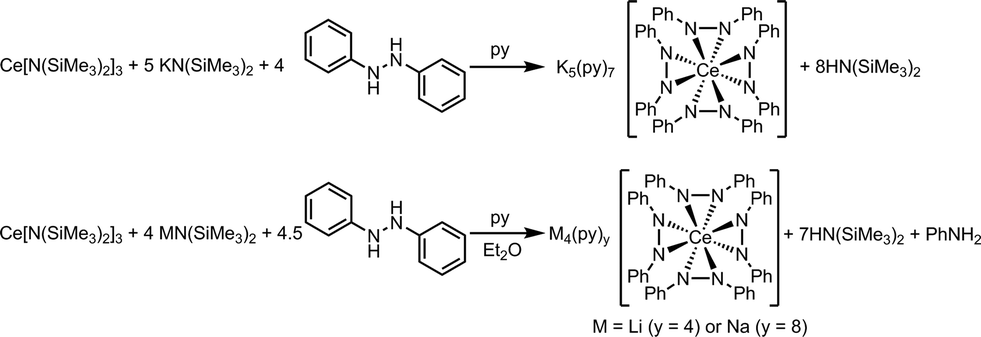 | ||
| Scheme 3 The balanced chemical equation for the formation of 3 (top), and 1 and 2 (bottom). | ||
The reduction of 1,2-diphenylhydrazine by Ce(III) was surprising as the reduction potential of 1,2-diphenylhydrazine was thermodynamically inaccessible.3,51 However, coordination of substrates to one or more Lewis acids had been shown to promote electron transfer.63–74 Fukuzumi and coworkers quantified the effect of Lewis acid coordination to O2 and its reduction with (TPP)Co (TPP = tetra-p-tolylporphyrin).73 Similarly, the reduction of 1,2-diphenylhydrazine by Ce(III) in the formation of complexes 1–3 was determined to be dependent on the Lewis acidity of the countercation, where K+ was not sufficiently Lewis acidic to promote the reduction of 1,2-diphenylhydrazine.
Magnetic, spectroscopic, and electrochemical characterization
To investigate the valences of complexes 1–3, magnetism and Ce LIII-edge XAS spectroscopy studies were performed. The oxidation state of complex 3 was corroborated with room temperature magnetic susceptibility measurements using Evans' method75,76 and solid state SQUID magnetometry (Table S3 and Fig. S5–S7 in the ESI,†Fig. 2). The room temperature χT products measured by both techniques, χT = 0.57 cm3 K mol−1 by Evans' method and χT = 0.70 cm3 K mol−1 by SQUID magnetometry, were similar to other reported Ce(III) complexes.77 Temperature dependent susceptibility plots of Ce(III) complexes typically show a decrease in the χT product at low temperatures due to thermal depopulation of crystal field levels,78–80 which was also observed in our data (Fig. 2). The low temperature χT product was also similar to previously reported Ce(III) complexes.77 Complexes 1 and 2 showed only a small paramagnetic shift in solution when Evans' method was applied (0.01 ppm compared to 0.1 ppm seen in complex 3, see ESI, Table S3†). Similarly, the room temperature χT products obtained by SQUID magnetometry were small, χT = 0.18 cm3 K mol−1 and χT = 0.089 cm3 K mol−1 for complexes 1 and 2 respectively (Fig. 2). The paramagnetic responses from complexes 1 and 2 were attributed to a small amount of paramagnetic impurity observed consistently over multiple measurements. Consistent with the magnetic data, the UV-Vis absorption spectra of 1 and 2 showed broad ligand-to-metal charge transfer bands, which are defining features in many Ce(IV) complexes (Fig. S26–S27, ESI†).36,81 Ce LIII-edge spectroscopic data were determined to be unreliable due to the instability of the complexes in the sample matrix (Fig. S8†).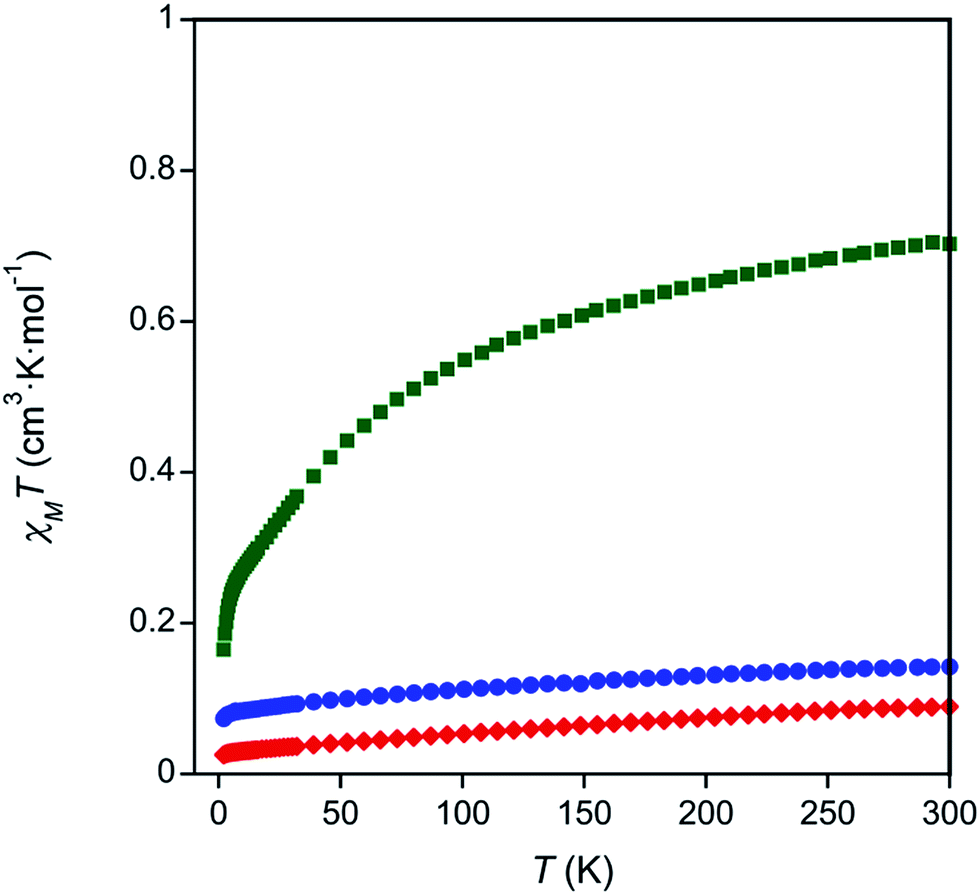 | ||
| Fig. 2 Temperature dependent magnetic data for complexes 1 (blue circles), 2 (red diamonds), and 3 (green squares). | ||
IR spectroscopy was performed to probe the change in N–N stretching frequency that was expected to accompany a change in formal oxidation state. We expected that the N–N stretching modes would increase in energy from complex 1 to 3 based on the Lewis acidity of both the Ce ion and the alkali metal cation in each complex. Compared to Ce(III) cations, the more Lewis acidic Ce(IV) cation was expected to accept more electron density through σ-bonding with the N–N units, reducing the electron density in the N–N bonds (see Fig. S38 in the ESI†). We also expected the alkali metal cations would similarly withdraw more electron density with increasing Lewis acidity. DFT calculations were used to identify the vibrational modes in the experimental spectra (see DFT section for further details). DFT computed vibrational spectra typically overestimate the energies of the vibrational modes,82–86 but apart from this overestimation, the calculated and experimental spectra were in good agreement (Table 2, see Fig. S23–S25 in the ESI†).
| Complex | N–N stretch (cm−1, sol = py) | N–N stretch (cm−1, sol = OMe2) |
|---|---|---|
| Li4(sol)[Ce(PhNNPh)4] (1) | 1255 | 1284 |
| Na4(sol)[Ce(PhNNPh)4] (2) | 1257 | 1289 |
| K4(OMe2)4[Ce(PhNNPh)4]− (3-OMe2) | — | 1303 |
| {K5(py)7[Ce(PhNNPh)4]}n (3) | 1265 | — |
The intense absorption band of the N–N stretch in complexes 1, 2, and 3 in both the experimental and calculated spectra allowed for assignment of that vibrational mode (see ESI, Fig. S23–S25†). The Ce–N vibrational modes were difficult to unambiguously assign because of their low intensities and overlap with ligand features. Complex 3 showed a ∼10 cm−1 increase in energy of the N–N stretching mode from complexes 1 and 2, consistent with a change in cerium oxidation state. Because the N–N stretching mode found in complexes 1 and 2 differed by only 2 cm−1 despite the change in Lewis acidity between Li+ and Na+, the more significant factor to the increase in energy between these complexes was evidently the oxidation state at the Ce cation. Overall, based on the spectroscopic and magnetic measurements, the oxidation state of the cerium metal centre was evidently impacted by the identity of the alkali metal cation in the secondary coordination sphere.
To better understand how the alkali metal cation in the secondary coordination sphere influenced the redox properties of the cerium metal centre, cyclic voltammetry was performed on complexes 1–3 (see Fig. S32–S37 in the ESI†). Because of the instability of complexes 1 and 2 in coordinating solvents and the insolubility of complex 3 in non-coordinating solvents, the electrochemistry of complexes 1 and 2 was performed in fluorobenzene, and in THF for complex 3. Beginning the scans from the rest potentials in each case, the CeIV/III reduction waves (in the cases of 1 and 2) and oxidation wave (in the case of 3) were reversible for complexes 1–3, where the difference in the anodic and cathodic waves were ΔE = 50 mV for complex 3 in THF, and ΔE = 60–70 mV for complexes 1 and 2 in fluorobenzene. The electrochemical reversibility of the CeIV/III redox event indicated that outer sphere electron transfer between cerium and the electrode was rapid in all three complexes, and that there was little ligand reorganization upon oxidation or reduction of the cerium cation in any geometry.
In order to compare the electrochemical potentials of complexes 1–3, we used 1,2-diphenylhydrazine, which was measured in both solvents, to normalize the influence of the solvent on the electron transfer between the electrode and the analyte. The E1/2 values for the CeIV/III reduction of complexes 1 and 2 were centred at −1.93 V and −1.88 V versus Fc/Fc+ respectively. Following the normalization for the difference in solvents, the E1/2 of the CeIII/IV oxidation of complex 3 was −2.02 V versus Fc/Fc+. The negative reduction potentials indicated that the Ce(IV) oxidation state was strongly stabilized by the hard, anionic PhNNPh2− ligand. Use of electron-rich anionic N-donors had been shown to support the Ce(IV) oxidation state previously.33 Thus, despite its measurement as a slightly more potent reductant than 1 and 2, complex 3 was isolated as a Ce(III) complex.
Ce(IV) and cation exchange reactions
Considering the oxidation potential of complex 3, we reasoned a potassium-supported Ce(IV) diphenylhydrazido analogue should be accessible. In an effort to isolate a Ce(IV) diphenylhydrazido complex with K+ cations in the secondary coordination sphere, we reacted 1,2-diphenylhydrazine with a formally Ce(IV) protonolysis starting material, Ce[N(SiHMe2)2]4.88,89 The addition of 1,2-diphenylhydrazine to the Ce(IV) starting material, however, resulted in immediate reduction (Scheme 4) and subsequent reaction with KN(SiMe3)2 formed complex 3, which was detected by 1H NMR spectroscopy. Adding 1,2-diphenylhydrazine to a mixture of KH and Ce[N(SiHMe2)2]4 also resulted in the formation of complex 3. Presumably, 0.5 equiv. azobenzene was the other product that formed in the course of the redox reaction, but this side product was not detected by 1H NMR spectroscopy (see Fig. S22, ESI†). Thus, the presence of alkali metal cations in the secondary coordination sphere were essential in stabilizing the Ce(IV) oxidation state. The alkali metals in solution structurally supported the resultant cerium diphenylhydrazido complex as well as modulated the electronics of the ligands in order to stabilize Ce(IV) and destabilize the ligand oxidation. These observations were consistent with those of Zdilla and coworkers, who disclosed that, upon removing the Li+ cations in the cluster Li4Mn4(μ3-NtBu)3(NtBu)(N), the Mn(V) cations reductively eliminated azo-tert-butane.90 | ||
| Scheme 4 Attempted synthesis of K4(sol)2[Ce(PhNNPh)4] starting from a Ce(IV) precursor. Instead, K5(py)7[Ce(PhNNPh)4] (3) formed as a result of this reaction (Fig. S22, ESI†). | ||
Based on the similar cerium electrochemistry of complexes 1 and 3 and the Ce–N and M–N (M = Li+ and K+) bond enthalpies, we expected that complex 3 should convert to complex 1 through a cation exchange reaction (Scheme 5). The successful metathesis of LiI with complex 3 in diethyl ether demonstrated that the oxidation state of cerium could be influenced by a change in the secondary coordination sphere (Fig. S15, ESI†). This reaction likely occurred through a Li+ promoted reduction of the 1,2-diphenylhydrazido ligand as well. By 1H NMR spectroscopy, the reaction appeared to proceed cleanly, but the percent conversion was consistently overestimated. Presumably the resonances of side-products present in the reaction overlapped with the desired products' resonances. Cation exchange reactions of 2 with KI were also successful, where complex 3 was the only product evident by 1H NMR spectroscopy (Fig. S20–S21 in the ESI†). In this case, the 1,2-diphenylhydrazido evidently acted as the reductant to form Ce(III) and azobenzene. A complex mixture formed when cation exchange of complex 1 with KI was attempted (Fig. S17–S19†). The complex mixture observed in this reaction could be as a result of the smaller lattice energy of LiI formation, the poorer solubility of KI, and the weaker M–N interaction with larger alkali metal cations.91,92
 | ||
| Scheme 5 Metathesis reaction of complex 3 with 4 equiv. of LiI. | ||
Conclusions
We have shown that the secondary coordination sphere about cerium 1,2-diphenylhydrazido complexes influenced both the primary coordination geometry and the oxidation state of the cerium metal centre, making these complexes the first example of a secondary coordination sphere influencing the oxidation state of f-element complexes. The alkali metals in the secondary coordination sphere both facilitated the reduction of the 1,2-diphenylhydrazine to produce a high-valent Ce(IV) complex, and structurally stabilized the high-valent Ce(IV) complexes. Furthermore, a cation exchange reaction with simple alkali metal salts had changed the oxidation state at the cerium ion. The use of multiple Lewis acids could be a general strategy to access high valent metal oxidation states. Electronic and steric changes to the primary coordination sphere in this system are currently under investigation.Experimental section
General methods
Unless otherwise indicated all reactions and manipulations were performed under an inert atmosphere (N2) using standard Schlenk techniques or in a Vacuum Atmospheres, Inc. Nexus II drybox equipped with a molecular sieves 13X/Q5 Cu-0226S catalyst purifier system. Glassware was oven-dried overnight at 150 °C prior to use. 1H, 13C, and 7Li NMR spectra were obtained on a Bruker DMX-300, on a Bruker DMX-360, or on a Bruker DRX-400 Fourier transform NMR spectrometer at 300, 360, and 400 MHz respectively. Chemical shifts were recorded in units of parts per million downfield from residual proteo solvent peaks (1H), or characteristic solvent peaks (13C). The 7Li spectra were referenced to external solution standards of LiCl in H2O. Evans' method was performed on a Bruker BioDRX-500 Fourier transform NMR spectrometer at 500 MHz. Hexamethylcyclotrisiloxane was used as the internal standard for Evans's method experiments. Elemental analyses were performed at the University of California, Berkeley Microanalytical Facility using a Perkin-Elmer Series II 2400 CHNS analyzer. UV-vis-NIR absorption measurements of complexes were performed using a PerkinElmer 950 UV-vis/NIR Spectrophotometer. One mm path length screw cap quartz cells were used with a blank measured before each run. The infrared spectra were obtained from 400–4000 cm−1 using a PerkinElmer 1600 series infrared spectrometer. FTIR solution spectra were first collected with a background of air, and then the solvent spectrum was subtracted using PerkinElmer software. GC/MS Spectrometry was performed using an Agilent 5937 GC/MS spectrometer with the CI method of ionization. Diethyl ether was used as the volatile solvent and He as the carrier gas.Materials
Tetrahydrofuran, diethyl ether, hexane, fluorobenzene, and pentane were purchased from Fisher Scientific. The solvents were sparged for 20 min with dry N2 and dried using a commercial two-column solvent purification system comprising columns packed with Q5 reactant and neutral alumina respectively (for hexane and pentane), or two columns of neutral alumina (for THF, diethyl ether, and fluorobenzene). Pyridine, also purchased from Fisher Scientific, was freeze–pump–thawed for 4 cycles and stored over 4 Å molecular sieves for three days before use. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc. Pyridine-d5 was stored over 4 Å molecular sieves for three days before use, and benzene-d6 was dried and stored over potassium for 2 days before use. Ce[N(SiMe3)2]3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 93 and Ce[N(SiHMe2)2]488,89 were prepared following published procedures. Li[N(SiMe3)2] (Acros) was recrystallized from hot pentane prior to use. K[N(SiMe3)2] (Acros) and Na[N(SiMe3)2] (Acros) were used as purchased. Hydrazobenzene (Sigma Aldrich, MP Biochemicals, Alfa Aesar) was sublimed under reduced pressure first at 60 °C to remove azobenzene, and then at 105 °C for further purification prior to use. Alternatively, the hydrazobenzene was purified by fractional recrystallization of hydrazobenzene dissolved in toluene and layered with pentane.
93 and Ce[N(SiHMe2)2]488,89 were prepared following published procedures. Li[N(SiMe3)2] (Acros) was recrystallized from hot pentane prior to use. K[N(SiMe3)2] (Acros) and Na[N(SiMe3)2] (Acros) were used as purchased. Hydrazobenzene (Sigma Aldrich, MP Biochemicals, Alfa Aesar) was sublimed under reduced pressure first at 60 °C to remove azobenzene, and then at 105 °C for further purification prior to use. Alternatively, the hydrazobenzene was purified by fractional recrystallization of hydrazobenzene dissolved in toluene and layered with pentane.
Electrochemistry
Voltammetry experiments (CV and DPV) were performed using a CH Instruments 620D Electrochemical Analyzer/Workstation and the data were processed using CHI software v 9.24. All experiments were performed in an N2 atmosphere drybox using electrochemical cells that consisted of a 4 mL vial, glassy carbon (3 mm diameter) working electrode, a platinum wire counter electrode, and a silver wire plated with AgCl as a quasi-reference electrode. The working electrode surfaces were polished prior to each set of experiments, and were periodically replaced to prevent the build-up of oxidized product on the electrode surfaces. Potentials were reported versus ferrocene (Fc), which was converted from cobaltocene for calibration at the end of each run.3 Solutions employed during CV studies were ∼3 mM in analyte. For electrochemistry collected in fluorobenzene, the solution was 100 mM in [nBu4N][B(3,5-(CF3)2-C6H3)4] ([nBu4N][BArF4]), and electrochemistry collected in THF had 100 mM in [nPr4N][B(3,5-(CF3)2-C6H3)4] ([nPr4N][BArF4]) as the electrolyte. All data were collected in a positive-feedback IR compensation mode. Scan rate dependences of 25–1000 mV s−1 were performed to determine electrochemical reversibility.Magnetism
Magnetic data were collected on a Quantum Design Multi-Property Measurement System (MPMS-7) with a Reciprocating Sample Option at 1 T from 2 to 300 K and at 2 K and 300 K from 0 to 7 T. Quartz wool was dried at 250 °C prior to use, plastic drinking straws were evacuated overnight prior to use. The plastic drinking straws themselves were used as the sample holders through heat sealing the plastic tubing. The plastic drinking straws were heat sealed at one end in a glovebox. Then a ground sample was loaded into the straw and capped with ∼10 mg of quartz wool. The other end of the plastic drinking straw was then sealed through application of heat, forming a pouch that contained the sample and the quartz wool. The sample and wool were weighed to the nearest 0.1 mg on a calibrated and levelled Mettler-Toledo AL-204 analytical balance. Corrections for the intrinsic diamagnetism of the samples were made using Pascal's constants.94 Data were collected on two independently prepared samples to ensure reproducibility.X-ray absorption spectroscopy
Ce LIII-edge XANES data were collected at the Stanford Synchrotron Radiation Lightsource, beamline 11-2, using a Si 220 (phi = 0) double monochromator that was detuned to 20% in order to reduce harmonic contamination. The resulting data have an energy resolution of 3.2 eV. Data were collected in transmission, using a CeO2 reference to calibrate the energy scale, setting the first inflection point of the CeO2 absorption to 5723 eV. A linear pre-edge background was subtracted and the data were subsequently normalized at 5800 eV.Since the compounds are extremely sensitive to oxygen, each sample was ground into a powder, mixed with dry boron nitride as a diluent, and then packed into the slots of a machined aluminum sample holder. Aluminized mylar was affixed to the holder with an indium-wire seal. After packaging, the samples were transported in dry nitrogen-filled containers to the beamline. Sample holders were quickly transferred to the vacuum chamber, exposing the sealed holders to air for less than thirty seconds before pumping out the chamber and collecting the data under vacuum.
X-ray crystallography
X-ray intensity data were collected on a Bruker APEXII CCD area detector employing graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at a temperature of 143(1) K. In all cases, rotation frames were integrated using SAINT,95 producing a listing of unaveraged F2 and σ(F2) values which were then passed to the SHELXTL96 program package for further processing and structure solution on a Dell Pentium 4 computer. The intensity data were corrected for Lorentz and polarization effects and for absorption using TWINABS97 or SADABS.98 The structures were solved by direct methods (SHELXS-97).99 Refinement was by full-matrix least squares based on F2 using SHELXL-97.99 All reflections were used during refinements. The weighting scheme used was w = 1/[σ2(Fo2) + (0.0907P)2 + 0.3133P] where P = (Fo2 + 2Fc2)/3. Non-hydrogen atoms were refined anisotropically and hydrogen atoms were refined using a riding model. Complex 1 (Li4(py)4[Ce(PhNNPh)4] C68H60N12Li4Ce), crystallizes in the orthorhombic space group Fddd with a = 11.4483(7) Å, b = 43.060(2) Å, c = 24.5220(14) Å, V = 12088.5(12) Å3, Z = 8, and dcalc = 1.333 g cm−3. A total of 74593 reflections were measured yielding 3485 unique reflections (Rint = 0.0381). Complex 2 (Na4(py)8[Ce(PhNNPh)4] C88H80N16Na4Ce) crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a = 13.9504(7) Å, b = 16.2467(9) Å, c = 19.0053(11) Å, α = 83.136(3)°, β = 83.696(3)°, γ = 84.993(3)°, V = 4238.8(4) Å3, Z = 2, and dcalc = 1.249 g cm−3. A total of 147398 reflections were measured, 19328 of them unique reflections (Rint = 0.0680). Complex 3 (K5(py)7[Ce(PhNNPh)4]) C83H75N15K5Ce, crystallizes in the triclinic space group P with a = 15.2003(12) Å, b = 15.4668(12) Å, c = 18.0353(15) Å, α = 93.849(5)°, β = 101.333(5)°, γ = 107.292(5)°, V = 3933.7(5) Å3, Z = 2, and dcalc = 1.366 g cm−3. A total of 95153 reflections were measured yielding 17890 unique reflections (Rint = 0.0544).
with a = 13.9504(7) Å, b = 16.2467(9) Å, c = 19.0053(11) Å, α = 83.136(3)°, β = 83.696(3)°, γ = 84.993(3)°, V = 4238.8(4) Å3, Z = 2, and dcalc = 1.249 g cm−3. A total of 147398 reflections were measured, 19328 of them unique reflections (Rint = 0.0680). Complex 3 (K5(py)7[Ce(PhNNPh)4]) C83H75N15K5Ce, crystallizes in the triclinic space group P with a = 15.2003(12) Å, b = 15.4668(12) Å, c = 18.0353(15) Å, α = 93.849(5)°, β = 101.333(5)°, γ = 107.292(5)°, V = 3933.7(5) Å3, Z = 2, and dcalc = 1.366 g cm−3. A total of 95153 reflections were measured yielding 17890 unique reflections (Rint = 0.0544).
Computational details
All calculations were performed with Gaussian 09 Revision D.01100 with the B3LYP hybrid DFT method. A 28-electron small core effective core potential was applied to cerium with published segmented natural orbital basis set incorporating quasi-relativistic effects,101–103 while the 6-31 G* basis set was applied to all other atoms. Geometry optimizations of 1 and 2 were based on their crystal structures, while the geometry optimization of the Ce(IV) structure of 3 was based off of the crystal structure coordinates of complex 2. The coordinates that were input for the geometry optimizations of the anionic Ce(III) analogues were based on the optimized geometry found for the Ce(IV) calculations 1, 2, and 3 with elongated Ce–N bonds. Geometry optimization was attempted with pyridine solvating the alkali metals. However, convergence could not be reached with this system. No other restrictions were placed on the systems besides the spin. All frequency calculations found no negative frequencies, except for the calculation of Li4(OMe2)4[Ce(PhNNPh)4] which had 4 small negative frequencies less than −17 cm−1, indicating that the optimized structures found were at an energy minimum. Molecular orbitals were rendered using Chemcraft v. 1.6.104
Synthetic details and characterization
868.66, 1582.74 (vs), 1547.75 (s), 1468.83 (vs), 1440.44 (s), 1296.41 (s), 1289.19 (s), 1256.80 (vs), 1162.99 (s), 1148.41, 1118.74, 1069.80, 1033.85, 1018.29, 987.09 (vs), 860.03 (s), 793.83, 746.86 (s), 702.93 (vs), 613.74, 599.51. Elemental analysis calculated for Na4(py)5(Et2O)[Ce(PhNNPh)4] C77H75CeNa4N13O: C, 64.65; H, 5.28; N, 12.73. Found: C, 64.58; H, 5.08; N, 12.70%.
Acknowledgements
We thank the U.S. Department of Energy, Office of Science, Early Career Research Program (Grant DE-SC0006518) and the University of Pennsylvania for financial support of this work. We also would like to thank the U.S. National Science Foundation for supporting our X-ray diffractometer (CHE-0840438) and for providing us with calculations resources on the Extreme Science and Engineering Discovery Environment (XSEDE) (grant OCI-1053575). We thank Prof. Jay Kikkawa (UPenn) for assistance with the magnetic measurements. We are grateful to Dr. Corwin Booth, Lawrence Berkeley National Laboratory, for his efforts in XAS measurements.Notes and references
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley and Sons Inc., Hoboken, NJ, 2006 Search PubMed.
- A. W. G. Platt, The Rare Earth Elements: Fundamentals and Applications, John Wiley & Sons Ltd., Chichester, United Kingdom, 2012 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- A. K. Das, Coord. Chem. Rev., 2001, 213, 307–325 CrossRef CAS.
- T. Imamoto, Y. Koide and S. Hiyama, Chem. Lett., 1990, 19, 1445–1446 CrossRef.
- T. Kreuzer, E. S. Lox, D. Lindner and J. Leyrer, Catal. Today, 1996, 29, 17–27 CrossRef CAS.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS.
- V. Sridharan and J. C. Menendez, Chem. Rev., 2010, 110, 3805–3849 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- G. A. Molander, Chem. Rev., 1992, 92, 29–68 CrossRef CAS.
- V. Nair, L. Balagopal, R. Rajan and J. Matthew, Acc. Chem. Res., 2004, 37, 21–30 CrossRef CAS.
- V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862–1891 CrossRef CAS.
- T.-L. Ho, Synthesis, 1973, 347–354 CrossRef CAS.
- S. V. More, M. N. V. Sastry and C.-F. Yao, Green Chem., 2006, 8, 91–95 RSC.
- K. Eguchi, T. Setoguchi, T. Inoue and H. Arai, Solid State Ionics, 1992, 52, 165–172 CrossRef CAS.
- W. Ruttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef PubMed.
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS PubMed.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- E. P. Murray, T. Tsai and S. A. Barnett, Nature, 1999, 400, 649–651 CrossRef CAS PubMed.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ionics, 2000, 129, 63–94 CrossRef CAS.
- J. Kaspar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419–449 CrossRef CAS.
- J. Kaspar and P. Fornasiero, J. Solid State Chem., 2003, 171, 19–29 CrossRef CAS.
- R. Burch, Catal. Rev.: Sci. Eng., 2004, 46, 271–333 CAS.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Y. Li, Q. Fu and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2000, 27, 179–191 CrossRef CAS.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Perez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
- L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS PubMed.
- L. Xu and J. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CAS.
- A. Kawashima, K. Matsubara and K. Honda, Bioresour. Technol., 2008, 99, 3439–3443 CrossRef CAS PubMed.
- M. Dejhosseini, T. Aida, M. Watanabe, S. Takami, D. Hojo, N. Aoki, T. Arita, A. Kishita and T. Adschiri, Energy Fuels, 2013, 27, 4624–4631 CrossRef CAS.
- N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS PubMed.
- J. A. Bogart, A. J. Lewis, M. A. Boreen, H. B. Lee, S. A. Medling, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2015, 54, 2830–2837 CrossRef CAS PubMed.
- J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS PubMed.
- J. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, Angew. Chem., Int. Ed., 2012, 51, 10159–10163 CrossRef CAS PubMed.
- J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed.
- C. R. Hamilton, R. A. Baglia, A. D. Gordon and M. J. Zdilla, J. Am. Chem. Soc., 2011, 133, 4208–4211 CrossRef CAS PubMed.
- S. K. Kondaveeti, S. Vaddypally, J. D. McCall and M. J. Zdilla, Dalton Trans., 2012, 41, 8093–8097 RSC.
- S. Vaddypally, S. K. Kondaveeti and M. J. Zdilla, Chem. Commun., 2011, 47, 9696–9698 RSC.
- P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and Z. Li, Dalton Trans., 2004, 129–136 RSC.
- W. J. Evans, D. J. Drummond, L. R. Chamberlain, R. J. Doedens, S. G. Bott, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 4983–4994 CrossRef CAS.
- A. A. Trifonov, M. N. Bochkarev, H. Schumann and J. Loebel, Angew. Chem., Int. Ed. Engl., 1991, 30, 1149–1151 CrossRef PubMed.
- K. L. Miller, B. N. Williams, D. Benitez, C. T. Carver, K. R. Ogilby, E. Tkatchouk, W. A. Goddard III and P. L. Diaconescu, J. Am. Chem. Soc., 2010, 132, 342–355 CrossRef CAS PubMed.
- Structural chemistry, ed. R. Allmann, John Wiley & Sons, New York, 1975 Search PubMed.
- C.-L. Pan, W. Chen and J. Song, Organometallics, 2011, 30, 2252–2260 CrossRef CAS.
- E. L. Muetterties and C. M. Wright, Q. Rev., Chem. Soc., 1967, 21, 109–194 RSC.
- E. L. Muetterties and L. J. Guggenberger, J. Am. Chem. Soc., 1974, 96, 1748–1756 CrossRef CAS.
- J. L. Hoard and J. V. Silverton, Inorg. Chem., 1963, 2, 235–242 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- T. S. Calderwood, C. L. Johlman, J. L. Roberts Jr., C. L. Wilkins and D. T. Sawyer, J. Am. Chem. Soc., 1984, 106, 4683–4687 CrossRef CAS.
- M. J. Zdilla, A. K. Verma and S. C. Lee, Inorg. Chem., 2008, 47, 11382–11390 CrossRef CAS PubMed.
- M. J. Zdilla, A. K. Verma and S. C. Lee, Inorg. Chem., 2011, 50, 1551–1562 CrossRef CAS PubMed.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS.
- W. J. Evans, G. Kociok-Kohn, V. S. Leong and J. W. Ziller, Inorg. Chem., 1992, 31, 3592–3600 CrossRef CAS.
- B. Gemund, H. Noth, H. Sachdev and M. Schmidt, Chem. Ber., 1996, 129, 1335–1344 CrossRef PubMed.
- Y. Ohki, Y. Takikawa, T. Hatanaka and K. Tatsumi, Organometallics, 2006, 25, 3111–3113 CrossRef CAS.
- F. Alonso, G. Radivoy and M. Yus, Tetrahedron, 2000, 56, 8673–8678 CrossRef CAS.
- J. M. Hoover, A. DiPasquale, J. M. Mayer and F. E. Michael, Organometallics, 2007, 26, 3297–3305 CrossRef CAS.
- Y. Nakajima and H. Suzuki, Organometallics, 2003, 22, 959–969 CrossRef CAS.
- A. F. Heyduk, R. A. Zarkesh and A. I. Nguyen, Inorg. Chem., 2011, 50, 9849–9863 CrossRef CAS PubMed.
- R. A. Zarkesh and A. F. Heyduk, Organometallics, 2011, 30, 4890–4898 CrossRef CAS.
- D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Chem.–Eur. J., 2014, 20, 4426–4438 CrossRef CAS PubMed.
- A. Sen, H. A. Stecher and A. L. Rheingold, Inorg. Chem., 1992, 31, 473–479 CrossRef CAS.
- J. R. Robinson, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem.–Eur. J., 2013, 19, 5996–6004 CrossRef CAS PubMed.
- D. H. Kim, U. S. Shin and C. E. Song, J. Mol. Catal. A: Chem., 2007, 271, 70–74 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubu, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS.
- S. Fukuzumi, N. Satoh, T. Okamoto, K. Yasui, T. Suenobu, Y. Seko, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 7756–7766 CrossRef CAS PubMed.
- S. Fukuzumi, Y. Fujii and T. Suenobu, J. Am. Chem. Soc., 2001, 123, 10191–10199 CrossRef CAS PubMed.
- S. Fukuzumi, J. Synth. Org. Chem., Jpn., 2003, 61, 1046–1055 CrossRef CAS.
- S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 CAS.
- K. Ohkubo, T. Suenobu, H. Imahori, A. Orita, J. Otera and S. Fukuzumi, Chem. Lett., 2001, 978–979 CrossRef CAS.
- K. Ohkubo, S. C. Menon, A. Orita, J. Otera and S. Fukuzumi, J. Org. Chem., 2003, 68, 4720–4726 CrossRef CAS PubMed.
- J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2014, 53, 3618–3628 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- M. D. Walter, R. Fandos and R. A. Andersen, New J. Chem., 2006, 30, 1065–1070 RSC.
- A. Panagiotopoulos, T. F. Zafiropoulos, S. P. Perlepes, E. Bakalbassis, I. Masson-Ramade, O. Kahn, A. Terzis and C. P. Raptopoulou, Inorg. Chem., 1995, 34, 4918–4920 CrossRef CAS.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2388 CrossRef CAS PubMed.
- H. An, H. Zhang, Z. Chen, Y. Li, X. Liu and H. Chen, Dalton Trans., 2012, 41, 8390–8400 RSC.
- C. A. Smith, E. W. Ainscough, H. M. Baker, A. M. Brodie and E. N. Baker, J. Am. Chem. Soc., 1994, 116, 7889–7890 CrossRef CAS.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- K. K. Irikura, R. D. Johnson III and R. N. Kacker, J. Phys. Chem. A, 2005, 109, 8430–8437 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Organometallics, 2012, 31, 535–544 CrossRef CAS.
- X. Wang, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Inorg. Chem., 2011, 50, 3826–3831 CrossRef CAS PubMed.
- X. Wang, H.-G. Cho, L. Andrews, M. Chen, D. A. Dixon, H.-S. Hu and J. Li, J. Phys. Chem. A, 2011, 115, 1913–1921 CrossRef CAS PubMed.
- D. D. Weis and G. E. Ewing, Anal. Chem., 1998, 70, 3175–3183 CrossRef CAS PubMed.
- A. R. Crozier, A. M. Bienfait, C. Maichle-Mossmer, K. W. Tornroos and R. Anwander, Chem. Commun., 2013, 49, 87–89 RSC.
- U. J. Williams, D. Schneider, W. L. Dorfner, C. Maichle-Mossmer, P. J. Carroll, R. Anwander and E. J. Schelter, Dalton Trans., 2014, 43, 16197–16206 RSC.
- S. Vaddypally, S. K. Kondaveeti, J. H. Roudebush, R. J. Cava and M. J. Zdilla, Chem. Commun., 2014, 50, 1061–1063 RSC.
- D. Quane, J. Chem. Educ., 1970, 47, 396–398 CrossRef CAS.
- D. F. C. Morris, Acta Crystallogr., 1956, 9, 197–198 CrossRef CAS.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- SHELXTL, Bruker ASX Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, TWINABS, University of Gottingen, Gottingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, SADABS, University of Gottingen, Gottingen, Germany, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, V. Voth, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Institute for Theoretical Chemistry, University of Cologne, http://www.tc.uni-koeln.de/PP/clickpse.en.html.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2002, 581, 139–147 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS PubMed.
- Chemcraft, http://www.chemcraftprog.com.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, UV-Vis spectra, FTIR spectra, Evans' method data, field dependence data, XAS spectra, electrochemical data, DFT coordinates and rendered molecular orbitals. CCDC 1404761–1404763. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02607e |
| This journal is © The Royal Society of Chemistry 2015 |