
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManaging the retro-pathway in direct catalytic asymmetric aldol reactions of thioamides†
Youmei
Bao
a,
Naoya
Kumagai
*a and
Masakatsu
Shibasaki
*ab
aInstitute of Microbial Chemistry (BIKAKEN), Tokyo, 3-14-23 Kamiosaki, Shinagawa-ku, Tokyo 141-0021, Japan. E-mail: nkumagai@bikaken.or.jp; mshibasa@bikaken.or.jp
bJST, ACT-C, 3-14-23 Kamiosaki, Shinagawa-ku, Tokyo 141-0021, Japan
First published on 7th August 2015
Abstract
Thioamides are the preferred pronucleophiles for direct catalytic asymmetric aldol reactions in the context of soft Lewis acid/hard Brønsted base cooperative catalysis. In-depth investigation of this proton-transfer catalysis, which is virtually in equilibrium, revealed that the prominence of the retro-aldol reaction depended on the substrate combination. The retro-aldol reaction is a serious issue in direct aldol reactions because the product distribution, including enantiomers and diastereomers, is governed by thermodynamic parameters, and the aldol products are obtained in much lower stereoselectivity compared with the kinetically controlled process. Herein we report the beneficial effect of an additive with a functional group architecture similar to that of the aldol adduct that suppresses the retro-aldol reaction by competitively binding to the catalyst. Strategic use of the additive led to high stereoselectivity, even when the combination of substrates was prone to the retro-aldol reaction.
Introduction
The direct catalytic asymmetric aldol reaction has gained popularity as an expeditious and straightforward method of accessing enantioenriched β-hydroxycarbonyl compounds.1,2 Chemo- and stereoselective access to this class of important chiral building blocks is a highly-discussed topic in the chemical community and the Mukaiyama-aldol reaction has been widely utilized since its initial discovery in 1973.3,4 The Mukaiyama-aldol reaction enables reliable coupling of aldehydes and enol silyl ethers at the expense of the preformation of active nucleophiles using a stoichiometric amount of reagents. The direct catalytic asymmetric aldol reaction was developed as an alternative to produce the aldol adduct in a “direct” manner without requiring activating reagents; the direct use of aldol donors eliminates the pre-activation process to generate active nucleophiles, providing an operationally simple synthetic protocol with minimal coproduction of reagent-derived waste. Although only limited aldol donors, e.g., ketones and aldehydes for facile enolization, were used in the early stage of the development, recent advances in this field have broadened the scope of available aldol donors, including those in the carboxylic acid oxidation state.5,6 The difficulty in using these aldol donors, however, originates from their reluctant enolization due to the low acidity of their α-protons, which is an initial trigger of the direct aldol reaction, and the low efficiency of the enolization impedes the overall process.We previously reported the particular utility of thioamides 1 (ref. 7 and 8) as aldol donors in the context of soft Lewis acid/hard Brønsted base cooperative catalysis.9,10 Thioamides have two advantageous features: (1) soft Lewis basicity and (2) divergent transformation into various functional groups for elaboration into biologically active compounds.11,12 The soft Lewis basicity enables chemoselective recognition/enolization in the presence of enolizable aldol acceptors (aldehydes 2), which ensures a smooth aldol reaction without self-condensation of aldehydes. We documented two catalytic systems for this aldol process, [Cu(CH3CN)4]PF6/(R,R)-Ph-BPE/LiOAr (1st generation; 1st-Gen)9a,b,12a,c and mesitylcopper/(R,R)-Ph-BPE/ArOH (2nd generation; 2nd-Gen) (Scheme 1a).9c,12b,d,e In particular, for the reaction of thiopropionamide, the catalytic performance of the 2nd-Gen catalyst is significantly better than that of the 1st-Gen catalyst. The 2nd-Gen catalyst is operationally simple and readily prepared by mixing commercial sources,13 and has been applied to enantioselective syntheses of key fragments of atorvastatin and thuggacin B.12b,d,e The high catalytic activity of the 2nd-Gen catalyst, however, may be highly problematic depending on the substrate sets used. In contrast to the high stereoselectivity that is stably and reliably obtained in the reaction of α-nonbranched aldehydes and thioacetamides/thiopropionamides, the combination of α-branched aldehydes and thiopropionamides produces various stereochemical outcomes that are highly sensitive to the reaction conditions e.g., catalyst loading and reaction time (Scheme 1b). The direct aldol reaction is a bimolecular coupling reaction that proceeds through a proton-transfer between substrates, and it is inherently prone to equilibration with the retro-aldol reaction. For catalytic enantioselective processes, the isolation of kinetic products is crucial for obtaining highly enantioenriched products, and the enantiopurity of the thermodynamic products after partial equilibration is significantly eroded over the course of the reaction. Although the aldol products from an unfavorable substrate combination are obtained in high stereoselectivity under specific conditions, systematic optimization of the reaction conditions is generally required in every single case, and even a slightly extended reaction time or higher catalyst loading leads to significantly decreased stereoselectivity. Furthermore, the prominence of the retro reaction depends on other parameters: the purity of the mesitylcopper and solvent,14,15 the purity and reactivity of each aldehyde, and the experimental manipulations under anhydrous conditions. Therefore, a more user-friendly protocol that allows for reliable production of the enantioenriched aldol adducts within a reasonable range of reaction conditions is desirable. Herein we report the significant effect of an additive on the aldol reaction to suppress the retro-aldol reaction, that contributed to establish a robust and reliable direct aldol protocol.
 | ||
| Scheme 1 Synthetic utility and problem of the direct catalytic asymmetric aldol reaction of thioamides. | ||
Results and discussion
Isobutyraldehyde (2a) was selected as a representative achiral α-branched aldehyde and subjected to a direct catalytic asymmetric aldol reaction with N,N-(diallyl)thiopropionamide (1a) under the previously optimized reaction conditions with the 2nd-Gen catalyst comprising mesitylcopper/(S,S)-Ph-BPE/ArOH (ArOH = 2,2,5,7,8-pentamethyl-6-chromanol).12b,d,e The reaction was traced over a 48 h period with 1.5 mol% of catalyst loading and the profile is summarized in Table 1. The reaction proceeded rapidly and more than 50% conversion was observed in 1 h at −70 °C. At this stage, the stereoselectivity was mostly determined under kinetic control and the syn-adduct was formed almost exclusively with 96% ee (entry 1). Although the conversion reached a plateau at nearly quantitative yield after 6 h (entry 2), the enantioselectivity decreased over time to 48% ee at 48 h, and the diastereoselectivity decreased to syn/anti = 11/1 (entries 3–6). Higher catalyst loading (3 mol%) led to a more drastic decrease in stereoselectivity and a virtually racemic product was quantitatively obtained with lower diastereoselectivity (entry 7). The proposed catalytic cycle is shown in Fig. 1. A mixture of three catalyst components in THF gave CuOAr/(S,S)-Ph-BPE complex 4, which formed an enolate from thioamide 1 as a soft Lewis acid/hard Brønsted base cooperative catalyst. Due to steric repulsion with amide substituents, Z-enolate 5 was likely formed and the subsequent aldol addition with aldehyde 2 through a six-membered transition state 6 afforded Cu-aldolate complex 7. Proton exchange with ArOH liberated the aldol adduct 3 and regenerated the initial catalyst complex 4. The Cu-aldolate 7 potentially served as a soft Lewis acid/hard Brønsted base cooperative catalyst to promote enolate formation to drive the catalytic cycle (blue arrow), because the reaction proceeded in the absence of ArOH. A higher reaction rate, however, was observed in the presence of ArOH, presumably because ArOH facilitated faster proton exchange and deprotonation of thioamide 1 from complex 4 due to lower steric demand. The entire process shown in Fig. 1 is reversible (black and red arrows). The sharp drop in stereoselectivity shown in Table 1 is indicative of the rapid retro-aldol process, in which the desired enantiomer of the syn-3aa isomer preferentially re-entered the catalytic cycle and reproduced the substrate mixture. The consistently high conversion indicated that the aldol adduct 3aa was thermodynamically favored over the substrate mixture 1a and 2a. The retro reaction, however, was sufficiently rapid compared to the forward reaction in this substrate set and the stereoselectivity steadily decreased over the course of the reaction. Although lowering the catalyst loading appeared to be the simplest option to attenuate the rapid decrease in enantioselectivity, the reaction with less than 1 mol% of catalyst had a significantly lower conversion. A catalyst loading of 1.5 mol% and reaction temperature of −70 °C were the best possible combination to control the reaction profile under facile operational reaction conditions, and it was difficult to quench the reaction at the appropriate stage to produce the aldol adduct 3aa in >90% yield and >90% ee.|
|
|||||
|---|---|---|---|---|---|
| Entry | x | Time (h) | Yieldb (%) | syn/antic | eed (%) (syn) |
| a 1a: 0.24 mmol, 2a: 0.2 mmol, ArOH = 2,2,5,7,8-pentamethyl-6-chromanol. b Determined by 1H NMR analysis of the crude mixture. c Determined by 1H NMR analysis of the crude mixture. d Determined by chiral stationary-phase HPLC analysis. | |||||
| 1 | 1.5 | 1 | 52 | >20/1 | 96 |
| 2 | 1.5 | 6 | 98 | >20/1 | 90 |
| 3 | 1.5 | 12 | 98 | >20/1 | 81 |
| 4 | 1.5 | 24 | 97 | 20/1 | 76 |
| 5 | 1.5 | 36 | >99 | 15/1 | 66 |
| 6 | 1.5 | 48 | 98 | 11/1 | 48 |
| 7 | 3 | 48 | >99 | 3.5/1 | 0 |

 | ||
| Fig. 1 Proposed catalytic cycle. Black arrows represent the forward pathway of the desired aldol reaction. Blue arrows represent a bypass route in which Cu(I)-aldolate 7 functioned as a catalyst. Red arrows represent retro-aldol pathways. | ||
Given the difficulties in reliably obtaining the aldol adduct with high stereochemical integrity due to the retro-aldol process,16 we envisioned that a dummy product that shares similar functional group architecture with the aldol adduct might competitively suppress re-entry of the product into the catalytic cycle (3 → 7 in Fig. 1). We designed dummy products 8 and 9 bearing a thioamide functionality and hydroxyl group linked through a two-carbon spacer as aldol adduct 3aa (Scheme 2a).17 To minimize the steric bias, methyl groups were selected as N-substituents for the thioamide and a hydroxyethyl group was linked to the thiocarbonyl unit for 8. For 9, a thiosalicylamide unit was adopted for the rigidity and acidity of the phenolic hydroxyl group to enhance the catalyst binding. The reaction of 1a and 2a was conducted with 1.5 mol% of catalyst in the presence of a two-fold excess of 9 relative to the catalyst (3 mol%), but the reaction was completely stopped and no product was obtained (Scheme 2b). This was likely due to the excessive binding ability of 9 to the Cu(I) complex, which arrested the overall catalysis. On the other hand, the reaction with 3 mol% of 8 under otherwise identical conditions proceeded to afford syn-3aa almost exclusively in 96% yield with 96% ee (Scheme 2b). Reaction profiles in the absence and presence of 8 clearly illustrate the beneficial effect of the additive (Fig. 2). As shown in Table 1, the retro-aldol process was sufficiently rapid even at the early stage of the reaction, and isolation of the aldol adduct 3aa in >95% with >95% ee was intractable. In stark contrast, both diastereo- and enantioselectivity were uniformly high during the steady progress of the aldol reaction with 8, providing a more reliable direct aldol protocol for substrates with which an extensive retro-aldol reaction was expected. Although additive 8 coordinated to Cu(I) complex 4 to slow down the forward reaction, the reaction rate was within an operationally acceptable level and the uniformity of the stereoselectivity was more beneficial to obtain the highly enantioenriched product. These results suggested that 8 would competitively suppress both the 3 → 7 and 4 → 5 processes (Fig. 1), and suppression of the former process was more influential in retarding the retro-aldol reaction.
 | ||
| Scheme 2 Design and effect of dummy product additives 8 and 9. | ||
 | ||
| Fig. 2 Reaction profile in the absence and presence of additive 8. Blue keys represent the result without 8; filled square: yield, open square: ee. Red keys represent the result with 8; filled circle: yield, open circle: ee. The numbers associated with the yield curves represent syn/anti ratio at the specified points. ArOH = 2,2,5,7,8-pentamethyl-6-chromanol. | ||
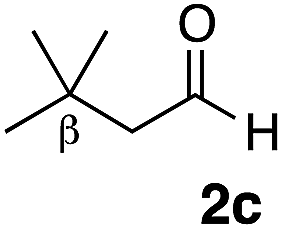
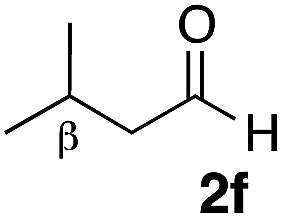
With the effective additive in hand, we investigated the generality of the revised aldol protocol (Table 2). Cyclohexanecarboxaldehyde (2b), an α-branched aldehyde, was also a noxious aldehyde in the reaction with thiopropanamide 1a. With 1 mol% of catalyst loading without 8, aldol adduct 3ab was obtained after 24 h in acceptable yield and stereoselectivity without a significant retro-aldol reaction (entries 1 and 2). Extending the reaction time (48 h) decreased the enantioselectivity (entry 3). Of particular note is that a slight increase in the catalyst loading to 1.5 mol% led to a substantially worse stereochemical outcome in the same reaction time (entry 4). The 0.5 mol% range of the catalyst loading could be offset by a marginal amount of impurities in the substrates, catalyst precursors, and solvents, making it difficult to consistently reproduce similar stereoselectivity without suppressing the retro-aldol reaction. With the aid of additive 8, the desired aldol adduct was reliably obtained with high stereoselectivity over a wide range of reaction times (entries 5–7). The aldol reaction of isobutyraldehyde (2a) and thioamide 1b derived from butyric acid was also a retro-prone combination and a beneficial effect of 8 was clearly observed (entries 8–11). It is noteworthy that the prominence of the retro-aldol process was observed not only for α-branched aldehydes, but also for α-non-branched aldehydes bearing a substituent at the β-position. 3,3-Dimethylbutanal (2c), having a quaternary stereogenic center at the β-position, also exhibited a time-dependent decrease in stereoselectivity under the standard 2nd-Gen catalyst conditions (entries 12 and 13). Although the reaction rate was retarded to some extent, high stereoselectivity remained uniformly high over the course of the reaction with the addition of 8, indicating that the product 3ac was prone to the retro-aldol reaction (entries 14 and 15). O-Functionalized aldehyde 2d with a sufficient steric bias at the β-position exhibited a similar tendency and a substantial additive effect was observed (entries 16–19). An extensive retro-aldol reaction was also evident with aldehyde 2e bearing ketal functionality at the β-position, indicating that the susceptibility to the retro-aldol reaction would be largely governed by steric factors (entries 20–23). Less sterically demanding isovaleraldehyde (2f), having a tertiary carbon at the β-position, also exhibited a retro-prone nature and additive 8 was effective for obtaining the aldol adduct with high stereochemical integrity (entries 24–27). In sharp contrast, an aldol reaction of aldehydes 2g–i having no branching substituents at either the α- or β-position afforded the desired aldol adducts with high stereoselectivity over a range of reaction times, irrespective of the use of 8 (entries 28–39), suggesting that the retro-reaction was sufficiently slow compared with the forward reaction. 4,4-Dimethylpentanal (2g), which is similar in structure to 2c but has substituents on the γ carbon, afforded the corresponding aldol adduct 3ag in uniformly high stereoselectivity with or without 8 (entries 28–31). A similar tendency was observed with aldehydes 2h–j bearing a trigonal aromatic sp2 carbon at the γ position and three bulky substituents on the Si atom at the γ or δ positions, and excellent stereoselectivity was observed, even under additive-free 2nd-Gen conditions (entries 32–43). The substructure of the thioamide was also responsible for the retro-aldol process as exemplified by the reaction with thioamide 1c derived from acetic acid (R2 = H) (entries 44 and 45). Whereas the combination of isobutyraldehyde (2a) and 1a underwent a substantial retro-aldol process, the reaction of 2a and 1c afforded the product 3ca with stable enantioselectivity, even in the absence of additive 8, indicating that the retro-aldol process did not occur in this case. Together, these findings suggested that the kinetic barrier for the retro-aldol reaction is closely related to the steric effects between the R1 and R2 substituents of the aldol adduct 3. When a significant retro-aldol reaction occurred, characterized by a time-dependent erosion of stereoselectivity, additive 8 effectively suppressed the retro-aldol reaction at the slight expense of the forward reaction rate. For the substrate sets in which the retro-aldol reaction was expected, the catalyst conditions with additive 8 offer a safer option for obtaining a highly enantioenriched product without laborious optimization of the reaction time and other reaction parameters.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 2 | 1 | x | y | t [h] | 3 | Yieldb [%] | syn/antic | eed [%] |
| R2 = | |||||||||
| a 1: 0.24 mmol, 2: 0.2 mmol, ArOH = 2,2,5,7,8-pentamethyl-6-chromanol. b Determined by 1H NMR analysis of the crude mixture. c Determined by 1H NMR analysis of the crude mixture. d ee of the syn diastereomer. Determined by chiral stationary-phase HPLC analysis. | |||||||||
| 1 |

|
Me 1a | 1 | 0 | 1 | 3ab | 66 | >20/1 | 92 |
| 2 | 1 | 0 | 24 | 89 | >20/1 | 90 | |||
| 3 | 1 | 0 | 48 | 95 | >20/1 | 72 | |||
| 4 | 1.5 | 0 | 48 | 97 | 3.3/1 | 0 | |||
| 5 | 1 | 2 | 1 | 40 | >20/1 | 96 | |||
| 6 | 1 | 2 | 24 | 94 | >20/1 | 94 | |||
| 7 | 1 | 2 | 48 | 96 | >20/1 | 95 | |||
| 8 |

|
Et 1b | 1.5 | 0 | 2 | 3ba | 62 | >20/1 | 95 |
| 9 | 1.5 | 0 | 48 | 97 | 12/1 | 79 | |||
| 10 | 1.5 | 3 | 2 | 26 | >20/1 | 97 | |||
| 11 | 1.5 | 3 | 48 | 96 | >20/1 | 94 | |||
| 12 |
|
Me 1a | 1.5 | 0 | 2 | 3ac | 56 | >20/1 | 89 |
| 13 | 1.5 | 0 | 48 | 98 | >20/1 | 80 | |||
| 14 | 1.5 | 3 | 2 | 10 | >20/1 | 92 | |||
| 15 | 1.5 | 3 | 48 | 94 | >20/1 | 90 | |||
| 16 |

|
Me 1a | 3 | 0 | 2 | 3ad | 96 | 10/1 | 81 |
| 17 | 3 | 0 | 48 | 99 | 3/1 | 20 | |||
| 18 | 3 | 6 | 2 | 22 | >20/1 | 93 | |||
| 19 | 3 | 6 | 48 | 96 | >20/1 | 89 | |||
| 20 |

|
Me 1a | 3 | 0 | 2 | 3ae | 89 | >20/1 | 92 |
| 21 | 3 | 0 | 48 | >99 | 13/1 | 85 | |||
| 22 | 3 | 3 | 2 | 69 | >20/1 | 93 | |||
| 23 | 3 | 3 | 48 | 97 | >20/1 | 93 | |||
| 24 |
|
Me 1a | 1.5 | 0 | 2 | 3af | 81 | >20/1 | 96 |
| 25 | 1.5 | 0 | 48 | 99 | 12/1 | 84 | |||
| 26 | 1.5 | 3 | 2 | 45 | >20/1 | 97 | |||
| 27 | 1.5 | 3 | 48 | 98 | >20/1 | 96 | |||
| 28 |

|
Me 1a | 7.5 | 0 | 2 | 3ag | 75 | >20/1 | 97 |
| 29 | 7.5 | 0 | 48 | 97 | >20/1 | 97 | |||
| 30 | 7.5 | 7.5 | 2 | 37 | >20/1 | 97 | |||
| 31 | 7.5 | 7.5 | 48 | 90 | >20/1 | 97 | |||
| 32 |

|
Me 1a | 3 | 0 | 2 | 3ah | 91 | >20/1 | 97 |
| 33 | 3 | 0 | 48 | >99 | >20/1 | 94 | |||
| 34 | 3 | 3 | 2 | 56 | >20/1 | 98 | |||
| 35 | 3 | 3 | 48 | >99 | >20/1 | 98 | |||
| 36 |

|
Me 1a | 5 | 0 | 2 | 3ai | 47 | >20/1 | 97 |
| 37 | 5 | 0 | 48 | 96 | >20/1 | 97 | |||
| 38 | 5 | 5 | 2 | 15 | >20/1 | 97 | |||
| 39 | 5 | 5 | 72 | 83 | >20/1 | 97 | |||
| 40 |

|
Me 1a | 3 | 0 | 2 | 3aj | 93 | >20/1 | 93 |
| 41 | 3 | 0 | 48 | >99 | >20/1 | 91 | |||
| 42 | 3 | 3 | 2 | 77 | >20/1 | 94 | |||
| 43 | 3 | 3 | 48 | 97 | >20/1 | 94 | |||
| 44 |

|
H 1c | 1.5 | 0 | 0.5 | 3ca | 24 | — | 89 |
| 45 | 1.5 | 0 | 24 | 81 | — | 89 | |||

The tendency toward the retro-aldol reaction based on the steric properties of the aldol adduct was further confirmed by a crossover experiment (Scheme 3). 3aa (94% ee) was a retro-prone aldol adduct and when subjected to the 2nd-Gen catalyst conditions in the presence of aldehyde 2j, 3aa readily underwent a retro-aldol reaction to provide isobutyraldehyde (2a) and thioamide 1a, which reacted with either 2a or 2j (Scheme 3a). At 24 h, the distribution of the aldol adducts was 40% of 3aa and 60% of 3aj, with 82% ee and 93% ee, respectively. The higher fraction of 3aj and high enantiopurity can be ascribed to the much slower retro-aldol process for 3aj. 3aa constantly underwent a rapid retro-aldol process and the mole fraction in the mixture was decreased with the decrease in the enantiopurity. After a prolonged aging time of 48 h, the mole fraction of 3aj reached 71% with a constant enantiopurity of 93% ee. The reluctant retro-aldol process was further evidenced by the analogous crossover experiment starting from 3aj (93% ee) and isobutyraldehyde (2a), which remained almost unchanged after 24 h under identical reaction conditions (Scheme 3b). Only a marginal amount of cross-over product 3aa was detected and the enantiopurity of 3aj remained the same.
 | ||
| Scheme 3 Crossover experiment using (a) retro-prone product 3aa and sterically α,β-nonbranched aldehyde 2j and (b) non-retro-prone product 3aj and α-branched aldehyde 3a. | ||
The catalytic conditions with additive 8 were also advantageous in a catalyst-controlled aldol reaction using a chiral aldehyde. The present direct aldol reaction preferentially afforded aldol adducts with all-syn configurations. Hence, the reaction of (S)-2k derived from L-lactic acid afforded all-syn (2R,3R,4S)-3ak exclusively in high yield with the catalyst prepared from (R,R)-Ph-BPE, irrespective of the presence or absence of additive 8 (Table 3).18 Notably, (S)-2k was configurationally stable under catalytic conditions; subjecting (S)-2k to the identical conditions without thioamide 1a afforded enantiomerically pure (S)-2k, providing clear evidence that no racemization (enolization) occurred. On the other hand, the combination of (S)-2k and the catalyst derived from (S,S)-Ph-BPE constituted a mismatched pair of chirality, leading to lower stereoselectivity (Table 4, entries 1–3). The desired diastereomer under catalyst-controlled stereoselection was (2S,3S,4S)-3ak, which was obtained as the major diastereomer,18 but a gradual decrease in stereoselectivity was observed in the absence of additive 8. This was presumably due to the inherently lower stereoselectivity and associated retro-aldol process, which was suggested by the beneficial effect of additive 8. The newly developed reaction conditions with additive 8 exhibited almost constant diastereoselectivity greater than 10/1 over 48 h (entries 4–6). Although the diastereoselectivity remained below appreciable levels, this observed diastereoselectivity was close to the highest predicted value of this mismatched pair under perfect kinetic control. Additive 8 competitively suppressed the retro-aldol reaction, but had no effect on the transition state energy for each isomer responsible for the inherent stereoselectivity. Another limitation of additive 8 is related to the equilibrium of the direct aldol reaction. The reaction of α-fully substituted aldehyde, e.g., pivalaldehyde (2l), and thioamide 1a gave aldol adduct 3al in less than 20% yield, irrespective of the use of 8, probably because this substrate mixture would be thermodynamically favored over the aldol adduct in this reaction (Scheme 4a and b). Racemic syn-3al, prepared using lithium diisopropylamide, readily underwent the retro-aldol reaction with the 2nd-Gen catalyst at −70 °C to give 84% of thioamide 1a, producing a similar substrate/product ratio that is an apparently equilibrated mixture (Scheme 4c).

|
|
|||||
|---|---|---|---|---|---|
| Entry | x | Time (h) | Yieldb (%) | Diastereomeric ratioc | |
| [(2S,3S,4S)-3ak/((2R,3R,4S)-3ak + others)] | |||||
| a 1a: 0.24 mmol, (S)-2k: 0.2 mmol, ArOH = 2,2,5,7,8-pentamethyl-6-chromanol. b Combined yield of diastereomers. Determined by 1H NMR analysis of the crude mixture. c (2R,3R,4S)-3ak was the second abundant diastereomer. | |||||
| 1 | 0 | 12 | 96 | 10.1/1 |

|
| 2 | 0 | 24 | 97 | 6.7/1 | |
| 3 | 0 | 48 | 99 | 6.7/1 | |
| 4 | 5 | 12 | 92 | 11.5/1 | |
| 5 | 5 | 24 | 91 | 10.1/1 | |
| 6 | 5 | 48 | 94 | 10.1/1 | |

 | ||
| Scheme 4 Forward and retro reaction using α-fully substituted aldehyde 2l. | ||
To quantify the tendency of the retro-aldol reaction, we performed a computational analysis of the aldol adducts. Although several nonbonding interactions need to be taken into account for precise discussion, bond length generally reflects the steric properties of the substituent on the atoms of interest and might provide clues to assess the steric effects observed in Table 2.19 Substituents on the nitrogen at the thioamide moiety of the aldol adduct 3 were simplified from N-diallyl to N-dimethyl to mitigate the computational load, and the structures of a series of simplified product 3′ were optimized using a 6-31G(d,p) basis set at the MP2 level of theory. Two C–C single bonds close to the R1 and R2 substituents were selected and the sum of their bond lengths L1 and L2 for each aldol adduct 3′ is plotted in Fig. 3.203al′, derived from pivalaldehyde (2l), had the largest value, likely due to the steric repulsion of the tert-butyl group. This value is an outlier in Fig. 3 (blue region) and consistent with the unfavourable equilibrium shown in Scheme 4. Products bearing α-branching substituents 3aa′, 3ab′, and 3ba′ gave shorter bond lengths than 3al′, followed by β-disubstituted (3ac′) and β-monosubstituted adducts (3af′). These are all retro-prone adducts and the sum of the bond lengths was larger (green region, except for 3af′) than that of the retro-free adducts. 3ag′ and 3ah′, having no substituents at the α and β positions, were free from retro-aldol process and shorter bond lengths were calculated. 3ca′ obtained from α-branching isobutyraldehyde (2a) and thioacetamide (R2 = H) was also a retro-free product due to smaller steric bias, and the sum of the bond lengths was similar to that of 3ag′ and 3ah′. Whereas a similar value of 3af′ and 3ag′ was unsatisfactory, this analysis based on the bond length can provide a determinant for predicting the likelihood of the retro-aldol reaction.
 | ||
| Fig. 3 Sum of the calculated bond lengths (L1 and L2) of the simplified (N-dimethyl) aldol adducts 3′. | ||
Conclusions
A significant additive effect to suppress the undesired retro reaction was revealed in the direct catalytic asymmetric aldol reaction of thioamide. The retro-aldol process significantly worsens the stereochemical outcome and is an important issue to be solved. The likelihood of the retro-aldol reaction is closely related to the steric factor, which was systematically investigated in both an experimental and computational context. A dummy product that shared the partial structure of the aldol-adduct effectively suppressed the retro-aldol process, presumably because the additive competitively bound to the catalyst to kinetically retard re-entry of the product into the retro-aldol cycle. The thus-developed improved direct aldol protocol provides a reliable catalytic system to afford aldol adducts from branched aldehydes with high stereochemical integrity.Acknowledgements
This work was financially supported by JST, ACT-C, and KAKENHI (No. 25713002) from JSPS. YB thanks JSPS for a postdoctoral fellowship for foreign researchers. Ms Yasuko Asada is gratefully acknowledged for her technical support on this project.Notes and references
- Seminal works on the direct aldol reaction, see: (a) Y. M. A. Yamada, N. Yoshikawa, H. Sasai and M. Shibasaki, Angew. Chem., 1997, 109, 1942–1944 ( Angew. Chem., Int. Ed. Engl. , 1997 , 36 , 1871–1873 ) CrossRef PubMed; (b) N. Yoshikawa, Y. M. A. Yamada, J. Das, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 4168–4178 CrossRef CAS; (c) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS; (d) B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003–12004 CrossRef CAS.
- For reviews of direct aldol reactions, see: (a) B. Alcaide and P. Almendros, Eur. J. Org. Chem., 2002, 1595–1601 CrossRef CAS; (b) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed; (c) Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Berlin, 2004 Search PubMed; (d) S. Mukherjee, W. J. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed; (e) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC; (f) S. M. Yliniemelä-Sipari and P. M. Pihko, in Science of Synthesis: Stereoselective Synthesis, ed. G. A. Molander, Thieme, Stuttgart, 2010, vol. 2, pp. 621–676 Search PubMed.
- Seminal work on the Mukaiyama-aldol reaction, see: T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 1011–1014 CrossRef CAS.
- For recent comprehensive reviews of Mukaiyama-aldol reactions, see: (a) G. L. Beutner and S. E. Denmark, Angew. Chem., Int. Ed., 2013, 52, 9086–9096 CrossRef CAS PubMed; (b) S. B. J. Kan, K. K.-H. Ng and I. Paterson, Angew. Chem., Int. Ed., 2013, 52, 9097–9108 CrossRef CAS PubMed; (c) J. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109–9118 CrossRef CAS PubMed . See also ref. 1c.
- For direct catalytic asymmetric aldol (-type) reactions using aldol donors in the carboxylic acid oxidation state without electron-withdrawing α-substituents: alkylnitriles: (a) Y. Suto, R. Tsuji, M. Kanai and M. Shibasaki, Org. Lett., 2005, 7, 375–377 CrossRef PubMed; Activated amides: (b) S. Saito and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 8704–8705 CrossRef CAS PubMed; β,γ-Unsaturated ester: (c) A. Yamaguchi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 10842–10843 CrossRef CAS PubMed; 5H-oxazol-4-ones: (d) T. Misaki, G. Takimoto and T. Sugimura, J. Am. Chem. Soc., 2010, 132, 6286–6287 CrossRef CAS PubMed; Direct catalytic asymmetric aldol reaction of thiazolidinethiones requiring the use of a stoichiometric amount of silylating reagent: (e) D. A. Evans, C. W. Downey and J. L. Hubbs, J. Am. Chem. Soc., 2003, 125, 8706–8707 CrossRef CAS PubMed.
- There are numerous examples of direct aldol reactions using aldol donors bearing electron-withdrawing α-substituents that are readily enolized under mild basic conditions. For a pioneering study using α-isocyanoacetates, see: Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405–6406 CrossRef CAS.
- For the use of thioamide as a pronucleophile in C–C bond-forming reactions under stoichiometric conditions, see: (a) Y. Tamaru, T. Harada, S. Nishi, M. Mizutani, T. Hioki and Z. Yoshida, J. Am. Chem. Soc., 1980, 102, 7806–7808 CrossRef CAS; (b) C. Goasdoue, N. Goasdoue, M. Gaudemar and M. Mladenova, J. Organomet. Chem., 1981, 208, 279–292 CrossRef CAS; (c) C. Goasdoue, N. Goasdoue and M. Gaudemar, Tetrahedron Lett., 1983, 24, 4001–4002 CrossRef CAS; (d) C. Goasdoue, N. Goasdoue and M. Gaudemar, J. Organomet. Chem., 1984, 263, 273–281 CrossRef CAS.
- For a pioneering study on the use of thioamide as pronucleophile in asymmetric reactions under stoichiometric conditions, see: N. Iwasawa, T. Yura and T. Mukaiyama, Tetrahedron, 1989, 45, 1197–1207 CrossRef CAS.
- (a) M. Iwata, R. Yazaki, Y. Suzuki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 18244–18245 CrossRef CAS PubMed; (b) M. Iwata, R. Yazaki, I.-H. Chen, D. Sureshkumar, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2011, 133, 5554–5560 CrossRef CAS PubMed; (c) D. Sureshkumar, Y. Kawato, M. Iwata, N. Kumagai and M. Shibasaki, Org. Lett., 2012, 14, 3108–3111 CrossRef CAS PubMed.
- Recent reviews of cooperative catalysis. Lewis acid/Brønsted base: (a) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed; (b) N. Kumagai and M. Shibasaki, Angew. Chem., 2011, 123, 4856–4868 ( Angew. Chem., Int. Ed. , 2011 , 50 , 4760–4772 ) CrossRef PubMed; Lewis acid/Lewis base: (c) M. Kanai, N. Kato, E. Ichikawa and M. Shibasaki, Synlett, 2005, 1491–1508 CrossRef CAS; (d) D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655–663 CrossRef CAS PubMed; Lewis acid/Brønsted acid and Lewis acid/Lewis acid (e) H. Yamamoto and K. Futatsugi, Angew. Chem., 2005, 117, 1958–1977 ( Angew. Chem., Int. Ed. , 2005 , 44 , 1924–1942 ) CrossRef PubMed; (f) H. Yamamoto and K. Futatsugi, in Acid Catalysis in Modern Organic Synthesis, ed. H. Yamamoto and K. Ishihara, Wiley-VCH, Weinheim, 2008 Search PubMed.
- For a general review of the transformation of thioamide functionality, see: T. S. Jagodziński, Chem. Rev., 2003, 103, 197–228 CrossRef PubMed.
- (a) M. Iwata, R. Yazaki, N. Kumagai and M. Shibasaki, Tetrahedron: Asymmetry, 2010, 21, 1688–1694 CrossRef CAS PubMed; (b) Y. Kawato, M. Iwata, R. Yazaki, N. Kumagai and M. Shibasaki, Tetrahedron, 2011, 67, 6539–6546 CrossRef CAS PubMed; (c) Y. Suzuki, M. Iwata, R. Yazaki, N. Kumagai and M. Shibasaki, J. Org. Chem., 2012, 77, 4496–4500 CrossRef CAS PubMed; (d) Y. Kawato, S. Chaudhary, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2013, 19, 3802–3806 CrossRef CAS PubMed; (e) A. Matsuzawa, C. Opie, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2014, 20, 68–71 CrossRef CAS PubMed.
- LiOAr must be prepared in a different flask and used immediately for a reliable reaction.
- For pioneering work on mesitylcopper, see: T. Tsuda, T. Yazawa, K. Watanabe, T. Fujii and T. Saegusa, J. Org. Chem., 1981, 46, 192–194 CrossRef CAS.
- For a comprehensive review of mesitylcopper, see: M. Stollenz and F. Meyer, Organometallics, 2012, 31, 7708–7727 CrossRef CAS.
- For representative cases of the organocatalyzed direct aldol reaction involving a significant retro-aldol reaction, see: (a) G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944–7947 CrossRef CAS PubMed; (b) G. Rulli, K. A. Fredriksen, N. Duangdee, T. Bonge-Hansen, A. Berkessel and H. Gröger, Synthesis, 2015, 45, 2512–2519 Search PubMed.
- For a similar strategy used for the direct aldol reaction of 7-azaindoline amide, see: K. Weidner, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2014, 53, 6150–6154 CrossRef CAS PubMed.
- Configuration of the aldol adduct was determined by derivatization. See ESI† for details.
- For a representative discussion of the C–C bond length and steric repulsion, see: (a) H. Kawai, T. Takeda, K. Fujiwara, T. Inabe and T. Suzuki, Cryst. Growth Des., 2005, 5, 2256–2260 CrossRef CAS; (b) P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef CAS PubMed.
- All the aldol adducts were oils, and direct comparison of the calculated bond lengths and crystallographic data was not conducted.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds. See DOI: 10.1039/c5sc02218e |
| This journal is © The Royal Society of Chemistry 2015 |