
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron chemistry in a new light
Guillaume
Duret
,
Robert
Quinlan
,
Philippe
Bisseret
* and
Nicolas
Blanchard
*
Laboratoire de Chimie Moléculaire, Université de Strasbourg, CNRS UMR 7509, ECPM, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: philippe.bisseret@unistra.fr; n.blanchard@unistra.fr
First published on 31st July 2015
Abstract
Photocatalysis has recently opened up new avenues for the generation of radical species under visible light irradiation conditions. A particularly fascinating class of photocatalyzed transformations relies on the activation of stable boron species with visible-light since it allows the creation of boryl and/or carbon radicals through single electron transfer or energy transfer without the need for specific and costly equipment. This new paradigm has found numerous applications in synthetic organic chemistry, catalysis, and macromolecular chemistry. In this minireview, the concepts underlying photoactivation of boron-species as well as applications to the creation of C–H, C–C, C–O, B–C and B–S bond are discussed.
1. Introduction
In the early days of photochemistry, Ciamician warned mankind about the long term environmental consequences of an eager exploitation of fossil fuels, notably for the organic chemical industry.1 He proposed to instead utilize sunlight as an energy source. Ciamician's dream remained for many years quite out of reach, being thwarted by the inability of the majority of organic compounds to absorb the photons of either the visible-light or the infra-red wavelengths that together constitute more than 95% of the solar spectrum. This situation has changed since the discovery in the 70's that tris-chelated ruthenium or iridium salts, typified by the intense red tris-2,2′-bipyridyl Burstall's complexes of ruthenium(II) such as Ru in Fig. 1 were able to absorb light in the visible region to give long-lived triplet photoexcited states endowed with both strong oxidizing or reducing properties depending on the conditions.2 In the presence of common sacrificial oxidants or reductants, these complexes turned out to be excellent photoredox catalysts able to enact single electron transfers under visible light irradiation. Although organic chemists were slow to exploit their potential, in particular compared to inorganic chemists, the situation has evolved radically since the seminal studies in 2008 and early 2009 by the groups of MacMillan,3 Yoon,4 and Stephenson5 that together put in the spotlight the productive merger of visible light and photocatalysts for the generation at room temperature of organic radicals without the need for specific and costly equipment. Their research served as a springboard and led to a rejuvenated interest in photocatalyzed organic transformations triggered by metal-based sensitizers or even purely organic dyes, such as those outlined in Fig. 1.6 The rapidly developing field of what is commonly referred to nowadays as visible-light photoredox catalysis has already impacted many areas of organic chemistry.6 In this review, the recent spinoffs of the method on boron chemistry are highlighted, first focussing the attention on the photogeneration of carbon radicals from organoboron derivatives (Section 2) and on the formation of boryl radicals mainly from N-heterocyclic carbene (NHC) boryl precursors (Section 3). Next, the recently developed photoredox-mediated oxidative hydroxylation of arylboronic acids (Section 4) will be discussed before considering visible-light-mediated processes aiming at utilizing as catalysts boron-dipyrromethene dye derivatives (BODIPYs) characterized with long-lived triplet excited states (Section 5). Finally, the most recent developments that have impacted visible-light organoboron chemistry will be exposed (Section 6).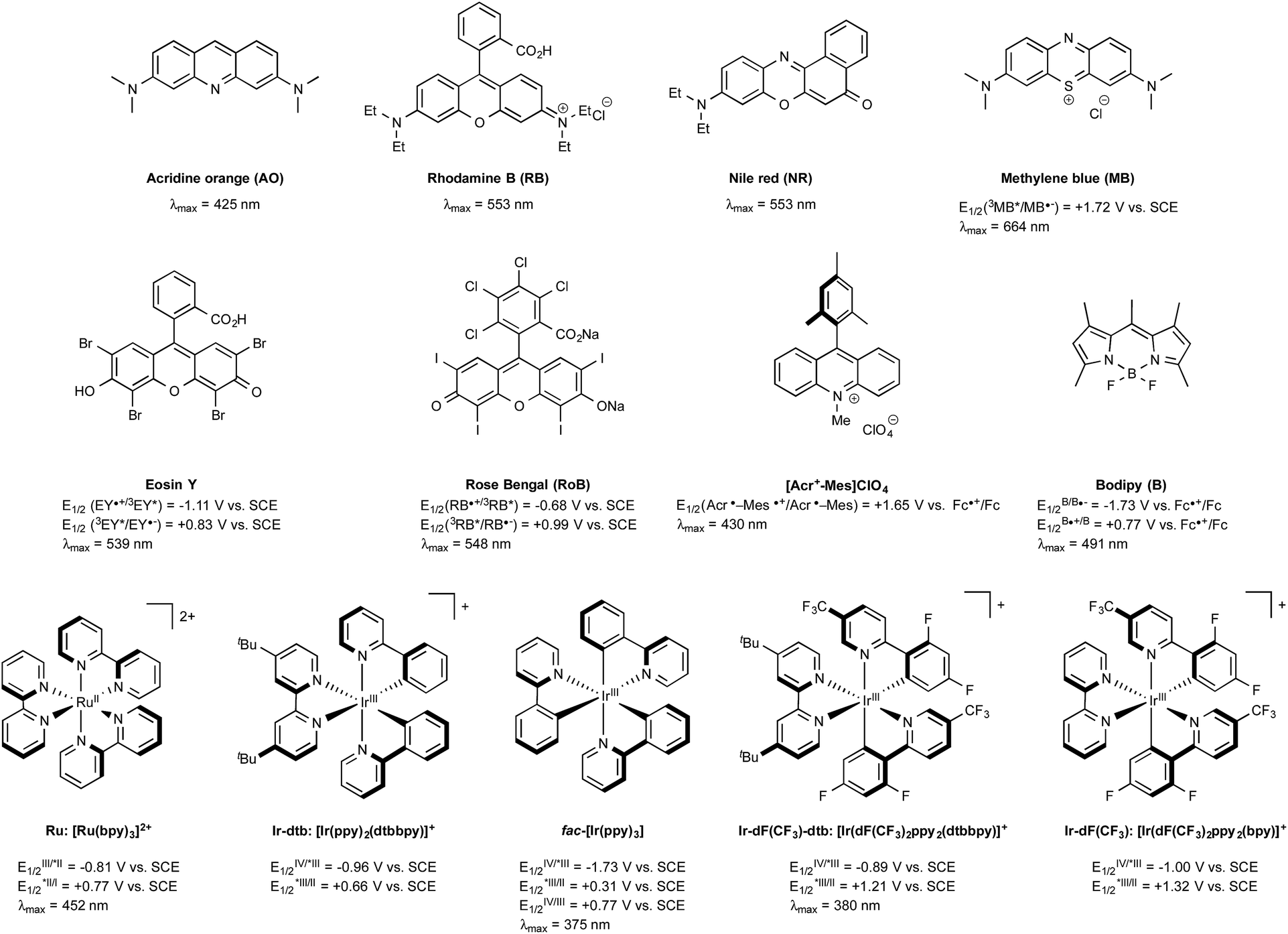 | ||
| Fig. 1 Selected examples of catalysts used in visible-light photoredox chemistry and their excited-state potentials. | ||
2. Visible-light-mediated photogeneration of carbon-centred radicals from organoboron derivatives
For already a few decades, organoboron derivatives have been employed as a convenient source of alkyl radicals in order notably to offer a green alternative to standard processes based on tin chemistry.Whilst most of the early studies dealt with the formation of alkyl radicals by autoxidation of trialkylboranes, even at low temperatures,7 further development spurred by the Renaud group incentive brought to the forefront the utilisation of B-alkylcatecholboranes as radical precursors (Scheme 1).8
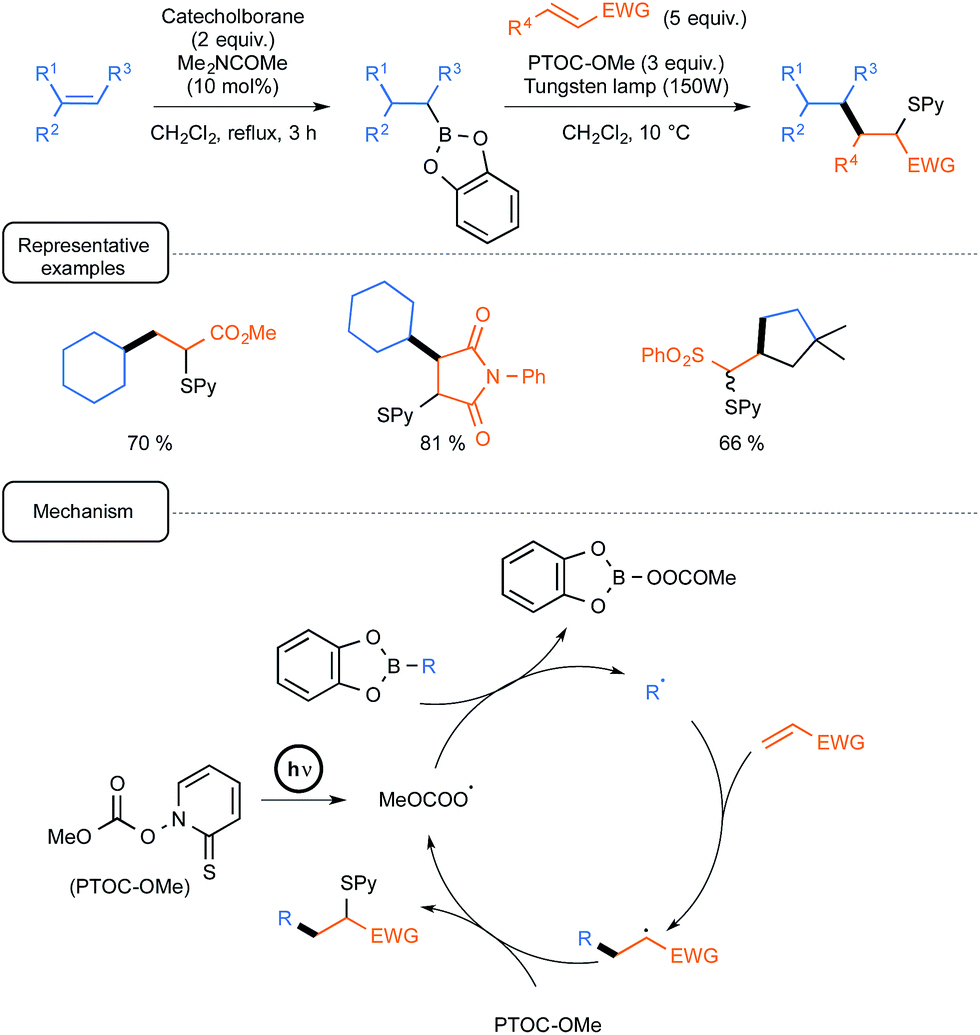 | ||
| Scheme 1 Conjugate addition of B-alkylcatecholboranes to olefins (EWG = electron withdrawing group). | ||
These derivatives, which could be easily prepared from alkenes via hydroboration with catecholborane, could be efficiently employed in conjugate additions to enones or enals, highlighting in the initiation step a homolytic substitution at boron with triplet oxygen.9 By contrast, under these conditions, the reaction could not be extended to other unsaturated partners such as ester or amide derivatives. This limitation was skilfully addressed, starting again from alkenes, by using a one-pot tandem sequence, conducted this time under the exclusion of oxygen and visible light irradiation, pinpointing the introduction in the condensation step of the Barton methylcarbonate PTOC-OMe {1-[(methoxycarbonyl)oxy]pyri-dine-2(1H)-thione} as a radical chain transfer reagent.10 As outlined in Scheme 1, the mechanistic pathway underpinning the radical transformation assigns a dual participation of the Barton carbonate. In the initiation step, the latter derivative would generate under simple irradiation by a tungsten lamp a methoxycarbonyloxyl radical (and/or eventually the corresponding decarboxylated methoxy radical) that would react by homolytic substitution with the organoboron to deliver the nucleophilic radical R˙. In the propagation step, this transient species would then add to the conjugated olefin to form a radical adduct that would react ultimately with the Barton carbonate to yield the S-pyridinylated product and regenerate the key-oxygenated radical. It should be noted in passing that this mechanistic scenario is inherently smartly regulated by differences in the properties of the three radical species involved, in term of polarity and/or affinity to boron(III), highlighting notably the lack of reactivity of carbon-centred radicals at boron, as opposed to oxygen-centred ones.11 As shown in Scheme 1, the sequence was quite efficiently amenable to a wide array of radical traps, such as unsaturated esters, amides and sulfones, not only in an intermolecular fashion but also in one case, in an intramolecular way from a dienic precursor. In a subsequent report, the same group further exploited the latter version, targeting precursors of bicyclic α-methylenelactones of biological interest.12
Further refinement of the method by the Renaud group includes a retooled hydroboration step based on the employment of a rhodium(I) catalyst, in the presence or not of a chiral ligand, enabling the preparation of enantiomerically enriched products and/or the chemoselective generation of radical from dienic precursors.13
Shortly after the seminal contribution of Renaud and co-workers, Dalko reported a closely related one-pot protocol, highlighting the utilization of the Barton phenylester PTOC-Ph in place of PTOC-OMe as well as the use of a 300 W halogen lamp as a source of visible light.14 Under such conditions, these authors extended the scope of the strategy to the utilization of unsaturated dialkylphosphonates as radicophilic partners.
It ought to be noted at this stage that, in spite of their performance, the tin-free procedures we just mentioned rely as a start on a hydroboration step and are thus restricted to the generation of alkyl radicals only. By contrast, organoboron derivatives such as trifluoroborate salts have recently emerged as powerful precursors not only for alkyl but also aryl radicals via oxidative carbon–boron cleavage.15
Up to three years ago, most of the oxidizing conditions described for this purpose included electrochemical methods and the use of stoichiometric or excess amount of oxidants such as copper salts or potassium persulfate.16 On rare occasions, the generation of radicals has also been performed using borates under photochemical conditions. If, as in most cases, irradiation under high-energy UV light was then required,17 cyanine borates such as n-butyltriphenylborate were found in contrast to be able to generate alkyl radicals, either in the solid state or in solution, simply by exposure to visible light.18 As outlined in Scheme 2, irradiation of such a borate with visible light at 532 nm resulted in efficient conversion to its short-lived excited singlet state, which led to the production of butyl and cyanine radicals that finally combine into the alkylated cyanine with concomitant bleaching of the cyanine dye.
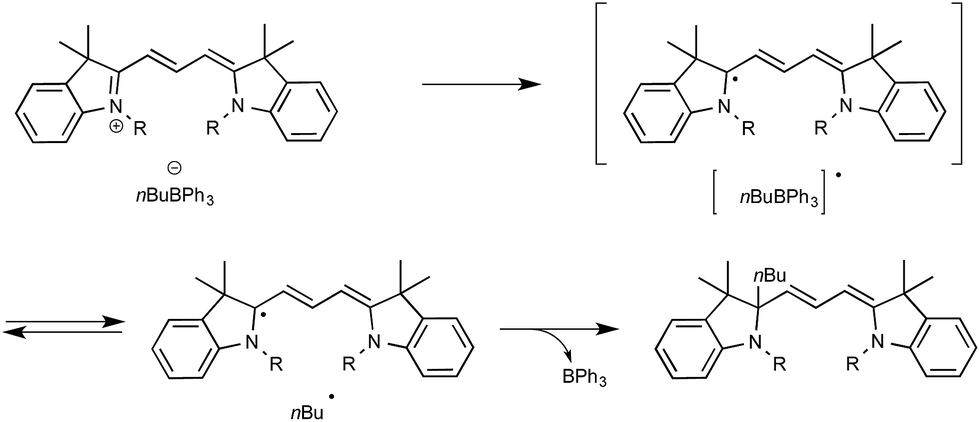 | ||
| Scheme 2 Photobleaching of cyanine borate. | ||
Evidence for the formation of cyanine and borate radicals in this process came not only from EPR studies but also from the observation of polymer formation when the irradiation was performed in acrylate or styrene-derived monomers solutions. In spite of their potential in radical chemistry and notably as photoinitiators for radical polymerization, cyanine borates have not received much attention since the initial forays from the groups of Filipovich and Schuster in the 80's. It is only in the past three years that the possibility of generating radicals from borates under visible light irradiation has come up again following the investigations of the Akita group. As a continuation of their research in visible-light-driven photoredox processes catalyzed by ruthenium or iridium-based complexes,19 these authors explored the possibility of oxidizing organoborates to generate alkyl, allyl, benzyl, and aryl radicals.20 The authors considered in a first set of experiments the Ir-dF(CF3)-catalyzed oxidative coupling of a range of trifluoroborates and triolborates, M[RB(OCH2)3CMe] with the archetypal radicophile 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO). As shown in Scheme 3, the photocatalytic C–O bond formation proceeded readily from benzylic and allylic trifluoroborate derivatives at room temperature using acetone as the solvent, and under continuous irradiation with cheap 425 nm blue LEDs. By contrast, the reaction failed with secondary and primary alkyltrifluoroborates, requiring instead the involvement of the more reactive triolborates. It is worth noting that the choice of the catalyst was essential here, as the utilisation of other complexes of lower oxidation potential at the photoexcited state, such as Ir-dF(CF3)-dtb or more markedly Ru, resulted in much less efficient conversions.
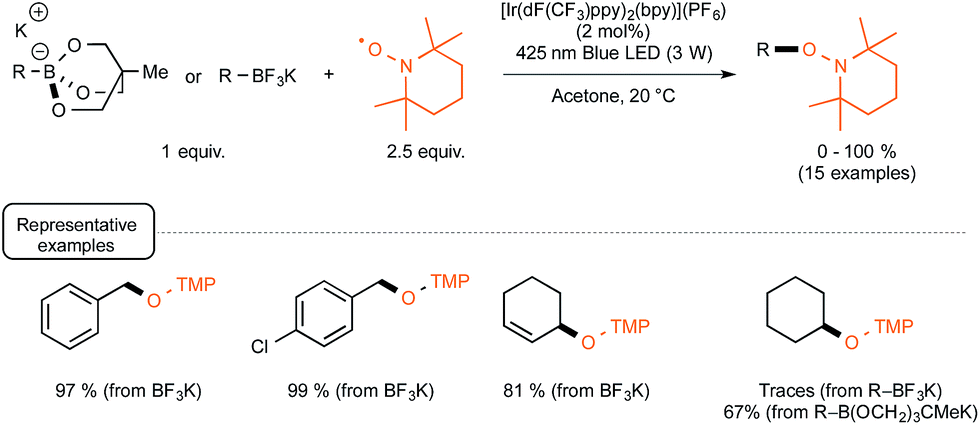 | ||
| Scheme 3 Photocatalytic C–O bond formation from borates and TEMPO. | ||
It is also worthy to note that the reactivity of aryl- or vinylborates was not considered in this transformation.
The postulated mechanism for the oxidative C–O bond formation involves, as outlined in Scheme 4, the initial excitation by visible light irradiation of the Ir(III) complex into its excited triplet state, which undergoes a single electron transfer from the organoborate, to yield the carbon-centred radical which ultimately combines with TEMPO. An excess of this latter derivative serves in addition as a sacrificial oxidant to regenerate the Ir(III) complex.
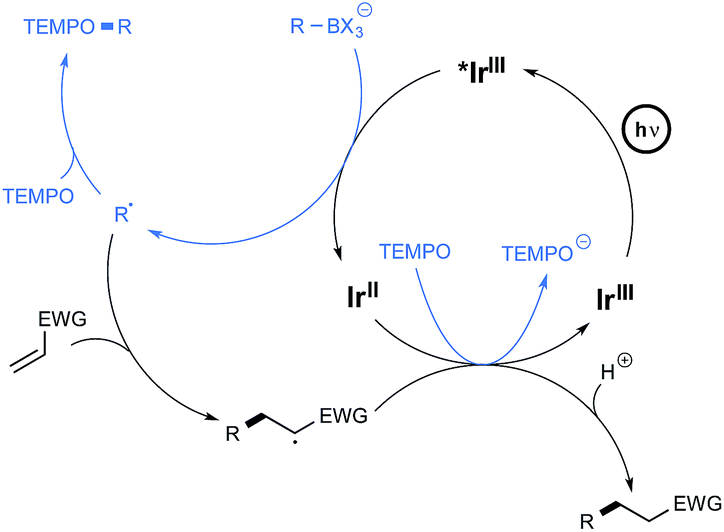 | ||
| Scheme 4 Postulated reaction mechanisms for the C–O and C–C bond formations. | ||
These promising preliminary experiments paved the way for the implementation of a series of C–C coupling experiments involving electron-deficient alkenes as the radicophilic partners and, once again, Ir-dF(CF3) as the photocatalyst. As highlighted in Scheme 5, under the conditions selected for the C–O couplings, a range of benzyl-, alkyl-, and even (hetero)aryl-borates could be used in the photocatalytic deboronative coupling experiments, although the yields remained modest with the latter borates, even as triol esters derivatives. Quite remarkably, in the reaction with a methylacrylate derivative equipped with a boronic acid ester moiety (Bpin), the coupling product with the preserved ester unit could be isolated in good yield, highlighting the selective reactivity of borates compared to boronic esters under Akita's photocatalytic conditions.
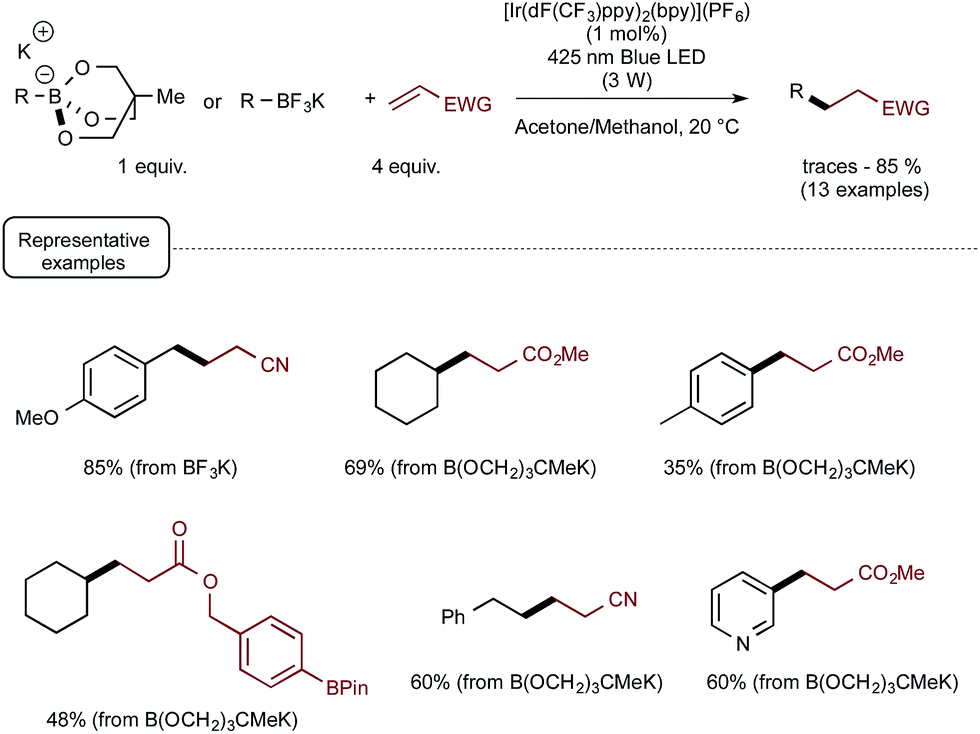 | ||
| Scheme 5 Photocatalytic C–C bond formation from borates and electron-deficient alkenes. | ||
The postulated redox-neutral mechanism for the C–C bond formation is outlined Scheme 4. The same pathway as for the C–O bond formation stands for the production of the carbon-centred radical from organoborates. This nucleophilic species then adds in a Michael pattern to the electron-deficient alkene to yield an alkyl radical which, itself, oxidizes the iridium complex back to its original Ir(III) form. The resulting carbanion is finally protonated by the solvent to generate the C–C coupling product.
Capitalizing on these encouraging results, the Akita group turned its attention to the use of trifluoroborate potassium salts to generate, for the first time by a photoredox process, alkoxymethyl-,21 and alkyl or aryl-thiomethyl radicals.22 They also studied in the same way the production of aminomethyl radicals from the corresponding trifluoroborates23 in order to offer an attractive alternative to the previously known methods to form aminoalkyl radicals via photoredox processes.24 As indicated in Scheme 6, a wide range of α-heteroatom methylations of electron-deficient alkenes could be quite successfully accomplished under the conditions previously reported for the photoredox C–C bond formations. They are in agreement with the postulated redox-neutral mechanism presented in Scheme 4.
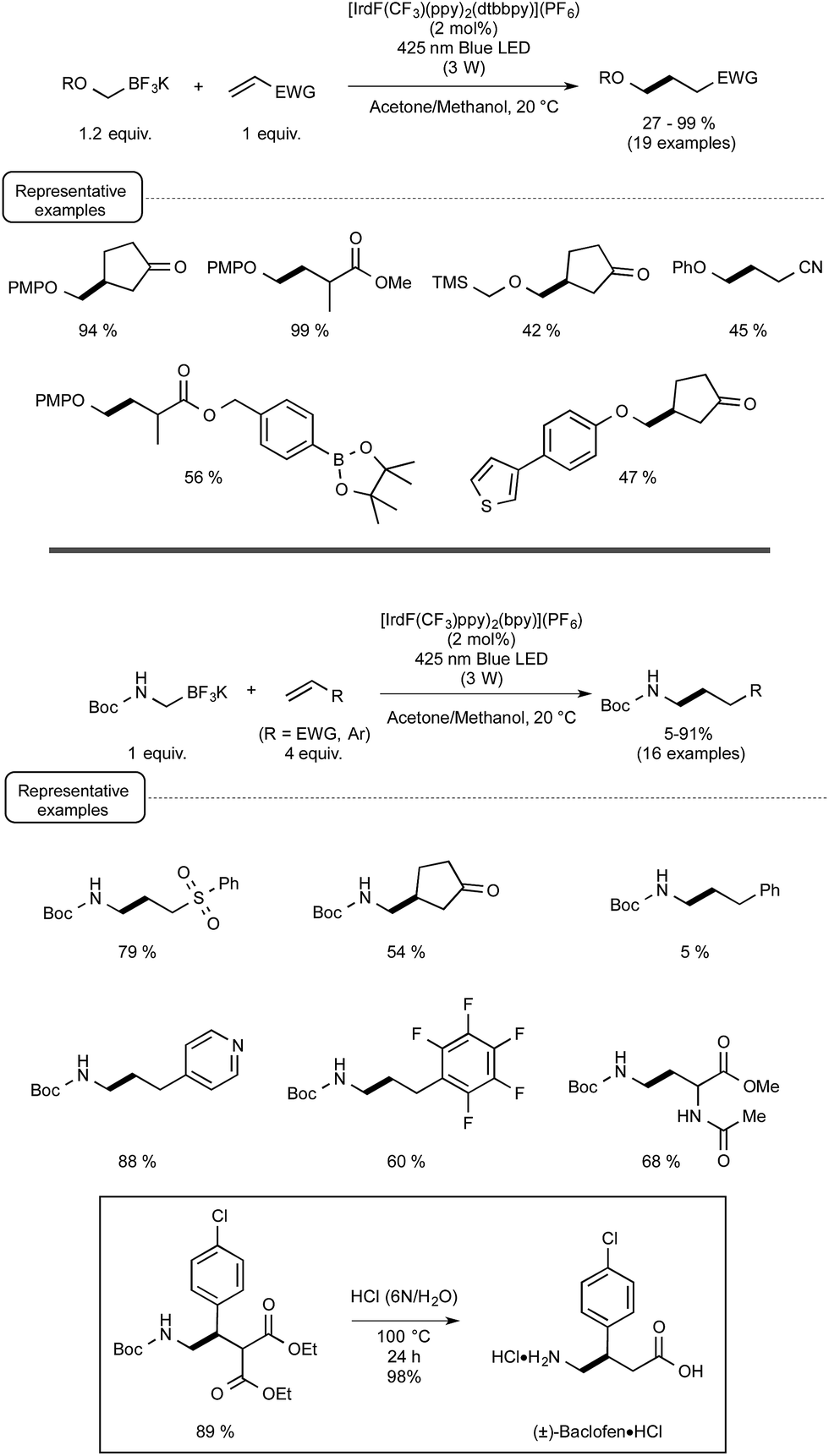 | ||
| Scheme 6 Redox-neutral α-heteroatom methylations of olefins (PMP: para-methoxyphenyl). | ||
More recently, the Akita group highlighted the utilization of 9-mesityl-10-methyl acridinium perchlorate25[Acr+-Mes]ClO4 in place of iridium-based photocatalyst for forging C–C bonds from potassium trifluoroborates and electron-deficient alkenes.26 As shown in Scheme 7, the organophotocatalytic protocol was amenable to a wide range of trifluoroborates encompassing notably secondary and tertiary alkyl derivatives that did not react satisfactorily with Ir-based photocatalysts of lower oxidation potential in their excited state. The postulated mechanism is related to the redox-neutral C–C bond formation outlined in Scheme 4, the organocatalyst in its photoexcited state serving as the oxidant in place of the Ir(III) photoexcited species.
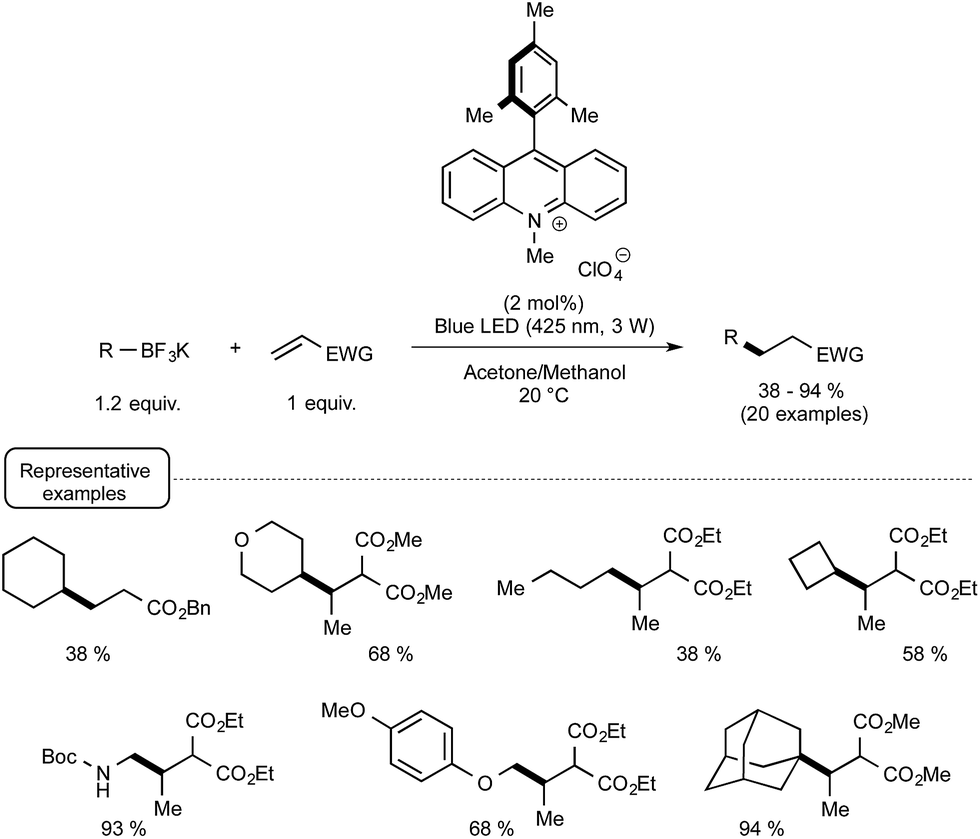 | ||
| Scheme 7 Methyl-acridinium-mediated photocatalytic C–C bond formation from potassium trifluoroborates and electron-deficient alkenes. | ||
In spite of its novelty, Akita's method of generating radicals under visible light irradiation has already attracted the attention of two research groups. Last year, the Chen group reported a redox-neutral deboronative alkynylation reaction compatible with a wide range of alkyl trifluoroborates and even boronic acids as coupling partners, which provides a remarkable alternative to the already known procedures to perform C(sp3)–C(sp) coupling reactions.27 After quite extensive scouting experiments run using potassium phenylethyltrifluoroborate in the presence of alkynes under visible light irradiation, making use of Ru as the photocatalyst, the optimized conditions outlined in Scheme 8 were found. The latter pinpointed the utilization of alkynylbenziodoxole (BI = benziodoxole) derivatives as well as the key-role of hydroxybenziodoxole (BI-OH) as a mild co-oxidant to mediate the reaction with suppression of side reactions. It was also found that trifluoroborates could be substituted by boronic acids with just little decrease in reaction yield.
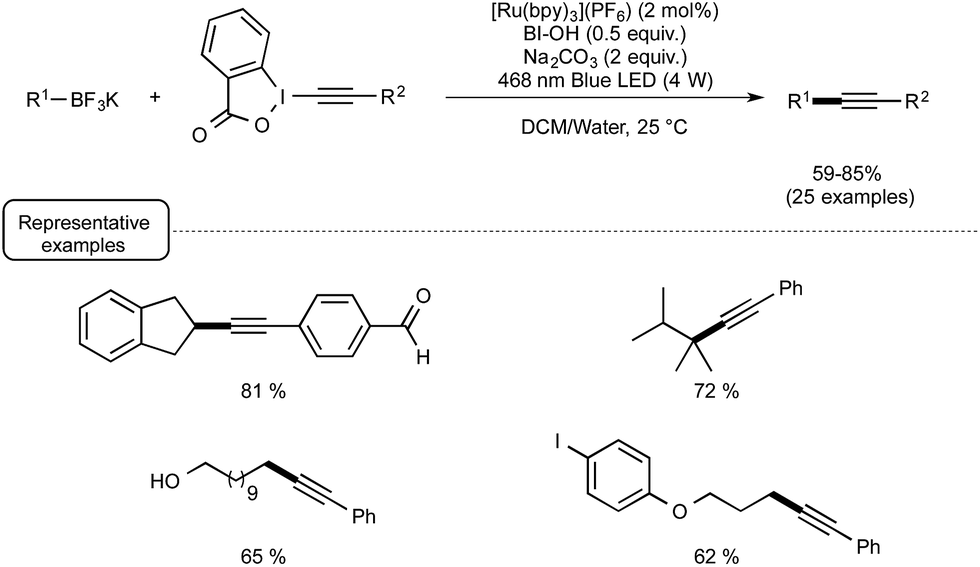 | ||
| Scheme 8 Visible-light-mediated deboronative alkynylation reaction. | ||
While no arylboron derivatives were considered as coupling partners here, the reaction performed on the whole quite well with primary, secondary, and tertiary alkyl compounds bearing a variety of substituents, such as a free hydroxyl group or a para-iodophenyl residue. In order to shed light on the mechanistic pathway of this unprecedented coupling reaction, a few control experiments were performed, notably by making use of a 13C-labeled BI-alkyne. As indicated in Scheme 9, the mechanistic proposal highlights the key-interaction of BI-OH with the photoexcited Ru(II) to generate Ru(III) for the reaction initiation. This complex of high oxidation potential would then be reductively quenched by the organoborate to yield the alkyl R radical. This radical would then perform α-addition to the BI-alkyne derivative generating the C(sp3)–C(sp) coupling product after elimination of BI radical.
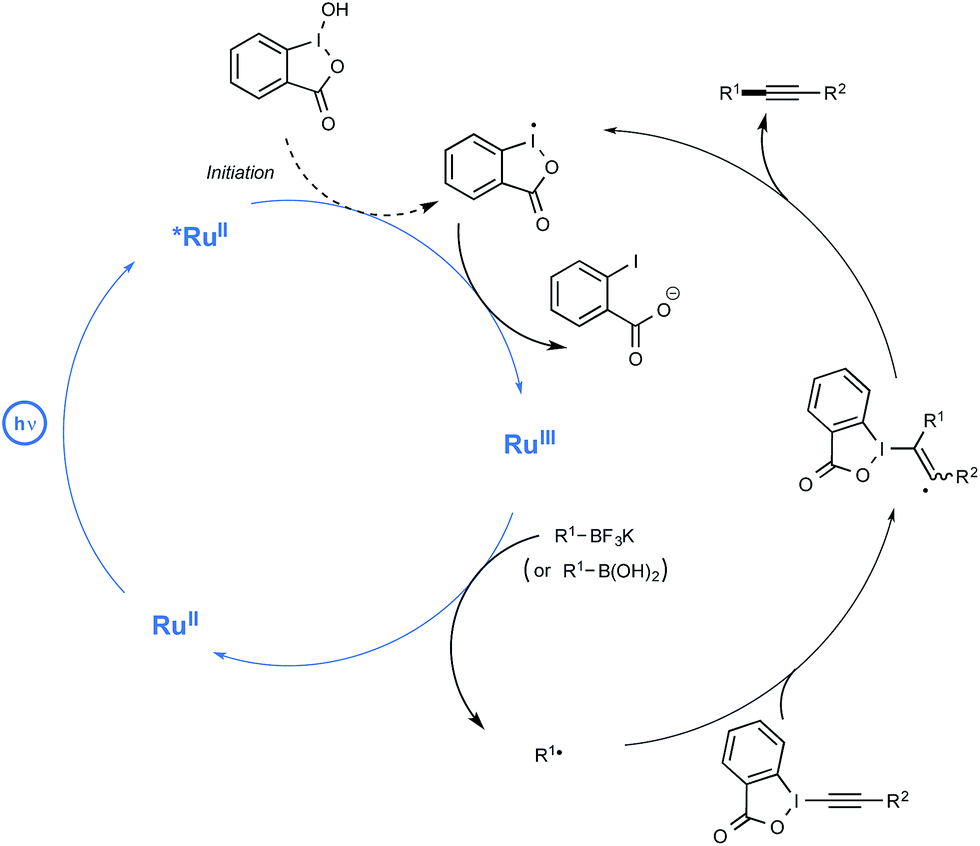 | ||
| Scheme 9 Mechanistic pathway of the deboronative alkynylation. | ||
Even more recently, the Chen group again afforded another demonstration of the proficiency of the Ru/BIOH combination to establish unprecedented visible-light-induced C–C coupling reactions.28 By choosing substituted acrylic acids as the coupling partner, they described a very mild C(sp3)–C(sp2) novel construction based on a deboronative/decarboxylative alkenylation process. As outlined in Scheme 10, the optimized reaction conditions were found to be very close to those selected for their previously described alkynylation reaction. The most significant variation stemmed for the utilisation of 1.5 equivalents of BI-OAc in place of only 0.5 equivalent of BIOH. The reaction conditions were found to tolerate the presence of a variety of functional groups on both coupling partners, such as bromide, keto, and esters functions.
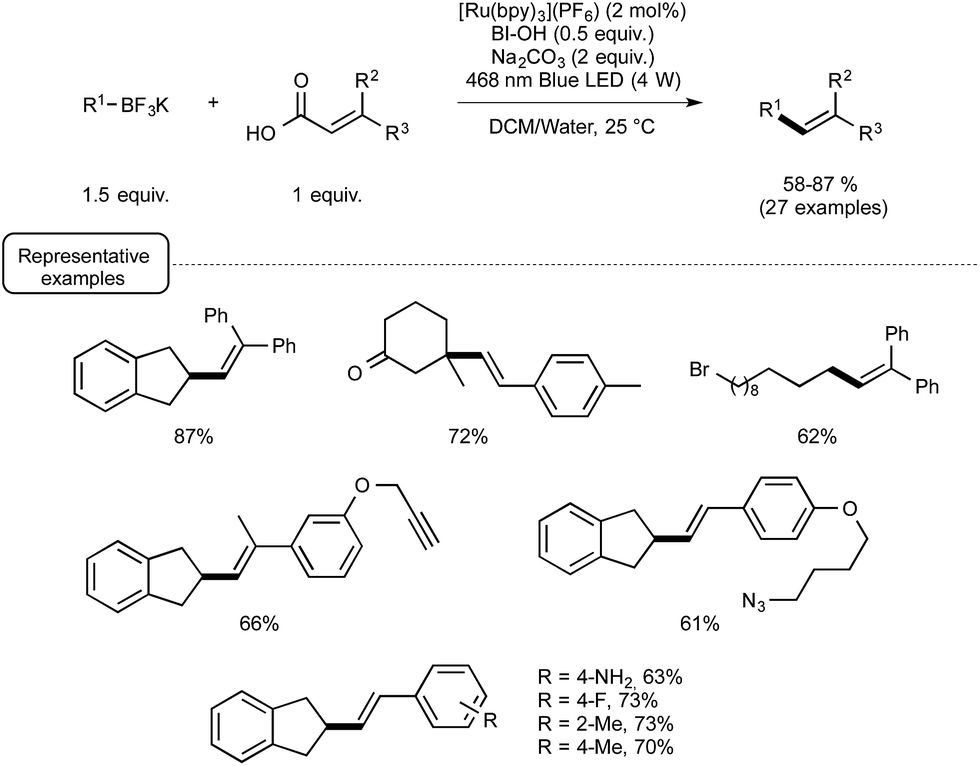 | ||
| Scheme 10 Deboronative/decarboxylative alkenylation reaction. | ||
In order to decipher the mechanism of this new reaction, a set of experiments were performed which assigned a pivotal role to BIOAc as an oxidant able not only to transform the Ru(II) photoexcited state into the corresponding Ru(III), but also to convert the starting carboxylic acid into the key benziodoxole intermediate. The authors even managed to isolate on one occasion the latter intermediate, which was characterized by X-ray crystallography. As shown in Scheme 11, this intermediate would serve in part as the reaction initiator being able to convert the photoexcited Ru(II) complex into its Ru(III) oxidation state. This would oxidize then the borate, generating thus the alkyl radical R. This radical would add subsequently to the sp2 carbon atom of the α,β-unsaturated carbonyl to give an adduct that would undergo a decarboxylation step to release the BI radical and yield the alkene product.
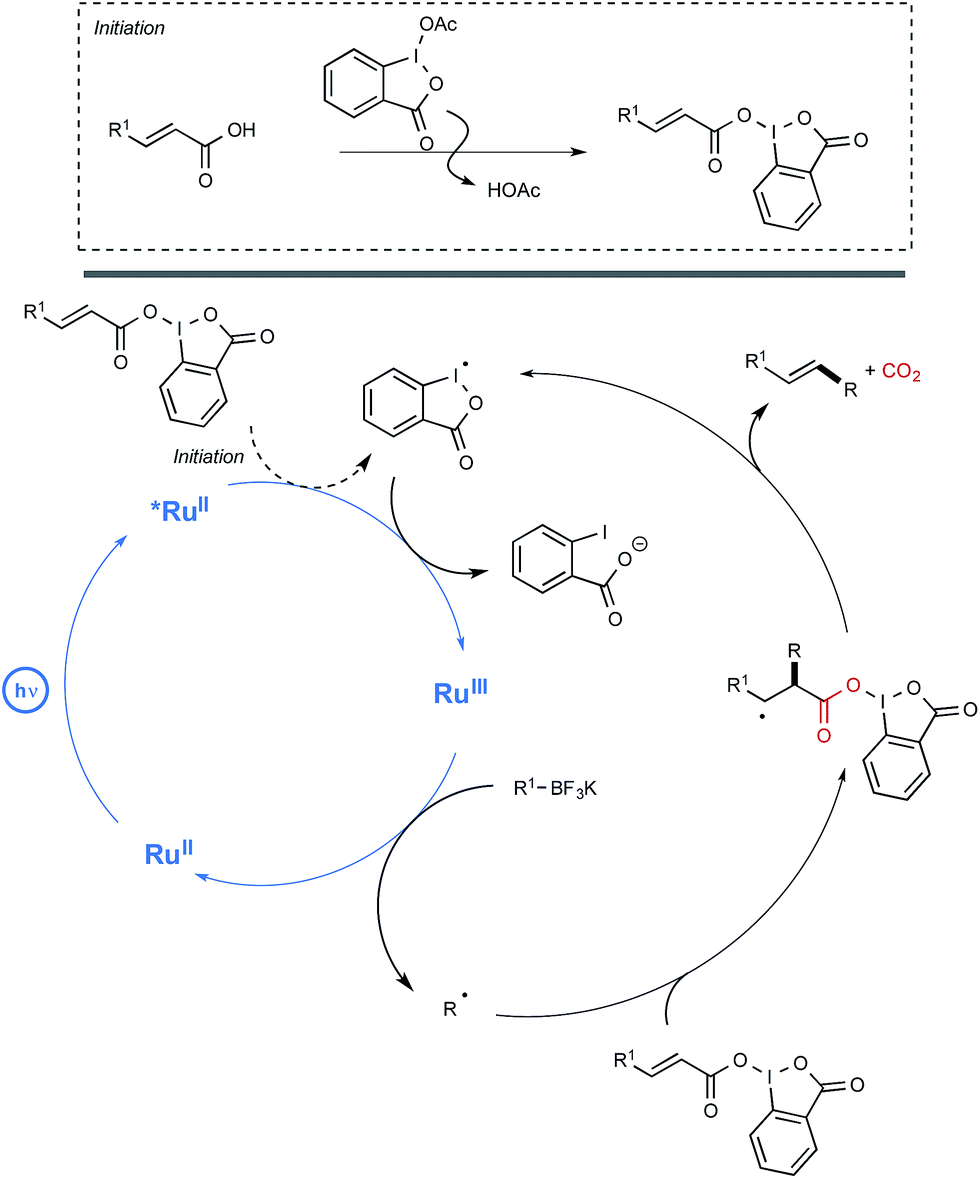 | ||
| Scheme 11 Mechanistic pathway accounting for the deboronative/decarboxylative alkenylation reaction. | ||
The Molander group meanwhile tackled the possibility of forging Csp3–Csp2 bonds, under a visible-light-driven process.29 In using aryl bromide and alkyltrifluoroborate derivatives, they hope to apply the newly developed paradigm of dual catalysis based on the productive merger of two different transition metals, each in charge of a catalytic cycle.30 Their aim was to offer an alternative to the conventional transmetalation step in Suzuki-type reactions, which is often quite challenging notably in the case of sterically hindered Csp3-hybridized organoboron electrophiles, as a result of its inherent two-electron nature. The insightful strategy described by the authors to address this limitation is shown in Scheme 12 and highlights the utilisation of an iridium photoredox catalyst in tandem with a nickel catalyst in order to enact a transmetalation pathway based on single-electron transfer chemistry. It was anticipated that the alkyl radical arising from the iridium-based oxidative deboronation of the starting alkylborate would combine to Ni(0) to yield a Ni(I) species (Scheme 12, red pathway). Oxidative addition of the starting arylbromide to the latter would generate a high-valent Ni(III) intermediate, which was expected to deliver the desired cross-coupled product after a reductive elimination step. It is worthy to note that an alternative scenario where oxidative addition at Ni(0) precedes radical combination is also a possibility (Scheme 12, orange pathway), but both pathways converge on common Ni(III) intermediate.29c
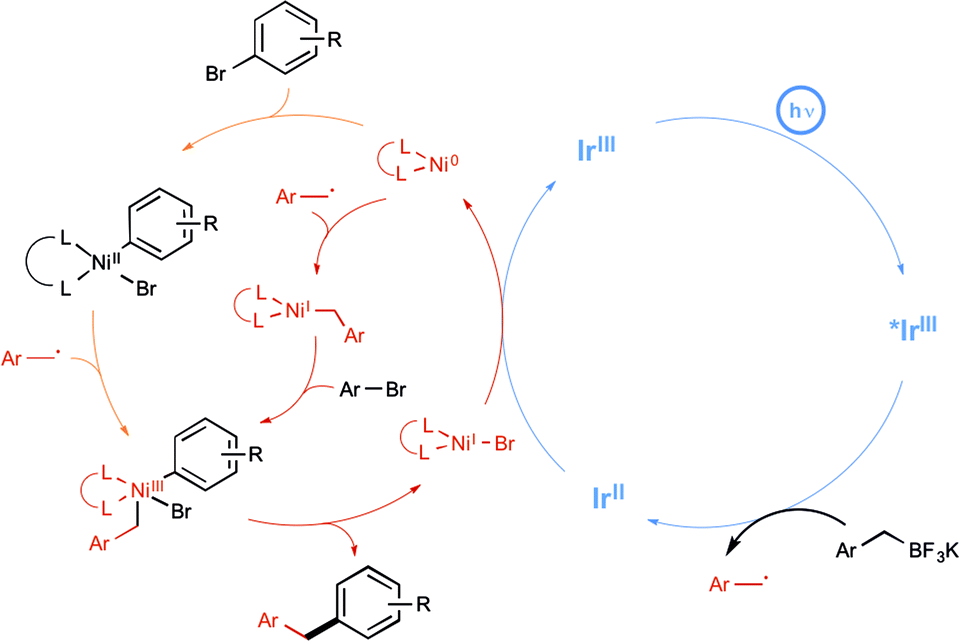 | ||
| Scheme 12 Hypothetical mechanism of the photoredox/nickel cross-coupling. | ||
As a proof of concept, the dual catalytic approach was first applied to benzylic trifluoroborates, known to be easily oxidized by the visible-light-induced excited state of Ir-dF(CF3).29a As shown in Scheme 13, by making use of Ni(COD)2 as the catalyst and 2,6-lutidine as an additive, the reaction proceeded quite well at room temperature and a high level of functional group tolerance was observed. The reaction manifold was in particular found compatible with functional groups potentially reactive towards the radical intermediates.
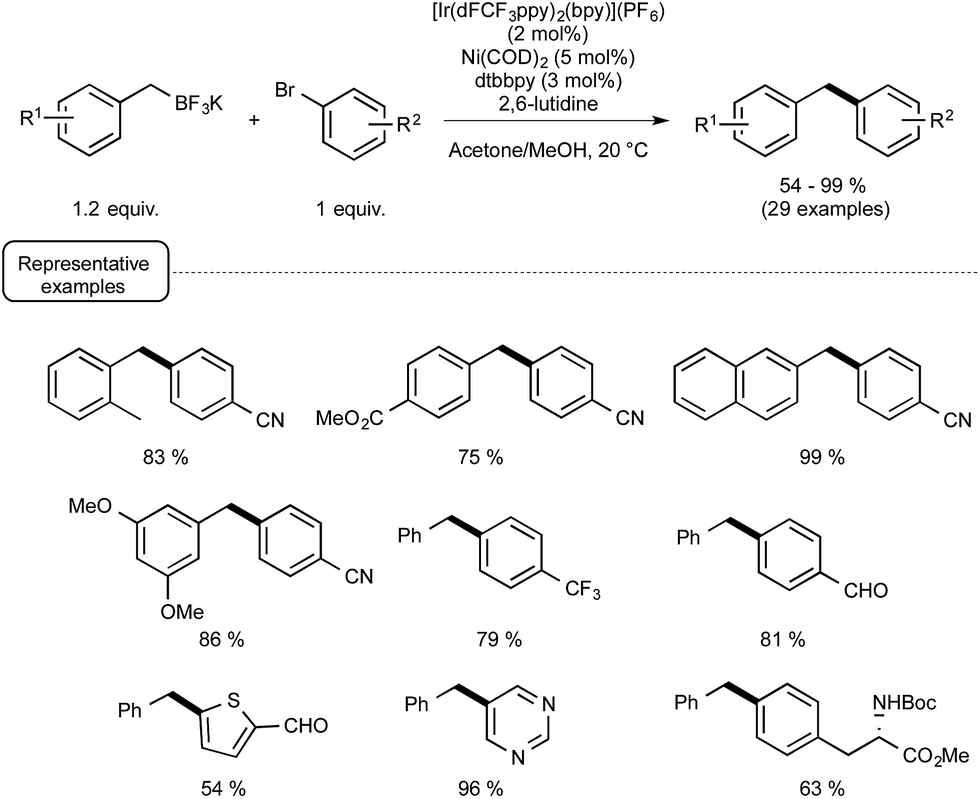 | ||
| Scheme 13 Photoredox cross-coupling of benzylic trifluoroborates and aryl bromides. | ||
Although they were found quite reluctant to the Ir-dF(CF3) catalyzed oxidation conditions under Akita's seminal experiments, secondary alkyl trifluoroborates were then considered in place of benzylic derivatives.29b As outlined in Scheme 14, by making use of reacting conditions closely derived from those employed for benzylic boron derivatives, the expected cross-coupling products could be obtained generally in very good yield, highlighting the great potential of the synergistic dual catalysis strategy.
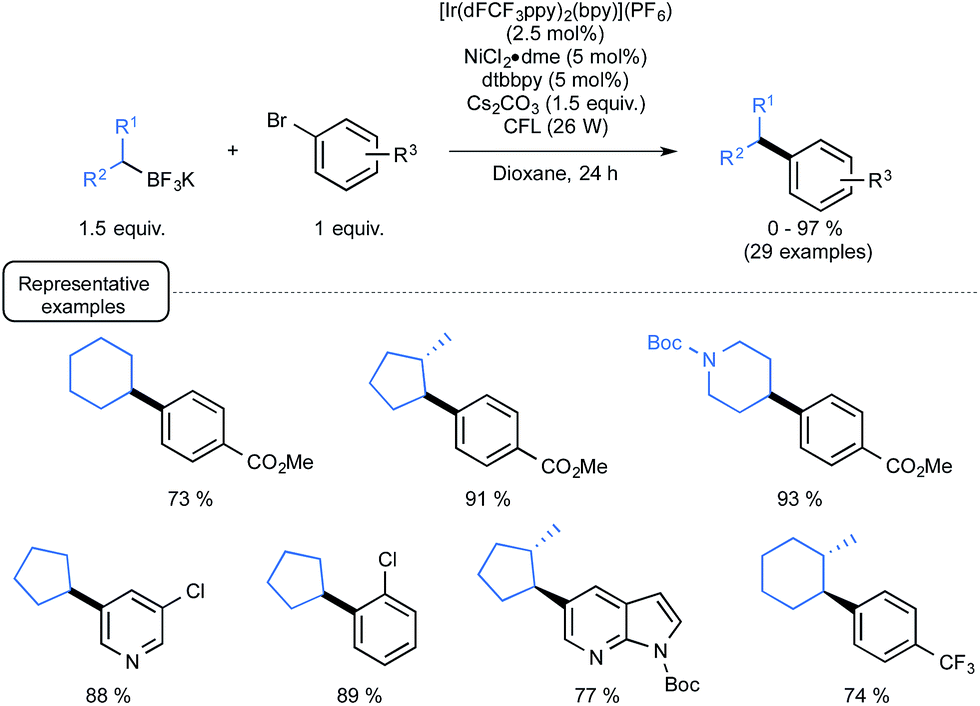 | ||
| Scheme 14 Photoredox cross-coupling of secondary alkyl trifluoroborates and aryl bromides. | ||
3. Visible-light-mediated formation of boryl radicals
Let us now turn attention to the recent research that highlights the formation of boryl radicals from stable boranes, such as NHC– or pyrazole–borane complexes, with the assistance of sunlight or visible light. Five years ago, the groups of Curran, Lacôte and Malacria reported the generation under the control of a sunlamp of boryl radicals from a palette of NHC–borane complexes, highlighting the use of a nonyl radical as a hydrogen atom abstractor.31 Their aim was to evaluate the rate constants of hydrogen abstraction in these complexes in order to compare them with those reported for well-known hydrogen atom donors such as tin or silyl-hydride derivatives. As shown in Scheme 15, the transient nonyl radical could be easily obtained by a Barton-type decarboxylation from the corresponding PTOC ester under sunlamp irradiation at room temperature. This nonyl radical subsequently abstracted a hydrogen atom from the NHC–borane, thus leading to the NHC-boryl radical, which finally added back in a propagation step to the starting PTOC ester with formation of NHC–BH2Py and regeneration of the alkyl radical.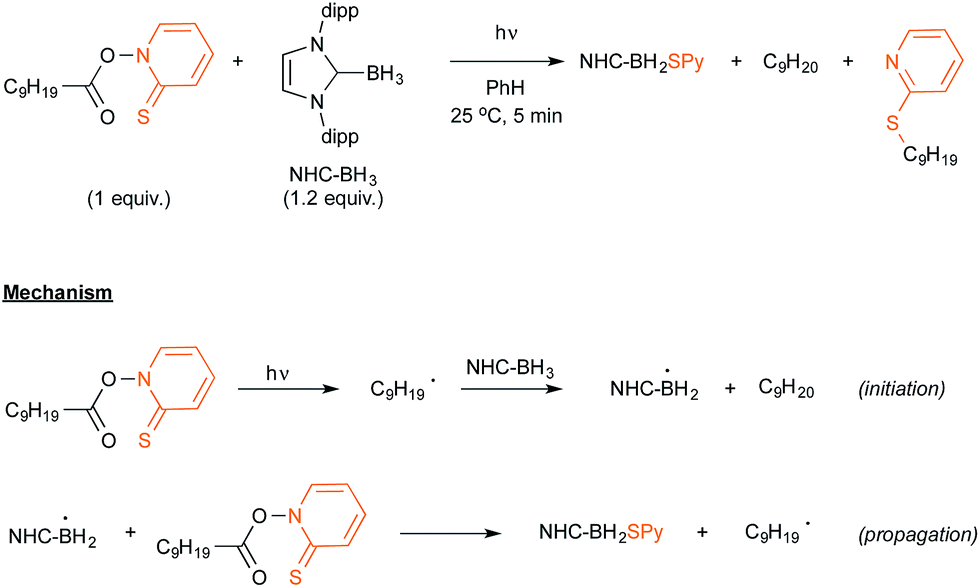 | ||
| Scheme 15 Barton-type decarboxylative experiment with dipp-Imd-BH3 as a hydrogen donor. | ||
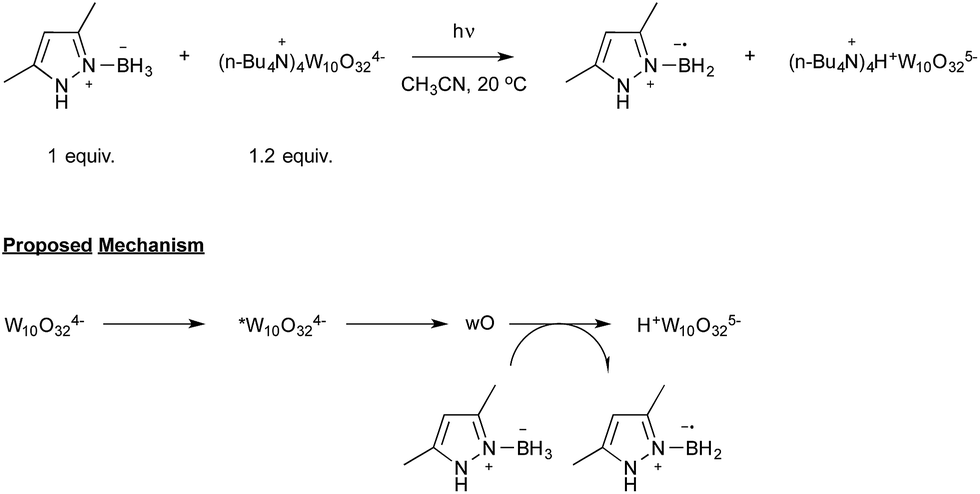
Aiming at discovering new initiating systems operative under the control of visible light irradiation, Lalevée reported that polyoxometalates32 were suitable photocatalysts for the cationic ring opening photopolymerization of epoxides.33 Tetrabutylammonium decatungstate (nBu4N+)4 W10O324− in combination with silane or ligated borane derivatives were selected as a source of respectively silyl and boryl radicals, that could be further oxidised to the corresponding cationic species. By selecting the easily prepared 3,5-dimethylpyrazole-borane, Blanchard and Lalevée reported the first generation of a boryl radical under visible light irradiation (Scheme 16).33 The experiment run at room temperature in acetonitrile as the solvent yielded the typical blue H+W10O325− reduced form of the polytungstate, accounting for the formation of the pyrazolyl-boryl radical. The formation of this radical was also confirmed through EPR experiments.
 | ||
| Scheme 16 Visible-light-mediated formation of 3,5-dimethylpyrazole-boryl radical. | ||
The proposed mechanism, as shown in Scheme 16, relies on the known photochemistry of polytungstate salts.32 Illumination of the tetra-anion would lead to a short-lived excited state that rapidly decays to a longer-lived transient, which has been referred to as wO. The latter would finally oxidize the boryl complex to yield the boryl radical as well as H+W10O325−.
Along the same lines, Lalevée explored the radical polymerisation of benchmark acrylate monomers, by utilizing initiation systems including NHC-boryl radicals generated by hydrogen atom abstraction from the parent NHC complexes under visible light irradiation.34 The aim was to first of all offer a mild, cheap and safe alternative to the UV light sources that had so far been utilized for that purpose, which required expensive photochemical equipment.35
Furthermore, as it has been recognized that visible light penetrates thick polymerization samples better than the more energetic UV light,36 it was thought that this process could improve the efficiency of polymer synthesis itself.
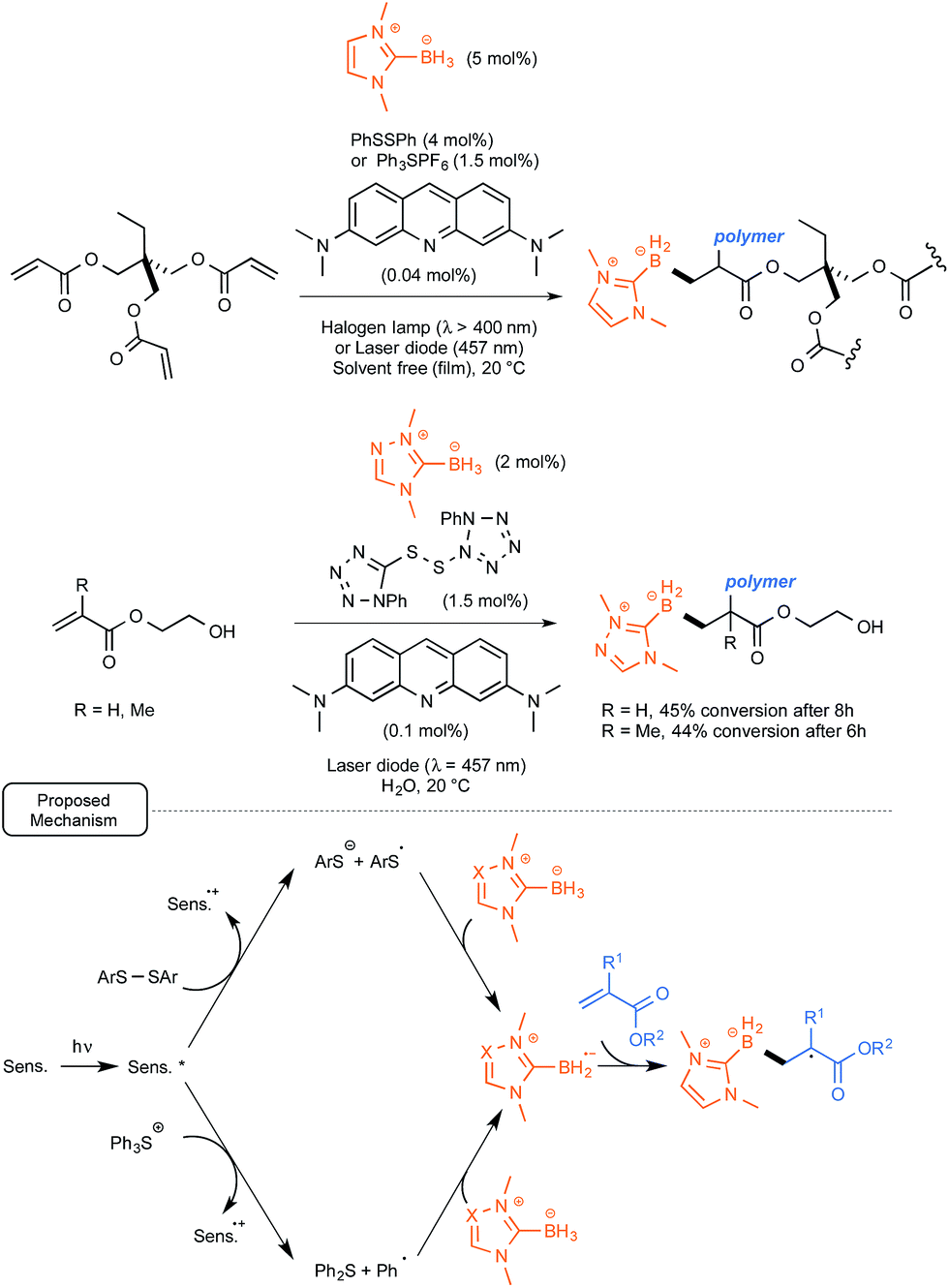
Rather than relying on the utilization of polyoxometalates, the Lalevée and Lacôte groups took advantage of their recent findings pinpointing the great hydrogen-atom-abstracting potential of thiyl radicals in the case of NHC-boryl complexes.37 As these sulfur radicals had been obtained upon UV irradiation of the corresponding disulfides, the possibility of introducing into the initiation system acridine orange as a sensitizer, in order to help and trigger the cleavage of the disulfide bond under less energetic conditions, was explored. Using a second strategy, the H-abstracting potential of phenyl radicals generated from the dye-sensitized reductive cleavage of triphenylsulfonium salts under visible light irradiation was tested, as originally proposed by Kampmeier and co-workers.38
As outlined in Scheme 17, both methods were tested in the photopolymerization of the archetypal monomer trimethylolpropane triacrylate. They proved to be quite rewarding, offering under solvent-free conditions at room temperature up to 50% polymerization after 3 min only of irradiation with a household halogen lamp (λ > 400 nm). As presented in Scheme 17, the scope of the method was extended to polymerization in water of hydroxymethyl- and methyl-acrylates, highlighting the use of a water-soluble triazolo-NHC–borane as well as a polar tetrazolo-disulfide derivative. For these conversions, the mechanism depicted in Scheme 17 was proposed. It highlights an electron transfer from the dye to either the disulfide derivative or the triphenylsulfonium salt in order to generate respectively the arylsulfide or phenyl radicals. These would be both endowed with the facilities to remove a hydrogen atom from the NHC–borane complexes.
 | ||
| Scheme 17 (a) Visible-light-driven polymerization of trimethylolpropane triacrylate; (b) visible-light-mediated polymerization in water of hydroxymethyl- and methyl-acrylates; (c) plausible mechanism of the NHC-boryl-assisted photopolymerization. | ||
In addition to the interest in NHC-boryl derivatives as a source of radicals for initiating polymerizations, the groups of Curran, Lalevée and Lacôte have also been intrigued by their potential as reducing agents of both organohalides and diaryldisulfides. Let us first consider the work devoted to the reduction of the latter compounds and which led to the synthesis of the first NHC-boryl mono- and disulfide derivatives.39
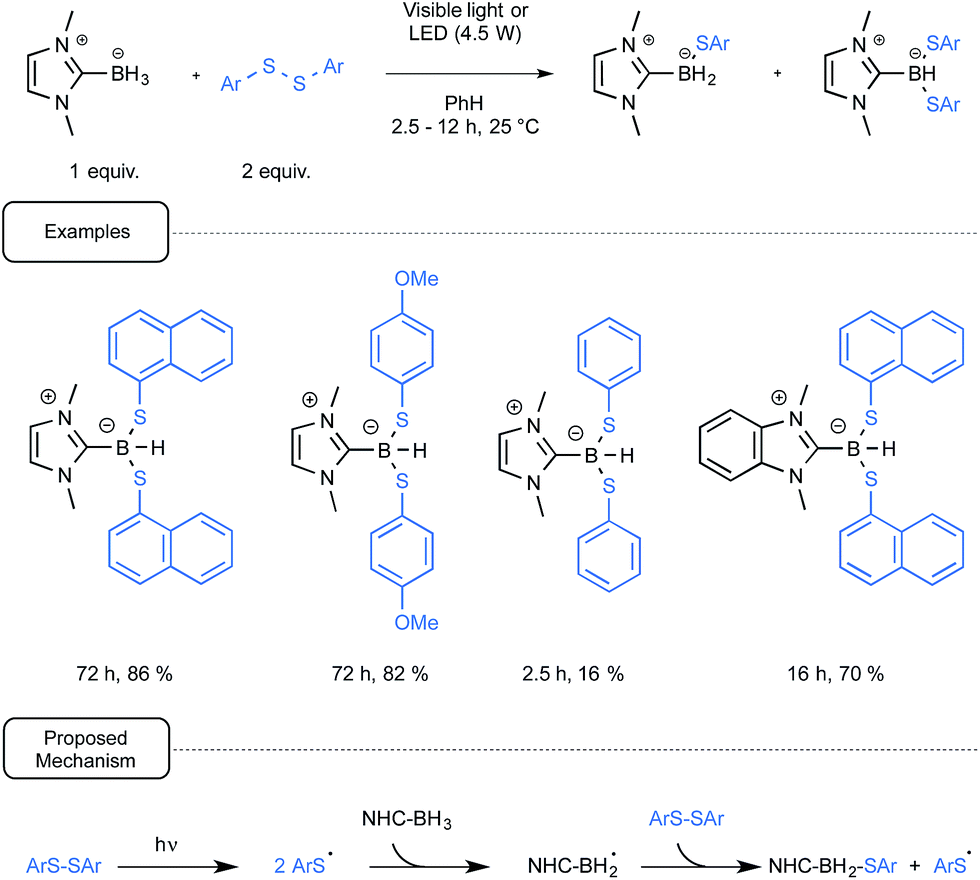
Gratifyingly, it was observed that in most cases the reduction of diaryldisulfides could be amenable at room temperature by using benzene as the solvent, simply by combining them with the NHC-boryl partner. The reaction performed at best under irradiation conditions, being quite slow when run in the dark. It was not necessary in that case to utilize UV-containing light sources as, under the control of simple household lamps, the reaction proceeded on the whole nicely, giving very good yields of the desired mixture of NHC boryl di- and mono sulfide products. Although these compounds could usually be separated by chromatography, the disulfide derivative could be synthesized as the sole compound by controlling the reaction time, as outlined in Scheme 18. The production of arylthiyl radicals, even at a low concentration in a visible-light-mediated initiation step would enact the generation by hydrogen atom abstraction of NHC-boryl radicals. The propagation of the reaction would then result from the homolytic radical substitution by the latter radicals of the starting diaryl disulfide.
 | ||
| Scheme 18 First synthesis of NHC-boryl sulfides and proposed mechanistic pathway. | ||
Contrary to the reduction of disulfides, the reduction of organohalides with NHC–boron derivatives proved to be more challenging,40 being amenable in the first experiments to only a small array of compounds that notably did not include aryl halides or sterically-hindered alkyl halides such as iodoadamantane. This situation recently changed with the recognition by the groups of Curran, Lalevée and Lacôte that phenylthiyl radicals, generated from thiophenol by the action of an initiator such as t-butylperoxide under UV irradiation, were able to significantly catalyse the reduction, highlighting the concept of “polarity-reversal catalysis”.41
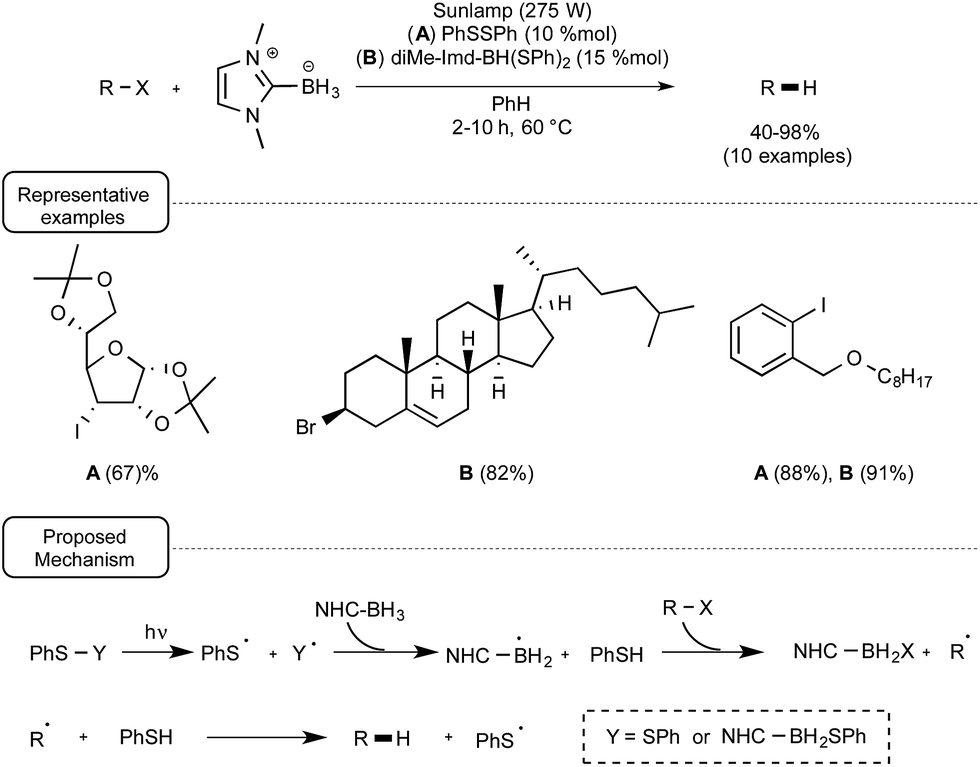
Anticipating that thiyl radicals could also be produced from the homolytical photocleavage of either the S–S or the B–S bond of respectively diphenyldisulfide or an NHC–boron mono- or disulfide derivative, these authors tested the reducing power of di-Me-Imd-BH3 on a palette of alkyl- and aryl iodides as well as bromides under the irradiation of a sunlamp in the presence of 10% of diphenyldisulfide or 15% of diMe-Imd-BH(SPh)2. As indicated in Scheme 19, at a temperature of 60 °C and by making use of benzene as the solvent, good yields of the reduction products were obtained. It ought to be noted that when the monosulfide derivative diMe-Imd-BH2SPh was used in place of PhSSPh or diMe-Imd-BH(SPh)2, the reaction did not proceed so well. The mechanistic proposal accounting for this transformation highlights a dual participation of diphenyl disulfide or boryl sulfide derivatives, both as initiators and as catalyst precursors.
 | ||
| Scheme 19 di-Me-Imd-BH3-mediated reduction of organohalides. | ||
4. Visible-light-mediated oxidation of arylboronic acids into phenols
Although a plethora of methods exist for the transformation of arylboronic acids into phenols, many of them suffer from a limited scope, as they require strong basic conditions and/or strong oxidizers.42 In addition, few among them are sustainable processes. In order to address these limitations, the Jørgensen and the Xiao groups developed a highly efficient visible-light-mediated photoredox strategy relying on the utilization of oxygen from ambient air as the terminal oxidant.43 As indicated in Scheme 20, by selecting Ru at low loadings as the catalyst, Hünig's base as a sacrificial reductant and DMF as the solvent, a wide range of electron-rich as well as electron-poor arylboronic acids could be smoothly converted into phenols in very good yields. It is worth noting that bubbling air into the medium was not required as the transformations proceeded quite well by simply keeping the medium opened to air. Interestingly, in the case of p-methoxyphenylboronic acid, when the non-metal sensitizer eosin Y was used in place of Ru, almost the same yield of phenol could be obtained, albeit after a longer reaction time.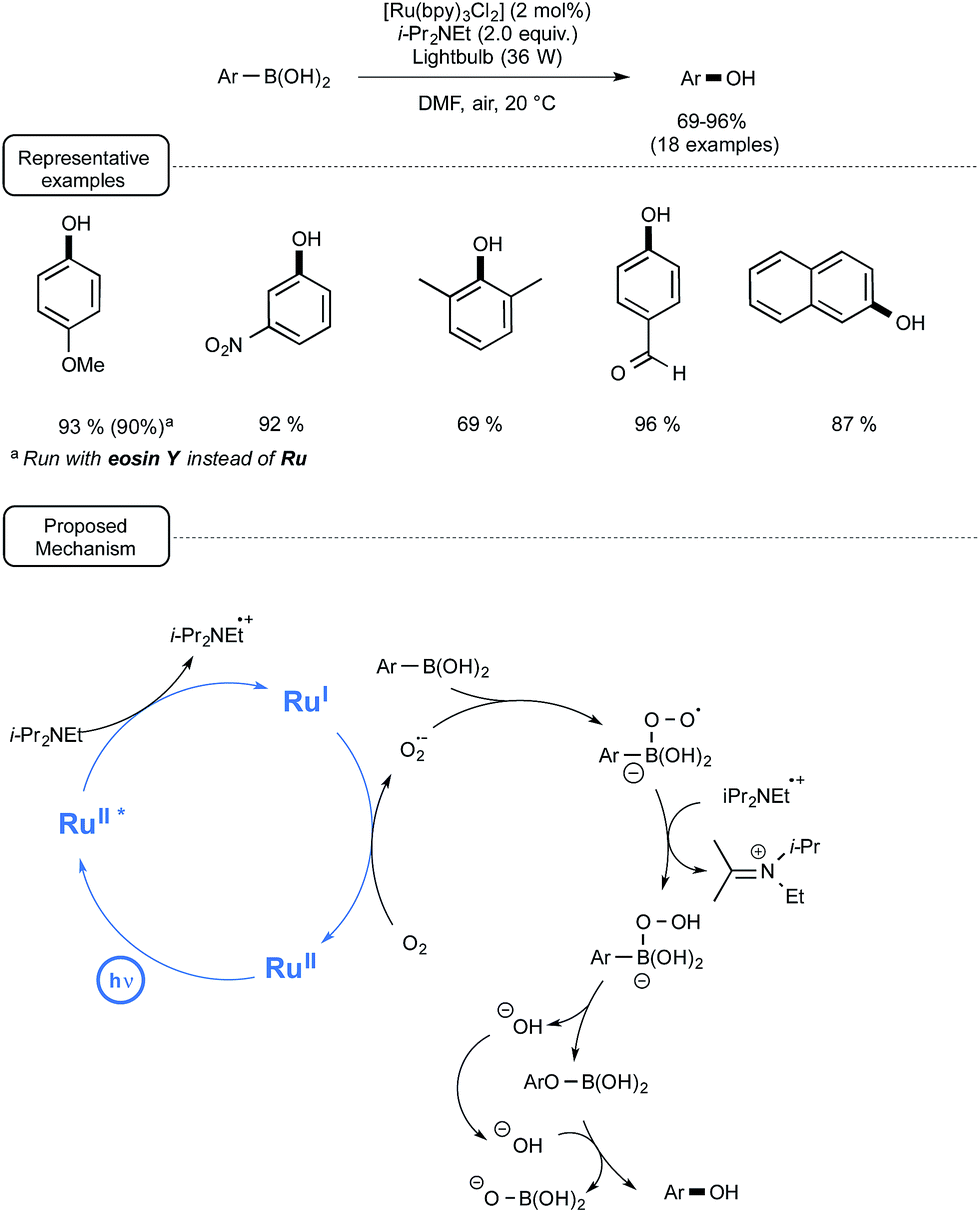 | ||
| Scheme 20 Ruthenium-catalyzed photoredox conversion of arylboronic acids into phenols. | ||
As outlined in Scheme 20, the proposed mechanism is based on the participation of the superoxide radical anion (O2˙−), which may originate from the single-electron reduction of molecular oxygen by Ru(I), itself generated via reductive quenching of the excited state of Ru by the Hünig's base. This superoxide species is supposed to transform the arylboronic acid into a boron peroxo key-intermediate, which would undergo a 1,2-aryl shift to give the phenol after hydrolysis. Although further studies are still needed in order to assess this mechanistic pathway, 18O labelling investigations have shown that the oxygen in the phenol arises from molecular oxygen.
This seminal report provided the incentive for other laboratories to develop further visible-light mediated deboronative hydroxylation processes. A year later, the Scaiano group reported the utilization of methylene blue MB as photosensitizer in place of Ru or eosin Y.44 As presented in Scheme 21 (Condition A), the utilization of this common dye was particularly rewarding, enabling the preparation at room temperature of a great variety of phenols in excellent yields. In a test reaction, MB was found more efficient as a catalyst than Ru.
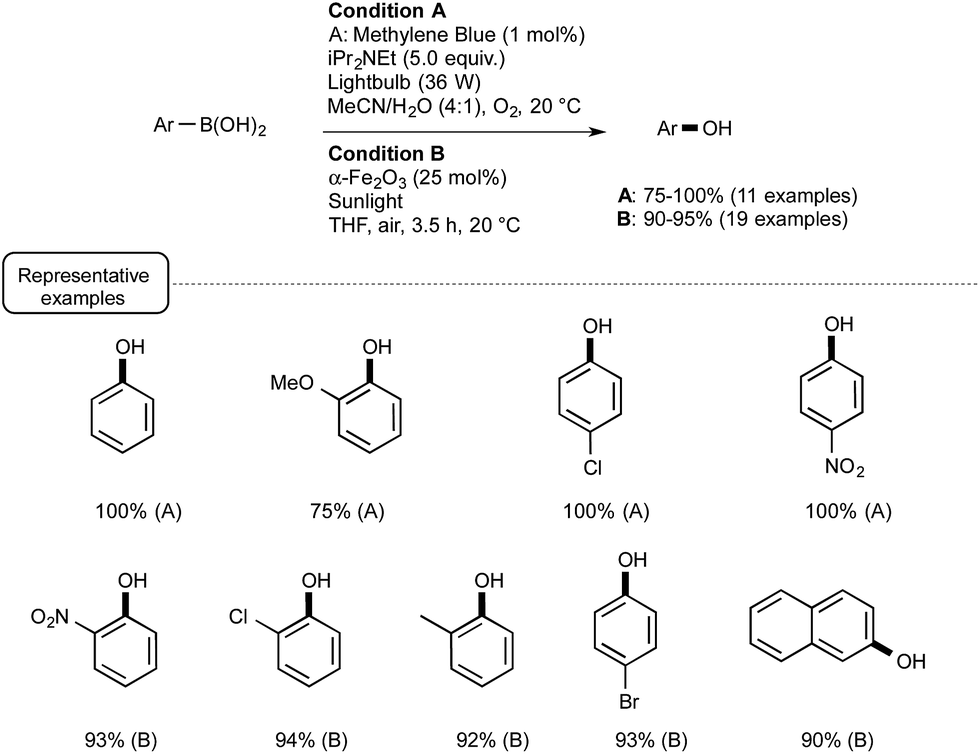 | ||
| Scheme 21 Methylene-blue- and iron-oxide-catalyzed photoredox conversion of arylboronic acids into phenols. | ||
In order to understand the role of MB, the authors performed a series of kinetic tests that supported a mechanism proceeding via the formation of a superoxide radical anion as in the case of Ru-mediated processes but also with the involvement of the semireduced form of methylene blue.
In the same vein, the Itoh group reported transformations into phenols that could be accomplished even in the absence of base such as Hünig's base.45 The trick in that case was to select 2-chloro-anthraquinone as a photosensitizer and isopropanol as the solvent. Under such conditions MB could not be used. Other recent modifications included the utilisation of continuous flow reactors in place of batch experiments46 as well as the employment, as a photosensitizer, of nanoparticles of a common iron oxide polymorph named α-Fe2O3.47 This last modification proposed by the Vishwakarma group in India, which lies in sharp contrast with the Ru- or non-metal-based processes, deserves special attention. It belongs to the field of heterogeneous photocatalysis that highlights the use of UV-activated semiconductors such as TiO2.48 As presented in Scheme 21 (Condition B), by making use of α-Fe2O3 under sunlight irradiation in ambient air, a wide range of phenols were obtainable in excellent yields from the parent boronic acids.
The mechanistic pathway still remains unclear, although experiments run with 18O2 and H218O suggested that the reaction uses oxygen for the oxidation process. As experiments run without catalyst or in the dark were not conclusive, the photo-induced electron/hole pairs in the semiconductor certainly play a key-role. This participation remains to be addressed as the conduction band electrons in α-Fe2O3 are supposed to be unable to react with oxygen, contrary to the electrons in TiO2, because of their relatively weak reducing power.
5. Visible-light-BODIPY-mediated transformations
Let us focus now on the recent studies from several laboratories aiming at using boron-dipyrromethene dyes (BODIPYs) as mediators and/or catalysts for visible-light-driven transformations in organic chemistry, including polymer chemistry. BODIPY derivatives have a rich pedigree in biochemistry and materials chemistry as fluorescent probes or sensors.49 By contrast, their utilization in organic chemistry has been impeded by their short triplet excited state lifetimes, which in many cases prevent them from interacting efficiently with organic molecules, notably through single electron transfer processes. In addition, limited data is available for their redox potentials in the excited state (see Fig. 1).6iLi and co-workers reported in 2011 the first utilisation in organic chemistry of BODIPY derivatives as photocatalysts under visible light irradiation.50 As a proof-of-concept, they studied the BODIPY-catalyzed photooxidation of thioanisole into the corresponding sulfoxide. As indicated in Scheme 22, by choosing one of the four polysubstituted BODIPYs at low loadings as the catalyst as well as methanol as the solvent, the reaction proceeded very efficiently under air at room temperature. By comparison, the utilization of Ru in place of a BODIPY derivative resulted in no improvement of the conversion. In addition, when the non-metal dyes Rhodamine B (RB) or Nile Red (NR) were used as the catalyst, the conversion in sulfoxide was much slower. As shown in Scheme 22, the proposed mechanism highlights the formation as an intermediate of singlet oxygen by energy transfer from BODIPY's excited triplet state generated under visible light irradiation. Singlet oxygen would then oxidize thioanisole into the corresponding sulfoxide.
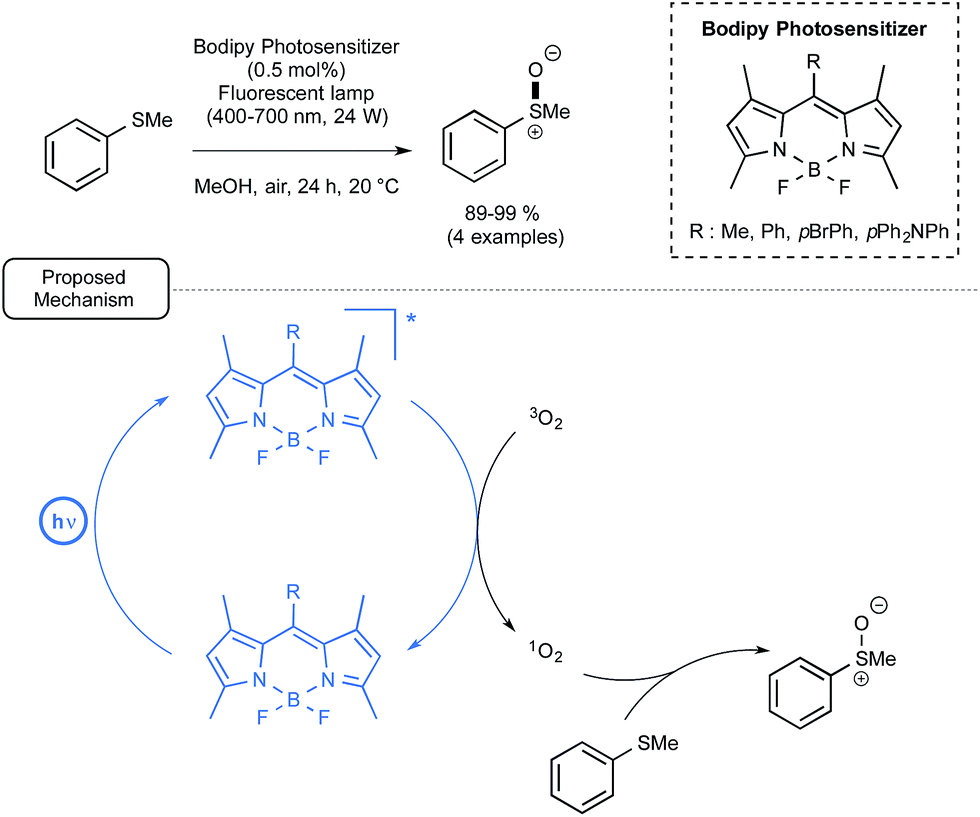 | ||
| Scheme 22 BODIPY photocatalyzed oxidation of thioanisole under visible light and proposed mechanism. | ||
In spite of its simplicity, the transformation described here deserves attention, not only because it implies, for the first time, BODIPYs as photocatalyst, but also because it highlights an energy transfer from the photocatalyst which contrasts with the widely encountered single electron transfer processes.51
In further studies, the same authors synthesized a BODIPY-based polymeric photocatalyst that could be easily removed after the reaction by simple filtration.52 They also prepared the four iodinated BODIPYs postulating that the presence of heavy atoms on the boron-dipyrromethene core would extend, under visible-light irradiation, the lifetime of the photocatalyst's triplet excited states and thus would enhance under air the production of singlet oxygen.53 The premise turned out to be satisfied and a shortening of the oxidation time of thioanisole into its sulfoxide was observed for selected BODIPY-based photocatalysts.
The Ramaiah group from India prepared a series of seven aza-BODIPY derivatives equipped with either iodine or bromine atoms, focusing again on their triplet excited state properties as well as their efficiency with regards to singlet-oxygen generation.54 Those bearing six heavy atoms were particularly effective. As presented in Scheme 23, one of them was tested for the photooxidation of 1-naphthol into 1,4-naphthoquinone under normal sunlight conditions using acetonitrile as the solvent and with a continuous stream of oxygen. After 8 h at room temperature under these conditions, the quinone could be obtained with a quantitative yield, whereas yields lower than 45% were obtained by utilizing common dyes such as methylene blue (MB) or Rose Bengal (RoB).
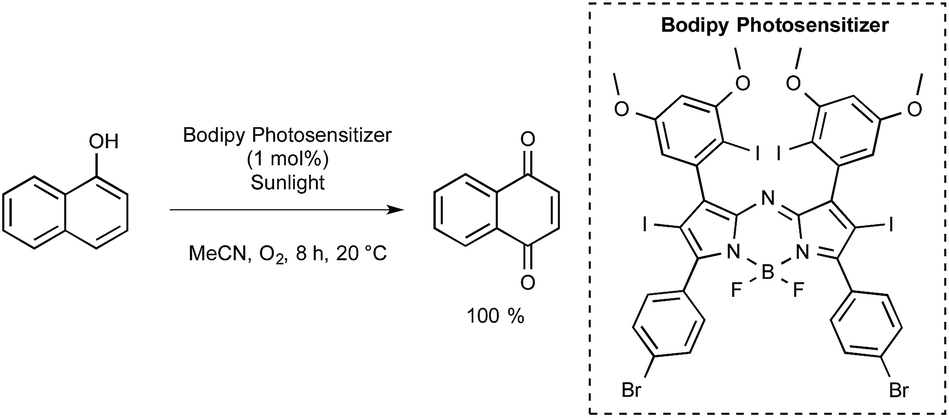 | ||
| Scheme 23 Aza-BODIPY photocatalyzed oxidation of 1-naphthol under sunlight. | ||
At the same time, the group of Zhao from China was exploring the possibility of employing iodo-BODIPYs as photocatalysts, not only for simple reactions, such as the previously mentioned oxidation of naphthols, but also, in place of common sensitizers, in recently reported more elaborated visible-light driven processes.55 In particular, as shown in Scheme 24, they tested an iodo-BODIPY as a surrogate for Ru in the newly described preparation by Xiao and co-workers of pyrrolo[2,1-a]-isoquinolines from N-substituted maleimides as dienophilic partners.56 By applying the conditions tailored for the Ru-catalyzed sequence, the utilization of the BODIPY-based photocatalyst proved rewarding as it led to the formation of isoquinolines in good yields but under significantly shortened reaction times. In order to decipher the mechanistic pathway accounting for the transformation, a series of test experiments were realized, which supported the mechanism originally proposed by Xiao.56 The BODIPY catalyst, once photoexcited, just played the role of oxidant previously imparted to Ru(II) in its excited form. Not only the key-role of the superoxide anion radical O2˙− rather than singlet oxygen could be assessed, but the formation of H2O2, as stated in Scheme 24, could also be confirmed.
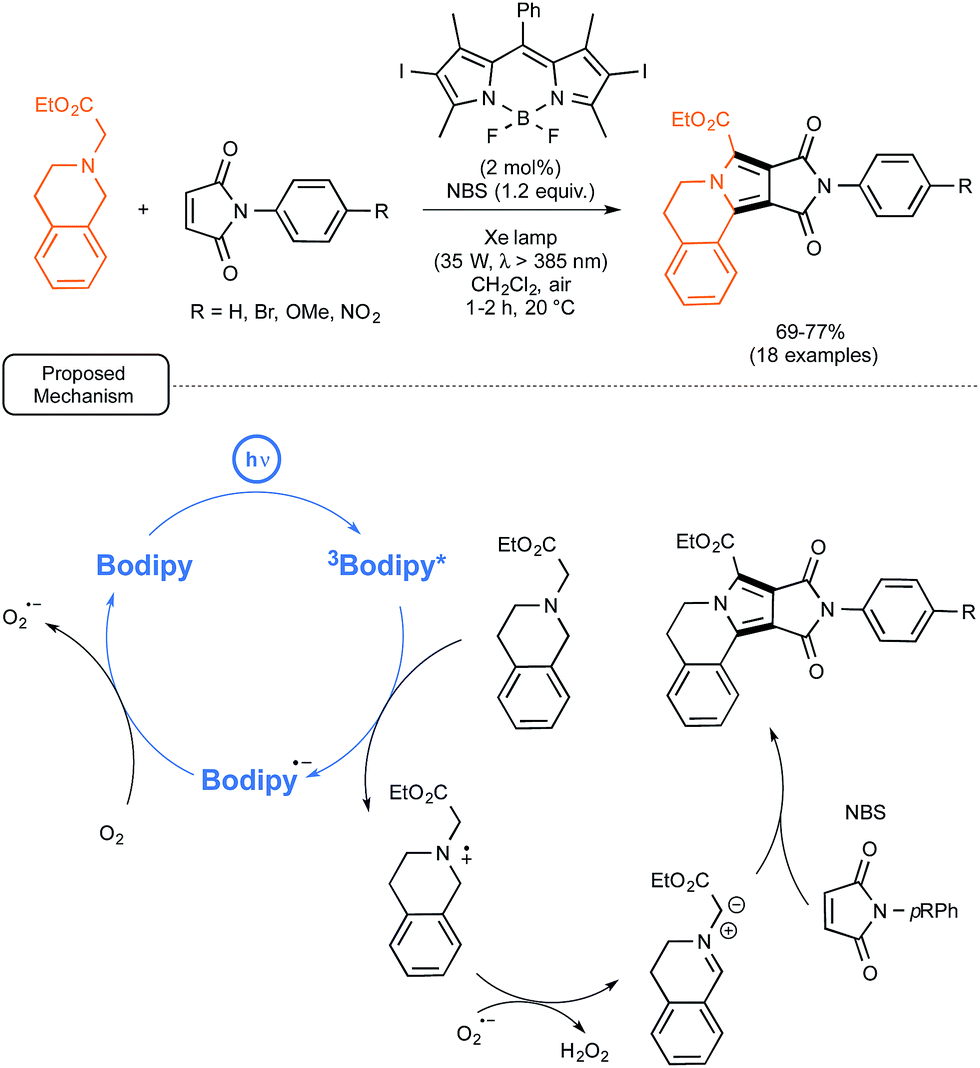 | ||
| Scheme 24 BODIPY-photocatalyzed preparation of pyrrolo[2,1-a]-isoquinolines from N-substituted maleimides and proposed mechanism. | ||
In a related work, the authors tested the same photocatalyzed synthesis by making use of more complex BODIPY-containing dyes such as fullerene–BODIPY dyads or BODIPY–iodo–aza-BODIPY triads.57 These modifications enabled shorter still reaction times.
In order to demonstrate that such BODIPY-based photocatalysts could also be involved as a reducing agent in photoredox catalytic reactions, they introduced it in place of eosin Y in the catalytic C–H arylation of heteroarenes with aryl diazonium salts recently reported by the König group.58 As outlined in Scheme 25, the reaction proceeded quite rapidly at room temperature with a photocatalyst at low loadings, not only with furan but also with thiophene as the heteroaryl partners.55b
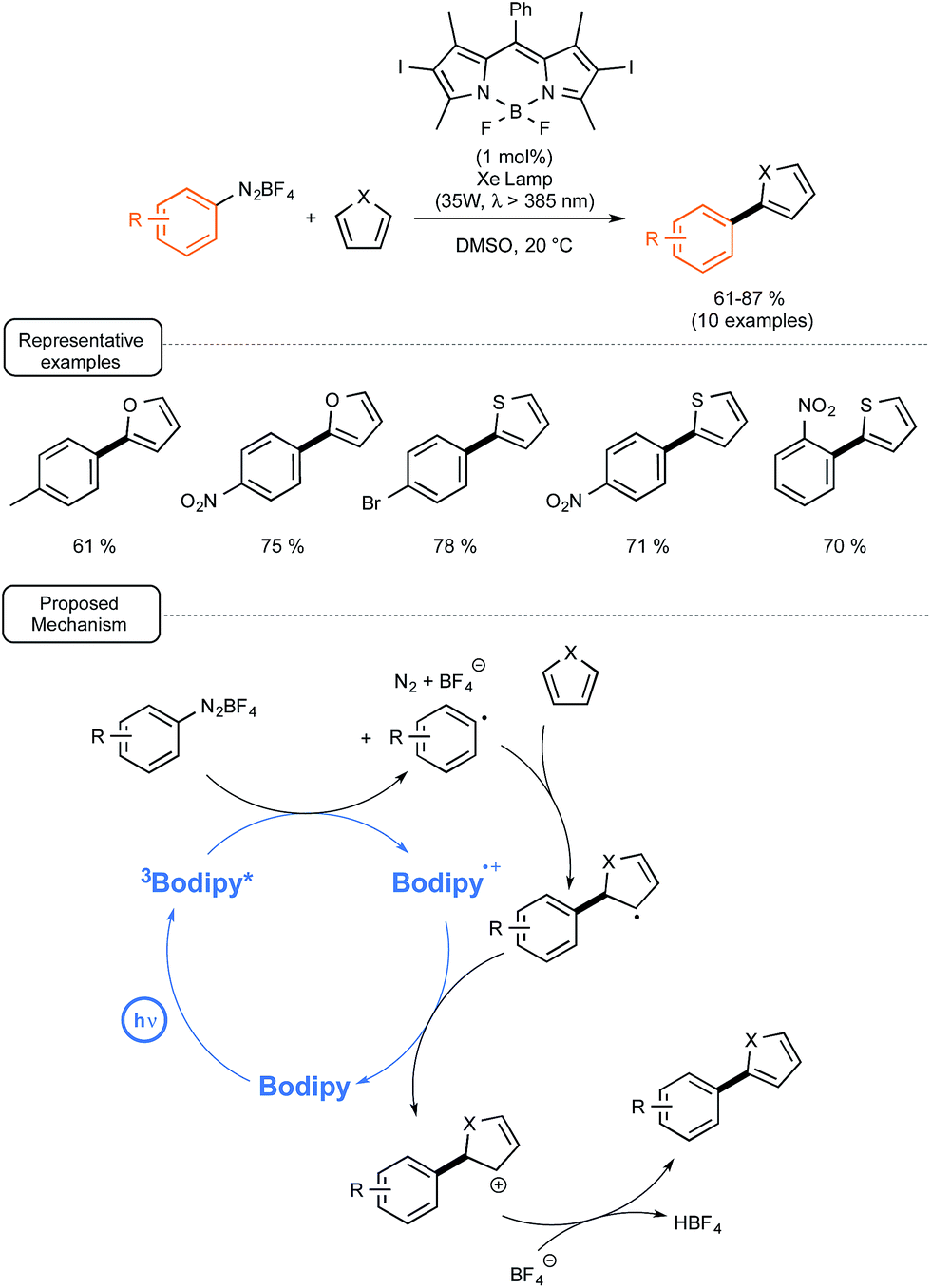 | ||
| Scheme 25 BODIPY-photocatalyzed arylation of furan and thiophene and proposed mechanistic pathway. | ||
Lalevée has also studied BODIPY derivatives as potential initiators or co-initiators for the cationic polymerization of divinylether as well as epoxy monomers upon visible-light exposure.59 The primary aim was to discover new formulations for the mild synthesis of the corresponding polymers in laminate that would not be affected by atmospheric oxygen.35c As outlined in Scheme 26, in the case of a representative divinylether monomer, by using a combination of BODIPY and diphenyliodonium hexafluorophosphate, a nearly full conversion into the desired polyvinyl polymer was observed after a brief irradiation time by a laser diode at 473 nm. The proposed mechanism in the case of two-component initiating system pinpoints, as shown in Scheme 26, the formation of a BODIPY cation radical as a primary initiator. This cation would be generated through oxidative quenching by the iodonium salt of the BODIPY's photogenerated excited state.
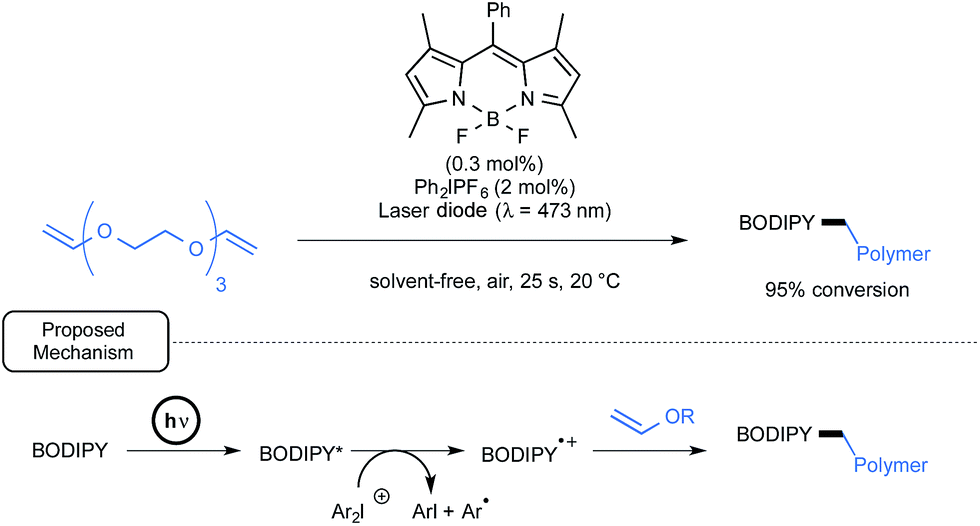 | ||
| Scheme 26 BODIPY-mediated photopolymerization of divinylether monomer and proposed mechanism. | ||
In the case of an epoxy monomer, an excellent polymer conversion could be achieved only by making use of a three-component initiating system that incorporated tris(trimethylsilyl)silane as the additional partner. The latter silane derivative would be able to deliver under the reaction conditions a tris(trimethylsilyl)silyl cation, which was able to initiate the cationic polymerization.
Let us close this section with findings from the last months highlighting the utilization as a new biological tool of BODIPY-containing protecting groups that could be easily removed under visible light irradiation. The group of Urano from the University of Tokyo was the first to discover by serendipity that, in some cases, 4-aryloxy BODIPY derivatives could release their tethered phenol moiety upon blue-green light irradiation.60 The biological potential of employing BODIPY as a caging group for phenol was exemplified by the photo-release of a relevant biogenic molecule in a two-step tandem process. As outlined in Scheme 27, BODIPY-caged histamine compound was shown to undergo first the photo-cleavage of its phenol part that further decomposed to generate the free biological amine alongside CO2 and quinone methide. As indicated, the latter intermediate readily hydrolyzed to yield 4-hydroxybenzyl alcohol.
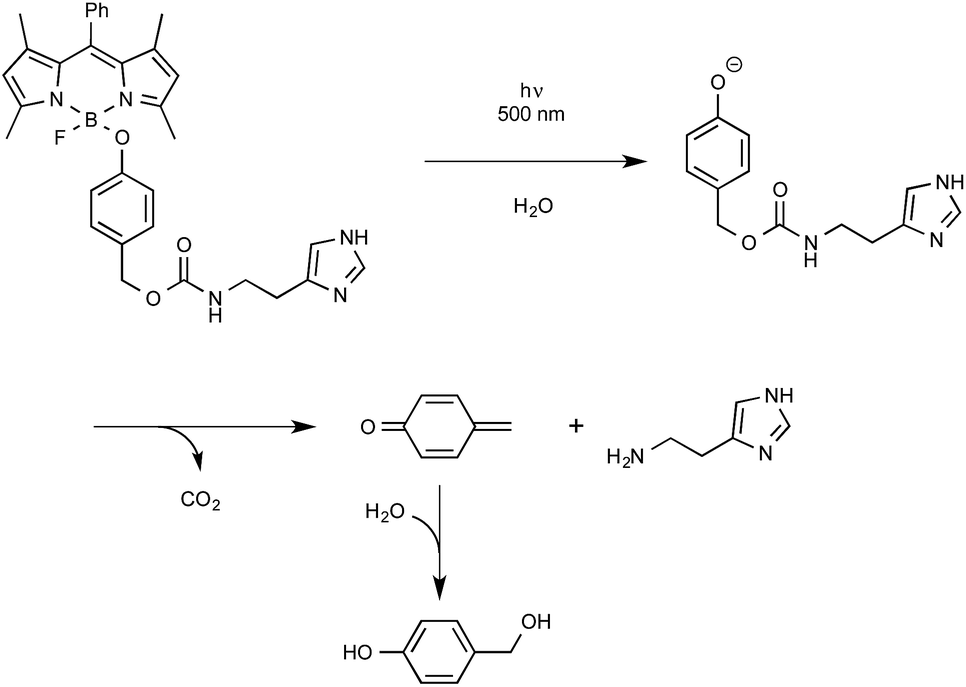 | ||
| Scheme 27 Uncaging reaction of BODIPY-caged histamine derivative. | ||
The Weinstain group from Israel employed for its part a source of green light to uncage a small series of model biogenic molecules linked to a BODIPY scaffold.61 As presented in Scheme 28, a carboxylic acid, a primary alkyl- or arylamine as well as a phenol could be freed from their parent BODIPY derivatives with the concomitant release of CO2.
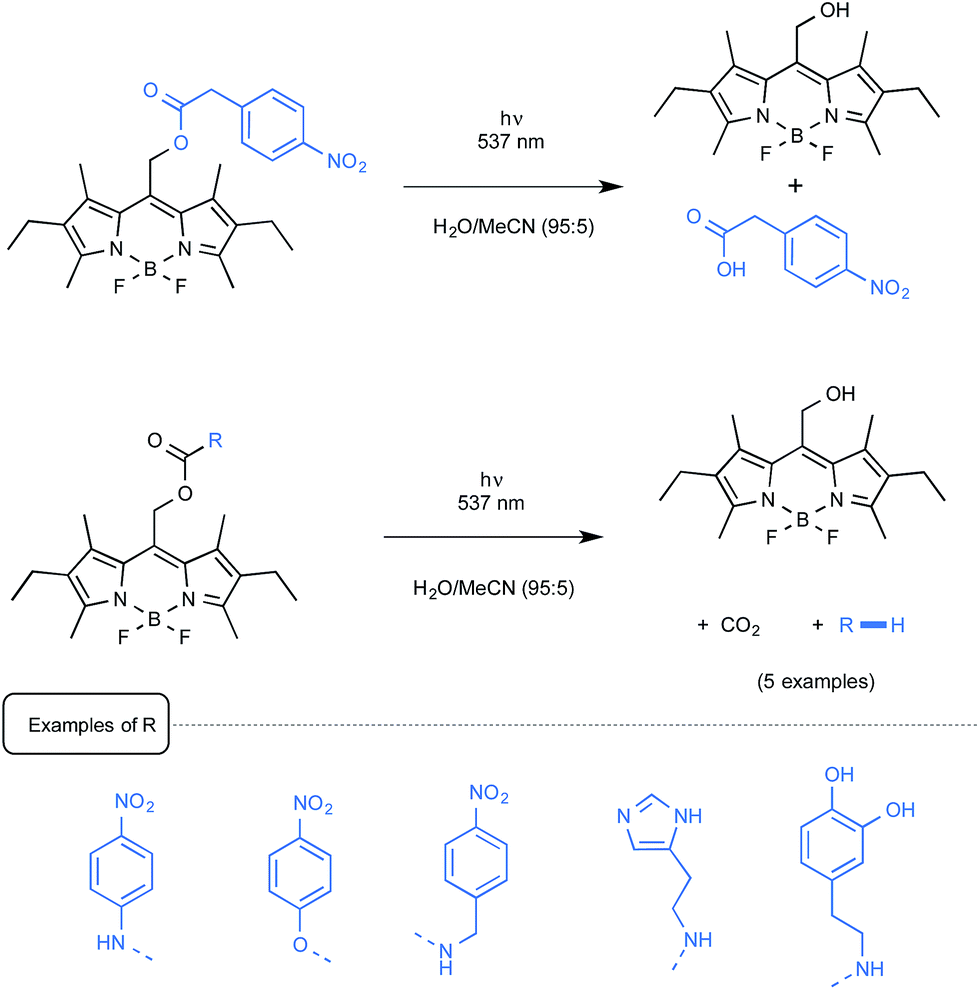 | ||
| Scheme 28 Uncaging reaction of model biogenic molecules. | ||
This photo-strategy to liberate free carboxylic acids from BODIPY-caged precursors has just been supported by an independent study performed by Winter and co-workers.62
6. Other transformations
Let us focus attention in a last section on a few other visible-light-mediated transformations that have recently impacted organoboron chemistry. Capitalising on seminal investigations from the MacMillan group demonstrating the Ru-mediated conversion of CF3I to CF3˙ under visible light irradiation,63 Ye and Sanford launched three years ago a novel strategy for the trifluoromethylation of boronic acids,64,65 which provided an early example of dual catalysis.30 As outlined in Scheme 29, the merger of visible-light photocatalysis with copper catalysis proved particularly rewarding here, enabling the preparation of a wide range of trifluoromethylated aryl- and heteroaryl derivatives at a quite forgiving temperature. The radical trifluoromethylation proceeded in high yield from boronic acids bearing either electron-donating or electron-withdrawing substituents. In addition, quite remarkably, 4-iodophenylboronic acid underwent selective trifluoromethylation to form the trifluoromethylated aryl compound with the preserved iodo atom in the para position. The plausible mechanistic pathway accounting for the transformation highlights, as shown in Scheme 29, two intertwined catalytic cycles.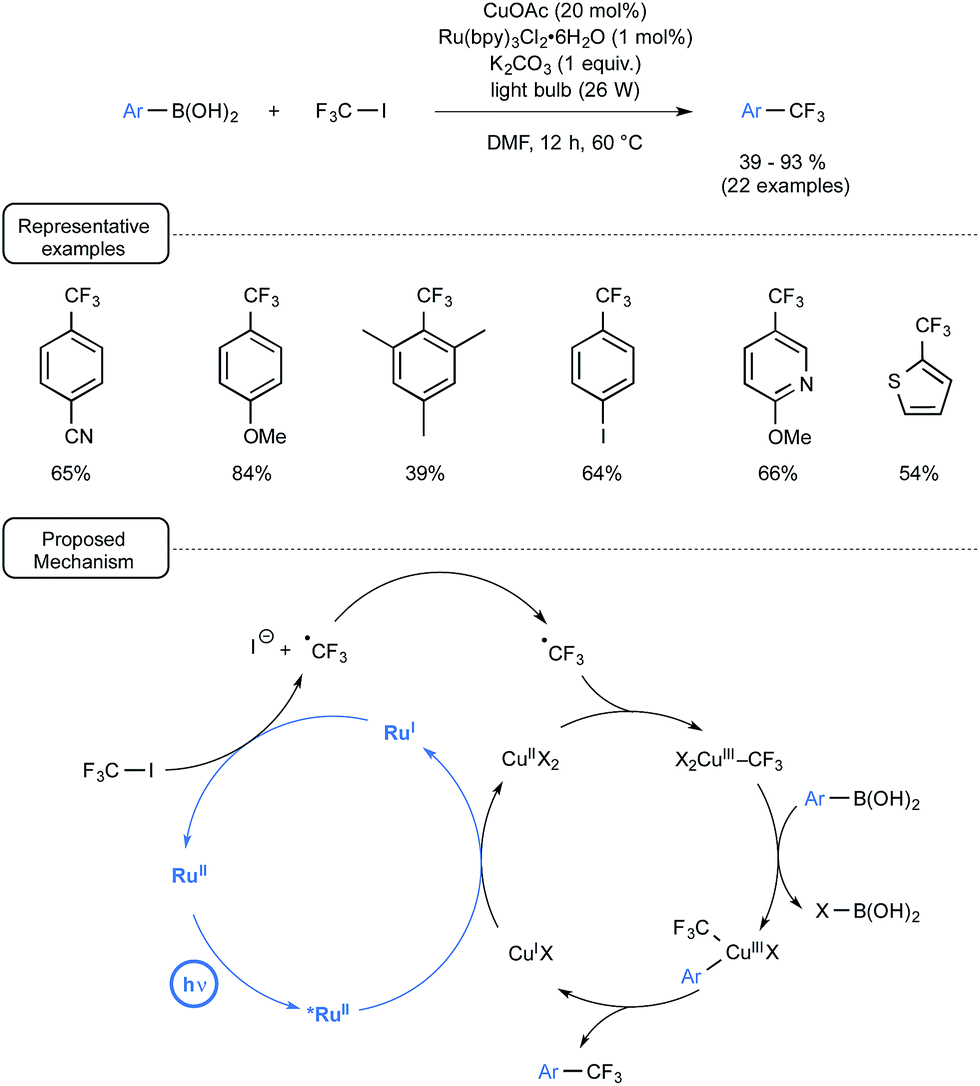 | ||
| Scheme 29 Cu/Ru-catalyzed trifluoromethylation of boronic acids and plausible mechanism. | ||
If the photocatalytic cycle, which accounts for the formation of the trifluoromethyl radical, stems from previous investigations from the MacMillan group and notably pinpoints the reducing power of Ru(I) species to generate CF3˙ from CF3I,63 the Cu catalytic cycle highlights for its part a transmetallation step between a Cu(III) trifluoromethylated key-intermediate and the arylboronic acid. It may be worthwhile to note at this stage that the proposed mechanism does not involve the formation of aryl radicals and stands thus in contrast with the pathways encountered in the second section. It tallies however perfectly with the lack of reactivity of boronic acids toward photoexcited Ru(II) observed in the deboronative alkynylation pathway disclosed in Scheme 8.
Another demonstration of the synergic interplay between catalytic cycles that involves aryl boronic acids has just been provided by Kobayashi and co-workers (Scheme 30).66 By merging Ir-photoredox catalysis and Cu catalysis, the scope of the Chan–Lam coupling reaction67 could be significantly expanded to aniline derivatives of low reactivity.
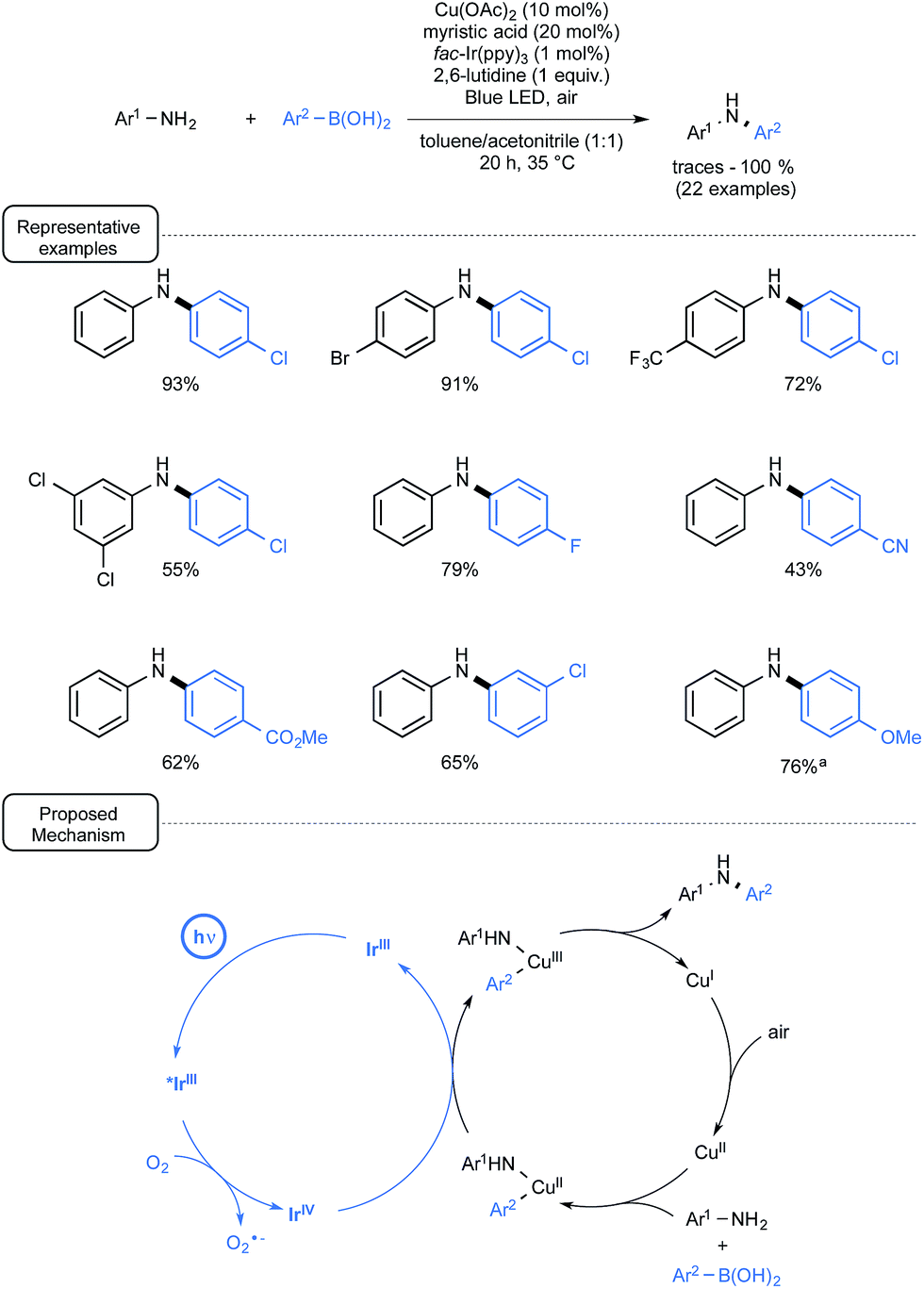 | ||
| Scheme 30 Cu/Ir-catalyzed Chan–Lam coupling reaction of anilines and boronic acids and plausible mechanism. aWith Cu(OAc)2 (5 mol%) and myristic acid (10 mol%). | ||
In that particular case, photoredox catalysis proved essential to address the difficulty in accessing the key-Cu(III) intermediate prevailing in Chan–Lam catalytic cycles.68
In closing, let us have a look from a different angle to the impact of visible light on boron chemistry and consider organoborons as reaction products rather than reagents. End 2012, the Yan group from Lishui University in China reported a visible-light-mediated synthetic route to arylboronates from the corresponding aryldiazonium salts.69 Whilst the synthesis of arylboronates already has a rich pedigree, most of the reported procedures suffer from limitations related to the use of toxic metals and/or harsh conditions.70 Capitalizing not only on the preparation by Wang of arylboronates from arylamines with B2pin2 in the presence of tert-butyl nitrite,71 but also on the visible-light-mediated recent chemistry of aryldiazonium salts,58,72 the authors successfully developed a mild and metal-free alternative to most of these methods. As presented in Scheme 31, under irradiation of a light bulb at room temperature, a palette of aryldiazonium tetrafluoroborates could be smoothly converted into the corresponding arylboronates by making use of B2pin2 as the borylating agent and eosin Y as the dye.
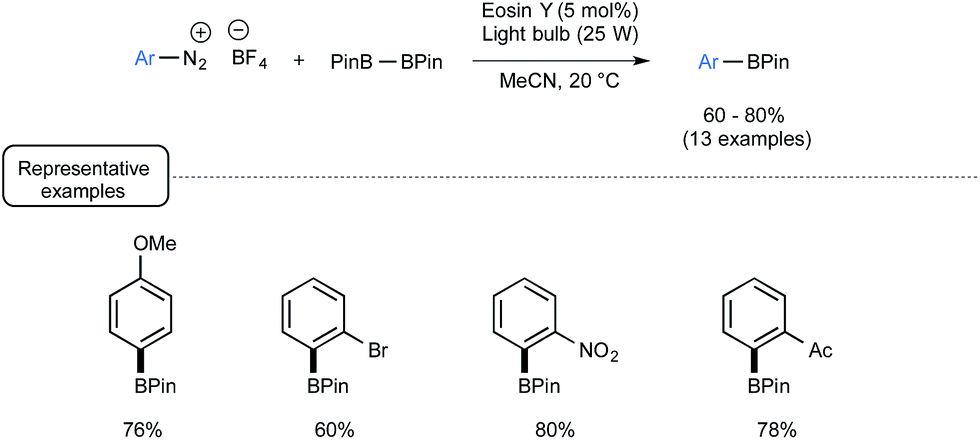 | ||
| Scheme 31 Visible-light-induced borylation of aryldiazonium salts. | ||
Taking into account its sustainability and its simplicity this reaction should deserve further studies notably to expand its scope and to comb through its mechanistic pathway.
7. Conclusions
For the past seven years or so, visible light photoredox catalysis has been spreading in organic chemistry like a wildfire, impacting the area of boron chemistry in the last three years. Although the field is obviously still in its infancy, it has already rekindled interest in radical boron chemistry, which, since its early developments in the seventies, has been essentially the preserve of trialkylborane derivatives. On the one hand the merger of visible light and photocatalysts has provided support to innovative strategies, such as the oxidative generation of alkyl- and aryl radicals from organoborate derivatives or the formation of NHC-boryl radicals by atom hydrogen abstraction from the corresponding NHC–borane complexes. In these cases, with simple fixtures and household visible light sources, mild and green alternatives to reported procedures were disclosed. On the other hand, even more interestingly, this merger has opened new vistas in cross-coupling reactions, encompassing notably a novel deboronative/decarboxylative alkenylation process and a Suzuki–Miyaura-derived procedure based on the newly developed paradigm of dual catalysis. In this case, the combination of two intertwined catalytic cycles highlighting the utilization of an iridium photoredox catalyst and a nickel-based catalyst proved particularly rewarding as it enabled the highly challenging coupling of sterically-hindered secondary-alkylboron derivatives. Because of its performance, it is anticipated that visible-light-driven dual catalysis will continue to fuel organoboron chemistry.Interesting perspectives in organic- and polymer chemistries are also expected from the utilisation of BODIPY derivatives as photosensitizers whose potential bespeaks their chemical robustness, their resistance to photobleaching under visible light irradiation as well as their readily tunable molecular structure. Scouting reports which focus on their utilisation, in place of conventional Ru(II) or Ir(III) complexes, as photoredox catalyst for the elaboration of highly functionalized compounds are particularly appealing. Last year, the serendipitous discovery that phenols could be released from a range of aryloxy BODIPY derivatives under visible light irradiation paved the way for exploring further the utilisation of BODIPY-containing protecting groups not only in chemistry but also in biology.
To conclude, we portend that, more than a mere fashion, visible-light-mediated processes will continue to shine over organoboron chemistry for many years to come.
Acknowledgements
The authors thank the University of Strasbourg and the CNRS for financial support.Notes and references
- G. Ciamician, Science, 1912, 36, 385–394 CAS
.
-
(a) F. H. Burstall, J. Chem. Soc., 1936, 173–175 RSC
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed
-
(a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed
-
(a) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC
-
(a) H. C. Brown and M. M. Midland, Angew. Chem., Int. Ed. Engl., 1972, 11, 692–700 CrossRef CAS PubMed
-
P. Renaud, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, 2012 Search PubMed
- A. Brecht-Forster, J. Fitremann and P. Renaud, Helv. Chim. Acta, 2002, 85, 3965–3974 CrossRef CAS
- C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415–3434 CrossRef CAS PubMed
- A.-P. Schaffner and P. Renaud, Eur. J. Org. Chem., 2004, 2291–2298 CrossRef CAS PubMed
- B. Becattini, C. Ollivier and P. Renaud, Synlett, 2003, 2003, 1485–1487 Search PubMed
- P. Renaud, C. Ollivier and V. Weber, J. Org. Chem., 2003, 68, 5769–5772 CrossRef CAS PubMed
- C. Cadot, J. Cossy and P. I. Dalko, Chem. Commun., 2000, 1017–1018 RSC
- For a review, see: G. Yan, M. Yang and X. Wu, Org. Biomol. Chem., 2013, 11, 7999–8008 CAS
-
(a) L. A. Shundrin, V. V. Bardin and H. J. Frohn, Z. Anorg. Allg. Chem., 2004, 630, 1253–1257 CrossRef CAS PubMed
- Y. Nishigaichi, T. Orimi and A. Takuwa, J. Organomet. Chem., 2009, 694, 3837–3839 CrossRef CAS PubMed
-
(a) M. C. Etter, B. N. Holmes, R. B. Kress and G. Filipovich, Isr. J. Chem., 1985, 25, 264–273 CrossRef CAS PubMed
-
(a) T. Koike and M. Akita, Chem. Lett., 2009, 38, 166–167 CrossRef CAS
- Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS PubMed
- K. Miyazawa, Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 7249–7251 RSC
- Y. Li, K. Miyazawa, T. Koike and M. Akita, Org. Chem. Front., 2015, 2, 319–323 RSC
- K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749–2755 CrossRef CAS PubMed
-
(a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed
- M. Hoshino, H. Uekusa, A. Tomita, S.-y. Koshihara, T. Sato, S. Nozawa, S.-I. Adachi, K. Ohkubo, H. Kotani and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4569–4572 CrossRef CAS PubMed
- T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 5, 21297–21300 RSC
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed
- H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881–1884 CrossRef CAS PubMed
-
(a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed
-
(a) E. Jahn and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 13326–13328 CrossRef CAS PubMed
- A. Solovyev, S.-H. Ueng, J. Monot, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, Org. Lett., 2010, 12, 2998–3001 CrossRef CAS PubMed
- For reviews, see:
(a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC
-
(a) J. Lalevée, N. Blanchard, M.-A. Tehfe and J. P. Fouassier, Macromol. Rapid Commun., 2011, 32, 838–843 CrossRef PubMed
- S. Telitel, S. Schweizer, F. Morlet-Savary, B. Graff, T. Tschamber, N. Blanchard, J. P. Fouassier, M. Lelli, E. Lacôte and J. Lalevée, Macromolecules, 2013, 46, 43–48 CrossRef CAS
-
(a) M.-A. Tehfe, M. Makhlouf Brahmi, J.-P. Fouassier, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte and J. Lalevée, Macromolecules, 2010, 43, 2261–2267 CrossRef CAS
- J.-P. Fouassier and J. Lalevée, RSC Adv., 2012, 2, 2621–2629 RSC
- X. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669–5674 CrossRef CAS PubMed
- X. Wang, F. D. Saeva and J. A. Kampmeier, J. Am. Chem. Soc., 1999, 121, 4364–4368 CrossRef CAS
- X. Pan, A.-L. Vallet, S. Schweizer, K. Dahbi, B. Delpech, N. Blanchard, B. Graff, S. J. Geib, D. P. Curran, J. Lalevée and E. Lacôte, J. Am. Chem. Soc., 2013, 135, 10484–10491 CrossRef CAS PubMed
- X. Pan, J. Lalevée, E. Lacôte and D. P. Curran, Adv. Synth. Catal., 2013, 355, 3522–3526 CrossRef CAS PubMed
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC
-
(a) K. Inamoto, K. Nozawa, M. Yonemoto and Y. Kondo, Chem. Commun., 2011, 47, 11775–11777 RSC
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 124, 808–812 CrossRef PubMed
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed
- K. Matsui, T. Ishigami, T. Yamaguchi, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, Synlett, 2014, 2613–2616 CAS
- I. G. T. M. Penders, Z. Amara, R. Horvath, K. Rossen, M. Poliakoff and M. W. George, RSC Adv., 2015, 5, 6501–6504 RSC
- S. D. Sawant, A. D. Hudwekar, K. A. Aravinda Kumar, V. Venkateswarlu, P. P. Singh and R. A. Vishwakarma, Tetrahedron Lett., 2014, 55, 811–814 CrossRef CAS PubMed
- Z. Zhang and P. A. Maggard, J. Photochem. Photobiol., A, 2007, 186, 8–13 CrossRef CAS PubMed
-
(a) V. Lakshmi, M. Rajeswara Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501–2517 RSC
- W. Li, Z. Xie and X. Jing, Catal. Commun., 2011, 16, 94–97 CrossRef CAS PubMed
- For other examples of photocatalysis highlighting energy transfers, see:
(a) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed
- W. Li, W. Zhang, X. Dong, L. Yan, R. Qi, W. Wang, Z. Xie and X. Jing, J. Mater. Chem., 2012, 22, 17445–17448 RSC
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC
- N. Adarsh, M. Shanmugasundaram, R. R. Avirah and D. Ramaiah, Chem.–Eur. J., 2012, 18, 12655–12662 CrossRef CAS PubMed
-
(a) L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed
- Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171–7175 CrossRef CAS PubMed
-
(a) L. Huang and J. Zhao, Chem. Commun., 2013, 49, 3751–3753 RSC
-
(a) D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed
- S. Telitel, J. Lalevée, N. Blanchard, T. Kavalli, M.-A. Tehfe, S. Schweizer, F. Morlet-Savary, B. Graff and J.-P. Fouassier, Macromolecules, 2012, 45, 6864–6868 CrossRef CAS
- N. Umeda, H. Takahashi, M. Kamiya, T. Ueno, T. Komatsu, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, ACS Chem. Biol., 2014, 9, 2242–2246 CrossRef CAS PubMed
- N. Rubinstein, P. Liu, E. W. Miller and R. Weinstain, Chem. Commun., 2015, 51, 6369–6372 RSC
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed
-
(a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed
- For selected copper-mediated trifluoromethylations of boronic acids up to 2011, see:
(a) L. Chu and F.-L. Qing, Org. Lett., 2010, 12, 5060–5063 CrossRef CAS PubMed
- W.-J. Yoo, T. Tsukamoto and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 6587–6590 CrossRef CAS PubMed
-
(a) D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS
-
(a) J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 2011, 829–856 CrossRef
- J. Yu, L. Zhang and G. Yan, Adv. Synth. Catal., 2012, 354, 2625–2628 CrossRef CAS PubMed
-
(a) H. C. Brown and T. E. Cole, Organometallics, 1983, 2, 1316–1319 CrossRef CAS
-
(a) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS PubMed
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |