
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ratiometric NMR pH sensing strategy based on a slow-proton-exchange (SPE) mechanism†
L. H.
Perruchoud
ab,
M. D.
Jones
a,
A.
Sutrisno
ab,
D. B.
Zamble
*ac,
A. J.
Simpson
*ab and
X.-a.
Zhang
*abd
aDepartment of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: xazhang@utsc.utoronto.ca; dzamble@chem.utoronto.ca; andre.simpson@utoronto.ca
bDepartment of Environmental and Physical Sciences, University of Toronto Scarborough, Toronto, ON M1C 1A4, Canada
cDepartment of Biochemistry, University of Toronto, Toronto, ON M5S 1A8, Canada
dDepartment of Biological Sciences, University of Toronto Scarborough, Toronto, ON M1C 1A4, Canada
First published on 20th July 2015
Abstract
Real time and non-invasive detection of pH in live biological systems is crucial for understanding the physiological role of acid–base homeostasis and for detecting pathological conditions associated with pH imbalance. One method to achieve in vivo pH monitoring is NMR. Conventional NMR methods, however, mainly utilize molecular sensors displaying pH-dependent chemical shift changes, which are vulnerable to multiple pH-independent factors. Here, we present a novel ratiometric strategy for sensitive and accurate pH sensing based on a small synthetic molecule, SPE1, which exhibits exceptionally slow proton exchange on the NMR time scale. Each protonation state of the sensor displays distinct NMR signals and the ratio of these signals affords precise pH values. In contrast to standard NMR methods, this ratiometric mechanism is not based on a chemical shift change, and SPE1 binds protons with high selectivity, resulting in accurate measurements. SPE1 was used to measure the pH in a single oocyte as well as in bacterial cultures, demonstrating the versatility of this method and establishing the foundation for broad biological applications.
Introduction
As a measure of proton activity, pH is a universally important parameter of our aqueous environment and biological milieu.1 In living organisms, acid–base homeostasis is essential for maintaining physiological functions and therefore requires tight regulation.2 A disrupted pH balance is associated with various abnormal states in biological systems. For example, low pH in humans has been linked to pathological conditions such as cystic fibrosis, ischemia and cancer,3 whereas elevated pH (alkalosis) may lead to hyperphosphatemia and hypocalcemia.4 The development of in vivo pH detection methods is currently of great importance for understanding the physiological roles of pH homeostasis as well as for disease diagnosis and therapeutic monitoring in cases where pH variation is a hallmark of the abnormality. It is possible to measure the pH in tissues by using conventional pH microelectrodes5 but the invasiveness and lack of spatial resolution is a major limitation. In contrast, fluorescence and bioluminescence imaging with optical pH sensors can report on pH with high spatial and temporal resolution,6 but they are restricted to superficial imaging depths due to light scattering and absorption. Although elegantly designed proof-of-principle methods based on other detecting techniques have emerged,7 noninvasive, accurate, and sensitive methods to measure the pH of living organisms remains an urgent challenge.Magnetic resonance (MR) based techniques can offer unlimited tissue penetration in a truly non-invasive manner, and versatile MR read-out methods are established for both spectroscopic and imaging purposes.8 The recent development of MRI contrast agents based on pH-dependent relaxivity9 and chemical exchange saturation transfer (CEST),10 which uses the saturation transfer of exchangeable protons to water, offer promise for in vivo pH mapping. These methods, however, often require specific calibration or external standards and high accuracy is difficult to achieve. The conventional and most widely used NMR and MR spectroscopic imaging (MRSI) methods for measuring pH rely on sensors that exhibit pH-dependent chemical shift changes, which can be monitored by 1H, 13C, 19F or 31P NMR signals.11 These pH sensors are typically small molecule acids or bases, such as phosphate12 or imidazole13 derivatives, with a pKa compatible with physiological conditions. They exist as a mixture of protonation states in vivo but exhibit only one set of NMR signals because the chemical exchange between these states is faster than the NMR time scale.14 The protons are highly mobile and rapid (de)protonation is facilitated by the hydrogen bond network of hydrated H+ in aqueous media, such that it exceeds the speed of diffusion.15 The observed average chemical shift of the conventional pH sensors is determined by the relative population of the protonated and unprotonated states and thus reflects the pH in solution. However, chemical shift is susceptible to artifacts caused by variations in ionic strength, local magnetic susceptibility, etc. In addition, the proton binding site (lone pair) of regular pH sensors will unavoidably be involved in interactions with metal ions, which will also induce pH-independent chemical shift changes and therefore experimental errors.16 An innovative strategy involving hyperpolarized 13C NMR techniques based on the pH-dependent equilibrium between carbon dioxide (CO2) and bicarbonate (HCO3−), which are in slow exchange in vivo, was recently explored.17 This approach, however, relies heavily on the carbonic anhydrase enzyme that catalyzes the interconversion between CO2 and HCO3−. These species are also components of pH-independent biomolecular processes, and the CO2 partial pressure is affected by the gas/solute equilibrium.18 In another strategy, a pilot study showed the possibility of 19F NMR pH sensing by ratio, when fast proton exchange is coupled with slow dissociation of intramolecular metal–ligand binding.19 The interaction of metal with other coordinative species in the aqueous media, such as HCO3−, however, perturbs the equilibrium between different protonation states.20 An ideal ratiometric MR pH sensor should have a slow proton exchange (SPE) on the NMR time scale, but still be fast enough for real time pH monitoring, and more importantly, its protonation equilibrium should not be affected by any factor other than pH.
In this paper, we report the first ratiometric 1H NMR pH sensing strategy to meet these criteria, based on a synthetic pH sensor, SPE1. This novel sensor is a cage-shaped urea cryptand with high proton selectivity and exhibits unusually slow interconversion rates between the different protonation states, which produce distinct NMR signals, allowing highly accurate ratiometric pH measurements. We demonstrate that this novel pH sensor is biocompatible and can be applied to monitor the pH in living biological systems, including fish oocytes and bacterial cultures.
Results and discussion
Principle and design of the SPE pH sensing strategy
The rapid chemical exchange between the non-protonated (B) and protonated (BH+) states of conventional pH probes makes it difficult to accurately measure the ratio of [B]/[BH+] directly by NMR, which is needed to calculate the pH value with the Henderson–Hasselbalch equation: pH = pKa + log[B]/[BH+].21 In contrast, SPE in protein structures is well documented.22 While amide or alcohol protons on the surface of a protein are in fast exchange with the surrounding aqueous solution, protons from similar groups deep in the protein core have restricted mobility due to the hydrophobicity of the local environment as well as their involvement in intramolecular hydrogen bonds.22 It is in principle possible to slow down proton exchange in synthetic molecules by introducing a sterically hindered hydrophobic environment and neighboring hydrogen bond acceptor groups that mimic protein structures. Small molecules with slow proton exchange however, are rare and have only been sporadically reported in the literature as unexpected findings.23 No systematic study has been conducted on exploring this unusual phenomenon.One molecule that displays such slow proton exchange properties is a tris-urea cryptand (1,4,6,9,12,14,19,21-octaazabicyclo[7.7.7]tricosane-5,13,20-trione)23b,c which we named SPE1 (Fig. 1). Both the bridgehead N-atoms in SPE1 adopt an endo conformation with the lone pair electrons pointing inside the molecular cavity. Upon protonation, the incoming protons are trapped inside the cage and stabilized in this position through intramolecular hydrogen bonding with the ureido oxygen atoms (Fig. 1).23c The proton transfer is sufficiently slow to allow direct NMR observation of both the protonated and the neutral forms of SPE1. The ratio between these two forms can therefore be used for accurate pH sensing. In addition, the size of the cryptand cavity is too small to bind any ions larger than H+, including Li+, the smallest metal cation.23b This minimizes the interaction with ions, which can perturb the chemical shift of conventional NMR pH sensors aforementioned. Other advantageous features of SPE1 include a pKa close to physiological pH and good water solubility. Moreover, because the molecule exhibits a mirror plane and C3 symmetry, the NMR spectrum is simple and unambiguous for peak assignment. Only 3 peaks are detected in the 1H NMR spectrum of neutral SPE1 in aqueous solution, one peak corresponding to the 6 urea protons and two peaks for 12 methylene protons each. Having more chemically-identical protons contributing to the intensity of a single peak in the spectrum increases sensitivity, which is one of the most common limitations of NMR.
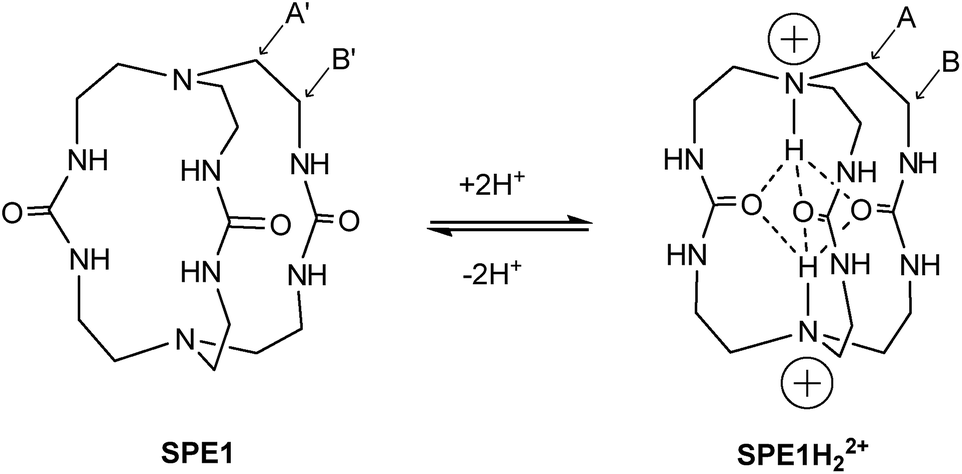 | ||
| Fig. 1 Structure and protonation states of the cage-shaped pH sensor SPE1. The protons attached to the bridgehead nitrogen atoms are trapped inside the cage due to hydrogen bonding with the ureido oxygen atoms, thereby allowing SPE between the two states. The 1H NMR signals of the labelled methylene positions are used for pH sensing. | ||
Synthesis of pH sensor
To test the applicability of the SPE strategy for pH sensing, a novel synthetic route was implemented to generate SPE1 in 3 steps, with a 38% overall yield (Scheme 1). SPE1 was synthesized from a tripodal amine, tris(2-aminoethyl)amine (tren), which was readily converted into an isothiocyanate derivative (3) upon treatment with carbon disulfide and N,N′-dicyclohexylcarbodiimide (DCC) in 78% yield, according to our previously published procedure.24 Rapid coupling of the isothiocyanate compound 3 with the trivalent amino counter partner, tren, under high dilution conditions generated the C3 symmetrical thiourea compound 2 in nearly quantitative yield. The thioureido groups in 2 were then converted to more water-soluble ureido analogs based on a reaction reported by Mikolajczyk in 1972,25 in which DMSO acts as the oxidant and solvent, in the presence of an acid catalyst. This convenient synthesis allows production of SPE1 in large scale, facilitating the following pH sensing studies.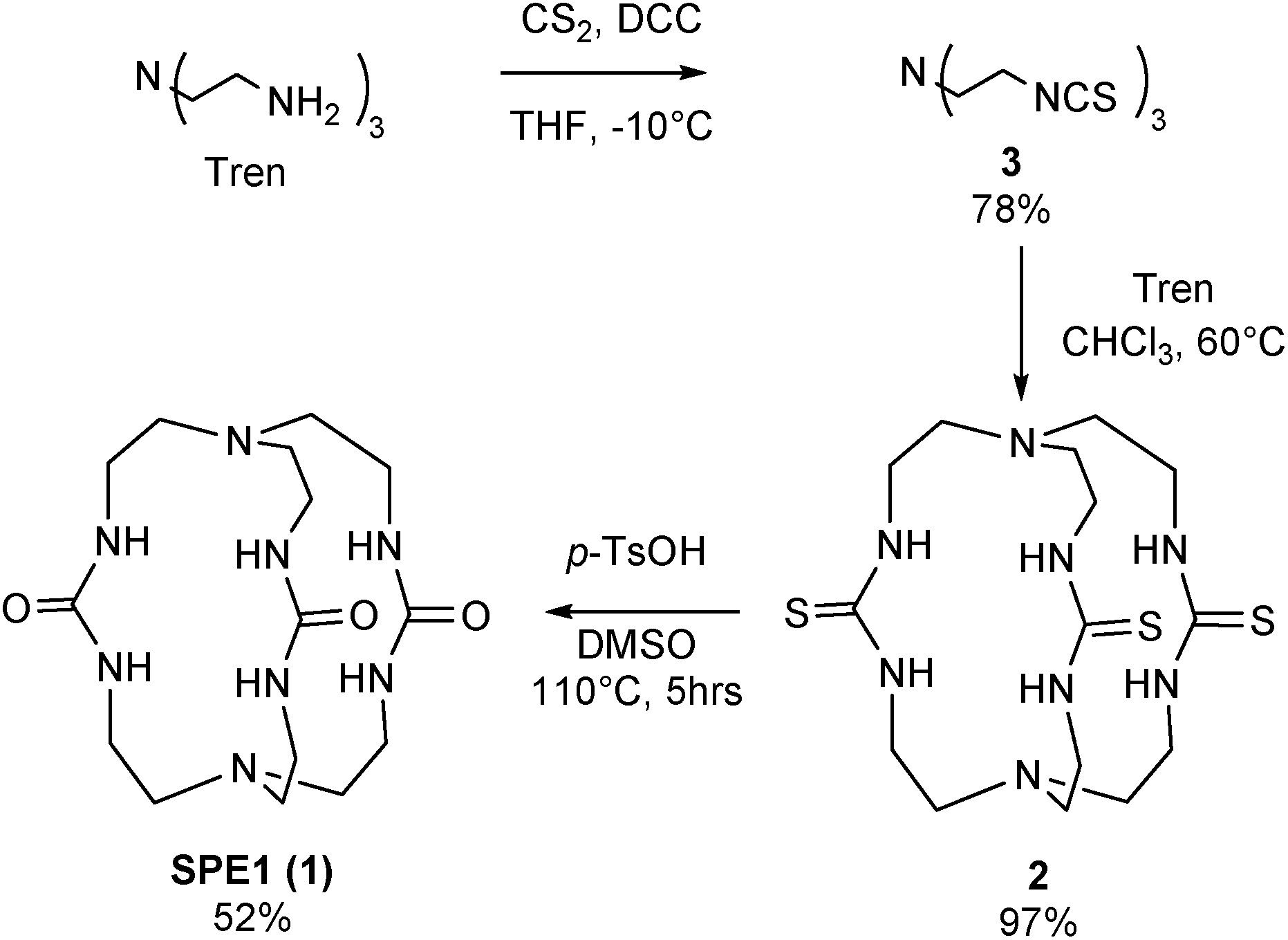 | ||
| Scheme 1 Synthesis of SPE1. | ||
Measurement of pH
Both bridgehead nitrogen atoms of SPE1 can be protonated under acidic conditions. Due to slow chemical exchange, the neutral SPE1 and its bis-protonated form (SPE1H22+) are simultaneously detected by 1H NMR as distinct species in aqueous solution. Notably, the mono-protonated form of SPE1 (SPE1H+) was not observed by NMR, due to the strong positive cooperativity in protonation (pKa2 > pKa1).23c This property greatly simplifies the NMR spectrum, as both neutral and bis-protonated SPE1 are highly symmetrical, enhancing the sensitivity of NMR signal detection. For pH calculations, a modified Henderson–Hasselbalch equation, which takes into account both protonation steps of SPE1, was used based on the ratio of neutral and bis-protonated SPE1: pH = pK′a + 1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log([SPE1]/[SPE1H22+]).
log([SPE1]/[SPE1H22+]).
In order to determine the apparent pKa, (pK′a = 1/2(pKa1 + pKa2)) and further demonstrate that SPE1 can be applied for accurate ratiometric pH sensing, a series of 1H NMR spectra of SPE1 dissolved in phosphate buffer at several pH values between 6.5 and 9.5 were collected (Fig. 2a). At room temperature, under basic conditions (pH ≥ 9), SPE1 is predominantly in the neutral form, producing two 1H NMR peaks at 2.58 and 3.13 ppm for both of the bridge methylene units (–CH2–CH2–). Upon gradual decrease in pH, the signals of SPE1H22+ emerge, as represented by new methylene peaks at 3.40 and 3.56 ppm, while the signals of neutral SPE1 remain detectable. As the pH decreases, the intensities of the SPE1 peaks diminish with simultaneous increase of the SPE1H22+ peaks, and the latter become dominant at pH 7 and below, confirming the relation between the solution pH and the SPE1/SPE1H22+ ratio. During the titration, a capillary with D2O was inserted into the NMR tube for a deuterium lock. Alternatively, a small amount of D2O can also be mixed directly with the NMR solution, since we observed that adding 10% D2O to the buffer solution did not change the SPE1/SPE1H22+ ratio significantly (Fig. S1†), indicating isotope impact from 10% D2O or less does not affect the accuracy of pH sensing. For quantitative analysis, pH was plotted against the fraction of neutral SPE1 (SPE1/[SPE1 + SPE1H22+]), obtained from the NMR integrals (Fig. 2b and ESI†). A similar titration curve was obtained at 37 °C. The apparent pKa for SPE1 was 8.00 ± 0.06 at 25 °C and 7.72 ± 0.07 at 37 °C.
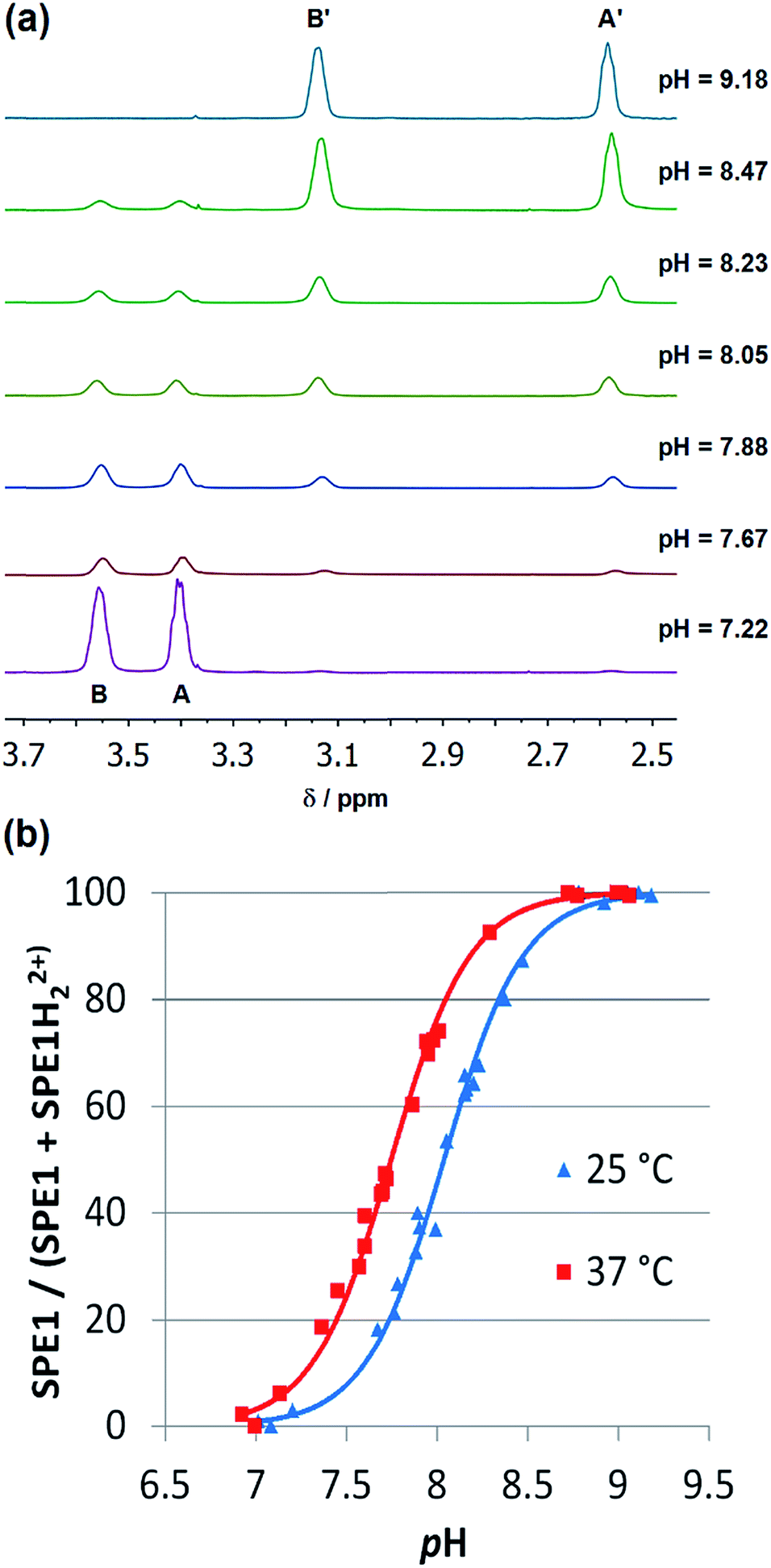 | ||
| Fig. 2 1H NMR pH titrations of SPE1 at 25 and 37 °C in phosphate buffer at 500 MHz. (a) Selected partial 1H NMR spectra of SPE1 at different pH values at 25 °C. Chemical shifts: (A) 3.40 ppm, (B) 3.56 ppm, (A′) 2.58 ppm, (B′) 3.13 ppm. (b) Ratiometric curves of 1H NMR pH titrations derived from the ratio of the different protonation states of SPE1. | ||
These numbers are in agreement with the pKa determined by potentiometric titration,23b confirming that the pH measured by the current ratiometric approach is comparable to the conventional pH electrode. At body temperature, SPE1 can operate as a pH sensor between pH 6.7 to 8.7, covering slightly acidified to mildly basic conditions. In addition, this method is very sensitive. Within a pH window close to the pKa of SPE1, differences as small as 0.02 pH units could be experimentally observed (Fig. 3).
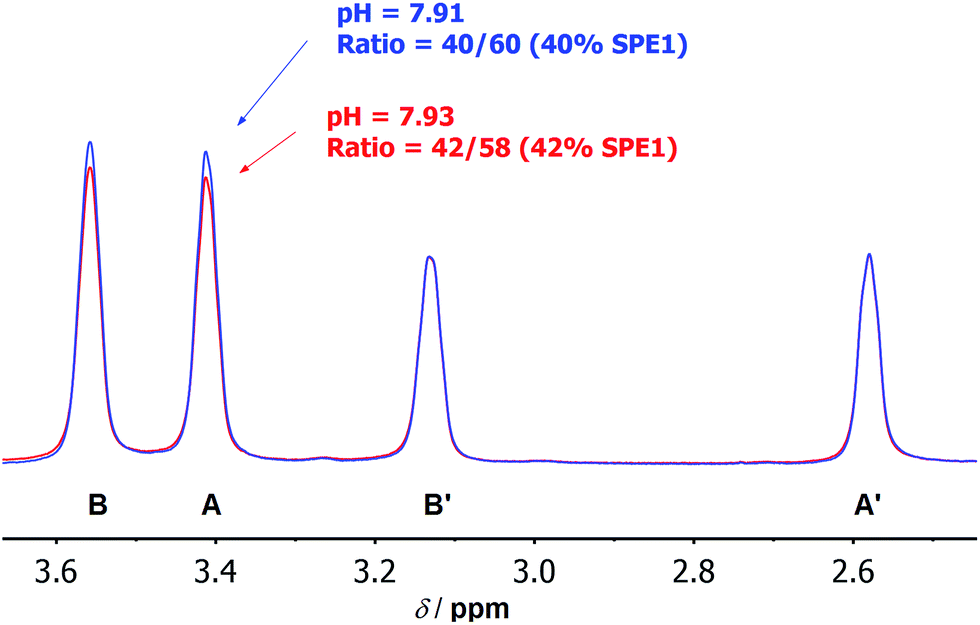 | ||
| Fig. 3 Selected 1H NMR spectra at 500 MHz showing the high accuracy of pH measurement by SPE1. Overlay of local 1H NMR spectra of SPE1 at pH 7.91 (blue) and 7.93 (red). The peak intensity was normalized to the signals of neutral SPE1 at 2.58 and 3.13 ppm. A difference in pH of 0.02 pH units can be detected. | ||
Biological applications of SPE1
Various microorganisms, including E. coli cells can grow in both aerobic and anaerobic culture, and are known to increase production of acidic metabolites in response to low oxygen stress.29 To monitor this process in real time by NMR, we conducted a kinetic study of concentrated E. coli culture (1 ml aliquot at OD600 = 1) in a sealed 4 mm NMR rotor at 37 °C and recorded the change in pH over time using SPE1 (1.8 mM, Fig. 4). An initial pH of 7.55 was determined from the intensity ratio 31/69 (31% for neutral SPE1). The solid NMR rotor insert remained sealed in the spectrometer and new 1H NMR spectra were acquired every 15 minutes. A continuous slow increase in the intensities of the SPE1H22+ peaks with a diminution of the peak intensities of SPE1 was observed. The high accuracy of the SPE-based method allowed precise measurements of small pH changes over 3 hours from pH 7.55 to 6.95 (Fig. 4). Interestingly, in conjunction with the gradual decrease of pH, two new sharp peaks appeared in the 1H NMR spectra and increased in intensity over the course of the experiment. The chemical shifts of 2.40 and 1.92 ppm of these singlet peaks are consistent with succinate and acetate, which are common metabolites observed in bacterial cultures growing with limited oxygen availability.30 It is known that bacteria modify their metabolism upon switching from aerobic to micro-aerobic or anaerobic conditions, by increasing the glycolysis rate with a concomitant decrease of acetyl-CoA degradation by the citric acid cycle.31 This adjustment causes an overall increase in proton concentration as well as other acidic metabolites such as acetate and succinate.32 Therefore our experiments confirmed that SPE1 was able to accurately monitor pH changes in real time in a biocompatible and reproducible manner and recorded the alteration of metabolism in live bacterial cultures deprived of oxygen. The current setup does not allow determination of the precise location of SPE1 within cells. Future work will involve the development of new SPE-based pH sensors with controllable cell-permeability and subcellular localization.
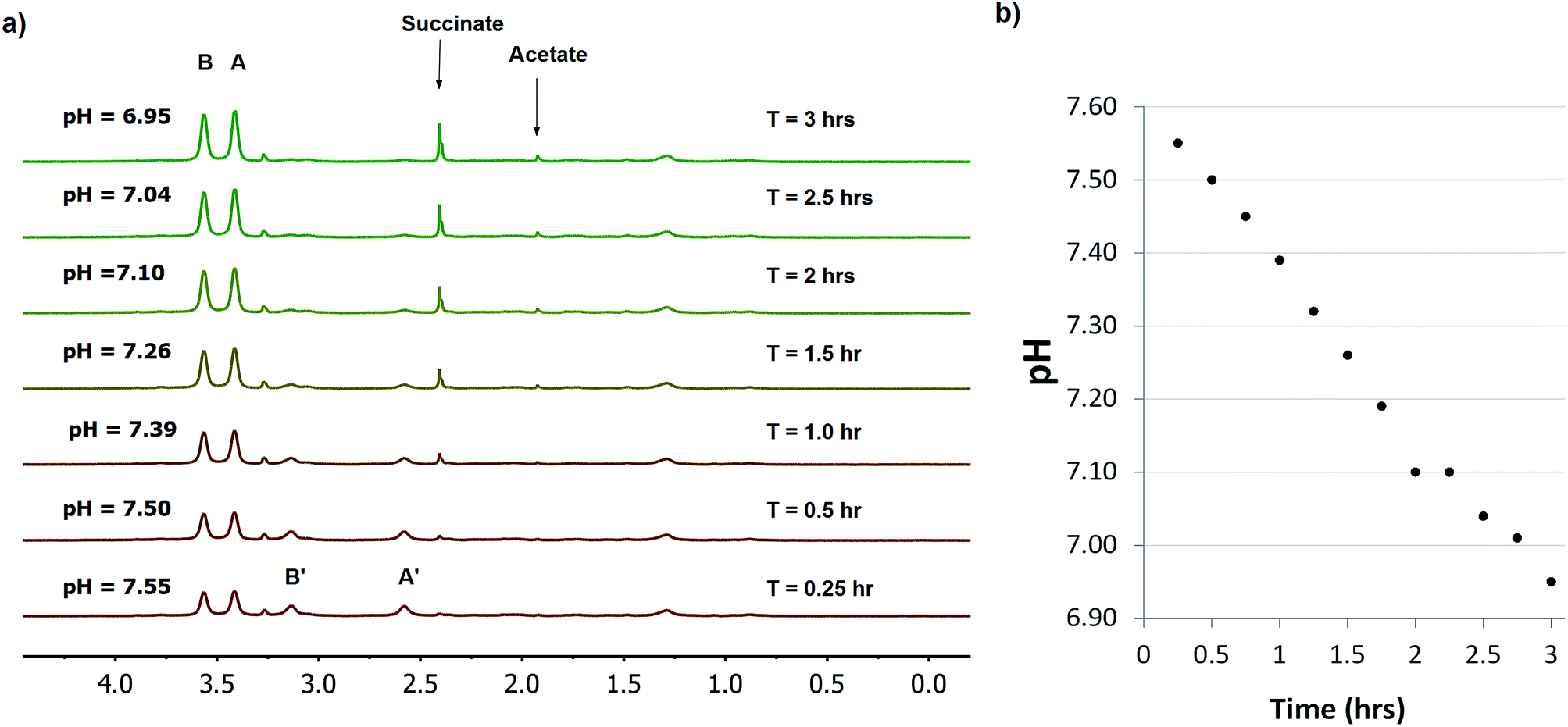 | ||
| Fig. 4 Monitoring pH in E. coli (OD600 = 1) using a 1.8 mM solution of SPE1 in phosphate buffer. (a) Selected stacked 1H NMR spectra of E. coli cells at 500 MHz in the presence of 1.8 mM SPE1. NMR measurements were taken continuously for 3 h using 256 scans (15 min intervals). (b) Graph of the decrease of pH over time of SPE1 treated E. coli cells. | ||
Experimental
Details for general experimental procedures, syntheses and characterization of all compounds can be found in the ESI.† All 1H NMR spectra were manually corrected for phase and baseline distortion using TopSpin™ 3.1 and MestReNova 8.1.4 and integral ratios were obtained by taking ±35 Hz around each peak. The chemical shifts were first calibrated to DSS as an internal standard, where the peaks of the neutral SPE1 appeared at 2.58 and 3.13 ppm. The chemical shifts were then referenced relative to the peaks of neutral SPE1.NMR monitored pH calibration of SPE1
A solution of SPE1 (2 mM) was dissolved in 10 mM phosphate buffer (pH = 7.4). Aliquots of 500 μl were prepared at different pH values by addition of HCl or NaOH, and the pH was measured using a calibrated pH electrode (Cole Parmer Thermo Scientific Orion pH microelectrode) and a VWR sympHony SB70P pH meter. The sample was placed in a 5 mm NMR tube with a sealed capillary filled with D2O. 1H NMR spectra were obtained at 25 °C or 37 °C with 64 scans per sample on a 500 MHz Bruker Avance spectrometer with presaturation of the water signal, a recycle time of 50 s and a 90° pulse width. The experimental data were fitted with MATLAB software using non-linear least square regression.Measurement of intracellular pH of Belonidae oocytes
768 time domain points were acquired for each spectrum with a spectral width of 20 ppm. Data were zero filled and multiplied by an exponential window function corresponding to a 1 Hz line broadening in the transformed spectrum.
Real time pH monitoring of E. coli culture
000g and re-suspended in 30 μl of a 1.8 mM solution of SPE1 in 10 mM phosphate buffer pH 8.0 containing 10% D2O. Another aliquot was collected and re-suspended in phosphate buffer to act as a blank for the NMR experiment and a control for cell viability over the course of the experiment. The sample was transferred to an NMR top insert made from Kel-F, sealed with a Kel-F sealing screw and cap, then inserted into a 4 mm o.d. zirconium rotor for the NMR experiment.
To test for viability of the sensor-free and sensor-treated cells, the cells were serially diluted 103 to 109 times in phosphate buffer after the NMR experiments and plated on LB-Amp plates to determine cell survival during the experiment.
768 time domain points with spectral widths of 20 ppm. Data were zero filled and multiplied by an exponential window function corresponding to a 1 Hz line broadening in the transformed spectrum.
Conclusions
We reported a novel and versatile strategy for ratiometric 1H NMR pH sensing based on a slow proton exchange (SPE) mechanism. A water-soluble small molecule cryptand SPE1 was prepared through a new synthetic route and was evaluated in vitro and in live cells for ratiometric NMR pH sensing. Slow chemical exchange between different protonation states and high proton selectivity of SPE1 were achieved by shielding the incoming protons inside the small molecular cavity and trapping them with intramolecular hydrogen bonding. Unlike typical small molecule acids or bases, which exhibit a single set of average NMR signals, SPE1 displays distinct peaks for the neutral and protonated forms due to unusual slow chemical exchange. It is therefore possible to use the ratio of NMR peak intensities to provide highly precise pH values of the aqueous media. The new approach is more robust, sensitive and accurate than conventional chemical-shift based methods, which are vulnerable to many pH-independent factors. SPE1 exhibits an apparent pKa value suitable for biological applications and shows no toxicity effects on cell cultures. Therefore the new method was applied to measure the pH in a single live fish oocyte, and to monitor the real time pH changes of a bacterial culture. Overall, SPE1 has great potential for measuring and mapping pH and pH changes in living systems. Next generation pH sensors based on the SPE mechanism are currently under development to cover different pH windows, which can further expand the scope of biological applications of this new strategy.Acknowledgements
This work was mainly supported by NSERC through a Discovery Grant (# 489075) and Connaught New Researcher Award to X.-a. Z., and a CIHR Operating Grant to D. B. Z. We are also grateful to the University of Toronto, Ontario Research Fund and Canada Foundation for Innovation. M. D. J. was supported in part by the Ontario Graduate Scholarship program. A. J. S. would like to thank the Mark Krembil and the Krembil Foundation for funding the hardware used in this research. We thank Prof. Nathan R. Lovejoy for providing us with the Belonidae oocytes.References
-
R. G. Bates, Determination of pH; Theory and Practice, Wiley, New York, 2nd edn, 1973 Search PubMed
.
-
(a) J. R. Casey, S. Grinstein and J. Orlowski, Nat. Rev. Mol. Cell Biol., 2010, 11, 50–61 CrossRef CAS PubMed
-
(a) A. A. Pezzulo, X. X. Tang, M. J. Hoegger, M. H. Abou Alaiwa, S. Ramachandran, T. O. Moninger, P. H. Karp, C. L. Wohlford-Lenane, H. P. Haagsman, M. van Eijk, B. Banfi, A. R. Horswill, D. A. Stoltz, P. B. McCray, M. J. Welsh and J. Zabner, Nature, 2012, 487, 109–113 CrossRef CAS PubMed
- R. Krapf, P. Jaeger, H. N. Hulter, C. Fehlman and R. Takkinen, Kidney Int., 1992, 42, 727–734 CrossRef CAS PubMed
-
(a) W. J. Waddell and R. G. Bates, Physiol. Rev., 1969, 49, 285–329 CAS
-
(a) J. Y. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS PubMed
- A. L. Vavere, G. B. Biddlecombe, W. M. Spees, J. R. Garbow, D. Wijesinghe, O. A. Andreev, D. M. Engelman, Y. K. Reshetnyak and J. S. Lewis, Cancer Res., 2009, 69, 4510–4516 CrossRef CAS PubMed
-
J. C. Lindon, G. E. Tranter and D. W. Koppenaal, in Encyclopedia of Spectroscopy and Spectrometry, Academic Press, Amsterdam, Boston, 2nd edn, 2010 Search PubMed
-
(a) S. R. Zhang, K. C. Wu and A. D. Sherry, Angew. Chem., Int. Ed., 1999, 38, 3192–3194 CrossRef CAS
-
(a) K. M. Ward and R. S. Balaban, Magn. Reson. Med., 2000, 44, 799–802 CrossRef CAS
-
(a) R. J. Gillies, N. Raghunand, M. L. Garcia-Martin and R. A. Gatenby, IEEE Eng Med Biol Mag., 2004, 23, 57–64 CrossRef
-
(a) R. B. Moon and J. H. Richards, J. Biol. Chem., 1973, 248, 7276–7278 CAS
- R. van Sluis, Z. M. Bhujwalla, N. Raghunand, P. Ballesteros, J. Alvarez, S. Cerdan, J. P. Galons and R. J. Gillies, Magn. Reson. Med., 1999, 41, 743–750 CrossRef CAS
- R. G. Bryant, J. Chem. Educ., 1983, 60, 933–935 CrossRef CAS
-
(a) D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS
-
(a) V. Asiago, G. A. Nagana Gowda, S. Zhang, N. Shanaiah, J. Clark and D. Raftery, Metabolomics, 2008, 4, 328–336 CrossRef CAS
-
(a) F. A. Gallagher, M. I. Kettunen, S. E. Day, D. E. Hu, J. H. Ardenkjaer-Larsen, R. Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–943 CrossRef CAS PubMed
-
(a) J. R. Casey, Biochem. Cell Biol., 2006, 84, 930–939 CrossRef CAS PubMed
- P. K. Senanayake, A. M. Kenwright, D. Parker and S. K. van der Hoorn, Chem. Commun., 2007, 2923–2925 RSC
- A. M. Kenwright, I. Kuprov, E. de Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC
- L. J. Henderson, Am. J. Physiol., 1908, 21, 173–179 CAS
- M. D. Finucane and O. Jardetzky, Protein Sci., 1996, 5, 653–662 CrossRef CAS PubMed
-
(a) J. Cheney and J. M. Lehn, J. Chem. Soc., Chem. Commun., 1972, 487–489 RSC
-
(a) X.-a. Zhang and W. D. Woggon, J. Am. Chem. Soc., 2005, 127, 14138–14139 CrossRef CAS PubMed
- M. Mikolajczyk and J. Luczak, Chem. Ind., 1972, 76 CAS
-
(a) Y. Zhu, C. D. Rice, Y. Pang, M. Pace and P. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2231–2236 CrossRef CAS PubMed
-
(a) Z. Serber, P. Selenko, R. Hansel, S. Reckel, F. Lohr, J. E. Ferrell, G. Wagner and V. Dotsch, Nat. Protoc., 2006, 1, 2701–2709 CrossRef CAS PubMed
-
(a) C. H. Johnson and D. Epel, Dev. Biol., 1982, 92, 461–469 CrossRef CAS
- G. Unden and J. Bongaerts, Biochim. Biophys. Acta, Bioenerg., 1997, 1320, 217–234 CrossRef CAS
-
(a) J. D. Partridge, G. Sanguinetti, D. P. Dibden, R. E. Roberts, R. K. Poole and J. Green, J. Biol. Chem., 2007, 282, 11230–11237 CrossRef CAS PubMed
-
D. Voet and J. G. Voet, Biochemistry, Wiley, Hoboken, NJ, 4th edn, 2011 Search PubMed
- Y. Matsuoka and K. Shimizu, Biotechnol. J., 2011, 6, 1330–1341 CrossRef CAS PubMed
- P. Selenko, Z. Serber, B. Gadea, J. Ruderman and G. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11904–11909 CrossRef CAS PubMed
- A. J. Simpson and S. A. Brown, J. Magn. Reson., 2005, 175, 340–346 CrossRef CAS PubMed
-
(a) M. L. Liu, X. A. Mao, C. H. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of 2, 3 and SPE1 (1), theoretical model of NMR pH titration and use of deuterium lock (Fig. S1), 1H NMR spectrum of sensor injected oocyte (Fig. S2), spectral editing of pH monitoring experiment (Fig. S3) and cell-permeability test (Fig. S4). See DOI: 10.1039/c5sc02145f |
| This journal is © The Royal Society of Chemistry 2015 |