
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed direct transformation of simple alkynes to alkenyl nitriles via aerobic oxidative N-incorporation†
Xiaoqiang
Huang
a,
Xinyao
Li
a and
Ning
Jiao
*ab
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 20th July 2015
Abstract
A novel direct transformation of aliphatic terminal alkynes to alkenyl nitriles through the incorporation of a nitrogen atom into the simple hydrocarbons has been reported. The usage of inexpensive copper catalyst, O2 as the sole oxidant, broad substrate scope as well as feasibility for “late-stage modification” make this protocol very promising. Mechanistic studies including DFT calculation demonstrate a novel 1,2-hydride shift process for this novel nitrogenation reaction.
Introduction
Direct transformation of simple and readily available hydrocarbons into complex and high value-added compounds is a long-standing topic in organic synthesis.1 Aliphatic alkynes, which are very common and easily accessible building blocks, provide chemists with a fertile testing ground for the construction of complex organic molecules.2–8 Many useful reactions of these simple hydrocarbons have been disclosed on the basis of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond transformation, such as coupling,3,4 addition,5 cyclization,6 and metathesis reactions.7 However, the transformation of simple aliphatic terminal alkynes involving the cleavage of the propargylic C(sp3)–H bond is still limited.8,9
C triple bond transformation, such as coupling,3,4 addition,5 cyclization,6 and metathesis reactions.7 However, the transformation of simple aliphatic terminal alkynes involving the cleavage of the propargylic C(sp3)–H bond is still limited.8,9
Recently, novel transformation of simple alkynes has been disclosed through the assistance of transition metals. Yamamoto's group significantly developed palladium/acid catalyzed alkylation and hydroamination reaction of internal alkynes with nucleophiles (Scheme 1, A).10 By using a Rh(I)/phosphine ligand/benzoic acid catalyst system, Breit and co-workers pioneeringly achieved the intermolecular coupling of aliphatic terminal alkynes with carboxylic acids and sulfonyl hydrazides under redox-neutral conditions (Scheme 1, B).11 Despite these breakthroughs, new catalytic systems and new strategies are highly desirable to disclose novel transformations of aliphatic terminal alkynes.
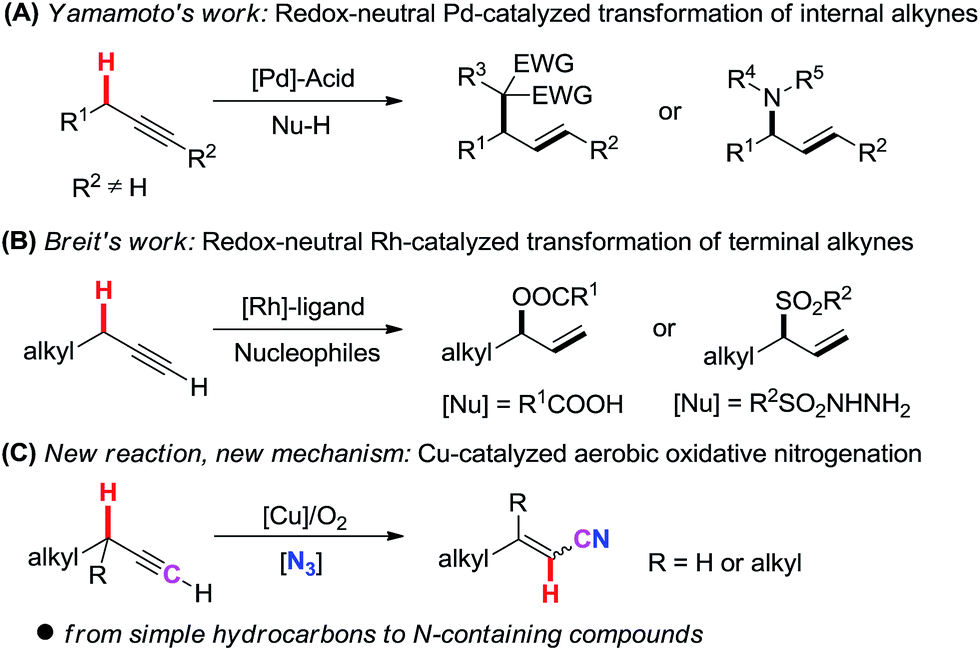 | ||
| Scheme 1 Direct transformation of simple alkynes involving the cleavage of a propargylic C(sp3)–H bond. | ||
Herein, we report a novel Cu-catalyzed aerobic oxidative transformation of simple terminal alkynes to alkenyl nitriles (Scheme 1, C). In this present chemistry: (1) a very simple hydrocarbon is successfully converted into an N-containing compound through the incorporation of a nitrogen atom into the substrate; (2) inexpensive Cu-catalyst, the green molecular oxygen oxidant, as well as the broad substrate scope make this protocol very attractive and low-cost; (3) a novel propargylic C(sp3)–H bond cleavage through 1,2-H shift mechanism is proved. (4) DFT calculation reasonably explains the mechanism and the stereoselectivity of products.
Results and discussion
We commenced this research by choosing commercially available 5-phenyl-1-pentyne 1a as the model substrate. To our delight, the nitrogenation product 2a was obtained with azidotrimethylsilane (TMSN3) as nitrogen source under copper-catalyzed aerobic conditions (Table 1). After extensive screening of different reaction parameters (see ESI† for more information), the direct functionalization of 1a gave 2a in 78% yield with a slight Z-selectivity (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E = 65:35) under the optimized conditions: TMSN3 (2.0 equiv.), CuBr (20 mol%), pyridine (2.0 equiv.) and NaOAc (1.0 equiv.), in PhCl at 90 °C under O2 (1.0 atm) for 48 h (entry 1, Table 1). As expected, only trace amount of 2a could be obtained under an argon atmosphere (entry 2). Copper catalyst is essential in this transformation, as no 2a was formed without copper salt or with other common metal salts (such as [Ag], [Fe], [Co], [Mn], see ESI†). Other copper salts showed lower efficiencies than CuBr (entries 4–5). Further studies indicated that the reaction did not work in the absence of pyridine (entry 7). DMEDA and L-proline led to no reaction (entries 8–9). Catalytic amount of pyridine only delivered 2a in 26% yield (entry 10). It is noteworthy that NaOAc, a very weak base, is the most effective additive while not indispensable for product formation (entries 11–13).12 However, great efforts to improve the Z:E ratio of the product did not reach a satisfying result (see ESI† for more information).
:E = 65:35) under the optimized conditions: TMSN3 (2.0 equiv.), CuBr (20 mol%), pyridine (2.0 equiv.) and NaOAc (1.0 equiv.), in PhCl at 90 °C under O2 (1.0 atm) for 48 h (entry 1, Table 1). As expected, only trace amount of 2a could be obtained under an argon atmosphere (entry 2). Copper catalyst is essential in this transformation, as no 2a was formed without copper salt or with other common metal salts (such as [Ag], [Fe], [Co], [Mn], see ESI†). Other copper salts showed lower efficiencies than CuBr (entries 4–5). Further studies indicated that the reaction did not work in the absence of pyridine (entry 7). DMEDA and L-proline led to no reaction (entries 8–9). Catalytic amount of pyridine only delivered 2a in 26% yield (entry 10). It is noteworthy that NaOAc, a very weak base, is the most effective additive while not indispensable for product formation (entries 11–13).12 However, great efforts to improve the Z:E ratio of the product did not reach a satisfying result (see ESI† for more information).
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
Z:Ec |
| a Standard conditions: 1a (0.40 mmol), TMSN3 (0.80 mmol), CuBr (0.08 mmol), pyridine (0.80 mmol) and NaOAc (0.40 mmol) in PhCl (2.0 mL) under O2 (balloon) was stirred at 90 °C for 48 h. b Isolated yields. c Determined by 1H NMR measurement of the crude mixture. DMEDA = N,N′-dimethyl-1,2-ethanediamine. | |||
| 1 | None | 78 |
63:35 |
| 2 | Ar instead of O2 | Trace | — |
| 3 | Without CuBr | 0 | — |
| 4 | Cu(OAc)2 instead of CuBr | 49 | 69:31 |
| 5 | CuBr2 instead of CuBr | 62 | 67:33 |
| 6 | 10 mol% CuBr was employed | 45 | 65:34 |
| 7 | Without pyridine | 0 | — |
| 8 | DMEDA instead of pyridine | 0 | — |
| 9 | L-proline instead of pyridine | 0 | — |
| 10 | 0.4 equiv. pyridine was employed | 26 | 69:31 |
| 11 | Without NaOAc | 48 | 64:36 |
| 12 | LiOAc instead of NaOAc | 48 | 64:36 |
| 13 | NaOMe instead of NaOAc | 47 | 67:33 |

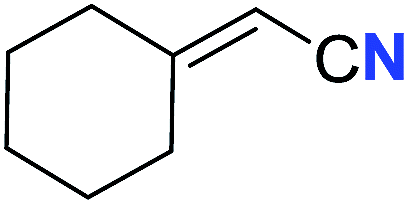
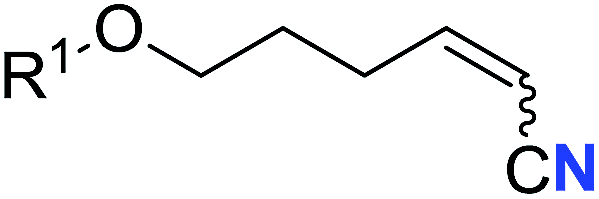
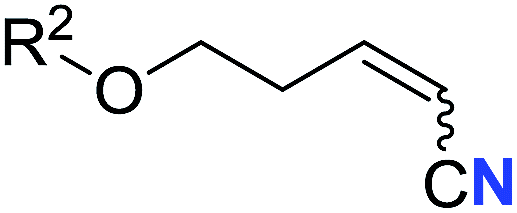
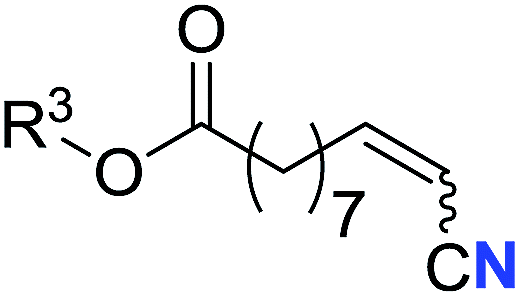
With the optimal conditions in hand, we next investigated the substrate scope of this transformation. This reaction exhibited a good functional group compatibility (Table 2). Long-chain-alkyl substituted alkynes were successfully transformed to the corresponding alkenyl nitriles in good yields (2a–2e). Notably, propargylic 3° C–H of 1f could be cleaved, giving 2f in 61% yield. To our satisfaction, terminal alkyne 1g, bearing a TBDMS protected hydroxyl group, worked well (2g, 68%). Remarkably, linkages, including ether bonds (2h–2n) and ester bonds (2o–2q), did not reduce effectiveness. Several functional groups (trifluoromethyl, chlorine, vinyl and thienyl) were well tolerated in the present catalytic system. Furthermore, reasonable yields were obtained for alkynes containing phthalimide and sulfonamide group, respectively (2r–2s). Interestingly, C–H bond adjacent to internal ethynyl group was inactive, which leads to the high regioselectivity.
|
|
||||
|---|---|---|---|---|
| Entry | 2 | Yield of 2b (%) |
Z:Ec |
|
| a Standard conditions: see entry 1, Table 1. b Isolated yields. c Determined by 1H NMR measurement of the crude mixture. d Two portions of TMSN3 (0.60 mmol) were added every 24 h. | ||||
| 1 |
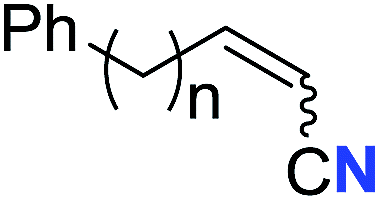
|
n = 3 | 78 (2a) | 65:35 |
| 2 | n = 2 | 44 (2b) | 67:33 |
|
| 3 |
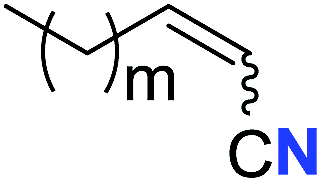
|
m = 7 | 76 (2c) | 61:39 |
| 4 | m = 6 | 62 (2d) | 66:34 |
|
| 5 |
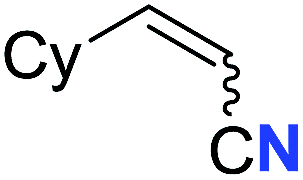
|
63 (2e) | 60:40 |
|
| 6 |
|
61 (2f) | — | |
| 7 |
|
R1 = TBDMS | 68 (2g) | 69:31 |
| 8 | R1 = n-C9H19 | 59 (2h) | 69:31 |
|
| 9 |
|
R2 = C6H5 | 60 (2i) | 68:32 |
| 10 | R2 = 4-MeOC6H4 | 50 (2j) | 69:31 |
|
| 11 | R2 = 4-CF3C6H4 | 66 (2k) | 63:37 |
|
| 12 | R2 = 2-ClC6H4 | 71 (2l) | 66:34 |
|
| 13 | R2 = 1-naphth | 60 (2m) | 64:36 |
|
| 14 | R2 = 2-naphth | 57 (2n) | 64:36 |
|
| 15 |
|
R3 = Me | 69 (2o) | 69:31 |
| 16d | R3 = 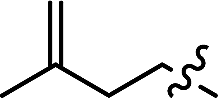 |
40 (2p) | 64:36 |
|
| 17 |
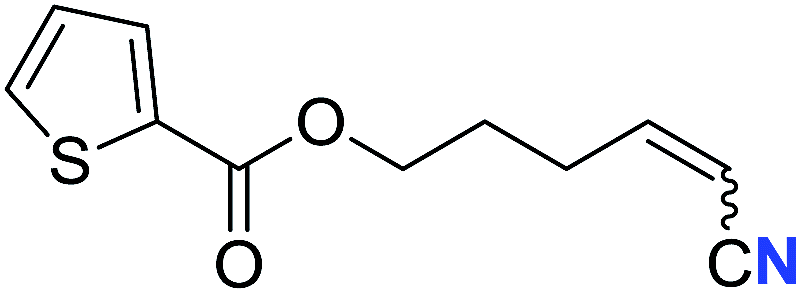
|
73 (2q) | 66:34 |
|
| 18 |
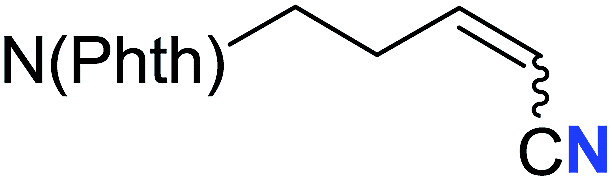
|
65 (2r) | 69:31 |
|
| 19 |
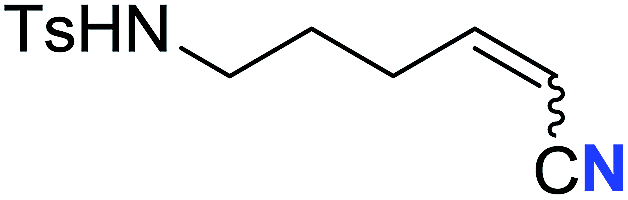
|
61 (2s) | 70:30 |
|
| 20 |
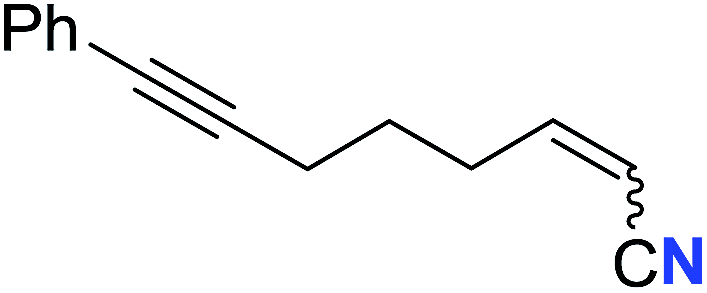
|
46 (2t) | 66:34 |
|

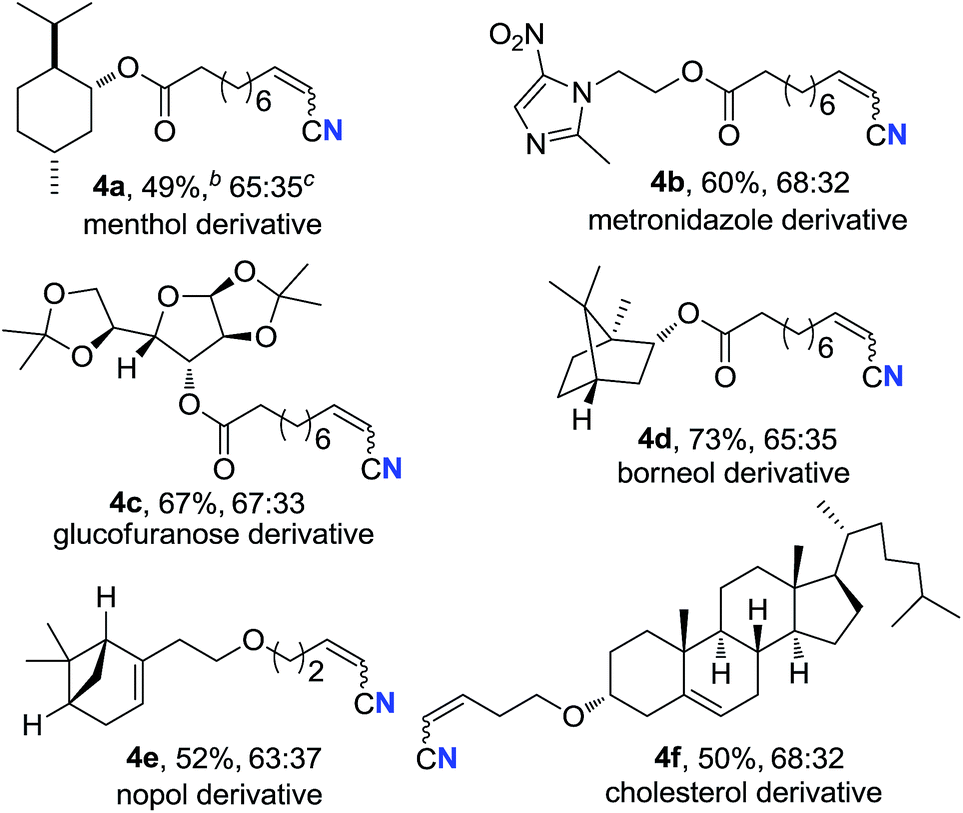
Alkenyl nitriles are not only useful building blocks in synthetic chemistry but also important structure motifs commonly found in drugs.13 Moreover, late-stage modification is a highly valuable strategy for medicinal chemistry research.14 Therefore, several complex bioactive molecule derivatives were submitted to the optimal conditions (Table 3). Natural alcohol derivatives containing ester or ether linkages, such as menthol, borneol, nopol and cholesterol, worked well in the current transformation, generating the corresponding alkenyl nitriles in 49–73% yield (4a, 4d–f), respectively. Alkyne 3b that was prepared from antibacterial metronidazole afforded alkyl alkenyl nitrile 4b in 60% yield. Besides, terminal alkyne with a protected sugar moiety selectively underwent aerobic oxidation, giving nitrogenation product 4c in 67% yield. These results demonstrate that the present protocol could be applied in late-stage bioactive compound modification.

To gain mechanistic insight into this transformation, some control experiments were conducted under the standard conditions. Allene 5, which could be generated from alkyne 1a, failed to afford nitriles under the present conditions (eqn (1)), indicating a novel mechanism different from Breit's works.11 In addition, propargylic azide 6 or allylic azide 7 could not furnish alkenyl nitriles either (eqn (2) and (3)). These results ruled out the possibility of 6 and 7 as intermediates of the transformation.15
 | (1) |
 | (2) |
 | (3) |
Considering that the current nitrogenation reaction could only be catalyzed by copper salt, and the Glaser–Hay homocoupling product3a could be detected in some cases, we postulated that copper acetylide might be an intermediate of this reaction. Although, no product was formed employing copper(I)-acetylide 8 as a substrate (eqn (4)), which might due to the aggregation of 8,162c could be obtained in comparative yield when 8 was used as a catalyst (eqn (5)). When C(sp)–H bond deuterated alkynes 1a-1-d1 was subjected to the reaction, no deuterium was detected in the product, which is in accordance with the existence of copper acetylide species (eqn (6)).
 | (4) |
 | (5) |
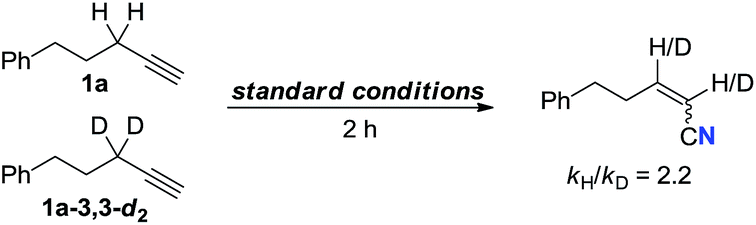
Furthermore, labeling experiment with propargylic C–H bond deuterated 1a-3,3-d2 was performed. To our surprise, nearly 100% incorporation of deuterium at the both α and β positions of the nitrile was observed (eqn (7)). Hence, the cleavage of propargylic C–H bond might proceed via a 1,2-hydride shift.17 Then, an intermolecular kinetic isotopic experiment was conducted giving the result of kH/kD = 2.2 (eqn (8)).18
 | (6) |
 | (7) |
 | (8) |
On the basis of all these results and previous reports, a proposed mechanism is depicted in Scheme 2. The reasonable first step is the formation of copper(I)-acetylide intermediate A.16 Then, copper triazolide B formed via Cu-catalyzed azide–alkyne cycloaddition (CuAAC)19 undergoes ring-opening reaction affording cuprated diazoimine C.20,21 The oxidation of C under aerobic conditions with assistance of pyridine gives α-diazonitrile D and regenerates the copper(I) catalyst.22 Subsequently, upon loss of dinitrogen D would afford carbene E or copper carbene F.23 Finally, 1,2-hydride shift of the carbene species generates the alkenyl nitrile.17 Alternatively, a mechanism with ethynyl azide could also be possible. Cu-catalyzed aerobic oxidative cross-coupling of terminal alkynes with TMSN3 might generate ethynyl azide G,3b,4 which is known to liberate dinitrogen leading to the formation of cyanocarbene species E.24
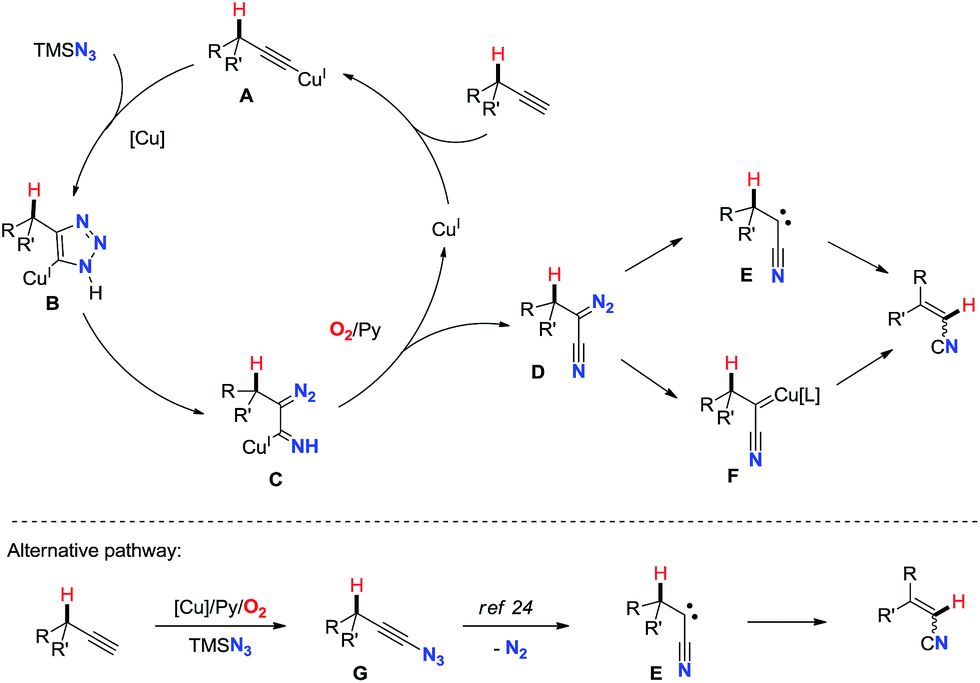 | ||
| Scheme 2 Proposed mechanism. | ||
To further explore the stereoselectivity of the reaction, density functional theory (DFT) calculation investigation was carried out (Fig. 1).25 After the sequential CuAAC19 and ring-opening process,20,21 the α-diazonitrile INT1 is generated (see ESI† for details). The pyrolysis of INT1 has two pathways. In pathway A, the thermal induced release of N2 through TS1 requires an activation free energy of 22.3 kcal mol−1 to give cyanocarbene carbene INT2. Alternatively, INT2 generated from ethynyl azide could not be excluded.24 The subsequent 1,2-hydride shift process17viaZ-TS2 and E-TS2 almost barrierlessly delivers Z-2 and E-2, respectively. It is noteworthy that the energy barrier gap between Z-TS2 and E-TS2 is insignificant (only 0.3 kcal mol−1), which might be due to the similar steric hindrance between hydrogen and cyano group. The calculated Z:E ratio of 2via pathway A is predicted to be 64:36, which is qualitatively consistent with the experimentally observed 66:34 Z:E ratio for this reaction.
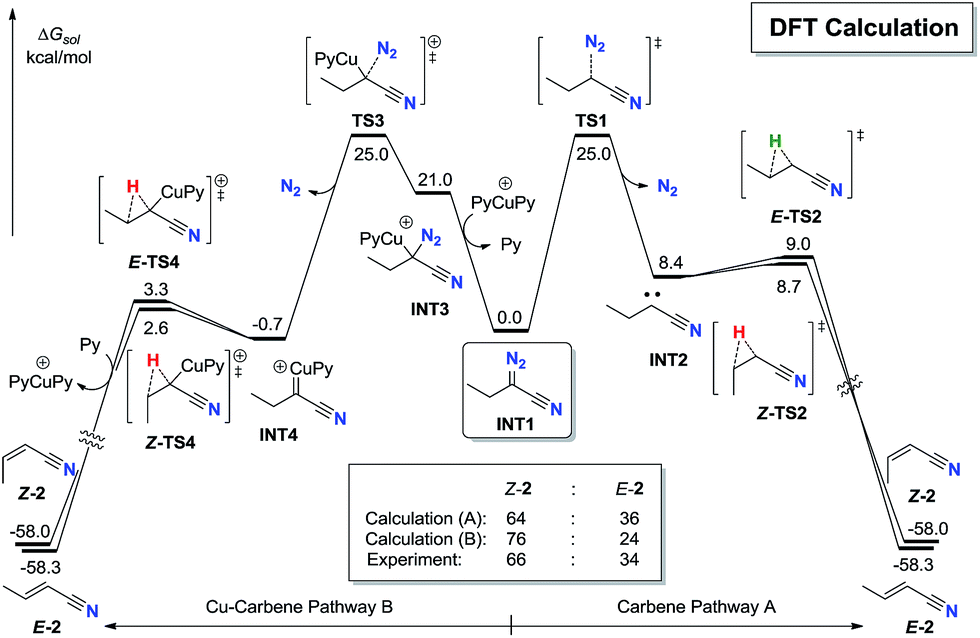 | ||
| Fig. 1 DFT-computed energy profiles. | ||
In alternative pathway B, Cu(I) catalyst can induce the Cu–C bond formation on INT3 with the release of N2 through TS3 in a stepwise manner, which requires an activation free energy of 22.3 kcal mol−1 to form Cu-carbene INT4.23 The subsequent 1,2-H shift process17viaZ-TS4 and E-TS4 also barrierlessly furnishes Z-2 and E-2, respectively. Notably, Z-TS4 is also only 0.7 kcal mol−1 lower in energy than E-TS4, which is corresponding to a 76:24 Z:E ratio of 2, in good agreement with the experimental observation.
Moreover, E-2 is only 0.3 kcal mol−1 lower in energy than Z-2, indicating that the Z to E isomerization of alkenyl nitriles 2 is short of driving force thermodynamically. These results could explain why the E:Z ratio of the products is so difficult to optimize whether by dynamic or thermodynamic means.
Conclusions
In conclusion, we have developed a novel copper-catalyzed aerobic oxidative nitrogenation of simple alkyl alkynes via propargylic C(sp3)–H bond cleavage. A variety of simple and easily accessible alkynes selectively undergo the transformation affording alkenyl nitriles. The late-stage modification of bioactive molecule derivatives makes this protocol very attractive. Mechanism studies indicate a 1,2-hydride shift might be the key step of this novel transformation. DFT calculation reasonably explains the stereoselectivity of products. Further studies on the mechanism and the application are ongoing in our group.Acknowledgements
Financial support from National Basic Research Program of China (973 Program) (grant no. 2015CB856600) and National Natural Science Foundation of China (nos 21325206, 21172006), and National Young Top-notch Talent Support Program are greatly appreciated. We thank Zun Wang in this group for reproducing the results of 2d, and 2s.Notes and references
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC.
- Acetylene Chemistry: Chemistry, Biology, and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2005 Search PubMed.
- For reviews on coupling reactions of terminal alkynes, see: (a) P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS; (b) Z. Shao and F. Peng, Angew. Chem., Int. Ed., 2010, 49, 9566 CrossRef CAS PubMed; (c) R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084 RSC; (d) C. Zhang, N. Li, X. Li, H. Chang, Q. Liu and W. Wei, Youji Huaxue, 2014, 34, 81 CrossRef CAS.
- For some recent C(sp)–H functionalizations of terminal alkynes, see: (a) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833 CrossRef CAS PubMed; (b) L. Zhao, O. Baslé and C.-L. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106 CrossRef CAS PubMed; (c) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (d) Y. Wei, H. Zhao, J. Kan, W. Su and M. Hong, J. Am. Chem. Soc., 2010, 132, 2522 CrossRef CAS PubMed; (e) M. Chen, X. Zheng, W. Li, J. He and A. Lei, J. Am. Chem. Soc., 2010, 132, 4101 CrossRef CAS PubMed; (f) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS PubMed; (g) C.-I. Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560 CrossRef CAS PubMed; (h) A. Harada, Y. Makida, T. Sato, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 13932 CrossRef CAS PubMed.
- For nucleophilic addition to alkynes, see: (a) A. Lumbroso, N. R. Vautravers and B. Breit, Org. Lett., 2010, 12, 5498 CrossRef CAS PubMed; (b) B. Xu, W. Wang, L.-P. Liu, J. Han, Z. Jin and G. B. Hammond, J. Organomet. Chem., 2011, 696, 269 CrossRef CAS; (c) T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 12418 CrossRef CAS PubMed; (d) Z. Liu, P. Liao and X. Bi, Org. Lett., 2014, 16, 3668 CrossRef CAS PubMed.
- For some reviews on Click chemistry and other cyclization reactions, see: (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252 RSC; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (d) K. Gilmore and I. V. Alabugin, Chem. Rev., 2011, 111, 6513 CrossRef CAS PubMed.
- For reviews on CC triple bond cleavage, see:
(a) A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC;
(b) H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55 RSC;
(c) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed.
- For the intramolecular cyclization of functionalized terminal alkynes via propargylic C(sp3)–H bond cleavage, see: (a) R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14, 280 CrossRef CAS PubMed; (b) H. Lebel, C. Trudel and C. Spitz, Chem. Commun., 2012, 48, 7799 RSC; (c) H. Lu, C. Li, H. Jiang, C. L. Lizardi and X. P. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7028 CrossRef CAS PubMed.
- For the propargylic oxidation of internal alkynes, see: (a) S. Sakaguchi, T. Takase, T. Iwahama and Y. Ishii, Chem. Commun., 1998, 2037 RSC; (b) E. C. McLaughlin and M. P. Doyle, J. Org. Chem., 2008, 73, 4317 CrossRef CAS PubMed; (c) Y. Zhao, A. W. T. Ng and Y.-Y. Yeung, Tetrahedron Lett., 2014, 55, 4370 CrossRef CAS.
- (a) I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 10262 CrossRef CAS; (b) I. Kadota, A. Shibuya, L. M. Lutete and Y. Yamamoto, J. Org. Chem., 1999, 64, 4570 CrossRef CAS PubMed; (c) M. Narsireddy and Y. Yamamoto, J. Org. Chem., 2008, 73, 9698 CrossRef CAS PubMed.
- (a) A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386 CrossRef CAS PubMed; (b) A. Lumbroso, N. Abermil and B. Breit, Chem. Sci., 2012, 3, 789 RSC; (c) U. Gellrich, A. Meissner, A. Steffani, M. Kahny, H. J. Drexler, D. Heller, D. A. Plattner and B. Breit, J. Am. Chem. Soc., 2014, 136, 1097 CrossRef CAS PubMed; (d) K. Xu, V. Khakyzadeh, T. Bury and B. Breit, J. Am. Chem. Soc., 2014, 136, 16124 CrossRef CAS PubMed; (e) P. Koschker, M. Kähny and B. Breit, J. Am. Chem. Soc., 2015, 137, 3131 CrossRef CAS PubMed.
- NaOAc is widely used in copper-catalyzed aerobic oxidation reactions to promote the efficiency: (a) D. Monguchi, T. Fujiwara, H. Furukawa and A. Mori, Org. Lett., 2009, 11, 1607 CrossRef CAS PubMed; (b) A. E. King, B. L. Ryland, T. C. Brunold and S. S. Stahl, Organometallics, 2012, 31, 7948 CrossRef CAS PubMed.
- (a) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappaport, Wiley, New York, 1983 Search PubMed; (b) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed; (c) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed; (d) J. J. Schafer and W. R. Short, Antiviral Ther., 2012, 17, 1495 CrossRef CAS PubMed.
- (a) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639 CrossRef CAS PubMed; (b) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767 CrossRef CAS PubMed; (c) X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed; (d) Z. Zhang and X. Jiang, Org. Lett., 2014, 16, 4400 CrossRef CAS PubMed.
- (a) K. Banert and M. Hagedorn, Angew. Chem., Int. Ed. Engl., 1989, 28, 1675 CrossRef , and references cited therein; (b) J. R. Fotsing and K. Banert, Eur. J. Org. Chem., 2005, 3704 CrossRef CAS.
- R. Buschbeck, P. J. Low and H. Lang, Coord. Chem. Rev., 2011, 255, 241 CrossRef CAS ; also see ref. 6c.
- For selected examples of carbene and metallocarbenoid 1,2-hydride shift, see: (a) D. F. Taber, R. J. Herr, S. K. Pack and J. M. Geremia, J. Org. Chem., 1996, 61, 2908 CrossRef CAS PubMed; (b) N. Jiang, Z. Qu and J. Wang, Org. Lett., 2001, 3, 2989 CrossRef CAS PubMed; (c) V. K. Aggarwal, C. G. Sheldon, G. J. Macdonald and W. P. Martin, J. Am. Chem. Soc., 2002, 124, 10300 CrossRef CAS PubMed; (d) J. A. May and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 12426 CrossRef CAS PubMed; (e) K. S. Graves, D. M. Thamattoor and P. R. Rablen, J. Org. Chem., 2011, 76, 1584 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- (a) M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389 CrossRef CAS; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (c) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed ; also see ref. 6.
- For reviews about reactions of metallocarbenes derived from 1,2,3-triazoles, see: (a) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371 CrossRef CAS PubMed; for selected examples catalyzed by Cu-catalyst, see: (b) I. Bae, H. Han and S. Chang, J. Am. Chem. Soc., 2005, 127, 2038 CrossRef CAS PubMed; (c) M. P. Cassidy, J. Raushel and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 3154 CrossRef CAS PubMed; (d) E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730 CrossRef CAS PubMed; (e) H.-D. Xu, Z.-H. Jia, K. Xu, M. Han, S.-N. Jiang, J. Cao, J.-C. Wang and M.-H. Shen, Angew. Chem., Int. Ed., 2014, 53, 9284 CrossRef CAS PubMed; (f) V. Helan, A. Gulevich and V. Gevorgyan, Chem. Sci., 2015, 6, 1928 RSC.
- For selected examples on Rh-catalyzed ring expansion and rearrangement involving azavinyl carbenes, see: (a) T. Miura, Y. Funakoshi, M. Morimoto, T. Biyajima and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17440 CrossRef CAS PubMed; (b) N. Selander, B. T. Worrell and V. V. Fokin, Angew. Chem., Int. Ed., 2012, 51, 13054 CrossRef CAS PubMed; (c) T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272 CrossRef CAS PubMed; (d) Y. Shi, A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2014, 53, 14191 CrossRef CAS PubMed; (e) T. Miura, T. Nakamuro, C.-J. Liang and M. Murakami, J. Am. Chem. Soc., 2014, 136, 15905 CrossRef CAS PubMed.
- L. M. Dornan, Q. Cao, J. C. A. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030 RSC.
- For formation of copper carbenes from diazo compounds, see: (a) W. Kirmse, Angew. Chem., Int. Ed., 2003, 42, 1088 CrossRef CAS PubMed; (b) Y. Liang, H. Zhou and Z.-X. Yu, J. Am. Chem. Soc., 2009, 131, 17783 CrossRef CAS PubMed; (c) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC.
- For references about alkynyl azides, see: (a) K. Banert, M. Hagedorn, J. Wutke, P. Ecorchard, D. Schaarschmidt and H. Lang, Chem. Commun., 2010, 46, 4058 RSC; (b) I. F. D. Hyatt and M. P. Croatt, Angew. Chem., Int. Ed., 2012, 51, 7511 CrossRef CAS PubMed; (c) K. Banert, R. Arnold, M. Hagedorn, P. Thoss and A. A. Auer, Angew. Chem., Int. Ed., 2012, 51, 7515 CrossRef CAS PubMed.
- All of the geometry optimizations and frequency calculations were performed at B3LYP(CPCM)/SDD-6-311+G(d,p)//B3LYP(gas)/SDD-6-31G(d) level in Gaussian 09 (M. J. Frisch, et al.). Computational details and references are given in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. See DOI: 10.1039/c5sc02126j |
| This journal is © The Royal Society of Chemistry 2015 |